PredPSD: A Gradient Tree Boosting Approach for Single-Stranded and Double-Stranded DNA Binding Protein Prediction


Abstract
:1. Introduction
2. Results
2.1. Analysis of mRMR Results
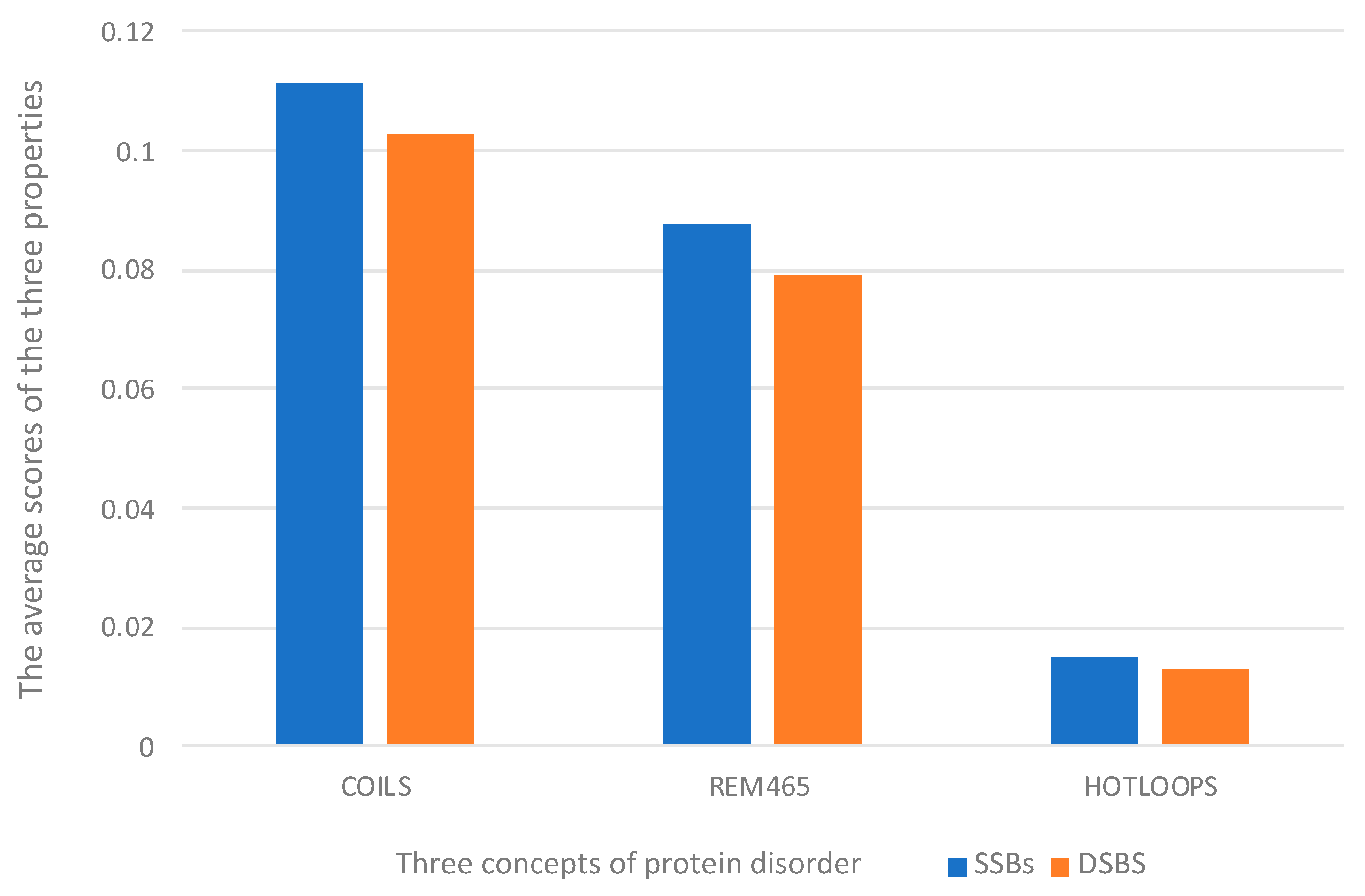
2.2. Feature Extraction Results Analysis
2.3. Predictive Performance of Features
2.4. Comparison with Previous Work
2.5. Comparative Analysis of Independent Test Results
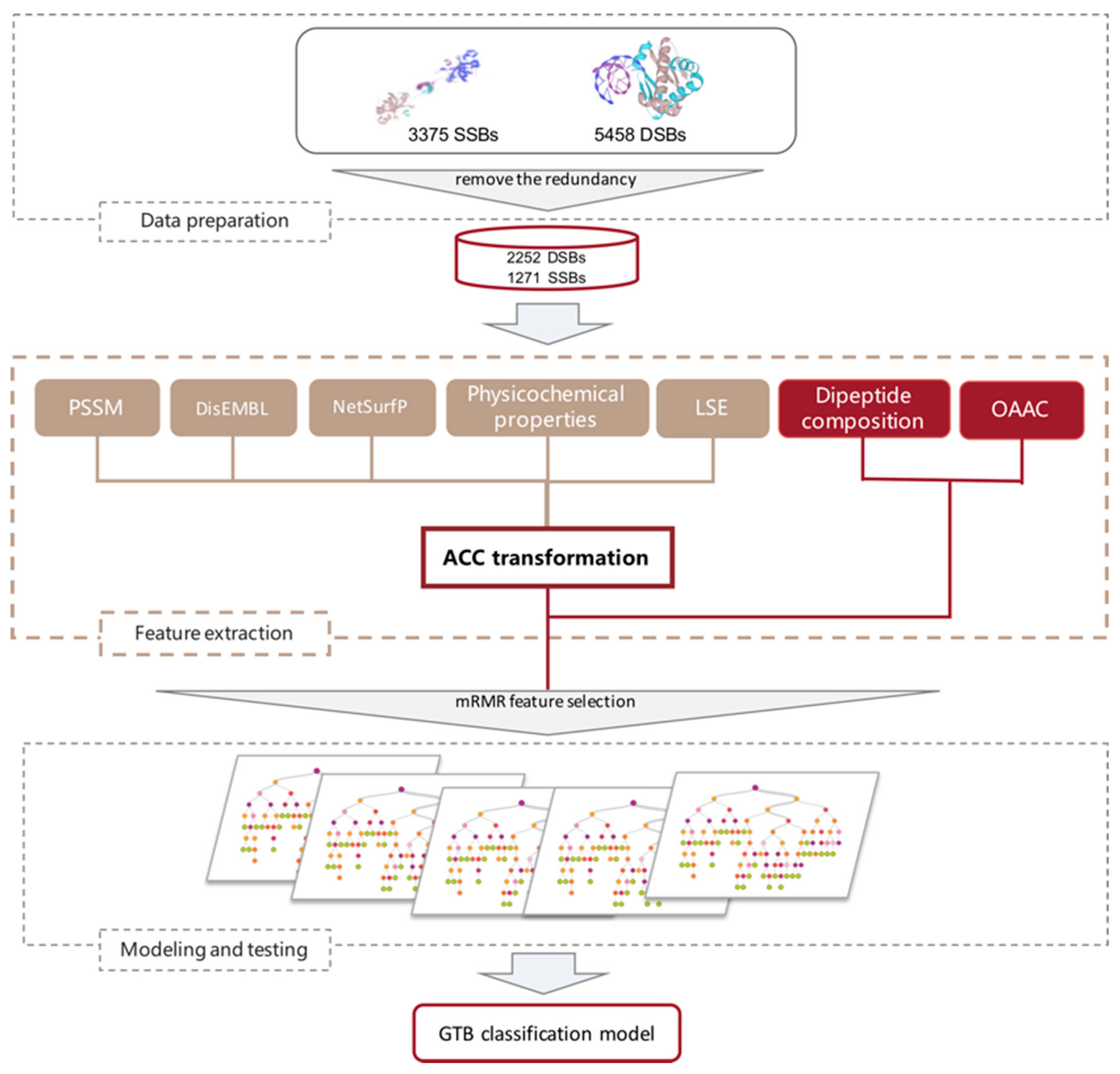
3. Materials and Methods
3.1. Datasets
3.2. Feature Extraction
3.2.1. Local Structural Entropy (LSE)
3.2.2. NetSurfP
3.2.3. DisEMBL
3.2.4. Overall Amino Acid Composition (OAAC)
3.2.5. Dipeptide Composition
3.2.6. PSSM
3.2.7. Physicochemical Properties
3.3. Feature Transformation
3.4. Feature Selection
3.5. Classification Model and Performance Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Laetitia, A.; Audrey, O.; Isabelle, M.B.; Anne-Lise, S.; Chantal, G.; Bernard, M.; Patrice, P.; Jean-Pierre, C. Role of the single-stranded DNA-binding protein SsbB in pneumococcal transformation: Maintenance of a reservoir for genetic plasticity. PLoS Genet. 2011, 7, e1002156. [Google Scholar]
- Edsö, J.R. Single- and double-stranded DNA binding proteins act in concert to conserve a telomeric DNA core sequence. Genome Integr. 2011, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Richard, D.J.; Emma, B.; Liza, C.; Wadsworth, R.I.M.; Kienan, S.; Sharma, G.G.; Nicolette, M.L.; Sergie, T.; Mcilwraith, M.J.; Pandita, R.K. Single-stranded DNA-binding protein hSSB1 is critical for genomic stability. Nature 2008, 453, 677–681. [Google Scholar] [CrossRef]
- Olga, S.N.; Lue, N.F. Telomere DNA recognition in Saccharomycotina yeast: Potential lessons for the co-evolution of ssDNA and dsDNA-binding proteins and their target sites. Front. Genet. 2015, 6, 162. [Google Scholar]
- Croft, L.V.; Bolderson, E.; Adams, M.N.; El-Kamand, S.; Kariawasam, R.; Cubeddu, L.; Gamsjaeger, R.; Richard, D.J. Human single-stranded DNA binding protein 1 (hSSB1, OBFC2B), a critical component of the DNA damage response. Semin. Cell Dev. Biol. 2018, 86, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Emmanuelle, D.; Amélie, H.M.; Giuseppe, B. Single-stranded DNA binding proteins unwind the newly synthesized double-stranded DNA of model miniforks. Biochemistry 2011, 50, 932–944. [Google Scholar]
