QSAR Model of Indeno[1,2-b]indole Derivatives and Identification of N-isopentyl-2-methyl-4,9-dioxo-4,9-Dihydronaphtho[2,3-b]furan-3-carboxamide as a Potent CK2 Inhibitor

 , , ,
, , ,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.2. Computational Methods
3.2.1. Computational Study
3.2.2. Data Set for QSAR
3.2.3. Molecular Descriptors
3.2.4. QSAR Study
3.2.5. Crystal Structure from PDB
3.2.6. Database Generation
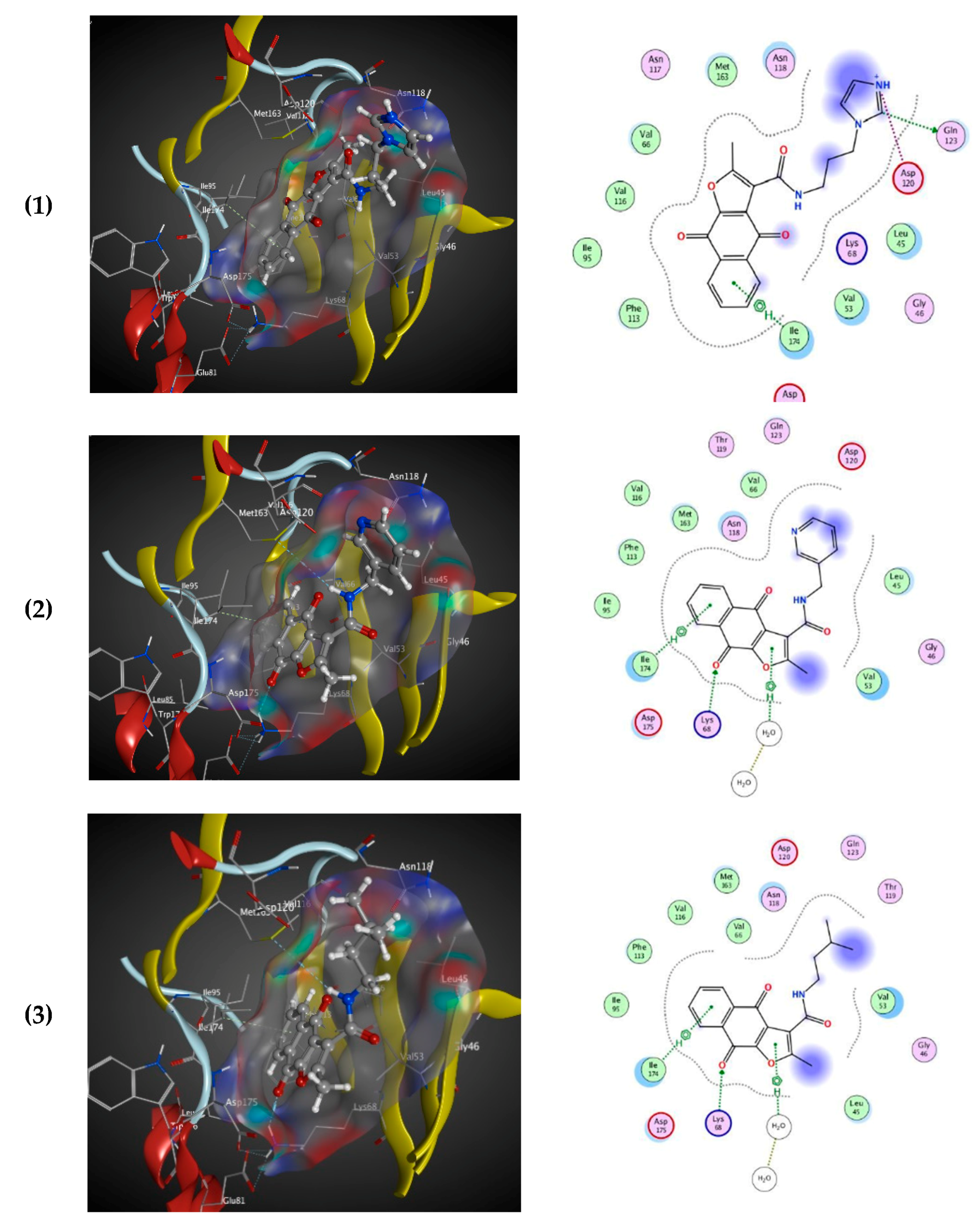
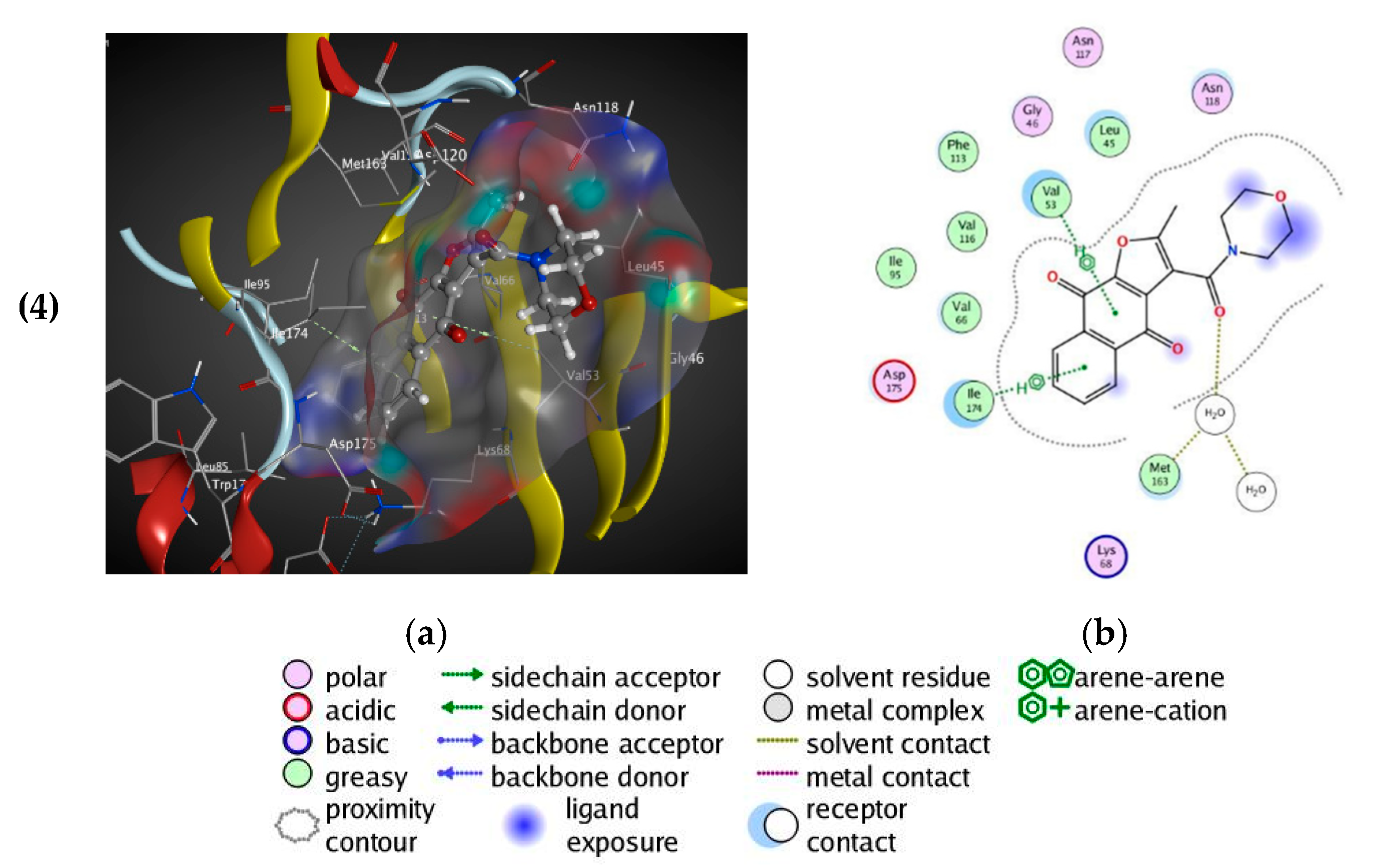
3.2.7. Docking Study
3.3. Biological Assays
3.3.1. Capillary Electrophoresis-Based Assay for Testing the Inhibitors of the Human CK2
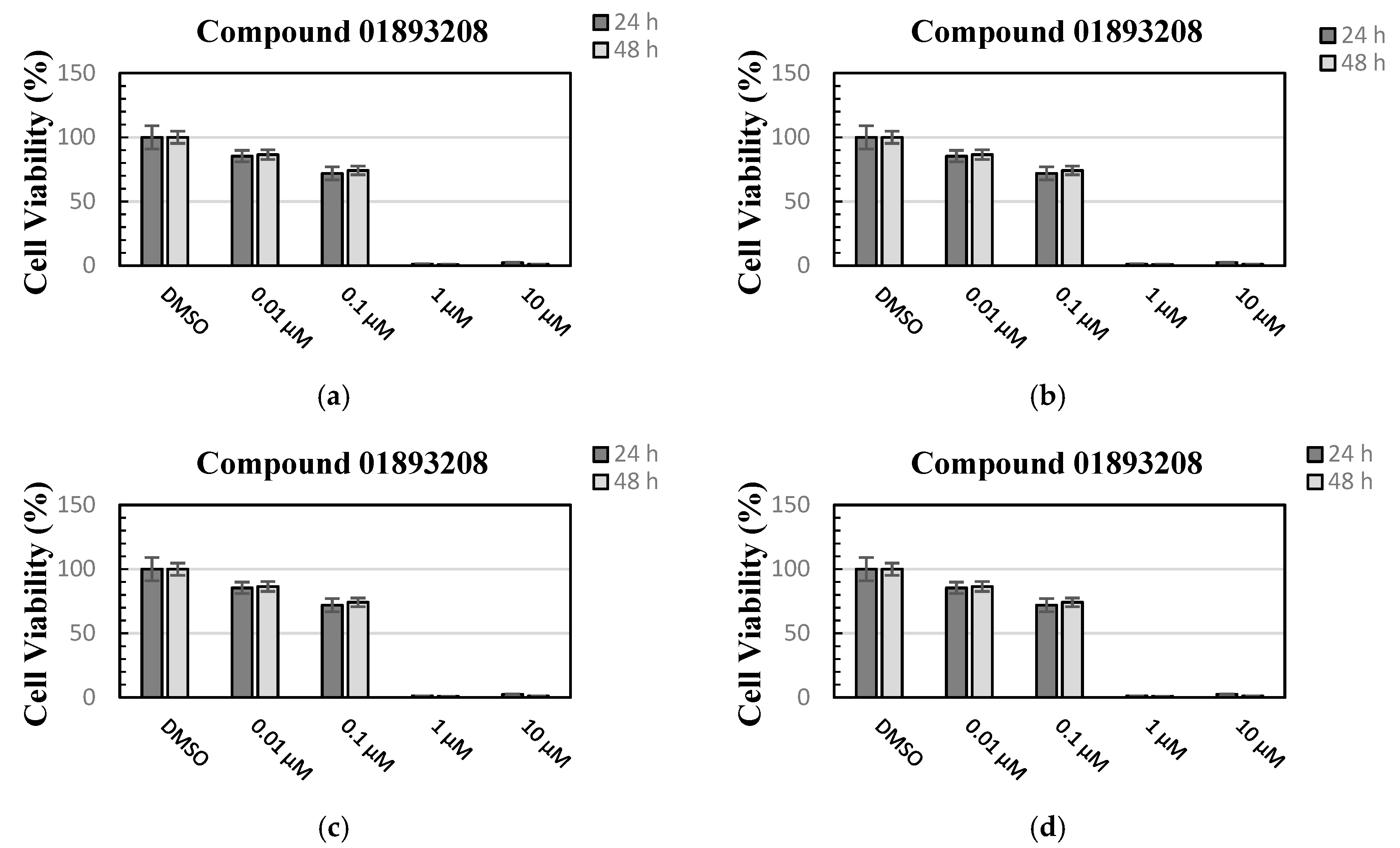
3.3.2. Cell Viability Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Burnett, G.; Kennedy, E.P. The enzymatic phosphorylation of proteins. J. Biol. Chem. 1954, 211, 969–980. [Google Scholar] [PubMed]
- Sarno, S.; Ghisellini, P.; Pinna, L.A. Unique activation mechanism of protein kinase CK2. The N-terminal segment is essential for constitutive activity of the catalytic subunit but not of the holoenzyme. J. Biol. Chem. 2002, 277, 22509–22514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristiani, A.; Costa, G.; Cozza, G.; Meggio, F.; Scapozza, L.; Moro, S. The role of the N-terminal domain in the regulation of the “constitutively active” conformation of protein kinase CK2alpha: insight from a molecular dynamics investigation. ChemMedChem 2011, 6, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Milito, A.; Spagnuolo, C.; Carbone, V.; Rosen, A.; Minasi, P.; Lauria, F.; Russo, G.L. CK2 and PI3K are direct molecular targets of quercetin in chronic lymphocytic leukaemia. Oncotarget 2017, 8, 42571–42587. [Google Scholar] [CrossRef] [Green Version]
- Chua, M.M.J.; Lee, M.; Dominguez, I. Cancer-type dependent expression of CK2 transcripts. PLoS ONE 2017, 12, e0188854. [Google Scholar] [CrossRef] [Green Version]
