
Structural Transformations in the Thermal Dehydration of [Cu2(bpa)(btec)(H2O)4]n Coordination Polymer

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Preliminary Characterization
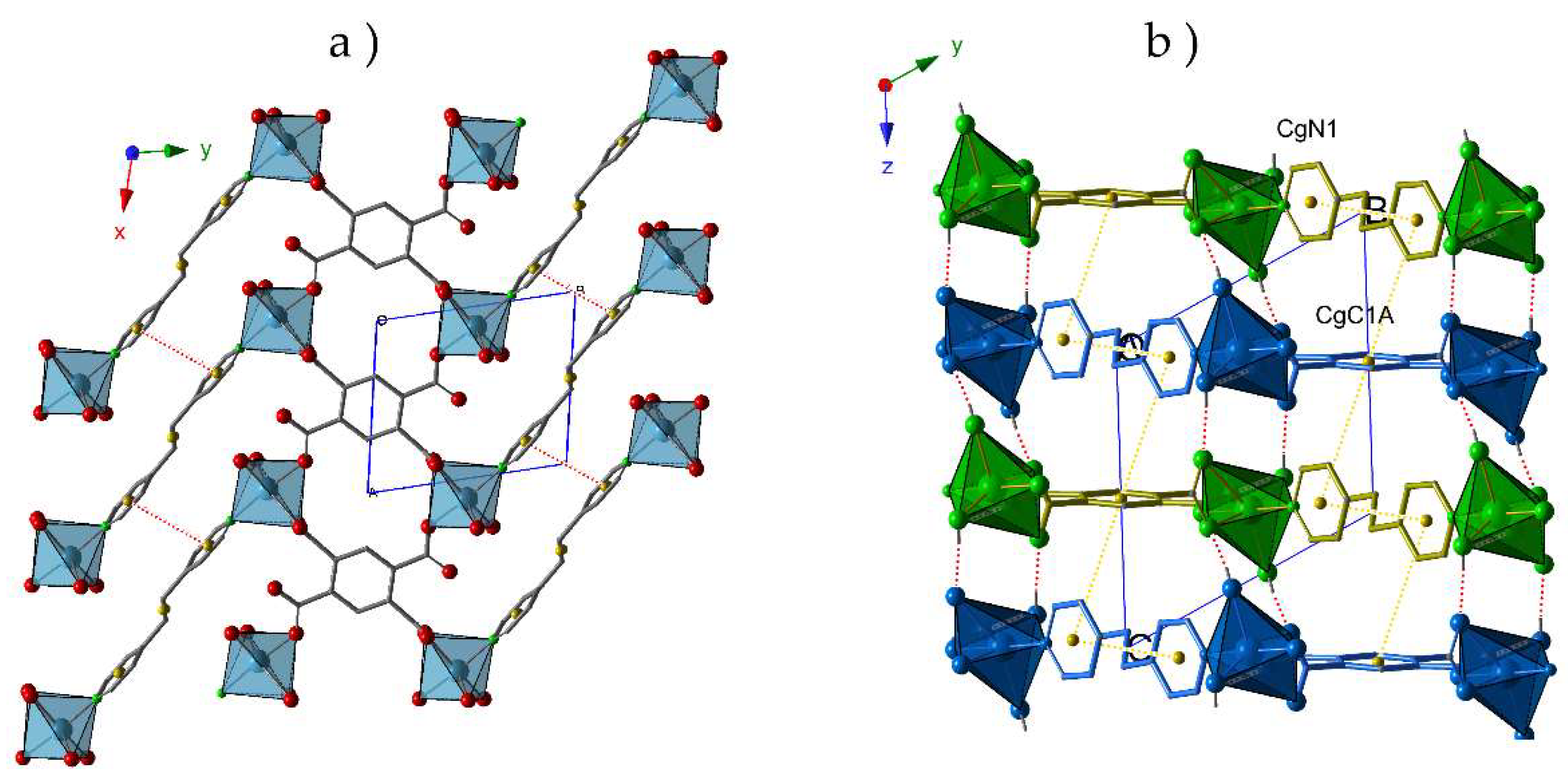
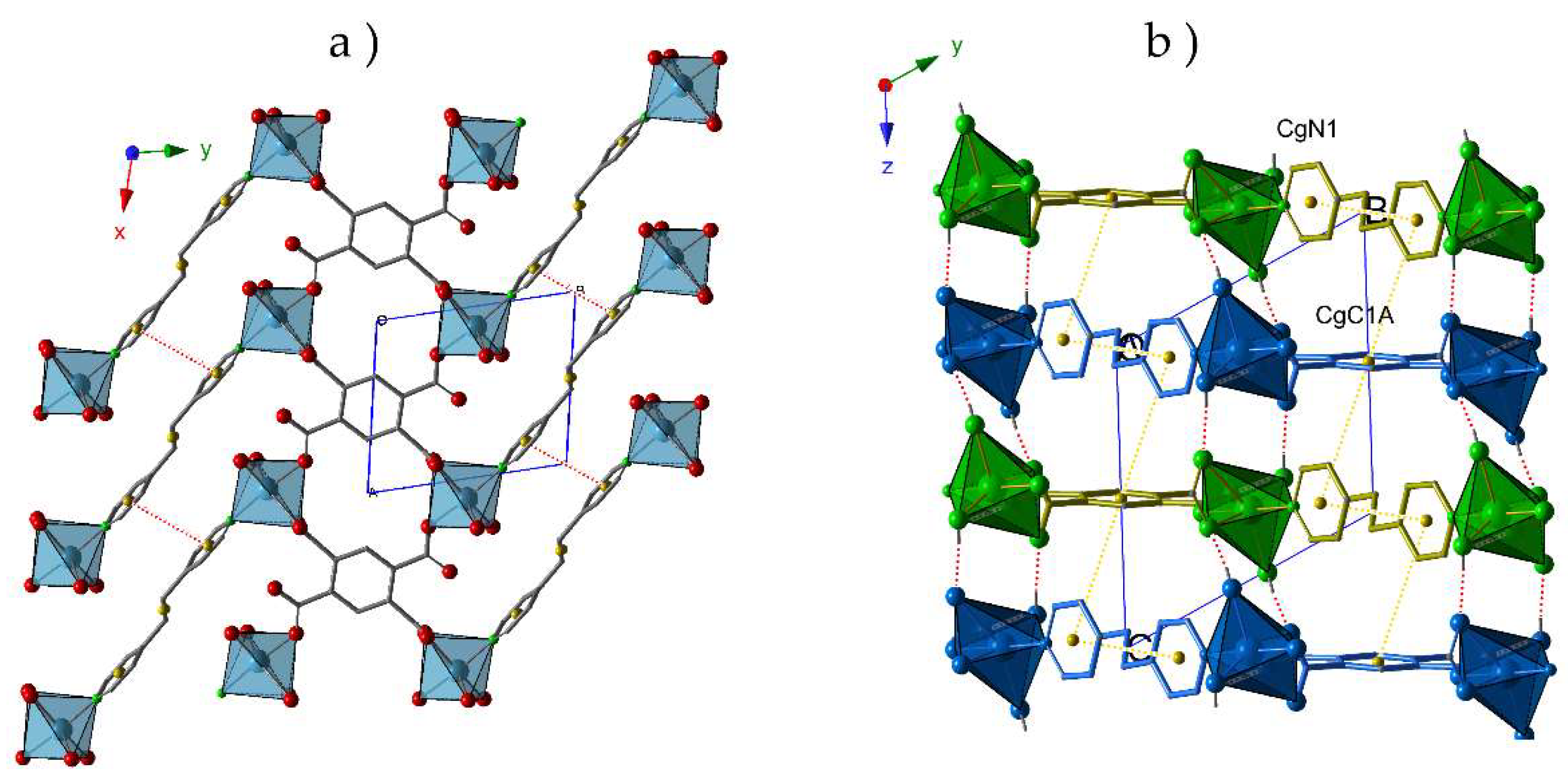
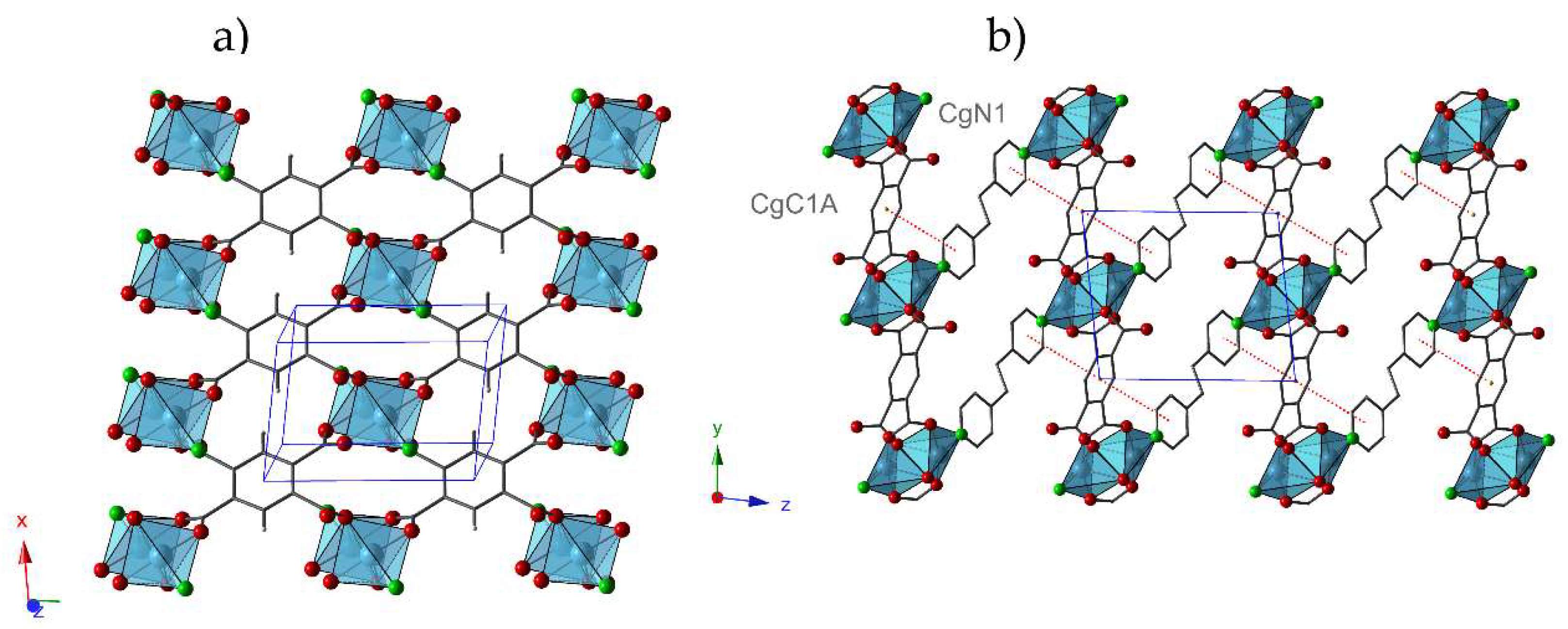
2.2. Crystal Structures
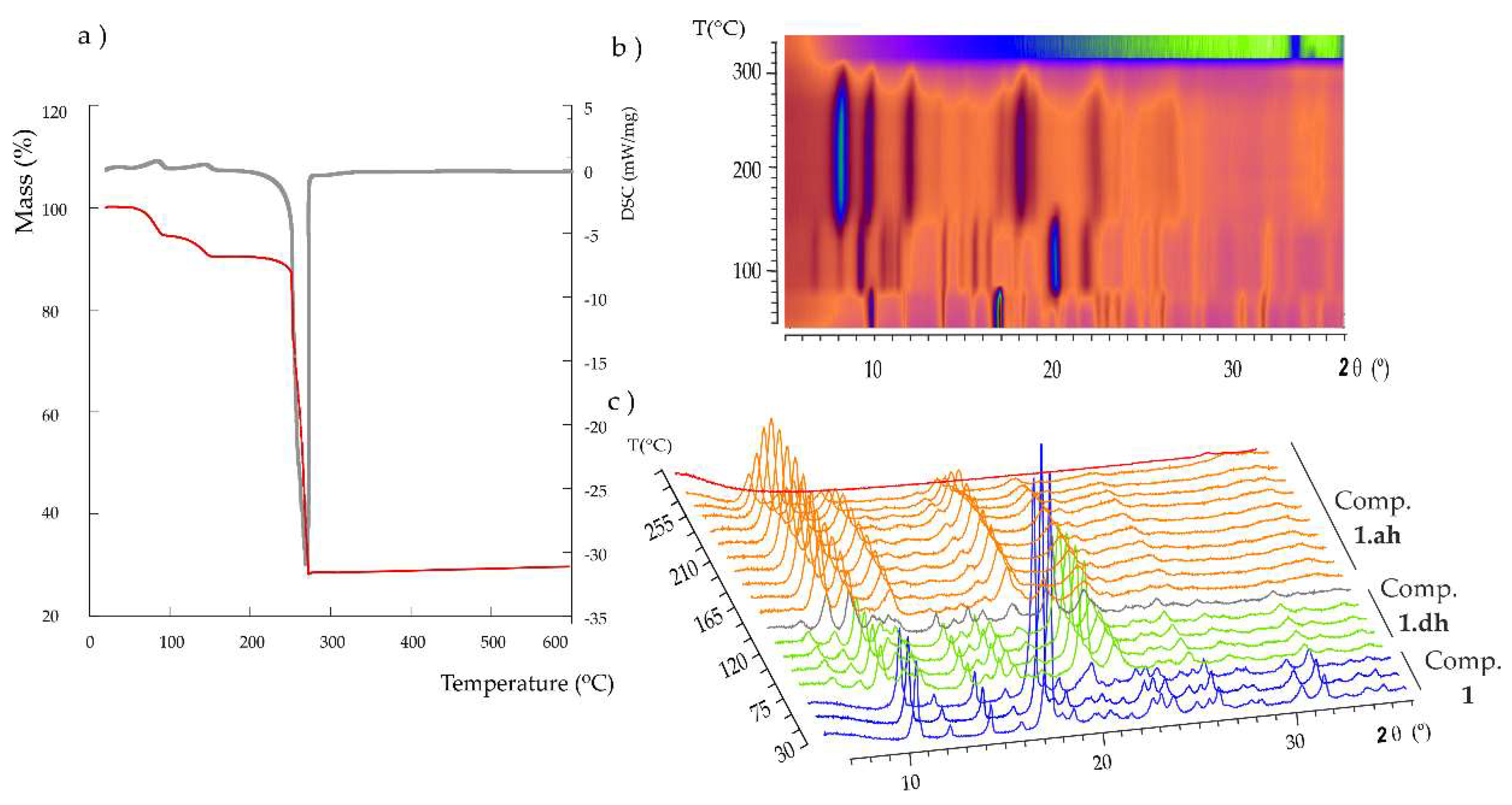
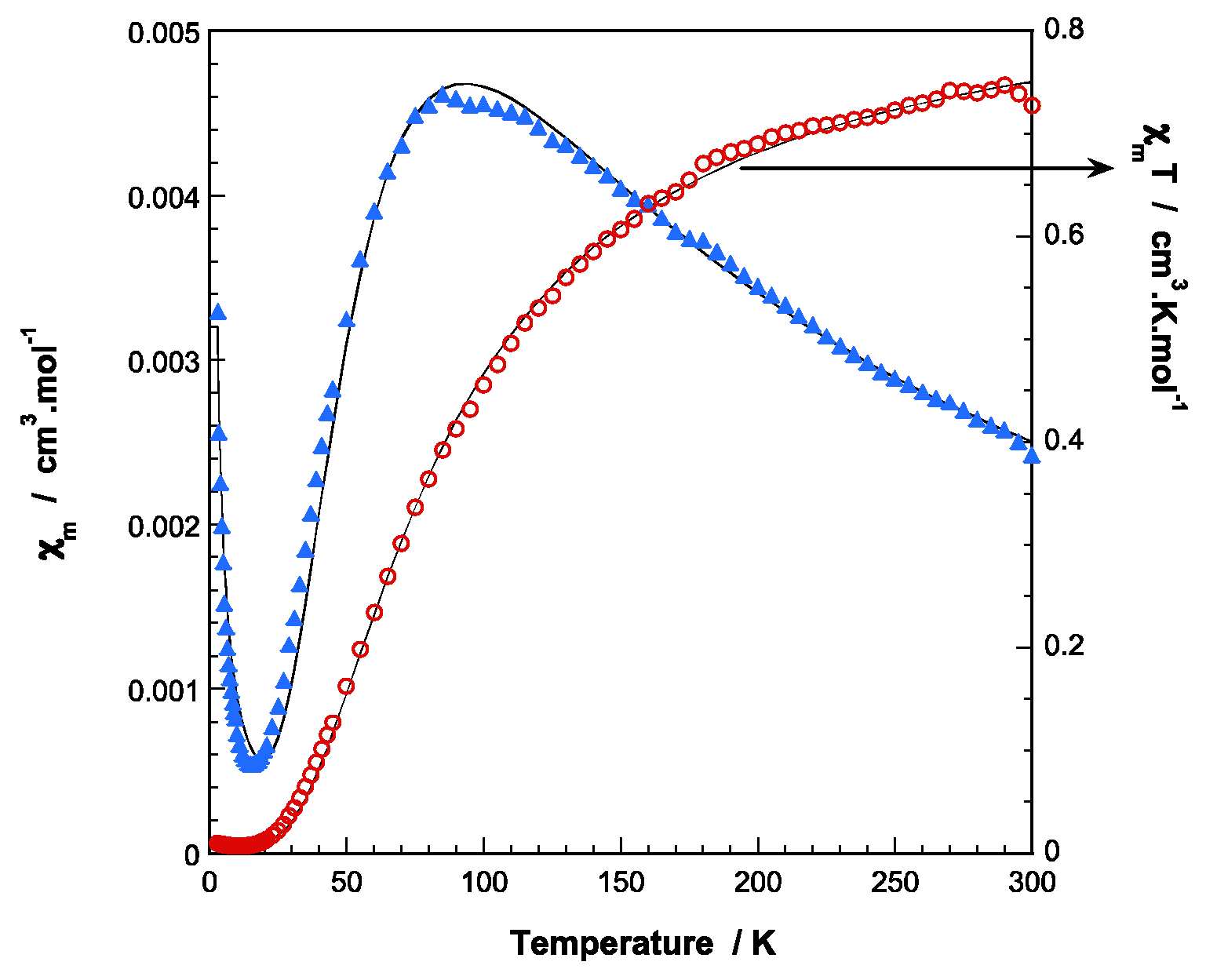
2.3. Thermal Behavior
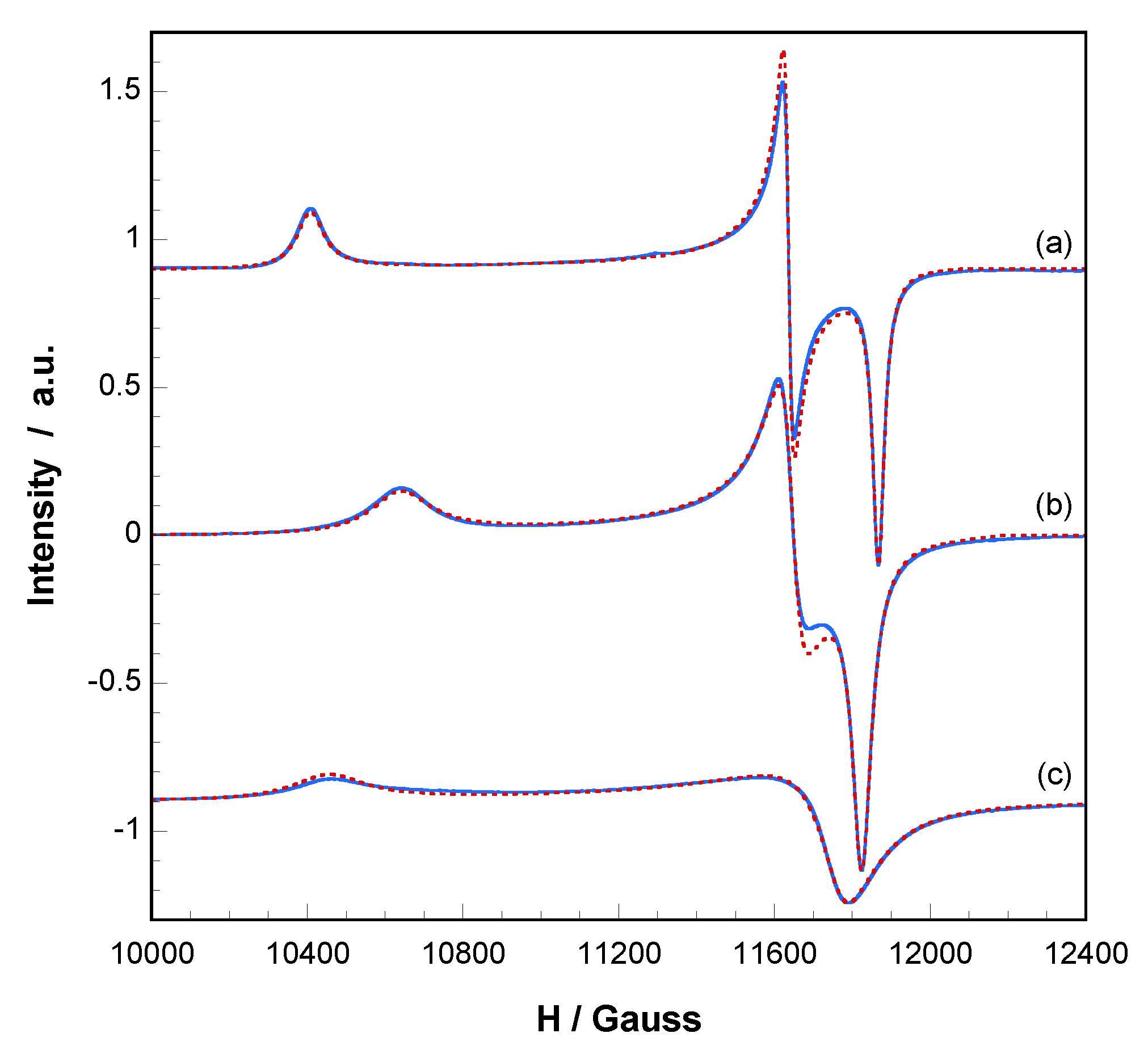
2.4. Reversibility of Thermally Triggered Phase Transformation and EPR Espectroscopy
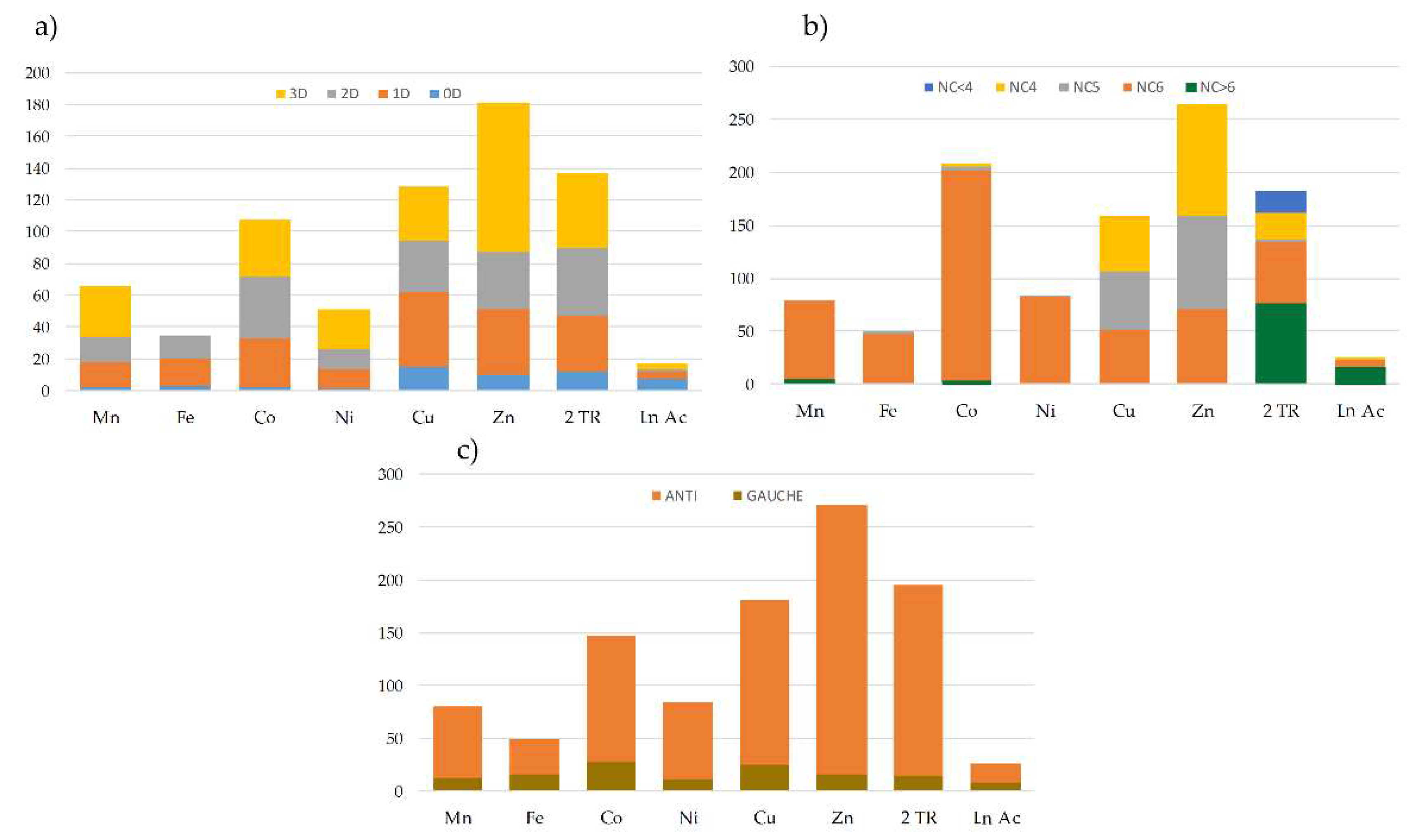
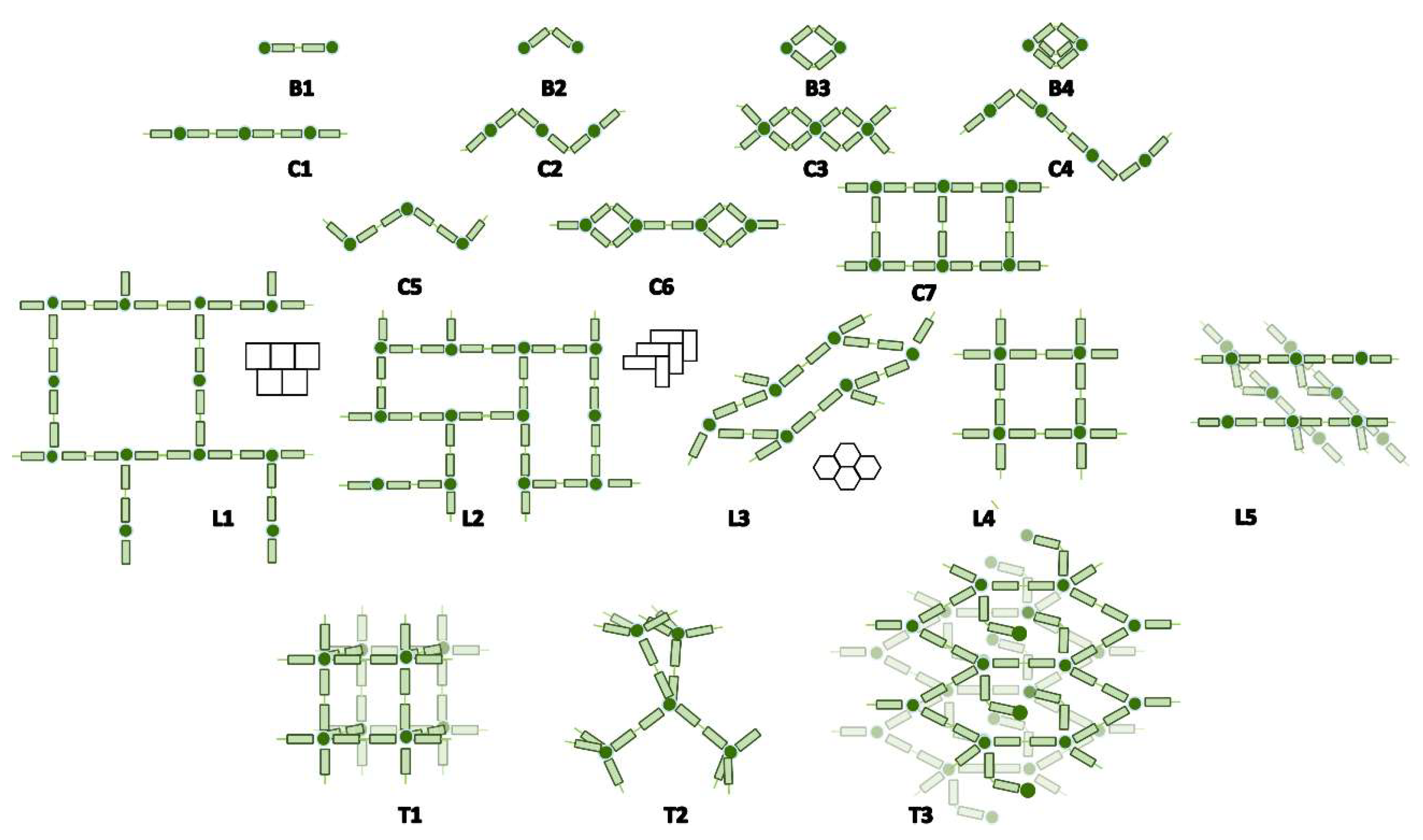
2.5. Topological Study of the bpa Ligand
3. Experimental
3.1. Materials and Methods
3.2. Synthesis of [Cu2(bpa)(btec)(H2O)4]n (1) and [Cu2(bpa)(btec)]n (1.ah)
3.3. Single-Crystal X-Ray Diffraction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.; Zheng, S.; Xue, H.; Pang, H. Metal-Organic Frameworks/Graphene-Based Materials: Preparations and Applications. Adv. Funct. Mater. 2018, 28, 1804950. [Google Scholar] [CrossRef]
- Wu, J.; Xu, F.; Li, S.; Ma, P.; Zhang, X.; Liu, Q.; Fu, R.; Wu, D. Porous Polymers as Multifunctional Material Platforms toward Task-Specific Applications. Adv. Mater. 2018, 31, 1802922. [Google Scholar] [CrossRef] [PubMed]
- Farha, O.K.; Hupp, J.T. Rational Design, Synthesis, Purification, and Activation of Metal-Organic Framework Materials. Acc. Chem. Res. 2010, 43, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zou, D.; Zhu, H.; Zhang, J. Mesoporous Metal-Organic Frameworks: Synthetic Strategies and Emerging Applications. Small 2018, 14, e1801454. [Google Scholar] [CrossRef]
- Ma, L.; Abney, C.; Lin, W. Enantioselective catalysis with homochiral metal-organic frameworks. Chem. Soc. Rev. 2009, 38, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wen, H.-M.; Chen, B.; Cui, Y.; Qian, G.; Zhou, W. Emerging Multifunctional Metal-Organic Framework Materials. Adv Mater 2016, 28, 8819–8860. [Google Scholar] [CrossRef] [PubMed]
- Bhanja, P.; Modak, A.; Bhaumik, A. Porous Organic Polymers for CO2 Storage and Conversion Reactions. ChemCatChem 2018, 11, 244–257. [Google Scholar] [CrossRef]
- Sosa, J.; Bennett, T.; Nelms, K.; Liu, B.; Tovar, R.; Liu, Y. Metal-organic framework hybrid materials and their applications. Crystals 2018, 8, 325. [Google Scholar] [CrossRef]
- Li, H.; Eddaoudi, M.; O’Keeffe, M.; Yaghi, M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. [Google Scholar] [CrossRef]
- Farha, O.K.; Oezguer Yazaydin, A.; Eryazici, I.; Malliakas, C.D.; Hauser, B.G.; Kanatzidis, M.G.; Nguyen, S.B.T.; Snurr, R.Q.; Hupp, J.T. De novo synthesis of a metal-organic framework material featuring ultrahigh surface area and gas storage capacities. Nat. Chem. 2010, 2, 944–948. [Google Scholar] [CrossRef]
