1. Introduction
Epinastine is a histamine H
1 receptor antagonist with high receptor selectivity. Epinastine cannot cross the blood-brain barrier in the body and may be classified as a second-generation antihistamine [
1]. Based on its polarity and cationic charge at physiological pH, it cannot easily diffuse into the central nervous system (CNS) [
2]. Thus, these physicochemical properties distinguish epinastine as a non-sedative antihistamine, and unlike other first-generation antihistamines acting on the CNS, it does not cause side effects, such as drowsiness or sedation [
3]. Epinastine has multiple effects that inhibit the allergic response in three ways: (1) stabilizes mast cells by preventing mast cell degranulation to control the allergic response, (2) prevents histamine binding to both the H
1- and H
2-receptors to stop itching and provide lasting protection, and (3) prevents the release of pro-inflammatory chemical mediators from the blood vessel to halt progression of the allergic response [
4]. In addition, Oshima et al. [
5] reported that epinastine suppresses the immune responses of Th-2 cell through functional modulation of dendritic cells, which play an essential role in the development of allergic immune responses, and thus has the results in favorable modification of the clinical status of allergic diseases. Epinastine has been used in the form of eye drops primarily in association with allergic conjunctivitis. In this regard, various safety information has been established through previous preclinical and clinical trials. Brar et al. [
6] reported that no significant ocular and systemic toxicity was observed in white rabbits and cynomolgus monkeys treated with 0.05–0.5% epinastine hydrochloride ophthalmic solution for 6 months. Yu et al. [
7] reported that epinastine rapidly reached high levels on the ocular surface without unwanted systemic side effects when 0.05% epinastine hydrochloride ophthalmic solution was administered to allergic conjunctivitis patients for 7 days. In addition to its use as eye drops, during the last 10−15 years, tablets of epinastine hydrochloride have been used for clinical application in asthma and urticaria, and patients are prescribed at a dose of 10−20 mg once daily. In vivo metabolism of epinastine occurs mainly in the liver, but the degree of metabolism is reported to be very low. That is, most of the epinastine administered into the body is excreted in the unchanged form [
8].
Analytical and pharmacokinetic (PK) reports of epinastine have been performed in the past. However, the reported methods had limitations and needed improvement. Most of all, there have been no reports on how PK characteristics can differ by analytical methods, and which PK parameters are significantly affected. Even in the same sample, there may be differences in the quantitative values depending on the analytical method, which may cause differences in the PK parameter values. Drug dose and usage settings based on the individual parameters for which the difference occurred may produce different results in the clinic. Therefore, in this study, we tried to identify the changes in PK parameters of the drug according to the analytical methods. For this study, epinastine hydrochloride tablets of 20 mg were orally administered to humans, and the subsequently collected plasma samples were analyzed by two newly developed assays (HPLC-UV and UPLC-MS/MS), which are most commonly used for quantification in biological samples. Our study also focused on comparing the performances of newly developed UPLC-MS/MS and HPLC-UV methods for the determination of epinastine in human plasma. All methods were fully validated according to international bioanalytical method guidelines and represent a useful tool for the characterization of PKs of 20 mg epinastine hydrochloride tablets in Korean subjects.
3. Discussion
Until now, few studies investigated PKs of epinastine, and analytical methods with limited validation information have been proposed. Some articles have reported bioanalytical methods for epinastine based on HPLC or specific analytical conditions [
14,
15,
16]. However, validation information could not be confirmed in those reports. Oiwa et al. [
14] reported that after oral and intravenous (IV) administration of
14C-epinastine hydrochloride in rats, there was dose-linearity, no gender difference on PKs, the largest distribution in the gastrointestinal tract, and a small amount excretion into the milk. Li et al. [
15] revealed that there was no difference in PKs between healthy Chinese and Tibetans after a single oral administration of 20 mg of epinastine hydrochloride tablets using an HPLC-UV method. Shi et al. [
16] also reported that test tablets had little difference in PK pattern from reference tablets in a bioequivalence study of epinastine hydrochloride in healthy Chinese subjects.
Several studies have used HPLC method for analysis of bulk drug [
11], formulation [
17], or dietary supplements [
9], but are rarely used for biological sample analysis. In addition, the LLOQ values they presented were so high that it was very limited for application in quantifying biological samples. Although some researchers analyzed epinastine in rat plasma using HPLC-UV, there were common limitations of high LLOQ and long run time [
12,
13]. Ahirrao et al. [
11] reported an HPLC-UV analysis of epinastine in bulk drug, and its LLOQ and limit of detection (LOD) were as high as 180 and 50 ng/mL, respectively. The HPLC-UV analysis of epinastine reported by Malakar et al. [
17] was also for the pharmaceutical dosage form, and the LLOQ was very high as 2 μg/mL. Do et al. [
9] reported the simultaneous analysis of 20 antihistamines, including epinastine, in dietary supplements, but it was very limited to be directly applied for the analysis of biological samples in consideration of different sample preparation and assay validation and long analysis time per sample. In addition, LLOQ was as high as 90 ng/mL and was not suitable for the analysis of biological samples. An HPLC-UV method, which analyzed rat plasma samples administered with 5–20 mg/kg epinastine, was introduced. However, the LLOQ of epinastine was very high, at 10 ng/mL [
12]. Also, the amount of organic solvent required for preparation per sample was more than 5 mL, and the required time was very long (more than 30 min). There was also a report of rat plasma analysis, although the LLOQ value was also as high as 20 ng/mL [
13]. The run-time per sample was very long, more than 16 min, and there was no assay validation, including stability, carry-over, etc.
There have also been some studies on human sample analysis using LC-MS/MS. However, they had limitations, such as high LLOQ [
10], lack of detailed information on methodology [
18], and much time and solvent [
19]. Bae et al. [
10] reported the LC-MS/MS method for epinastine in abstract form, but the LLOQ value was as high as 1 ng/mL, and specific experimental methods were lacked. In addition, Yu et al. [
18] also measured epinastine concentrations in tear samples after topical ophthalmic administration of epinastine eyedrops to humans. However, due to the lack of detailed description and validation information of the assay, it was limited to apply for epinastine analysis in other biological samples. Although the analytical method reported by Shi et al. [
19] was sensitive to 0.1 ng/mL of LLOQ, the required time for the analysis was very long, and the consumption of solvents was large due to HPLC method. Those previously reported contents on epinastine assay are summarized in
Table S1.
Because of the clinical importance and wide application of epinastine, studies on various analytical methods and pharmacological effects have been reported from the past to the present. However, as mentioned above, there were limitations in reported epinastine assays, and so we have been working to develop UPLC-MS/MS and HPLC-UV methods that complement these limitations. As shown in our previous report [
20], epinastine showed a very low C
max of 10–20 ng/mL. Therefore, it is essential to develop highly accurate and sensitive assay methods to obtain definite PK results, including the absorption and elimination phase, in drugs, such as epinastine.