- Murtaza, I.; Wang, H.X.; Feng, X.; Alenina, N.; Bader, M.; Prabhakar, B.S.; Li, P.F. Down-regulation of catalase and oxidative modification of protein kinase CK2 lead to the failure of apoptosis repressor with caspase recruitment domain to inhibit cardiomyocyte hypertrophy. J. Biol. Chem. 2008, 283, 5996–6004. [Google Scholar] [CrossRef] [Green Version]
- Pagano, M.A.; Arrigoni, G.; Marin, O.; Sarno, S.; Meggio, F.; Treharne, K.J.; Mehta, A.; Pinna, L.A. Modulation of protein kinase CK2 activity by fragments of CFTR encompassing F508 may reflect functional links with cystic fibrosis pathogenesis. Biochemistry 2008, 47, 7925–7936. [Google Scholar] [CrossRef] [Green Version]
- Vlajkovic, S.M.; Housley, G.D.; Greenwood, D.; Thorne, P.R. Evidence for alternative splicing of ecto-ATPase associated with termination of purinergic transmission. Brain Res. Mol. Brain Res. 1999, 73, 85–92. [Google Scholar] [CrossRef]
- De Bourayne, M.; Gallais, Y.; El Ali, Z.; Rousseau, P.; Damiens, M.H.; Cochet, C.; Filhol, O.; Chollet-Martin, S.; Pallardy, M.; Kerdine-Romer, S. Protein kinase CK2 controls T-cell polarization through dendritic cell activation in response to contact sensitizers. J. Leukoc. Biol. 2017, 101, 703–715. [Google Scholar] [CrossRef]
- Cozza, G.; Pinna, L.A.; Moro, S. Kinase CK2 inhibition: an update. Curr. Med. Chem. 2013, 20, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Protein kinase CK2 inhibitors: a patent review. Expert Opin. Ther. Pat. 2012, 22, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Haidar, S.; Meyers, A.; Bollacke, A.; Jose, J. Synthesis and biological evaluation of 2,6-di(furan-3-yl)anthracene-9, 10-dione as an inhibitor of human protein kinase CK2. Pharmazie 2015, 70, 772–776. [Google Scholar] [PubMed]