- Hillman, F.; Brito, J.; Jeong, H.K. Rapid One-Pot Microwave Synthesis of Mixed-Linker Hybrid Zeolitic-Imidazolate Framework Membranes for Tunable Gas Separations. ACS Appl. Mater. Interfaces 2018, 10, 5586–5593. [Google Scholar] [CrossRef]
- Adatoz, E.; Avci, A.K.; Keskin, S. Opportunities and challenges of MOF-based membranes in gas separations. Sep. Purif. Technol. 2015, 152, 207–237. [Google Scholar] [CrossRef]
- Chen, B.; Liang, C.; Yang, J.; Contreras, D.S.; Clancy, Y.L.; Lobkovsky, E.B.; Yaghi, O.M.; Dai, S. A microporous metal-organic framework for gas-chromatographic separation of alkanes. Angew. Chem. Int. Ed. 2006, 45, 1390–1393. [Google Scholar] [CrossRef]
- Gao, X.; Zhai, M.; Guan, W.; Liu, J.; Liu, Z.; Damirin, A. Controllable Synthesis of a Smart Multifunctional Nanoscale Metal-Organic Framework for Magnetic Resonance/Optical Imaging and Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2017, 9, 3455–3462. [Google Scholar] [CrossRef]
- Luo, Z.; Wang, R.; Gu, C.; Li, F.; Han, Y.; Li, B.; Liu, J. A metal-organic framework with unusual nanocages: Drug delivery. Inorg. Chem. Commun. 2017, 76, 91–94. [Google Scholar] [CrossRef]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sebrie, C.; Baati, T.; Eubank, J.F.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mater. 2010, 9, 172–178. [Google Scholar] [CrossRef]
- Kreno, L.E.; Leong, K.; Farha, O.K.; Allendorf, M.; Van Duyne, R.P.; Hupp, J.T. Metal-Organic Framework Materials as Chemical Sensors. Chem. Rev. 2012, 112, 1105–1125. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, L.; Xiao, Y.; Fronczek, F.R.; Xue, M.; Cui, Y.; Qian, G. A luminescent metal-organic framework with Lewis basic pyridyl sites for the sensing of metal ions. Angew. Chem. Int. Ed. 2009, 48, 500–503. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Applications of metal-organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Leus, K.; Sun, Z.; Li, X.; Depauw, H.; Wang, A.; Zhang, J.; Van Der Voort, P. Catalytic oxidative desulfurization of model and real diesel over a molybdenum anchored metal-organic framework. Microporous Mesoporous Mater. 2019, 277, 245–252. [Google Scholar] [CrossRef]
- Zhang, C.-L.; Lu, B.-R.; Cao, F.-H.; Wu, Z.-Y.; Zhang, W.; Cong, H.-P.; Yu, S.-H. Electrospun metal-organic framework nanoparticle fibers and their derived electrocatalysts for oxygen reduction reaction. Nano Energy 2019, 55, 226–233. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, H.; Tan, L.; Huang, Z.; Wu, Q.; Ren, X.; Ren, J.; Meng, X. Microwave-Activated Mn-Doped Zirconium Metal-Organic Framework Nanocubes for Highly Effective Combination of Microwave Dynamic and Thermal Therapies Against Cancer. ACS Nano 2018, 12, 2201–2210. [Google Scholar] [CrossRef]