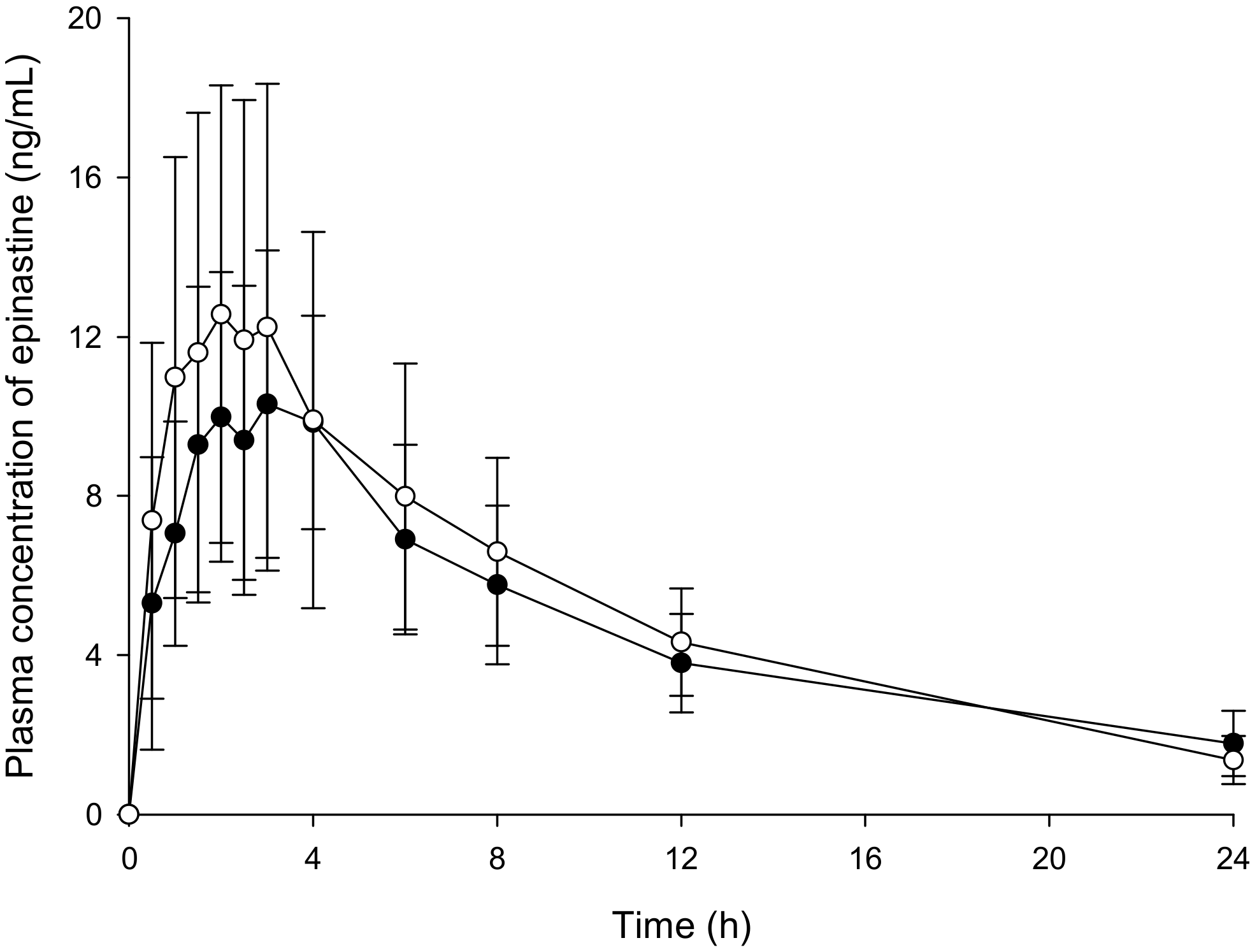
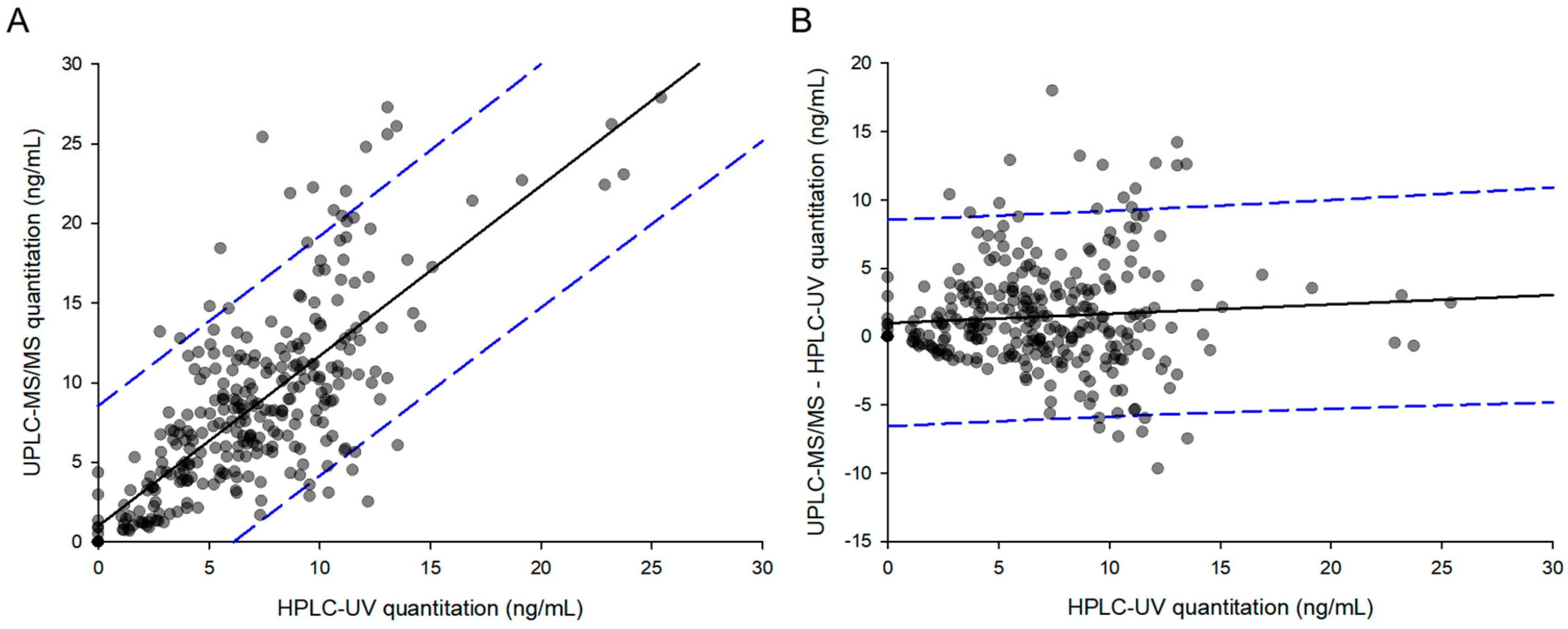
The use of UPLC columns, optimizing the mobile phase, the use of gradient elution program, enhancing the extraction efficiency and removing impurities through appropriate sample preparation, and sample concentration through evaporation could greatly improve the sensitivity than the previously reported methods. Our results demonstrated that the developed methods were reproducible, selective, precise, accurate, and relatively impervious to endogenous interference. UPLC-MS/MS was found to be 50 times more sensitive than HPLC-UV for the determination of epinastine. Therefore, UPLC-MS/MS method was more adequate for the analysis of clinical samples where the dosages were lower. Highly sensitive UPLC-MS/MS was required in vivo in order to quantify substantially low levels of analytes in large numbers of biological samples. Thus, we quantified epinastine concentration in plasma samples obtained from all individuals. As a result, the PK profile, showing the elimination and the absorption phases, was determined accurately. In addition, UPLC-MS/MS required substantially less organic solvent and less sample volume for sample preparation. Furthermore, the required time for the total sample analysis was significantly reduced. It has, therefore, a more economical modality and is environmentally friendly. However, UPLC-MS/MS is associated with a high operating cost and is more expensive than HPLC-UV. Therefore, HPLC-UV analysis is more economical if the PK differences in HPLC-UV and UPLC-MS/MS quantification results are not as large as determined in the epinastine quantitative assay. Although the run time was 15 min, and the HPLC-UV method lasted three times longer, the HPLC-UV method was shorter than the UPLC-MS/MS method in sample preparation, suggesting that HPLC-UV method was more economical for the analysis of a small number of total samples. Our results illustrated the fact that UPLC-MS/MS could be very sensitive in comparison with HPLC-UV, which typically goes down to 1 ng/mL (LLOQ of developed HPLC-UV method). However, for general clinical studies, HPLC-UV analysis could be used, and an adequate PK profile could be obtained with a method validated between 1 and 100 ng/mL. The developed methods could be applied to the analysis of epinastine in plasma, as well as other biological samples. The reason for the difference in plasma sample preparation between UPLC-MS/MS and HPLC-UV methods is that the extraction solvent composition for each analytical instrument was developed and applied. Extraction solvents applied to HPLC-UV suspected significant matrix effects in UPLC-MS/MS, and the recovery was not satisfactory. In other words, it was optimized and applied to UPLC-MS/MS with an extraction solvent different from HPLC-UV in consideration of the matrix effect and recovery in UPLC-MS/MS.
The ratios (
) presented in
Table 5 have a very important meaning. As mentioned above (in the
Section 4.5.3 Accuracy and precision), the accuracy and precision of analysis typically allowed for variability within ±15%. Variability within ±20% was allowed at the LLOQ. Nevertheless, the average ratio of V
d/F, C
max, T
max, and t
1/2 showed a difference of more than 20%. In other words, when comparing only the mean values of parameters (V
d/F, C
max, T
max, and t
