An Efficient Synthesis of Aryl-Substituted Pyrroles by the Suzuki–Miyaura Coupling Reaction of SEM-Protected Pyrroles


Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Typical Procedure to Synthesize Compounds 2a–c
3.3. Typical Procedure to Synthesize Products 3a–s
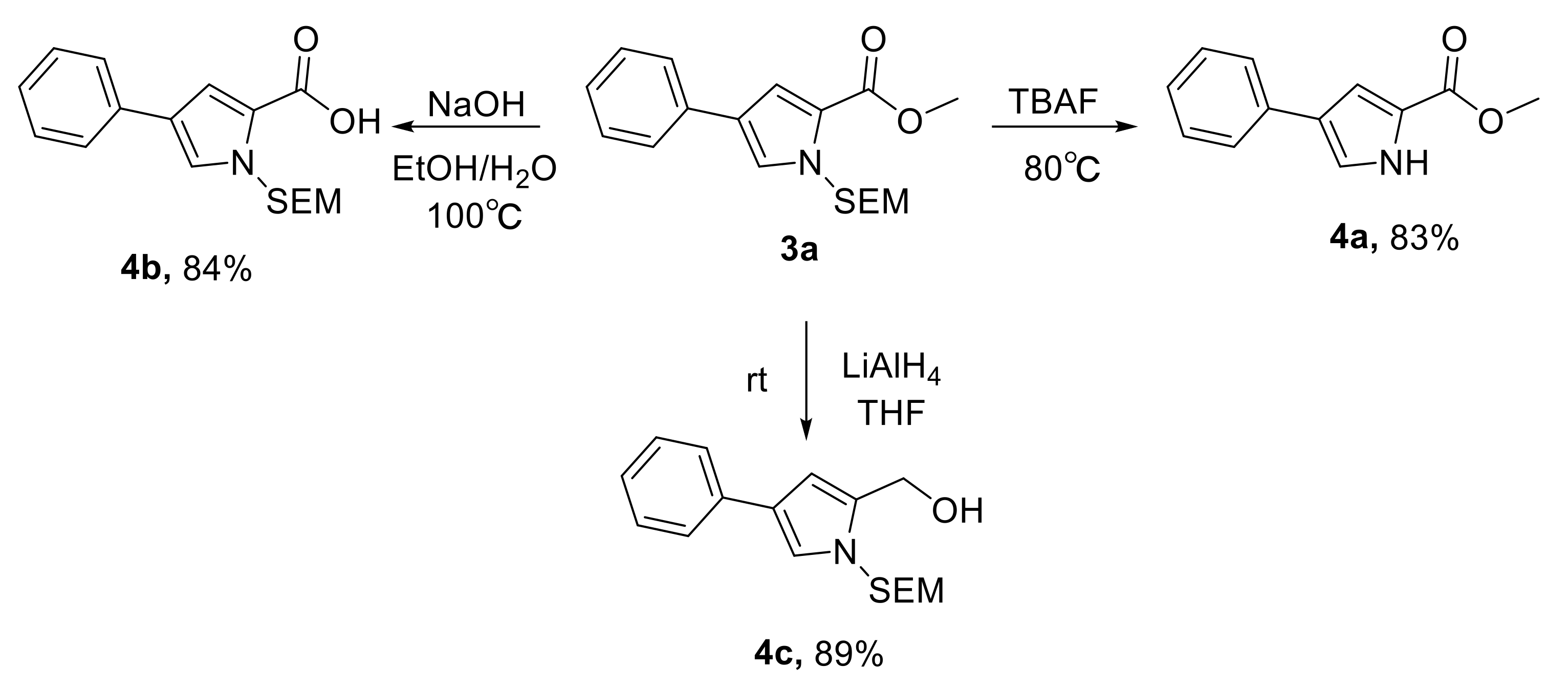
3.4. Procedure for the Preparation of Methyl 4-phenyl-1H-pyrrole-2-carboxylate (4a)
3.5. Procedure for the Preparation of 4-Phenyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrole-2-carboxylic acid (4b)
3.6. Procedure for the Preparation of (4-Phenyl-1-((2-(trimethylsilyl)ethoxy)methyl)-1H-pyrrol-2-yl)methanol (4c)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ma, Z.N.; Ma, Z.C.; Zhang, D.W. Synthesis of multi-substituted pyrrole derivatives through [3+2] cycloaddition with tosylmethyl isocyanides (TosMICs) and electron-deficient compounds. Molecules 2018, 23, 2666. [Google Scholar] [CrossRef] [PubMed]
- Hao, F.; Reddy, A.R.; Zhou, C.Y.; Che, C.M. Cobalt(II) porphyrin catalyzed cascade reaction of pyrrolyl ketones for construction of polysubstituted pyrrolizidines and pyrrolizines. Adv. Synth. Catal. 2018, 360, 1433–1438. [Google Scholar] [CrossRef]
- Guillon, J.; Le Borgne, M.; Rimbault, C.; Moreau, S.; Savrimoutou, S.; Pinaud, N.; Baratin, S.; Marchivie, M.; Roche, S.; Bollacke, A.; et al. Synthesis and biological evaluation of novel substituted pyrrolo[1,2-a]quinoxaline derivatives as inhibitors of the human protein kinase CK2. Eur. J. Med. Chem. 2013, 65, 205–222. [Google Scholar] [CrossRef] [Green Version]
- Desplat, V.; Moreau, S.; Gay, A.; Fabre, S.B.; Thiolat, D.; Massip, S.; Macky, G.; Godde, F.; Mossalayi, D.; Jarry, C.; et al. Synthesis and evaluation of the antiproliferative activity of novel pyrrolo[1,2-a]quinoxaline derivatives, potential inhibitors of Akt kinase. Part II. J. Enzyme. Inhib. Med. Chem. 2010, 25, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, K.M.; Jackson, P.J.; James, C.H.; Basu, B.P.; Hartley, J.A.; de la Fuente, M.; Schatzlein, A.; Robson, M.; Pedley, R.B.; Pepper, C.; et al. GC-targeted C8-linked pyrrolobenzodiazepine-biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J. Med. Chem. 2013, 56, 2911–2935. [Google Scholar] [CrossRef]
- Burnham, B.S.; Berkowitz, J.D.; Orr, Z.; McCranor, B.; Cooper, M.; McCann, K.; Pillitteri, K.; Hallback, J.; Bergh, L.-M.; Gofman, L.; et al. Lipid-lowering effects of polymers derived from halophenyl pyrroles. Lett. Drug Des. Discov. 2011, 8, 544–549. [Google Scholar] [CrossRef]