- Rieter, W.J.; Pott, K.M.; Taylor, K.M.L.; Lin, W. Nanoscale Coordination Polymers for Platinum-Based Anticancer Drug Delivery. J. Am. Chem. Soc. 2008, 130, 11584–11585. [Google Scholar] [CrossRef]
- Stock, N.; Biswas, S. Synthesis of Metal-Organic Frameworks (MOFs): Routes to Various MOF Topologies, Morphologies, and Composites. Chem. Rev. 2012, 112, 933–969. [Google Scholar] [CrossRef]
- Maity, D.K.; Bhattacharya, B.; Mondal, R.; Ghoshal, D. Five diverse bivalent metal coordination polymers based on benzene dicarboxylate and bent dipyridyl ligands: syntheses, structures, and photoluminescent properties. CrystEngComm 2014, 16, 8896–8909. [Google Scholar] [CrossRef]
- Haneda, T.; Kawano, M.; Kojima, T.; Fujita, M. Thermo-to-photo-switching of the chromic behavior of salicylideneanilines by inclusion in a porous coordination network. Angew. Chem. Int. Ed. 2007, 46, 6643–6645. [Google Scholar] [CrossRef]
- Sun, Q.-Z.; Liu, N.-W.; Pan, J.-Q.; Zhang, B.-G.; Liu, H. Synthesis, structures and magnetic properties of two 2D → 3D entangled Cobalt(II) coordination polymers. Transit. Met. Chem. 2017, 42, 517–524. [Google Scholar] [CrossRef]
- Kitagawa, S.; Kondo, M. Functional micropore chemistry of crystalline metal complex-assembled compounds. Bull. Chem. Soc. Jpn. 1998, 71, 1739–1753. [Google Scholar] [CrossRef]
- Humby, J.D.; Benson, O.; Smith, G.L.; Argent, S.P.; da Silva, I.; Cheng, Y.; Rudic, S.; Manuel, P.; Frogley, M.D.; Cinque, G.; et al. Host-guest selectivity in a series of isoreticular metal-organic frameworks: Observation of acetylene-to-alkyne and carbon dioxide-to-amide interactions. Chem. Sci. 2019, 10, 1098–1106. [Google Scholar] [CrossRef]
- Southon, P.D.; Liu, L.; Fellows, E.A.; Price, D.J.; Halder, G.J.; Chapman, K.W.; Moubaraki, B.; Murray, K.S.; Letard, J.-F.; Kepert, C.J. Dynamic Interplay between Spin-Crossover and Host-Guest Function in a Nanoporous Metal-Organic Framework Material. J. Am. Chem. Soc. 2009, 131, 10998–11009. [Google Scholar] [CrossRef]
- Manna, B.; Desai, A.V.; Ghosh, S.K. Neutral N-donor ligand based flexible metal-organic frameworks. Dalton Trans. 2016, 45, 4060–4072. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, N.; Masoomi, M.Y.; Morsali, A.; Junk, P.C.; Wang, J. Sonochemical synthesis and structural characterization of a new Zn(II) nanoplate metal-organic framework with removal efficiency of Sudan red and Congo red. Ultrason. Sonochem. 2018, 45, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, A.; Mohammad, A.; Mobin, S.M. Recent Advances in Single-Crystal-to-Single-Crystal Transformation at the Discrete Molecular Level. Cryst. Growth Des. 2017, 17, 2893–2910. [Google Scholar] [CrossRef]
- Neogi, S.; Sen, S.; Bharadwaj, P.K. Single-crystal to single-crystal transformations in metal-organic frameworks. Met.-Org. Framework Mater 2014, 473–522. [Google Scholar]