1/2), they were out of 15% or 20% (usual tolerance in analysis). In the case of V
d/F, the mean value of epinastine after a single oral dose could be calculated as low as 27% when analyzing plasma samples using UPLC-MS/MS and as short as 28% as for t
1/2. There also could be a difference in the accumulation index as 1.117 (by UPLC-MS/MS method) and 1.301 (by HPLC-UV method), when estimating the accumulation index by the following equation: 1/1−e
−k·τ, where τ is the 24 h as dosing interval. This might affect the dosing and use of epinastine in relation to a cumulative evaluation in the body at multiple doses. In the case of C
max, the mean value of epinastine after a single oral dose could be determined as high as 27% when analyzing plasma samples using UPLC-MS/MS and as short as 21% for T
max. This might affect the dosing and use of epinastine with regard to drug effects and toxicity. In addition, as shown in
Figure 6 and
Table 6, the results can be different when multiple-dose simulations are performed with single-dose PK parameters calculated based on data obtained by different assays. This could greatly affect PK evaluation. As shown in
Table 6, the ratio of C
max/C
min at steady-state was about 2 times greater in UPLC-MS/MS than HPLC-UV. This means that if we choose the HPLC-UV method for the calculation of epinastine PK parameters, we would be apt to underestimate the safety or toxicity of epinastine. Using the UPLC-MS/MS method, we could quantify the concentration of the epinastine terminal phase in the blood, resulting in shorter t
1/2 (than HPLC-UV), large clearance (than HPLC-UV), and large estimation of the difference between steady-state C
max and C
min in multiple-dose simulations. In other words, the LLOQ reduction in the analysis by UPLC-MS/MS caused some differences in the estimation of PK parameters compared to HPLC-UV by quantifying epinastine elimination phases that were not accurately quantified by HPLC-UV. Our findings suggested that the choice of the assay for biosamples is critical for PK analysis and clinical dose and regimen settings. Previously reported literature [
21,
22] has emphasized that there are differences in the results of an assay for tacrolimus concentrations in the blood by different analytical methods. In particular, the difference between the results of microparticle enzyme immunoassay (MEIA) and LC-MS/MS was significant at low concentrations and suggested that it could significantly affect the treatment of patients [
21]. In the study of Braun et al. [
22], the same patient samples (tacrolimus administered) were analyzed by various analytical methods. As a result, the drug concentrations were measured differently at 10.5, 7.92, and 2.93 ng/mL in MEIA, enzyme-linked immune-sorbent assay, and LC-MS/MS. In other words, when one sample is analyzed by LC-MS/MS, the concentration is measured to be low, and the administration dose should be increased. On the other hand, when the MEIA method is used, the concentration could be over-estimated, indicating that dose adjustment is not necessary. This suggests that inappropriate treatment may have been performed. Although the therapeutic range of epinastine is not as narrow as that of tacrolimus, dose adjustment of epinastine due to differences in assays should be carefully considered in the treatment of patients, as with tacrolimus.