- Guillon, J.; Le Borgne, M.; Rimbault, C.; Moreau, S.; Savrimoutou, S.; Pinaud, N.; Baratin, S.; Marchivie, M.; Roche, S.; Bollacke, A.; et al. Synthesis and biological evaluation of novel substituted pyrrolo[1,2-a]quinoxaline derivatives as inhibitors of the human protein kinase CK2. Eur. J. Med. Chem. 2013, 65, 205–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alchab, F.; Ettouati, L.; Bouaziz, Z.; Bollacke, A.; Delcros, J.G.; Gertzen, C.G.; Gohlke, H.; Pinaud, N.; Marchivie, M.; Guillon, J.; et al. Synthesis, Biological Evaluation and Molecular Modeling of Substituted Indeno[1,2-b]indoles as Inhibitors of Human Protein Kinase CK2. Pharmaceuticals 2015, 8, 279–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzi, G.J.; Bouaziz, Z.; Winter, E.; Daflon-Yunes, N.; Honorat, M.; Guragossian, N.; Marminon, C.; Valdameri, G.; Bollacke, A.; Guillon, J.; et al. Phenolic indeno[1,2-b]indoles as ABCG2-selective potent and non-toxic inhibitors stimulating basal ATPase activity. Drug Des. Devel. Ther. 2015, 9, 3481–3495. [Google Scholar] [PubMed] [Green Version]
- Hundsdorfer, C.; Hemmerling, H.J.; Gotz, C.; Totzke, F.; Bednarski, P.; Le Borgne, M.; Jose, J. Indeno[1,2-b]indole derivatives as a novel class of potent human protein kinase CK2 inhibitors. Bioorg. Med. Chem. 2012, 20, 2282–2289. [Google Scholar] [CrossRef]
- Hundsdorfer, C.; Hemmerling, H.J.; Hamberger, J.; Le Borgne, M.; Bednarski, P.; Gotz, C.; Totzke, F.; Jose, J. Novel indeno[1,2-b]indoloquinones as inhibitors of the human protein kinase CK2 with antiproliferative activity towards a broad panel of cancer cell lines. Biochem. Biophys. Res. Commun. 2012, 424, 71–75. [Google Scholar] [CrossRef]
- Rongved, P.; Kirsch, G.; Bouaziz, Z.; Jose, J.; Le Borgne, M. Indenoindoles and cyclopentacarbazoles as bioactive compounds: synthesis and biological applications. Eur. J. Med. Chem. 2013, 69, 465–479. [Google Scholar] [CrossRef]