- Chen, L.; Chen, Q.; Wu, M.; Jiang, F.; Hong, M. Controllable Coordination-Driven Self-Assembly: From Discrete Metallocages to Infinite Cage-Based Frameworks. Acc. Chem. Res. 2015, 48, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.-Y.; Wang, C.-C. Water-induced reversible SCSC or solid-state structural transformation in coordination polymers. CrystEngComm 2015, 17, 8776–8785. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, Y.; Zhang, B.; Zhu, D. Crystal-to-Crystal Transformation from Antiferromagnetic Chains into a Ferromagnetic Diamondoid Framework. J. Am. Chem. Soc. 2009, 131, 6934–6935. [Google Scholar] [CrossRef]
- Du, M.; Li, C.-P.; Wu, J.-M.; Guo, J.-H.; Wang, G.-C. Destruction and reconstruction of the robust [Cu2(OOCR)4] unit duringcrystal structure transformations between two coordination polymers. Chem. Commun. 2011, 47, 8088–8090. [Google Scholar] [CrossRef]
- Bravo-Garcia, L.; Barandika, G.; Fidalgo-Marijuan, A.; Bazan, B.; Urtiaga, M.K.; Lezama, L.; Arriortua, M.I. Thermal and Magnetic Diversity in the Behaviour of the CuII-bdc-bpa System: 1D, 2D and Interpenetrated 3D Frameworks. Eur. J. Inorg. Chem. 2016, 2016, 4783–4791. [Google Scholar] [CrossRef]
- Bravo-Garcia, L.; Barandika, G.; Bazan, B.; Urtiaga, M.K.; Arriortua, M.I. Thermal stability of ionic nets with CuII ions coordinated to di-2-pyridyl ketone: Reversible crystal-to-crystal phase transformation. Polyhedron 2015, 92, 117–123. [Google Scholar] [CrossRef]
- Lu, Y.; Miller, J.D. Carboxyl Stretching Vibrations of Spontaneously Adsorbed and LB-Transferred Calcium Carboxylates as Determined by FTIR Internal Reflection Spectroscopy. J. Colloid Interface Sci. 2002, 256, 41–52. [Google Scholar] [CrossRef]
- Pinsky, M.; Avnir, D. Continuous Symmetry Measures. 5. The Classical Polyhedra. Inorg. Chem. 1998, 37, 5575–5582. [Google Scholar] [CrossRef]
- Calos, N.J.; Forrester, J.S.; Schaffer, G.B. A Crystallographic Contribution to the Mechanism of a Mechanically Induced Solid State Reaction. J. Solid State Chem. 1996, 122, 273–280. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. FULLPROF: Program for Rietveld Refinement and Pattern Matching Analysis. In Abstracts of the Satellite Meeting on Powder Diffraction of the XV; Congress of the IUCr: Toulouse, France, 1990; p. 127. [Google Scholar]
- Hathaway, B.J.; Billing, D.E. Electronic properties and stereochemistry of mononuclear complexes of the copper(II) ion. Coord. Chem. Rev. 1970, 5, 143–207. [Google Scholar] [CrossRef]
- Hathaway, B.J. Correlation of the electronic properties and stereochemistry of mononuclear [CuN4–6] chromophores. J. Chem. Soc. Dalton Trans. 1972, 1196–1199. [Google Scholar] [CrossRef]
- McGarvey, B.R. Electron spin resonance of transition-metal complexes. Transit. Met. Chem. (N.Y.) 1966, 3, 89–201. [Google Scholar]