- Doan, P.H.; Pitter, D.R.G.; Kocher, A.; Wilson, J.N.; Iii, G. A New Design Strategy and Diagnostic to Tailor the DNA-Binding Mechanism of Small Organic Molecules and Drugs. ACS Chem. Biol. 2016, 11, 3202–3213. [Google Scholar] [CrossRef]
- Dasgupta, D.; Howard, F.B.; Sasisekharan, V.; Miles, H.T. Drug-DNA binding specificity: Binding of netropsin and distamycin to poly(d2NH2A-dT). Biopolymers 2010, 30, 223–227. [Google Scholar] [CrossRef]
- Gao, Y.G.; Priebe, W.; Wang, A.H. Substitutions at C2’ of daunosamine in the anticancer drug daunorubicin alter its DNA-binding sequence specificity. Eur. J. Biochem. 2010, 240, 331–335. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, W.; Zou, B.; Wang, J.; Deng, Y.; Deng, L. DrugCombDB: A comprehensive database of drug combinations toward the discovery of combinatorial therapy. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Wang, W.; Liu, J.; Xiong, Y.; Zhu, L.; Zhou, X. Analysis and classification of DNA-binding sites in single-stranded and double-stranded DNA-binding proteins using protein information. IET Syst. Biol. 2014, 8, 176. [Google Scholar] [CrossRef] [Green Version]
- Tchurikov, N.A.; Fedoseeva, D.M.; Sosin, D.V.; Snezhkina, A.V.; Melnikova, N.V.; Kudryavtseva, A.V.; Kravatsky, Y.V.; Kretova, O.V. Hot spots of DNA double-strand breaks and genomic contacts of human rDNA units are involved in epigenetic regulation. J. Mol. Cell Biol. 2015, 7, 366–382. [Google Scholar] [CrossRef]
- Zhu, X.; Ericksen, S.S.; Mitchell, J.C. DBSI: DNA-binding site identifier. Nucleic Acids Res. 2013, 41, e160. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Terribilini, M.; Wu, F.; Jernigan, R.L.; Dobbs, D.; Honavar, V. Predicting DNA-binding sites of proteins from amino acid sequence. BMC Bioinform. 2006, 7, 262. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, R.; Ahmad, S.; Gromiha, M.M. Novel approach for selecting the best predictor for identifying the binding sites in DNA binding proteins. Nucleic Acids Res. 2013, 41, 7606–7614. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Wei, L.; Zou, Q. A Review of DNA-binding Proteins Prediction Methods. Curr. Bioinform. 2019, 14, 246–254. [Google Scholar] [CrossRef]