- Hochscherf, J.; Lindenblatt, D.; Witulski, B.; Birus, R.; Aichele, D.; Marminon, C.; Bouaziz, Z.; Le Borgne, M.; Jose, J.; Niefind, K. Unexpected Binding Mode of a Potent Indeno[1,2-b]indole-Type Inhibitor of Protein Kinase CK2 Revealed by Complex Structures with the Catalytic Subunit CK2alpha and Its Paralog CK2alpha’. Pharmaceuticals (Basel Switz.) 2017, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui-Jain, A.; Drygin, D.; Streiner, N.; Chua, P.; Pierre, F.; O’Brien, S.E.; Bliesath, J.; Omori, M.; Huser, N.; Ho, C.; et al. CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res. 2010, 70, 10288–10298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chon, H.J.; Bae, K.J.; Lee, Y.; Kim, J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Front Pharmacol. 2015, 6, 70. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G. The Development of CK2 Inhibitors: From Traditional Pharmacology to in Silico Rational Drug Design. Pharmaceuticals 2017, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haidar, S.; Bouaziz, Z.; Marminon, C.; Laitinen, T.; Poso, A.; Le Borgne, M.; Jose, J. Development of Pharmacophore Model for Indeno[1,2-b]indoles as Human Protein Kinase CK2 Inhibitors and Database Mining. Pharmaceuticals 2017, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Haidar, S.; Aichele, D.; Birus, R.; Hielscher, J.; Laitinen, T.; Poso, A.; Jose, J. In Vitro and in Silico Evaluation of Bikaverin as a Potent Inhibitor of Human Protein Kinase CK2. Molecules 2019, 24, 1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, K.M.; Bui, Q.H.; Tran, T.D.; Huynh, T.N. QSAR modeling on benzo[c]phenanthridine analogues as topoisomerase I inhibitors and anti-cancer agents. Molecules 2012, 17, 5690–5712. [Google Scholar] [CrossRef]
- Tong, W.; Lowis, D.R.; Perkins, R.; Chen, Y.; Welsh, W.J.; Goddette, D.W.; Heritage, T.W.; Sheehan, D.M. Evaluation of quantitative structure-activity relationship methods for large-scale prediction of chemicals binding to the estrogen receptor. J. Chem. Inf. Comput. Sci. 1998, 38, 669–677. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE) 2013. Available online: https://www.chemcomp.com/index.htm (accessed on 22 October 2019).