- Bleaney, B.; Bowers, K.D. Anomalous paramagnetism of copper acetate. Proc. R. Soc. London, Ser. A 1952, 214, 451–465. [Google Scholar] [CrossRef]
- Rodríguez-Fortea, A.; Alemany, P.; Alvarez, S.; Ruiz, E. Exchange Coupling in Carboxylato-Bridged Dinuclear Copper(ii) Compounds: A Density Functional Study. Chem. Eur. J. 2001, 7, 627–637. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Lin, R.; Fu, Y.; Brock, C.P.; Guarr, T.F. Structural, spectroscopic, and electrochemical investigation of luminescent bimetallic complexes of rhenium(I). Inorg. Chem. 1992, 31, 4346–4353. [Google Scholar] [CrossRef]
- Malaestean, I.L.; Kravtsov, V.C.; Speldrich, M.; Dulcevscaia, G.; Simonov, Y.A.; Lipkowski, J.; Ellern, A.; Baca, S.G.; Kogerler, P. One-Dimensional Coordination Polymers from Hexanuclear Manganese Carboxylate Clusters Featuring a {MnII4MnIII2(μ4-O)2}.Core and Spacer Linkers. Inorg. Chem. 2010, 49, 7764–7772. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Nagao, S.; Iida, M.; Ogata, K.; Ogura, K. Palladium(II)-directed assembly of macrocyclic dinuclear complexes composed of (en)Pd2+ and bis(4-pyridyl)-substituted bidentate ligands. Remarkable ability for molecular recognition of electron-rich aromatic guests. J. Am. Chem. Soc. 1993, 115, 1574–1576. [Google Scholar] [CrossRef]
- Tudor, V.; Marin, G.; Lloret, F.; Kravtsov, V.C.; Simonov, Y.A.; Julve, M.; Andruh, M. New alkoxo-bridged mixed-valence cobalt clusters: Synthesis, crystal structures and magnetic properties. Inorg. Chim. Acta 2008, 361, 3446–3452. [Google Scholar] [CrossRef]
- Wang, S.; Xing, H.; Li, Y.; Bai, J.; Pan, Y.; Scheer, M.; You, X. 2D and 3D cadmium(II) coordination polymers from a flexible tripodal ligand of 1,3,5-tris(carboxymethoxy)benzene and bidentate pyridyl-containing ligands with three-, eight- and ten-connected topologies. Eur. J. Inorg. Chem. 2006, 3041–3053. [Google Scholar] [CrossRef]
- Ferbinteanu, M.; Marinescu, G.; Roesky, H.W.; Noltemeyer, M.; Schmidt, H.-G.; Andruh, M. {[Co(μ-bpe)(bpe)2(H2O)2](0.5bpe)(H2O)(ClO4)2}n: a transition metal-organo network with a novel supramolecular architecture (bpe = 1,2-bis(4-pyridyl)ethane). Polyhedron 1998, 18, 243–248. [Google Scholar] [CrossRef]
- Dalai, S.; Sarathi Mukherjee, P.; Zangrando, E.; Ray Chaudhuri, N. Synthesis, crystal structure and magnetic properties of two new dicyanamide bridged 2D and 1D complexes of Mn(II). New J. Chem. 2002, 26, 1185–1189. [Google Scholar] [CrossRef]
- Gunes, B.; Soylu, H.; Ozbey, S.; Tufan, Y.; Karacan, N. Bis(1,2-di-4-pyridylethane-N:N’)cadmium(II) tetracyanonickelate(II) m-xylene solvate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1996, C52, 2425–2427. [Google Scholar] [CrossRef]
- Wang, C.-C.; Lin, H.-W.; Yang, C.-H.; Liao, C.-H.; Lan, I.-T.; Lee, G.-H. Synthesis and structure of a novel two-dimensional bilayer framework of a [M(C5O5)(dpe)] coordination polymer. New J. Chem. 2004, 28, 180–182. [Google Scholar] [CrossRef]