4. Materials and Methods
4.1. Reagents and Chemicals
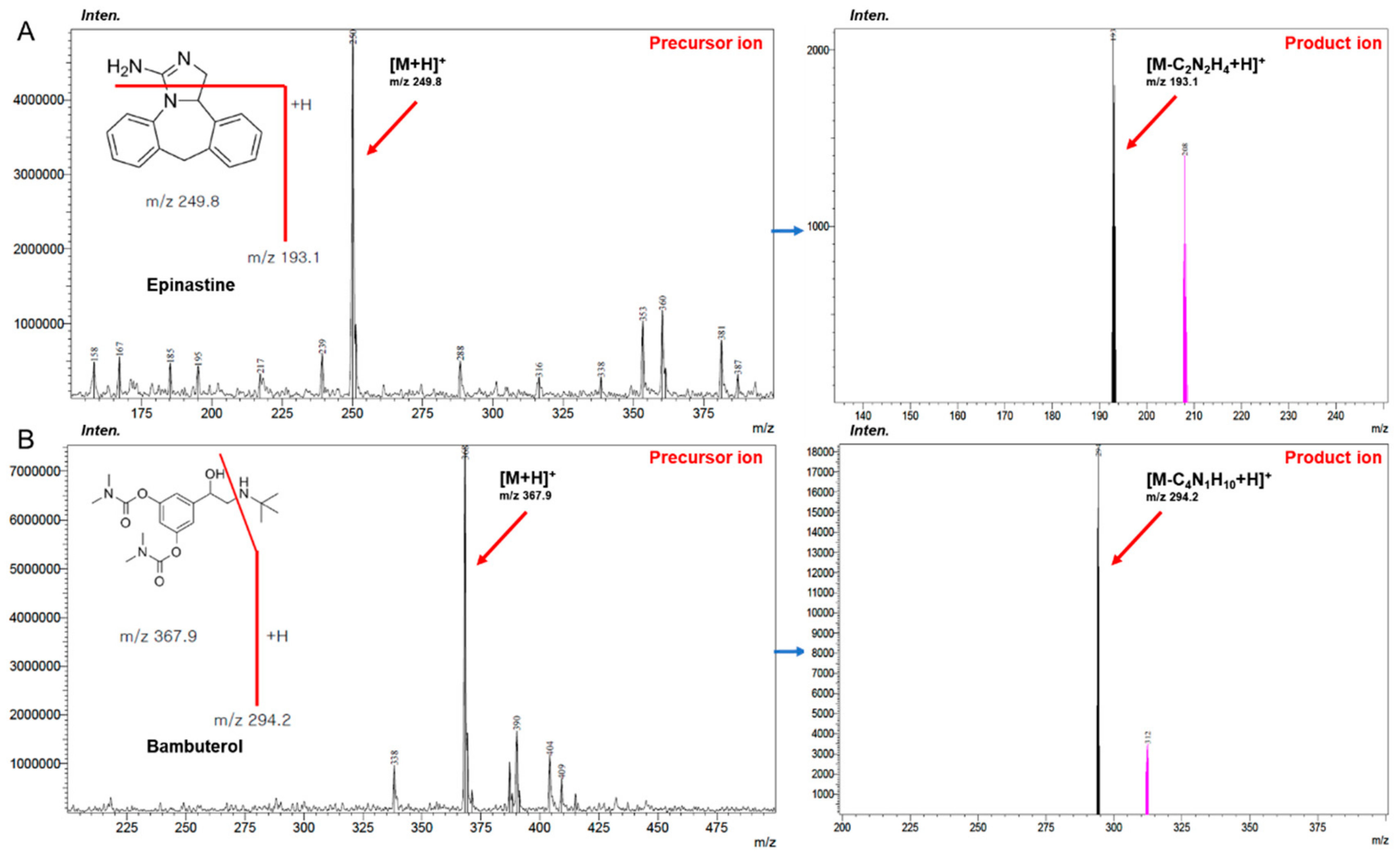
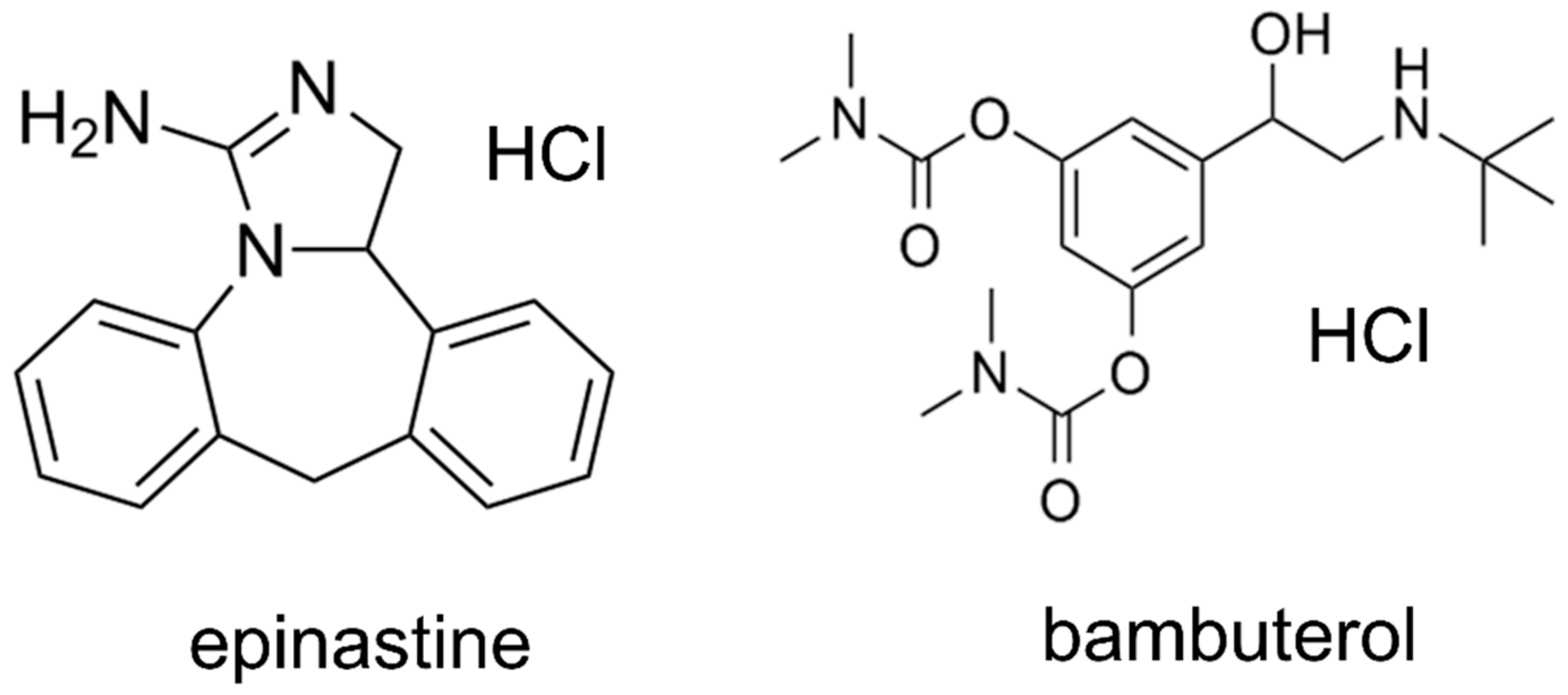
Epinastine hydrochloride (purity ≥ 99%) was obtained from Heumann Pharma GmbH & Co. Generica KG (Nuremberg, Germany), and bambuterol (purity ≥ 99%) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Bambuterol was used as an IS in both methods (HPLC-UV and UPLC-MS/MS).
Figure 7 presents the structures of epinastine and bambuterol. LC-MS/MS grade water (18.2 mΩ), methanol, ACN, and HPLC grade ethyl acetate were obtained from Fisher Scientific (Hampton, NH, USA). LC-MS/MS grade formic acid was purchased from DUKSAN Inc. (Ansan, Korea). All chemicals used in this study met the highest HPLC grade or quality available.