- Kong, Y.; Xuan, S.; Yan, A. Computational models on quantitative prediction of bioactivity of HIV-1 integrase 3’ processing inhibitors. SAR QSAR Environ. Res. 2014, 25, 729–746. [Google Scholar] [CrossRef]
- Alexander, D.L.; Tropsha, A.; Winkler, D.A. Beware of R(2): Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model 2015, 55, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Tropsha, A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inform. 2010, 29, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. Mol. Divers. 2002, 5, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Golbraikh, A.; Tropsha, A. Predictive QSAR modeling based on diversity sampling of experimental datasets for the training and test set selection. J. Comput. Aid. Mol. Des. 2002, 16, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Jabor Gozzi, G.; Bouaziz, Z.; Winter, E.; Daflon-Yunes, N.; Aichele, D.; Nacereddine, A.; Marminon, C.; Valdameri, G.; Zeinyeh, W.; Bollacke, A.; et al. Converting potent indeno[1,2-b]indole inhibitors of protein kinase CK2 into selective inhibitors of the breast cancer resistance protein ABCG2. J. Med. Chem. 2015, 58, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Olgen, S.; Gotz, C.; Jose, J. Synthesis and biological evaluation of 3-(substituted-benzylidene)-1,3-dihydro-indolin derivatives as human protein kinase CK2 and p60(c-Src) tyrosine kinase inhibitors. Biol. Pharm. Bull. 2007, 30, 715–718. [Google Scholar] [CrossRef] [Green Version]
- Espacenet Patent Search. Available online: https://worldwide.espacenet.com/publicationDetails/originalDocument?CC=WO&NR=2017151625A1&KC=A1&FT=D&ND=5&date=20170908&DB=&locale=en_EP# (accessed on 22 October 2019).