- Wang, Q.-M.; Guo, G.-C.; Mak, T.C.W. A coordination polymer based on twofold interpenetrating three-dimensional four-connected nets of 42638 topology, [CuSCN(bpa)] [bpa = 1,2-bis(4-pyridyl)ethane]. Chem. Commun. 1999, 1849–1850. [Google Scholar] [CrossRef]
- Fujita, M.; Kwon, Y.J.; Miyazawa, M.; Ogura, K. One-dimensional coordinate polymer involving heptacoordinate cadmium(II) ions. J. Chem. Soc. Chem. Commun. 1994, 1977–1978. [Google Scholar] [CrossRef]
- Hennigar, T.L.; MacQuarrie, D.C.; Losier, P.; Rogers, R.D.; Zaworotko, M.J. Supramolecular isomerism in coordination polymers: conformational freedom of ligands in [Co(NO3)2(1,2-bis(4-pyridyl)ethane)1.5]n. Angew. Chem. Int. Ed. Engl. 1997, 36, 972–973. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Zheng, X.-J.; Li, W.-J.; Wang, M.-W.; Fang, C.-Y. Assembly of supramolecular networks with the inclusion of water chains, cyclic hepta and octa water clusters. Z. Anorg. Allg. Chem. 2008, 634, 2663–2669. [Google Scholar] [CrossRef]
- Kim, S.H.; Park, B.K.; Song, Y.J.; Yu, S.M.; Koo, H.G.; Kim, E.Y.; Poong, J.I.; Lee, J.H.; Kim, C.; Kim, S.-J.; et al. Construction of crystal structures of metal(II)-benzoates (M = Mn, Ni, Co, Cu, Zn, and Cd) and 1,2-bis(4-pyridyl)ethane: Effects of metal coordination modes and their catalytic activities. Inorg. Chim. Acta 2009, 362, 4119–4126. [Google Scholar] [CrossRef]
- Dias de Souza, N.L.G.; Garcia, H.C.; de Souza, M.C.; Fernandes, A.L.d.A.; Pereira, G.C.; Diniz, R.; de Oliveira, L.F.C. Crystal architectures of copper and zinc metal complexes containing 2-thiophenepropionate and 1,2-bis(4-pyridyl)ethane building blocks. J. Mol. Struct. 2015, 1085, 21–27. [Google Scholar] [CrossRef]
- May, L.J.; Shimizu, G.K.H. Mutual structure-directing effects of a non-interpenetrated square grid coordination polymer and its complementary complex anion net. Chem. Commun. 2005, 1270–1272. [Google Scholar] [CrossRef]
- Plater, M.J.; Foreman, M.R.S.J.; Skakle, J.M.S. Synthesis of co-ordination networks from flexible bis-(4-pyridyl) ligands and cadmium salts. Cryst. Eng. 2001, 4, 293–308. [Google Scholar] [CrossRef]
- Lin, H.; Wu, X.; Maggard, P.A. Ligand-Based Modification of the Structures and Optical Properties of New Silver(I)-Rhenate(VII) Oxide/Organic Hybrid Solids. Inorg. Chem. 2009, 48, 11265–11276. [Google Scholar] [CrossRef]
- Dannenbauer, N.; Matthes, P.R.; Mueller-Buschbaum, K. Luminescent coordination polymers for the VIS and NIR range constituting LnCl3 and 1,2-bis(4-pyridyl)ethane. Dalton Trans. 2016, 45, 6529–6540. [Google Scholar] [CrossRef]
- Pascal, P. Magnetochemical researches. Ann. Chim. Phys. 1910, 19, 5–70. [Google Scholar]