- Lopez-Perez, A.; Robles-Machin, R.; Adrio, J.; Carretero, J.C. Oligopyrrole Synthesis by 1,3-dipolar cycloaddition of azomethine ylides with bissulfonyl ethylenes. Angew. Chem. Int. Ed. 2007, 46, 9261–9264. [Google Scholar] [CrossRef]
- Gao, M.; He, C.; Chen, H.; Bai, R.; Cheng, B.; Lei, A. Synthesis of pyrroles by click reaction: Silver-catalyzed cycloaddition of terminal alkynes with isocyanides. Angew. Chem. Int. Ed. 2013, 52, 6958–6961. [Google Scholar] [CrossRef] [PubMed]
- Farney, E.P.; Yoon, T.P. Visible-light sensitization of vinyl azides by transition-metal photocatalysis. Angew. Chem. Int. Ed. 2014, 53, 793–797. [Google Scholar] [CrossRef]
- O’Brien, A.G.; Levesque, F.; Seeberger, P.H. Continuous flow thermolysis of azidoacrylates for the synthesis of heterocycles and pharmaceutical intermediates. Chem. Commun. 2011, 47, 2688–2690. [Google Scholar] [CrossRef]
- Tiwari, D.K.; Pogula, J.; Sridhar, B.; Tiwari, D.K.; Likhar, P.R. Nano-copper catalysed highly regioselective synthesis of 2,4-disubstituted pyrroles from terminal alkynes and isocyanides. Chem. Commun. 2015, 51, 13646–13649. [Google Scholar] [CrossRef]
- Kudryavtsev, K.V.; Ivantcova, P.M.; Churakov, A.V.; Vasin, V.A. Phenyl α-bromovinyl sulfone in cycloadditions with azomethine ylides: An unexpected facile aromatization of the cycloadducts into pyrroles. Tetrahedron Lett. 2012, 53, 4300–4303. [Google Scholar] [CrossRef]
- Pasko, C.M.; Dissanayake, A.A.; Billow, B.S.; Odom, A.L. One-pot synthesis of pyrroles using a titanium-catalyzed multicomponent coupling procedure. Tetrahedron 2016, 72, 1168–1176. [Google Scholar] [CrossRef] [Green Version]
- Chemler, S.R.; Fuller, P.H. Heterocycle synthesis by copper facilitated addition of heteroatoms to alkenes, alkynes and arenes. Chem. Soc. Rev. 2007, 36, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Dowlut, M.; Mallik, D.; Organ, M.G. An efficient low-temperature Stille-Migita cross-coupling reaction for heteroaromatic compounds by Pd-PEPPSI-IPent. Chem. Eur. J. 2010, 16, 4279–4283. [Google Scholar] [CrossRef]
- Castro, M.C.R.; Raposo, M.M.M. Synthesis of π-conjugated systems bearing thiophene and pyrrole heterocycles through palladium catalyzed cross-coupling reactions. Tetrahedron 2016, 72, 1881–1887. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, N.; Goossen, L.J. Decarboxylative coupling reactions: A modern strategy for C-C-bond formation. Chem. Soc. Rev. 2011, 40, 5030–5048. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Souleé, J.-F.; Doucet, H. Functionalization of C−H Bonds via Metal-Catalyzed Desulfitative Coupling: An Alternative Tool for Access to Aryl- or Alkyl-Substituted (Hetero)arenes. ACS Catal. 2015, 5, 978–991. [Google Scholar] [CrossRef]