- Wei, L.; Tang, J.; Zou, Q. Local-DPP: An improved DNA-binding protein prediction method by exploring local evolutionary information. Inf. Sci. 2017, 384, 135–144. [Google Scholar] [CrossRef]
- Song, L.; Li, D.; Zeng, X.; Wu, Y.; Guo, L.; Zou, Q. nDNA-prot: Identification of DNA-binding proteins based on unbalanced classification. BMC Bioinform. 2014, 15, 298. [Google Scholar] [CrossRef] [Green Version]
- Shula, S.; Gershon, E.; Yael, M.G. From face to interface recognition: A differential geometric approach to distinguish DNA from RNA binding surfaces. Nucleic Acids Res. 2011, 39, 7390. [Google Scholar]
- Nimrod, G.; Szilágyi, A.; Leslie, C.; Ben-Tal, N. Identification of DNA-binding proteins using structural, electrostatic and evolutionary features. J. Mol. Biol. 2009, 387, 1040–1053. [Google Scholar] [CrossRef] [Green Version]
- Szabóová, A. Prediction of DNA-binding propensity of proteins by the ball-histogram method using automatic template search. BMC Bioinform. 2012, 13, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Wan, S.; Ju, Y.; Tang, J.; Zeng, X. Pretata: Predicting TATA binding proteins with novel features and dimensionality reduction strategy. BMC Syst. Biol. 2016, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G. DNA-Binding Specificities of Human Transcription Factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Wei-Zhong, L.; Jian-An, F.; Xuan, X.; Kuo-Chen, C. iDNA-Prot: Identification of DNA binding proteins using random forest with grey model. PLoS ONE 2011, 6, e24756. [Google Scholar]
- Morgan, H.; Estibeiro, P.; Wear, M.; Heinemann, U.; Cubeddu, L.; Gallagher, M.; Sadler, P.J.; Walkinshaw, M.D. Sequence specificity of single-stranded DNA-binding proteins: A novel DNA microarray approach. Nucleic Acids Res. 2007, 35, e75. [Google Scholar] [CrossRef] [Green Version]