- CrysAlis Pro CCD V38.2 and RED; Oxford Diffraction, Ltd.: Oxford, UK, 2009.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1 and 1.ah are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Lengths (Å) | Bond Angles (°) | |||||
|---|---|---|---|---|---|---|
| Cu1-N1 | 2.003(4) | N1-Cu1-O2Aii | 89.4(2) | O1Aii-Cu1-O1W | 108.7(1) | |
| Cu1-O3A | 1.970(3) | N1-Cu1-O1W | 90.6(2) | O1Aii-Cu1-N1 | 91.6(1) | |
| Cu1-O1W | 1.975(4) | O3A-Cu1-O2Aii | 86.0(2) | O2W-Cu1-N1 | 98.5(2) | |
| Cu1-O2Ai | 2.023(3) | O3A-Cu1-O1W | 93.2(2) | O2W-Cu1-O1Aii | 155.3(1) | |
| Cu1-O2W | 2.304(4) | O1Aii-Cu1-O2Aii | 55.9(2) | O2W-Cu1-O3A | 84.4(1) | |
| Cu1-O1A | 2.508(4) | O1Aii-Cu1-O3A | 84.2(2) | O2W-Cu1-O1W | 93.7(1) | |
| CSM Distortion | OC-6 | TPR-6 | ||||
| Cu1 | 3.814 | 14.855 | ||||
| D-H···A | D···A | <D-H···A | D-H···A | D···A | <D-H···A |
| O1W-H1WA···O1Ai | 2.655(5) | 170(5) | C2 -H2···O2Aii | 3.318(7) | 174 |
| O1W-H1WB···O4A | 2.590(6) | 156(6) | C6 -H6···O1Ai | 3.266(7) | 145 |
| O2W-H2WA···O2Aii | 2.755(5) | 172(6) | C7 -H7B···O1Wiii | 3.298(7) | 132 |
| Cgi-Cgj | DC | ANG | DZ | DZ’ | DXY |
| CgN1-CgN1iii | 3.843(3) | 0.0(3) | 3.605(2) | 3.605(2) | 1.331 |
| CgC1A-CgN1iv | 5.308(3) | 88.3(3) | 0.357(2) | 4.914(2) |
| Bond Length (Å) | Bond Angles (°) | ||||||
|---|---|---|---|---|---|---|---|
| Cu1—O1A | 1.930 (6) | N1-Cu1-O4A | 92.7 (3) | O1Ai-Cu1-N1 | 96.4 (3) | ||
| Cu1—O3Aii | 1.970 (6) | O4A-Cu1-O1A | 88.2 (3) | O1Ai-Cu1-O1A | 100.3 (3) | ||
| Cu1—O4Aiii | 1.985 (6) | O1A-Cu1-O3A | 88.9 (3) | O1Ai-Cu1-O3A | 82.7 (3) | ||
| Cu1—N1 | 1.978 (8) | O3A-Cu1-N1 | 95.1 (3) | O1Ai-Cu1-O4A | 80.2 (3) | ||
| Cu1—O1Ai | 2.442 (9) | ||||||
| CSM distortion | vOC-5 | TBPY-5 | SPY-5 | ||||
| Cu1 | 2.584 | 2.405 | 2.755 | ||||
| D-H···A | D···A | <D-H···A | D-H···A | D···A | <D-H···A |
| C2-H2···O4Ai | 2.979(11) | 111 | C6-H6···O3Aii | 2.998(12) | 118 |
| Cgi-Cgj | DC | ANG | DZ | DZ’ | DXY |
| CgN1-CgC1Aiii | 4.291(6) | 19.7(5) | 2.707(4) | 3.604(4) | 2.329 |
| Topology | No. | Description | CSD Refcode |
|---|---|---|---|
| B | 234 | 0D | |
| B1 | 205 | Bpa bridge (anti) between 2 metal centers | JUMPUC [51] |
| B2 | 6 | Bpa bridge (gauche) between 2 metal centers | UKIBIA [52] |
| B3 | 11 | Double bpa bridge (gauche) | WAFTEC [53] |
| B4 | 1 | Triple bpa bridge (gauche) | AGACER [54] |
| Bn(X) | 11 | Bpa bridge (anti o gauche) between X metal centers | LELMUL [55] |
| C | 450 | 1D | |
| C1 | 251 | Bpa chain (anti) | LADQOW [56] |
| C2 | 25 | Zig-zag de bpa chain (gauche) | WUJLUI [57] |
| C3 | 25 | Double bpa bridge chain (gauche) | TEJYUC [58] |
| C4 | 11 | Alternated zig-zag bpa chain (gauche and anti) | ISIKIE [59] |
| C5 | 118 | Zig-zag bpa chain (anti) | DAQFAC [60] |
| C6 | 5 | Bpa chain (anti) and double bpa bridge (gauche) | POGLEC [61] |
| C7 | 15 | Stairs bpa (anti) | NAMTOK [62] |
| L | 22 | 2D | |