- Gach, K.; Modranka, J.; Szymanski, J.; Pomorska, D.; Krajewska, U.; Mirowski, M.; Janecki, T.; Janecka, A. Anticancer properties of new synthetic hybrid molecules combining naphtho[2,3-b]furan-4,9-dione or benzo[f]indole-4,9-dione motif with phosphonate subunit. Eur. J. Med. Chem. 2016, 120, 51–63. [Google Scholar] [CrossRef]
- Jimenez-Alonso, S.; Guasch, J.; Estevez-Braun, A.; Ratera, I.; Veciana, J.; Ravelo, A.G. Electronic and cytotoxic properties of 2-amino-naphtho[2,3-b]furan-4,9-diones. J. Org. Chem. 2011, 76, 1634–1643. [Google Scholar] [CrossRef]
- Bannwitz, S.; Krane, D.; Vortherms, S.; Kalin, T.; Lindenschmidt, C.; Zahedi Golpayegani, N.; Tentrop, J.; Prinz, H.; Muller, K. Synthesis and structure-activity relationships of lapacho analogues. 2. Modification of the basic naphtho[2,3-b]furan-4,9-dione, redox activation, and suppression of human keratinocyte hyperproliferation by 8-hydroxynaphtho[2,3-b]thiophene-4,9-diones. J. Med. Chem. 2014, 57, 6226–6239. [Google Scholar] [CrossRef]
- RCSB PDB. Available online: https://www.rcsb.org/pdb/explore/explore.do?structureId=3C13 (accessed on 22 October 2019).
- Pauli, I.; dos Santos, R.N.; Rostirolla, D.C.; Martinelli, L.K.; Ducati, R.G.; Timmers, L.F.; Basso, L.A.; Santos, D.S.; Guido, R.V.; Andricopulo, A.D.; et al. Discovery of new inhibitors of Mycobacterium tuberculosis InhA enzyme using virtual screening and a 3D-pharmacophore-based approach. J. Chem. Inf. Model 2013, 53, 2390–2401. [Google Scholar] [CrossRef]
- Gratz, A.; Gotz, C.; Jose, J. A CE-based assay for human protein kinase CK2 activity measurement and inhibitor screening. Electrophoresis 2010, 31, 634–640. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nr. | Chemical Structure | Tested (IC50 nM) pIC50 | Pred. (IC50 nM) pIC50 | Nr. | Chemical Structure | Tested (IC50 nM) pIC50 | Pred. (IC50 nM) pIC50 |
|---|---|---|---|---|---|---|---|
| 4e |  | (1400) 5.8540 | (2745) 5.5610 | 4y |  | (610) 6.2150 | (600) 6.2178 |
| 4f | 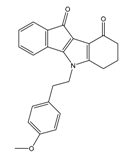 | (4100) 5.3870 | (2050) 5.6888 | 5d | 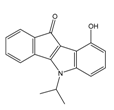 | (2000) 5.6980 | (2490) 5.6058 |
| 4g |  | (360) 6.4440 | (304) 6.5177 | 5g | 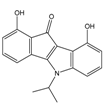 | (560) 6.2520 | (549) 6.2605 |
| 4h | 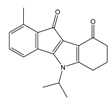 | (110) 6.9580 | (98) 7.0060 | 5h | 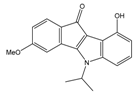 | (480) 6.3180 | (580) 6.2371 |
| 4p | 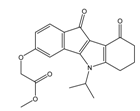 | (1520) 5.8180 | (1490) 5.8394 | 5j | 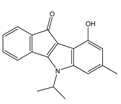 | (1270) 5.8960 | (1060) 5.9747 |