- Handy, S.T.; Bregman, H.; Lewis, J.; Zhang, X.; Zhang, Y. An unusual dehalogenation in the Suzuki coupling of 4-bromopyrrole-2-carboxylates. Tetrahedron Lett. 2003, 44, 427–430. [Google Scholar] [CrossRef]
- Smith, J.A.; Ng, S.; White, J. The regioselective synthesis of aryl pyrroles. Org. Biomol. Chem. 2006, 4, 2477–2482. [Google Scholar] [CrossRef]
- Handy, S.T.; Zhang, Y.; Bregman, H. A Modular synthesis of the lamellarins: Total synthesis of lamellarin G trimethyl ether. J. Org. Chem. 2004, 69, 2362–2366. [Google Scholar] [CrossRef]
- Muchowski, J.M.; Solas, D.R. Protecting groups for the pyrrole and indole nitrogen atom. the [2-(trimet hy1silyl)ethoxy]methyl moiety. lithiation of 1-[[2-(trimethylsilyl)ethoxy]methyl]pyrrole. J. Org. Chem. 1984, 49, 203–205. [Google Scholar] [CrossRef]
- Whitten, J.P.; Matthews, D.P.; McCarthy, J.R. [2-(Trimethylsilyl)ethoxy]methyl (SEM) as a novel and effective imidazole and fused aromatic imidazole protecting group. J. Org. Chem. 1986, 51, 1891–1894. [Google Scholar] [CrossRef]
- Katz, J.D.; Overman, L.E. Studies towards the total synthesis of palau’amine. Formation of 4,5-dihydropyrrole-2-carboxylate intermediates by alkene–enamide ring-closing metathesis. Tetrahedron 2004, 60, 9559–9568. [Google Scholar] [CrossRef]
- Toure, B.B.; Lane, B.S.; Sames, D. Catalytic C-H arylation of SEM-protected azoles with palladium complexes of NHCs and phosphines. Org. Lett. 2006, 8, 1979–1982. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Seiple, I.B.; Young, I.S.; O’Malley, D.P.; Maue, M.; Baran, P.S. Synthesis of 1,9-dideoxy-pre-axinellamine. Angew. Chem. Int. Ed. 2008, 47, 3578–3580. [Google Scholar] [CrossRef]
- Sadler, S.A.; Hones, A.C.; Roberts, B.; Blakemore, D.; Marder, T.B.; Steel, P.G. Multidirectional synthesis of substituted indazoles via iridium-catalyzed C-H borylation. J. Org. Chem. 2015, 80, 5308–5314. [Google Scholar] [CrossRef] [PubMed]
- Mccomas, C.C.; Serrano-wu, M.H.; Vacca, J.P. Fused quadracyclic compounds, compositions and uses thereof. U.S. Patent 20170190713A1, 6 July 2017. [Google Scholar]
Sample Availability: Samples of the compounds 3a–s and 4a–c are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