4.2. Chromatographic Conditions and Instrumentation
4.2.1. UPLC-MS/MS Method
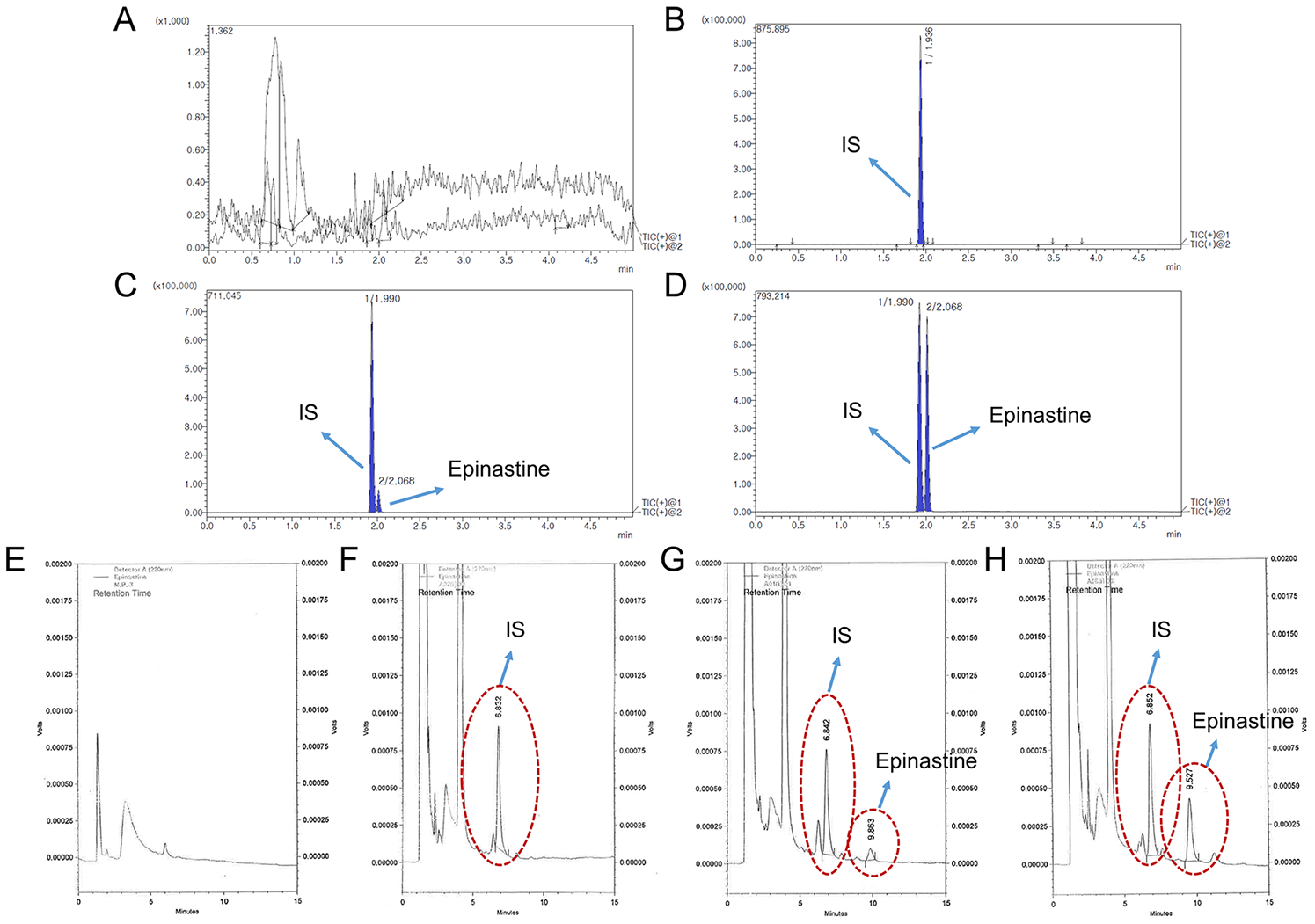
The UPLC-MS/MS method was conducted using an LC-30AD of Shimadzu Nexera X2 Series UPLC system (Shimadzu, Kyoto, Japan) coupled with a SIL-30AC autosampler and DGU-20A degassing unit with Shimadzu-8040 mass spectrometer. In order to obtain an optimum chromatogram, various condition tests were carried out on the mobile phase pH (0.01% formic acid in water (pH 2.7), 0.1% formic acid in water (pH 2.3), 0.05% formic acid in water (pH 2.5), 0.1% acetic acid in water (pH 2.9), 0.05% trifluoracetic acid in water (pH 2.1), and 0.3% triethylamine in water (pH 4.5), v/v), containing 1% (v/v) of 5 mM ammonium formate (pH 3.0) buffer, and column (Acquity UPLC® BEH C18 (50 mm × 2.1 mm, 1.7 μm), HALO-C18 (100 mm × 2.1 mm, 2.7 μm), and Phenomenex KINETEX core-shell C18 (50 mm × 2.1 mm, 1.7 μm) column). The optimized chromatographic separation of epinastine was obtained on a Phenomenex KINETEX core-shell C18 column at an oven temperature of 40 °C. The mobile phase configuration was ACN (mobile phase B) and 0.1% aqueous formic acid, containing 1% (v/v) of 5 mM ammonium formate (pH 3.0) buffer (mobile phase A, pH 2.3), with gradient elution and a flow rate of 0.3 mL/min. The gradient elution program was as follows: 0–0.5 min (20% B), 0.5–1.5 min (20–80% B), 1.5–3.5 min (80% B), 3.5–3.51 min (80–20% B), and 3.51–5.0 min (20% B). All analytical procedures were conducted with positive electrospray ionization, and quantification was achieved using MRM modes at m/z 249.80 → 193.10 for epinastine and at m/z 367.90 → 294.20 for IS, respectively. Acquisition and analysis of data were achieved using a LabSolutions program. The injection volume was 5 μL, and the collision energies of epinastine and IS were −36 and −20 eV, respectively.
4.2.2. HPLC-UV Method
The HPLC system consisted of a Shimadzu LC 10 AD series (Shimadzu, Kyoto, Japan) equipped with a photodiode array detector with LabSolutions software. A Nova-Pak C18 column (150 × 3.9 mm, 4 μm particle size) from Waters Inc. (Milford, MA, USA) was applied as a stationary phase, and the oven temperature was maintained at 40 °C. The mobile phase consisted of 20 mM phosphate buffer (pH 3.5) and methanol mixed with a small amount of ACN (64/30/6, v/v/v) at a flow rate of 0.8 mL/min. Chromatographic separation was conducted using an isocratic elution. The total run time was 15 min per sample. Injection volume was 50 μL using the Rheodyne injector, and the detection wavelength was 220 nm. Peaks were assigned by spiking the samples with standard compounds and comparison of the retention times and UV spectra.