| 4r | 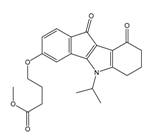 | (520) 6.2840 | (666) 6.1762 | 5k | 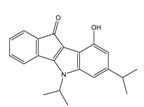 | (1650) 5.8380 | (1230) 5.9210 |
| 4s |  | (1510) 5.8210 | (1285) 5.8909 | 6b | 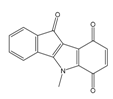 | (1120) 5.950 | (1050) 5.9779 |
| 4v | 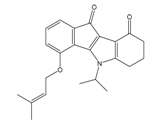 | (25) 7.6020 | (27) 7.5749 | 6d |  | (1350) 5.8690 | (1417) 5.8485 |
| 4w | 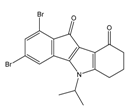 | (110) 6.9580 | (108) 6.9667 | 6f | 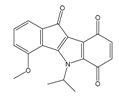 | (870) 6.0600 | (590) 6.2289 |
| 4x |  | (170) 6.7690 | (220) 6.6584 | 6g | 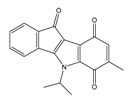 | (430) 6.3660 | (708) 6.1497 |
| Nr. | Chemical Structure | Tested (IC50 nM) pIC50 | Pred. (IC50 nM) pIC50 | Nr. | Chemical Structure | Tested (IC50 nM) pIC50 | Pred. (IC50 nM) pIC50 |
|---|---|---|---|---|---|---|---|
| 4d | 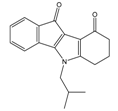 | (1090) 5.9620 | (116) 6.9346 | 5c | 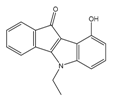 | (2760) 5.559 | (333) 6.4781 |
| 4i | 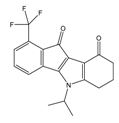 | (210) 6.6770 | (48) 7.3107 | 5f |  | (2930) 5.5330 | (475) 6.3235 |
| 4j |  | (140) 6.8540 | (74) 7.1318 | 6a | 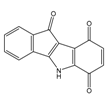 | (1780) 5.749 | (210) 6.6764 |
| 4q |  | (3210) 5.4930 | (201) 6.6959 | 6c | 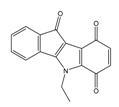 | (1580) 5.801 | (325) 6.4858 |
| 5a |  | (2220) 5.6530 | (560) 6.2513 | 6e | 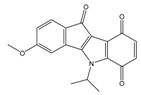 | (4160) 5.3810 | (524) 6.2800 |
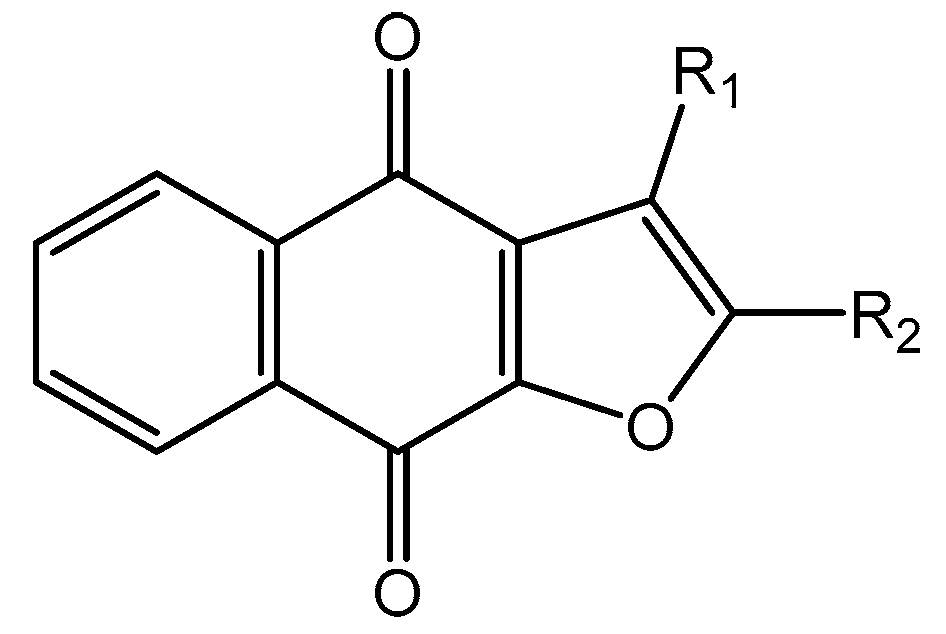
| ZINC Code | Chemical Structure | Pred. (IC50 nM)/pIC50 | S Score | ZINC Code | Chemical Structure | Pred. (IC50 nM)/pIC50 | S Score |
|---|---|---|---|---|---|---|---|
| 01893208 |  | (849) 6.0711 | −7.0598 | 002700665 | 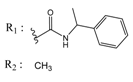 | (118.104) 2.9347 | −6.9187 |
| 37867960 |  | (4650) 5.3323 | −7.0005 | 00449156 | 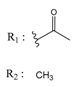 | (71) 7.1470 | −5.8886 |
| 02700659 | 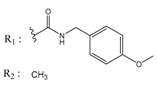 | (292) 6.5347 | −7.0460 | 0079314 | 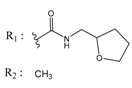 | (61) 7.2195 | −6.8687 |
| 01214105 |  | (2930) 5.5334 | −6.5439 | 04776566 | 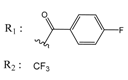 | (46600) 4.3312 | −6.2294 |
| 00082235 |  | (1145) 5.9413 | −6.7446 | 00720964 |  | (19) 7.7102 | −6.6344 |
| 00978111 |  | (245500) 3.6018 | −6.0406 | 10034949 | 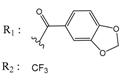 | (21) 7.6758 | −6.7250 |