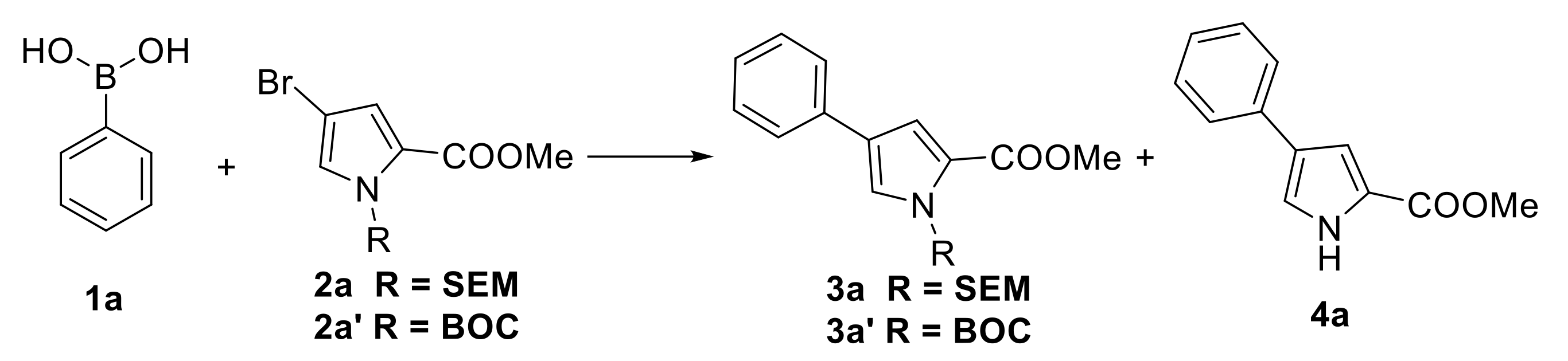
| Entry | R | Catalyst | T (°C) | Base (equiv.) | Solvent | Time (h) | Yield (%) b | |
|---|---|---|---|---|---|---|---|---|
| 3a (3a′) | 4a | |||||||
| 1 | SEM | Pd(PPh3)4 | 90 | Na2CO3 (2) | Dioxane/H2O (4:1) | 1 | 61 | 0 |
| 2 | SEM | Pd(PPh3)2Cl2 | 90 | Na2CO3 (2) | Dioxane/H2O (4:1) | 1 | 45 | 0 |
| 3 | SEM | Pd(AcO)2 | 90 | Na2CO3 (2) | Dioxane/H2O (4:1) | 6 | 13 | 0 |
| 4 | SEM | Pd(dppf)Cl2 | 90 | Na2CO3 (2) | Dioxane/H2O (4:1) | 6 | 28 | 0 |
| 5 | SEM | Pd(PPh3)4 | 110 | Na2CO3 (2) | DMF | 6 | trace | 0 |
| 6 c | SEM | Pd(PPh3)4 | 90 | Na2CO3 (2) | Dioxane/H2O (4:1) | 1 | 57 | 0 |
| 7 c | SEM | Pd(PPh3)4 | 90 | Na2CO3 (6) | Dioxane/H2O (4:1) | 1 | 57 | 0 |
| 8 | SEM | Pd(PPh3)4 | 90 | Na2CO3 (2) | Dioxane/H2O (4:0.1) | 6 | 5 | 0 |
| 9 | SEM | Pd(PPh3)4 | 90 | K2CO3 (2) | Dioxane/H2O (4:1) | 5 | 45 | 0 |
| 10 | SEM | Pd(PPh3)4 | 90 | KF (2) | Dioxane/H2O (4:1) | 6 | 32 | 0 |
| 11 | SEM | Pd(PPh3)4 | 90 | Cs2CO3 (2) | Dioxane/H2O (4:1) | 5 | 85 | 0 |
| 12 | SEM | Pd(PPh3)4 | 110 | Cs2CO3 (2) | Dioxane/H2O (4:1) | 3 | 75 | 0 |
| 13 | SEM | Pd(PPh3)4 | 60 | Cs2CO3 (2) | Dioxane/H2O (4:1) | 5 | 53 | 0 |
| 14 d | SEM | Pd(PPh3)4 | 90 | Cs2CO3 (2) | Dioxane/H2O (4:1) | 2 | 78 | 0 |
| 15 | BOC | Pd(PPh3)4 | 90 | Cs2CO3 (2) | Dioxane/H2O (4:1) | 2 | 76 | 5 |
| 16 | BOC | Pd(PPh3)4 | 90 | Na2CO3 (2) | Dioxane/H2O (4:1) | 2 | 64 | 11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, K.; Gao, M.; Zhao, H.; Zhang, D.; Yan, H.; Huang, H. An Efficient Synthesis of Aryl-Substituted Pyrroles by the Suzuki–Miyaura Coupling Reaction of SEM-Protected Pyrroles. Molecules 2019, 24, 1594. https://doi.org/10.3390/molecules24081594
Cui K, Gao M, Zhao H, Zhang D, Yan H, Huang H. An Efficient Synthesis of Aryl-Substituted Pyrroles by the Suzuki–Miyaura Coupling Reaction of SEM-Protected Pyrroles. Molecules. 2019; 24(8):1594. https://doi.org/10.3390/molecules24081594
Chicago/Turabian StyleCui, Keli, Meng Gao, Hongyi Zhao, Dongfeng Zhang, Hong Yan, and Haihong Huang. 2019. "An Efficient Synthesis of Aryl-Substituted Pyrroles by the Suzuki–Miyaura Coupling Reaction of SEM-Protected Pyrroles" Molecules 24, no. 8: 1594. https://doi.org/10.3390/molecules24081594
APA StyleCui, K., Gao, M., Zhao, H., Zhang, D., Yan, H., & Huang, H. (2019). An Efficient Synthesis of Aryl-Substituted Pyrroles by the Suzuki–Miyaura Coupling Reaction of SEM-Protected Pyrroles. Molecules, 24(8), 1594. https://doi.org/10.3390/molecules24081594