4.3. Preparation of Standard Solutions
Epinastine and IS stock solutions were prepared as follows: each of epinastine hydrochloride and IS was accurately weighed and dissolved in methanol at a concentration of 1 mg/mL as epinastine prior to obtaining working solutions. All stock solutions were stored at −20 °C. The standard working solutions of epinastine (10, 20, 50, 100, 200, 500, and 1000 ng/mL in HPLC-UV analytical procedure; 0.2, 1, 10, 50, 100, 500, and 1000 ng/mL in UPLC-MS/MS analytical procedure) and the IS (100 ng/mL) were diluted stepwise with 100% methanol from the standard stock solutions. In addition, calibration standards were determined by adding each diluted working solution into a blank human plasma to obtain the final concentrations of epinastine ranging from 1–100 ng/mL in HPLC-UV analytical method and 0.02–100 ng/mL in UPLC-MS/MS analytical method. In order to examine the accuracy and precision of the developed method, QC samples of four concentration levels (1, 5, 20, and 80 ng/mL in HPLC-UV; 0.02, 5, 20, and 80 ng/mL in UPLC-MS/MS) were prepared in a similar way. Preparation of QC samples and calibration curves were performed on the same day of analysis in both methods.
4.4. Sample Extraction
In the HPLC-UV analytical method, epinastine was extracted from human plasma by employing the LLE method. A total of 500 μL of human plasma samples were added to a 50 μL of the IS solution (100 ng/mL of bambuterol in 100% methanol), and 500 μL of 0.1 M sodium carbonate was added to human plasma samples. Then, 4 mL of dichloromethane was added to the mixed sample and vortexed for 7 min and centrifuged at 6000× g for 20 min. The supernatant was removed, and the organic layer was transferred to another test tube. A 200 μL of 25 mM sulfuric acid solution was added, and the mixture was back-extracted for 1 min and centrifuged at 6000 rpm for 5 min. A 150 μL of the upper layer was transferred to an Eppendorf tube (Axygen Scientific Inc., Union City, CA, USA), and an aliquot (50 μL) of this final sample solution was taken and injected into the HPLC-UV system. In UPLC-MS/MS analytical method, the sample extraction for epinastine was extensively tested using the PP method with ACN and methanol, as well as the LLE method using di-ethyl ether, ethyl acetate, methylene chloride, and MTBE. As a result, the samples were extracted by LLE using ethyl acetate, and the protein was precipitated by PP using methanol. A total of 100 μL of human plasma samples were added to a 10 μL of the IS solution (100 ng/mL of bambuterol in 100% methanol). Then, 1000 μL of ethyl acetate-methanol (1/2, v/v) was added to the mixed samples and vortexed for 6 min and centrifuged at 13,000× g for 6 min. Then, 1000 μL of the supernatant corresponding to the organic layer was dried gently with a centrifugal vacuum evaporator of the CVE-3000 model (EYELA Co., Tokyo, Japan) under nitrogen gas at 35 °C for 4 h. The dried matter was reconstituted with 50 μL of 100% methanol and vortexed for 6 min. After centrifugation for 6 min at 13,000× g, 5 μL of aliquots were injected into the UPLC-MS/MS system.
4.5. Method Validation
The validation of newly developed methods was performed in accordance with the Guidance for Industry: Bioanalytical Method Validation issued by the United States FDA [
23], in terms of recovery, accuracy, precision, selectivity, linearity, sensitivity, matrix effect, carryover, stability, and ISR.
4.5.1. Selectivity and Sensitivity
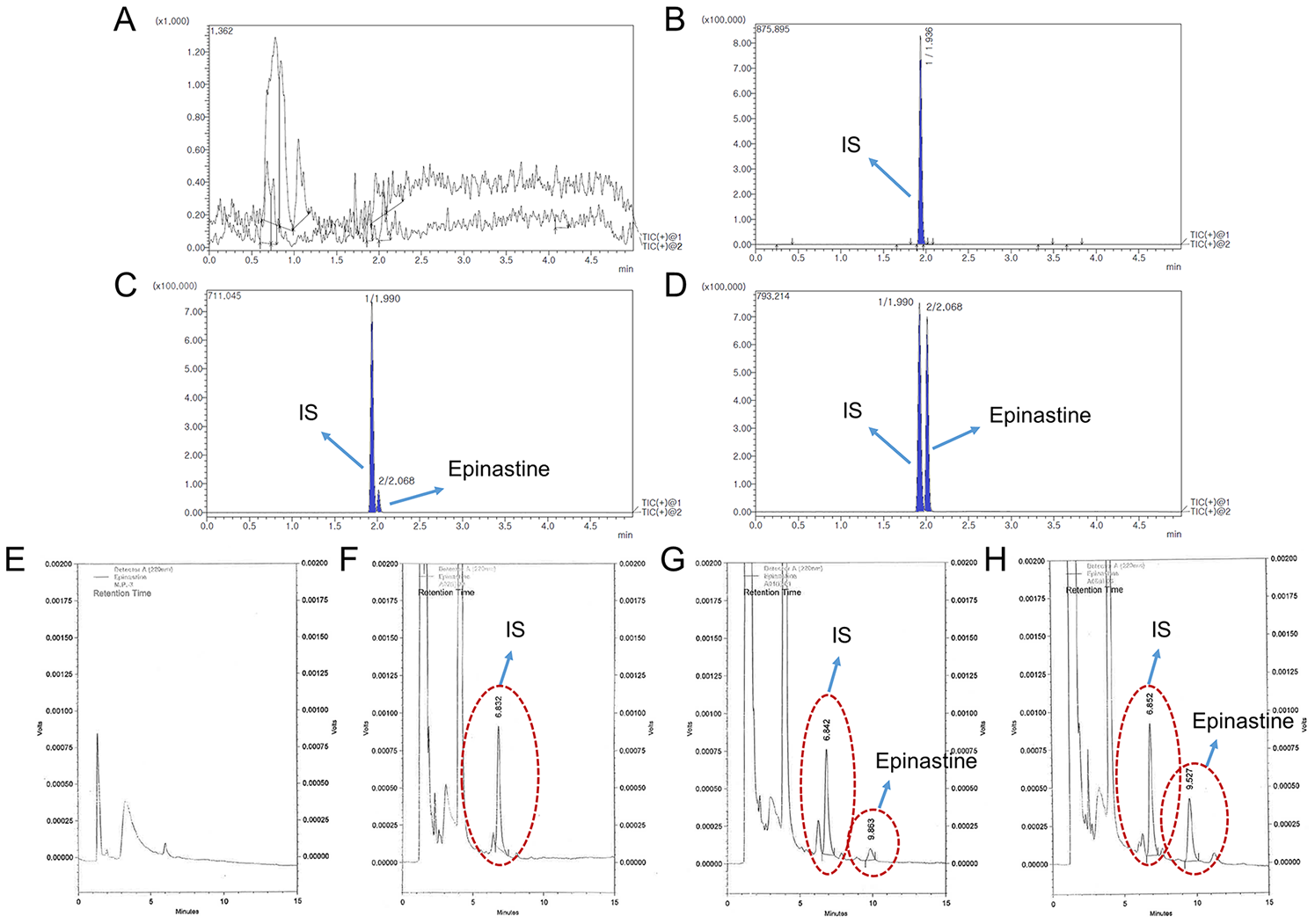
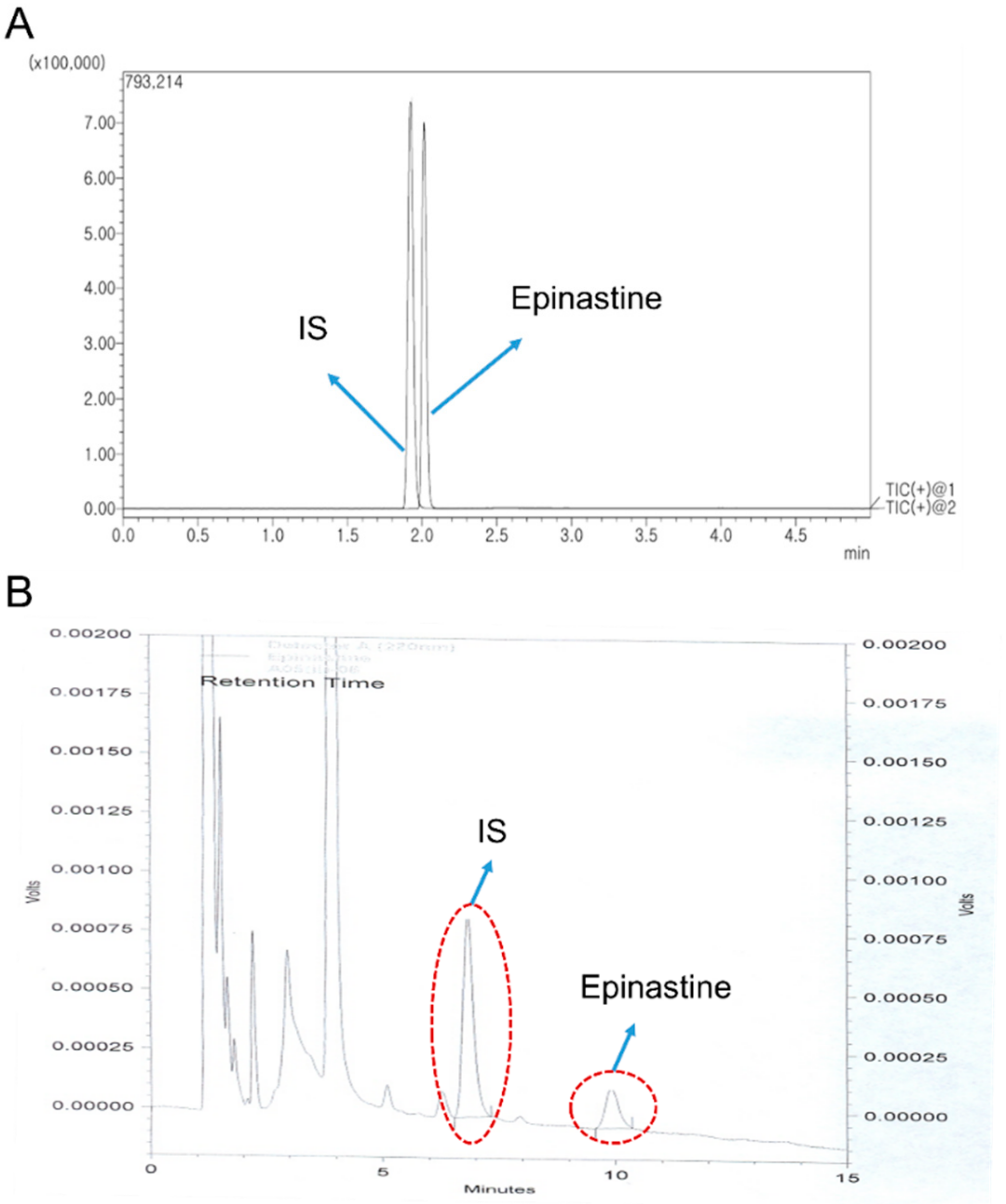