| L1 | 1 | Brick bpa layer (anti) | XOLCOR [63] |
| L2 | 6 | Herringbone bpa layer (anti) | NUHLOS [64] |
| L3 | 1 | Corrugated hexagonal bpa layer (anti) | TUBBAV [65] |
| L4 | 11 | Square bpa layer (anti) | PAKSOK [66] |
| L5 | 6 | Linear bpa layer (anti) connected by bpa bridge (anti or gauche) | NAMSEZ [62] |
| T | 12 | 3D | |
| T1 | 6 | Cubic bpa structure (anti) | IDAJUS [67] |
| T2 | 3 | Diamond like bpa structure (anti) | HUWDEJ [68] |
| T3 | 4 | Complex structure of bpa layers (anti) connected by bpa (gauche) | VAKDIX [69] |
| 1 | 1.ah | |
|---|---|---|
| Formula | C22H22Cu2N2O12 | C22H12Cu2N2O8 |
| FW (gmol−1) | 633.49 | 280.72 |
| Crystal System. | Triclinic | Triclinic |
| Space Group (n°) | P−1, (2) | P−1, (2) |
| a (Å) | 7.2481(11) | 5.6282(5) |
| b (Å) | 9.2514(11) | 8.6560(5) |
| c (Å) | 9.8569(14) | 10.0512(8) |
| α (°) | 115.68(1) | 95.739(6) |
| β (°) | 97.80(1) | 97.168(7) |
| γ (°) | 96.80(1) | 96.154(6) |
| V(Å3) | 578.4(1) | 479.84(6) |
| Z | 1 | 1 |
| F (000) | 322 | 282 |
| μ (mm−1) | 2.926 | 3.273 |
| ρcalc (gcm−3) | 1.819 | 1.943 |
| Crystal Size, mm | 0.05 × 0.04 × 0.03 | 0.12 × 0.07 × 0.06 |
| Limiting Indices | −8 ≤ h ≤ 8 | −6≤ h ≤ 6 |
| −10 ≤ k ≤10 | −10 ≤ k ≤6 | |
| −11 ≤ l ≤7 | −11 ≤ l ≤11 | |
| Reflections Collected | 3163, | 3185 |
| Unique (Rint) | 1831 (0.042) | 1693 (0.049) |
| Observed [I > 2σ(I)] a | 1560 | 1527 |
| Parameters/Restraints | 188/4 | 154/0 |
| R(F)/wR(F2) [I > 2σ(I)] a | 0.062/0.159 | 0.083/0.238 |
| R(F)/wR(F2) (all data) a | 0.071/0.168 | 0.088/0.241 |
| Goodness of Fit on F2 | 1.064 | 1.230 |
| L. Diff. Peak and Hole (e Å−3) | 1.194, −1.200 | 1.656, −1.082 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bravo-García, L.; Larrea, E.S.; Artetxe, B.; Lezama, L.; Gutiérrez-Zorrilla, J.M.; Arriortua, M.I. Structural Transformations in the Thermal Dehydration of [Cu2(bpa)(btec)(H2O)4]n Coordination Polymer. Molecules 2019, 24, 1840. https://doi.org/10.3390/molecules24091840
Bravo-García L, Larrea ES, Artetxe B, Lezama L, Gutiérrez-Zorrilla JM, Arriortua MI. Structural Transformations in the Thermal Dehydration of [Cu2(bpa)(btec)(H2O)4]n Coordination Polymer. Molecules. 2019; 24(9):1840. https://doi.org/10.3390/molecules24091840
Chicago/Turabian StyleBravo-García, Laura, Edurne S. Larrea, Beñat Artetxe, Luis Lezama, Juan M. Gutiérrez-Zorrilla, and María I. Arriortua. 2019. "Structural Transformations in the Thermal Dehydration of [Cu2(bpa)(btec)(H2O)4]n Coordination Polymer" Molecules 24, no. 9: 1840. https://doi.org/10.3390/molecules24091840
APA StyleBravo-García, L., Larrea, E. S., Artetxe, B., Lezama, L., Gutiérrez-Zorrilla, J. M., & Arriortua, M. I. (2019). Structural Transformations in the Thermal Dehydration of [Cu2(bpa)(btec)(H2O)4]n Coordination Polymer. Molecules, 24(9), 1840. https://doi.org/10.3390/molecules24091840