- Kresten, L.L.; Best, R.B.; Depristo, M.A.; Dobson, C.M.; Michele, V. Simultaneous determination of protein structure and dynamics. Nature 2005, 433, 128–132. [Google Scholar]
- Wang, W.; Liu, J.; Zhou, X. Identification of single-stranded and double-stranded dna binding proteins based on protein structure. Bioinformatics 2013, 15, S4. [Google Scholar] [CrossRef] [Green Version]
- Francesco, R.; Bonham, A.J.; Mason, A.C.; Reich, N.O.; Plaxco, K.W. Reagentless, electrochemical approach for the specific detection of double- and single-stranded DNA binding proteins. Anal. Chem. 2009, 81, 1608–1614. [Google Scholar]
- Cai, Y.D.; Doig, A.J. Prediction of Saccharomyces cerevisiae protein functional class from functional domain composition. Bioinformatics 2004, 20, 1292–1300. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Wang, F.; Lei, M.; Lue, N. A proposed OB-fold with a protein-interaction surface in Candida albicans telomerase protein Est3. Nat. Struct. Mol. Biol. 2008, 15, 985. [Google Scholar]
- Zasedateleva, O.A.; Mikheikin, A.L.; Turygin, A.Y.; Prokopenko, D.V.; Chudinov, A.V.; Belobritskaya, E.E.; Chechetkin, V.R.; Zasedatelev, A.S. Gel-based oligonucleotide microarray approach to analyze protein-ssDNA binding specificity. Nucleic Acids Res. 2008, 36, e61. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Liu, J.; Sun, L. Surface shapes and surrounding environment analysis of single- and double-stranded DNA-binding proteins in protein-DNA interface. Proteins-Struct. Funct. Bioinform. 2016, 84, 979–989. [Google Scholar] [CrossRef]
- Remo, R.; West, S.M.; Alona, S.; Peng, L.; Mann, R.S.; Barry, H. The role of DNA shape in protein-DNA recognition. Nature 2009, 461, 1248–1253. [Google Scholar]
- Rim, K.I.W.; Cubeddu, L.; Blankenfeldt, W.; Naismith, J.H.; White, M.F. Insights into ssDNA recognition by the OB fold from a structural and thermodynamic study of Sulfolobus SSB protein. EMBO J. 2014, 22, 2561–2570. [Google Scholar]
- Yi, X.; Juan, L.; Dong-Qing, W. An accurate feature-based method for identifying DNA-binding residues on protein surfaces. Proteins-Struct. Funct. Bioinform. 2011, 79, 509–517. 79 2011, 79, 509–517. [Google Scholar]