Selectivity was evaluated to ensure the effect of endogenous substances located in the closed retention time of the analytes. Therefore, the blank plasma, zero plasma, plasma spiked with epinastine hydrochloride of LLOQ level, and real plasma samples collected after oral administration of two tablets of epinastine (epinastine hydrochloride 20 mg) to Korean subjects were used to prove and demonstrate selectivity. The used blank plasma was collected from six different individuals. The sensitivity of the method was expressed as LLOQ, LLOQ and LOD were determined as the lowest concentration of the standard samples within the range of quantification with a signal-to-noise ratio of at least 10:1 and 3:1, respectively, with an acceptable precision of less than 20% and accuracy within ±20%. All of these were evaluated using five replicate samples.
4.5.2. Linearity
Calibration curves were determined using the seven calibration points by linear regression with the weighting factor of 1/concentration2. The linearity of the calibration curve was conducted by plotting the epinastine/IS peak area versus the theoretical concentration of epinastine. A correlation coefficient (r2) value with its linear calibration equation was obtained.
4.5.3. Accuracy and Precision
Both intra-day accuracy and precision were assessed by analyzing the QC samples at five different times on the same day. In addition, inter-day evaluations were similarly conducted on five consecutive days. The concentration of each QC sample was quantified using the calibration standards prepared on that day. The precision was assessed by determining the CV value for the analysis of QC samples. The CV value at each concentration level was not allowed to deviate by more than ±15%. However, it was limited to 20% in LLOQ. The accuracy was determined based on the criteria, which are the mean value, not exceeding 15% of the nominal concentration. As with precision standards, it was limited to 20% in LLOQ.
4.5.4. Matrix Effect and Recovery
The recovery efficiencies of epinastine were assessed for the QC samples at low, medium, and high concentrations in five replicates. In addition, the recovery efficiency of the IS was determined. The extraction recovery of the assay from human plasma was evaluated by comparing the detector (UV and MS/MS) response of extracted samples (A) with those of the samples added at the same concentration after extracting the blank plasma (B). In addition, the matrix effect for UPLC-MS/MS method was assessed by comparing the peak area of analyte post-extraction (B) from blank plasma with the absolute standard (C) of the same analyte. The matrix effect and recovery efficiency were determined by the following formula: Recovery = (A)/(B) × 100%; Matrix effect = (B)/(C) × 100%.
4.5.5. Stabilities
The stabilities of epinastine in human plasma were assessed under various conditions, including freeze-thaw, long-term, and short-term stability. Two different levels of QC samples, low (5 ng/mL) and high (80 ng/mL) concentrations, were used for all stability tests. The short-term stability test was conducted by maintaining the QC samples for 24 h at 25 °C, and the long-term stability was determined by analysis of QC samples frozen for 4 weeks at −80 °C. The QC samples were stored for 24 h at −80 °C and then thawed completely at 25 °C for the freeze and thaw stability test. This cycle was repeated in succession, and analysis was conducted after the third cycle. Stabilities of epinastine and IS stock solution were measured after storage for 4 weeks at −20 °C. In addition, for post-preparative stability, the processed QC samples were placed in autosampler for 24 h at 15 °C (at UPLC-MS/MS method), and on the table for 24 h at 25 °C (at HPLC-UV method). Stabilities were determined as the % ratio of measured epinastine concentration to initial epinastine concentration (n = 5). Samples were considered stable if the test values at each level were within ±15% of the sample nominal concentration, and the precision was less than 15%.