| 53166374 | 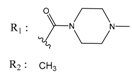 | (82) 7.0870 | −6.4533 | 10034950 | 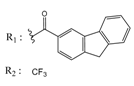 | (35.105) 2.4537 | −6.2979 |
| 02700667 | 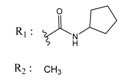 | (8550) 5.0684 | −6.6746 | 00065262 | 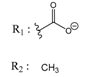 | (11) 7.9146 | −5.9976 |
| 01236034 |  | (0.08) 10.0672 | −6.4042 | 01214623 |  | (16.106) 1.7974 | −6.4351 |
| 01236036 | 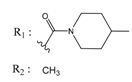 | (3900) 5.4007 | −6.3467 | 05776381 | 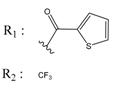 | (8550) 5.0608 | −6.2530 |
| 01236042 | 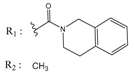 | (5700) 5.2432 | −6.8031 | 10034955 | 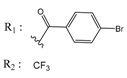 | (108000) 3.9677 | −6.1499 |
| 01236043 |  | (28500) 4.5408 | −6.8501 | 00721112 | 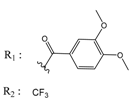 | (450) 6.333 | −6.6130 |
| 01236046 |  | (9.105) 3.0440 | −7.0466 |
| ZINC Code | Chemical Structure | % Inhibitory a (IC50 b) nM | Pred. (IC50 nM)/pIC50 |
|---|---|---|---|
| 01893208 | 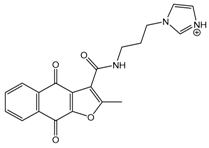 | 39 (12000) c | (845) 6.0711 |
| 37867960 | 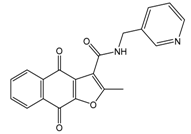 | 35 (14000) c | (4650) 5.3323 |
| 00082235 |  | 75 (2330) | (1145) 5.9413 |
| 01236034 |  | 45 (11000) c | (0.08) 10.0672 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haidar, S.; Marminon, C.; Aichele, D.; Nacereddine, A.; Zeinyeh, W.; Bouzina, A.; Berredjem, M.; Ettouati, L.; Bouaziz, Z.; Le Borgne, M.; et al. QSAR Model of Indeno[1,2-b]indole Derivatives and Identification of N-isopentyl-2-methyl-4,9-dioxo-4,9-Dihydronaphtho[2,3-b]furan-3-carboxamide as a Potent CK2 Inhibitor. Molecules 2020, 25, 97. https://doi.org/10.3390/molecules25010097
Haidar S, Marminon C, Aichele D, Nacereddine A, Zeinyeh W, Bouzina A, Berredjem M, Ettouati L, Bouaziz Z, Le Borgne M, et al. QSAR Model of Indeno[1,2-b]indole Derivatives and Identification of N-isopentyl-2-methyl-4,9-dioxo-4,9-Dihydronaphtho[2,3-b]furan-3-carboxamide as a Potent CK2 Inhibitor. Molecules. 2020; 25(1):97. https://doi.org/10.3390/molecules25010097
Chicago/Turabian StyleHaidar, Samer, Christelle Marminon, Dagmar Aichele, Abdelhamid Nacereddine, Wael Zeinyeh, Abdeslem Bouzina, Malika Berredjem, Laurent Ettouati, Zouhair Bouaziz, Marc Le Borgne, and et al. 2020. "QSAR Model of Indeno[1,2-b]indole Derivatives and Identification of N-isopentyl-2-methyl-4,9-dioxo-4,9-Dihydronaphtho[2,3-b]furan-3-carboxamide as a Potent CK2 Inhibitor" Molecules 25, no. 1: 97. https://doi.org/10.3390/molecules25010097