4.5.6. Carryover
The carryover test was conducted by injecting a blank sample after injecting the sample with the highest concentration (100 ng/mL for epinastine) in the standard curve. The acceptance criterion of the carryover is that the peak in the blank sample should be less than 20% of the peak in the LLOQ sample.
4.5.7. Incurred Sample Reanalysis
ISR was conducted to ensure the reproducibility of newly developed methods for the analysis of epinastine. Sample selection (10% of the analyzed samples) was accomplished using a computerized random method for ISR. The selection criterion involved samples near the elimination phase in the PK profile of epinastine and Cmax. Thirty-two human plasma samples were selected and compared with initial analyzed values. The results satisfied the acceptable criteria that the variability between the mean value of the initial analysis and that of the reanalysis was within ±15%. In addition, reanalysis values for 67% of all samples were within 20% of their initial values.
4.6. Pharmacokinetic Studies
Twenty-six healthy males of Korean subjects (22–25 years of age, bodyweight 60–85 kg, height 165–195 cm) were recruited for a clinical trial (as bioequivalence test). The Institutional Review Board of the Institute of Bioequivalence and Bridging Study (Chonnam National University, Gwangju, Korea) approved this study protocol (Bioequivalence Test No. 324; 02.04.2005). The clinical trial was conducted according to the revised Declaration of Helsinki for biomedical research involving human subjects and the rules of Good Clinical Practice. The written consent was obtained prior to participating from all participants. In addition, all participants received laboratory, physical, and medical tests prior to a clinical trial. As a result, this study included only well-healthy participants. All the participants fasted more than 10 h before receiving 20 mg epinastine hydrochloride tablets followed by fasting for 4 h. In addition, they avoided drinks or foods containing caffeine or xanthine ingredients during this study. All participants received two tablets of epinastine (epinastine hydrochloride 20 mg) with water of 240 mL. The blood samples were taken from the forearm vein before the administration (0 h) and 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, and 24 h after oral administration. The blood samples were transferred into Vacutainer® tube (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) of 10 mL capacity and were centrifuged (10,000 × g) immediately for 10 min at 4 °C. The supernatant plasma samples were transferred to polyethylene tube and stored at −80 °C until further analysis. Tmax, Cmax, and Cmin were determined using the plasma drug concentration curve over time. The AUC0–∞ was integrated by a linear trapezoidal rule to the final measured concentration (Clast) and extrapolated to infinity by adding the area from Clast (AUC0–t) to infinity (Clast/k); k means the elimination rate constant at terminal phase. The CL/F was determined by dividing the dose of epinastine by the AUC0–∞, where F is the bioavailability of oral administration. The t1/2 was determined as 0.693/k and Vd/F as dose/k·AUC0–∞. All PK parameters were determined via noncompartmental analysis using the WinNonlin® software (version 8.1, Pharsight®, a Certara™ Company, Princeton, NJ, USA).
4.7. Pharmacokinetic Parameters Comparison
In addition to those mentioned (in the
Section 4.8 Statistical analysis) to compare the differences in PK parameter values according to the analytical methods, the parameter values calculated using HPLC-UV and UPLC-MS/MS were divided as follows:
. Closer values to 1 mean that there is little effect on the PK parameters between the two methods.
4.8. Statistical Analysis
Statistical analysis was performed using the Statistical Package for the Social Sciences (SPSS) software version 23 (IBM, Armonk, NY, USA) on the PK parameters calculated by HPLC-UV and UPLC-MS/MS methods. In other words, all PK parameters determined by each quantification method were analyzed for statistical significance by Student’s t-test with p < 0.05, indicating a significant difference.
5. Conclusions
The purposes of this study were to compare the PK parameter values of epinastine obtained using the two methods and to discuss their meanings, as mentioned in the introduction. In this study, we provided the newly developed analysis methods—UPLC-MS/MS and HPLC-UV. Our study focused on comparing the performances of newly developed UPLC-MS/MS and HPLC-UV methods for the determination of epinastine in human plasma.
No published report has compared the differences in analytical methods, such as HPLC-UV and UPLC-MS/MS, in biosamples for epinastine. Generally, UPLC-MS/MS methods show higher throughput, selectivity, and sensitivity compared to HPLC-UV methods [
24]. However, because MS detectors are expensive, analyzing biosamples using relatively less expensive UV detectors would be a great economic advantage. Numerous studies comparing LC-MS and LC-UV methods in the analysis of biosamples for other drugs have been reported from the past [
24,
25,
26,
27,
28,
29,
30]. The purpose of the inter-comparison of these methods is perhaps to ensure that the results are identical even if one of the two methods is used. In other words, by choosing the appropriate analytical method for a specific situation, it would be possible to derive the optimal PK result. In general, the use of LC-UV is more appropriate where rapid turn round of data is not required, and plasma concentrations are high. However, if sensitivity is an issue, limited amounts of plasma are available, and/or where tight deadlines are pivotal, LC-MS is the method of choice [
30].
Both methods were fully validated according to international bioanalytical method guidelines and represented a useful tool for the characterization of PKs of 20 mg epinastine hydrochloride tablets in Korean subjects. Furthermore, comparison (especially focused on PK parameters) of two commonly used analytical methods (HPLC-UV and UPLC-MS/MS) for in vivo analysis of epinastine was performed for the first time. As a result, this method comparison study confirmed that different dose and usage settings might be possible based on PK parameters calculated by other methods. Therefore, if careful consideration is required for dose and regimen settings of drugs, it may be necessary to consider differences in interpretation of PKs according to the assays reported in this study. Finally, our conclusion was that the choice of the assay was very important for the analysis of biological samples (especially where the concentration of drug in the plasma sample is relatively low, as in this study). The UPLC-MS/MS method, which could provide a lower LLOQ than the HPLC-UV method, quantified the concentrations of the drug’s initial absorption and elimination phases in the blood, resulting in more clear PK profiles in case of epinastine. However, this conclusion could not be generalized for some drugs. Due to the presence of the matrix effect or the reduction of ionization sites, there might be some drugs that are more sensitively quantified in LC-UV than LC-MS/MS. These results might affect the calculation of PK parameters (such as t1/2 and clearance) and simulation results at multiple doses, although there were no significant differences in concentration values. In the case of drugs requiring tight therapeutic drug monitoring (TDM), very careful attention should be paid to the dosage and regimen based on these PK parameters. As a result, this study suggested that UPLC-MS/MS was better for PK studies on substances with low plasma concentrations despite the maximum daily dose of the drug, such as epinastine. Our findings are very new and have not been reported previously and are expected to be very important data for PK studies and interpretation of results.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}