- Taisuke, W.; Yoshiaki, K.; Yutaro, K.; Noriko, N.; Seiki, K.; Ryoji, M. Structure of RecJ exonuclease defines its specificity for single-stranded DNA. J. Biol. Chem. 2010, 285, 9762–9769. [Google Scholar]
- Wang, W.; Sun, L.; Zhang, S.; Zhang, H.; Shi, J.; Xu, T.; Li, K. Analysis and prediction of single-stranded and double-stranded DNA binding proteins based on protein sequences. BMC Bioinform. 2017, 18, 300. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Jensen, L.J.; Diella, F.; Bork, P.; Gibson, T.J.; Russell, R.B. Protein Disorder Prediction: Implications for Structural Proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef] [Green Version]
- Dickey, T.H.; Altschuler, S.E.; Wuttke, D.S. Single-stranded DNA-binding proteins: Multiple domains for multiple functions. Structure 2013, 21, 1074–1084. [Google Scholar] [CrossRef] [Green Version]
- Li, W. Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences. Bioinformatics 2006, 22, 1658. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.J.; Feng, C.Q.; Lai, H.Y.; Chen, W.; Lin, H. Predicting protein structural classes for low-similarity sequences by evaluating different features. Knowl. Based Syst. 2019, 163, 787–793. [Google Scholar] [CrossRef]
- Tan, J.X.; Li, S.H.; Zhang, Z.M.; Chen, C.X.; Chen, W.; Tang, H.; Lin, H. Identification of hormone binding proteins based on machine learning methods. Math. Biosci. Eng. 2019, 16, 2466–2480. [Google Scholar] [CrossRef]
- Chan, C.H.; Liang, H.K.; Hsiao, N.W.; Ko, M.T.; Lyu, P.C.; Hwang, J.K. Relationship between local structural entropy and protein thermostabilty. Proteins Struct. Funct. Bioinform. 2004, 57, 684–691. [Google Scholar] [CrossRef]
- Deng, L.; Guan, J.; Wei, X.; Yi, Y.; Zhang, Q.C.; Zhou, S. Boosting prediction performance of protein-protein interaction hot spots by using structural neighborhood properties. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2013, 20, 878–891. [Google Scholar] [CrossRef] [Green Version]
- Agnew, H.D.; Coppock, M.B.; Idso, M.N.; Lai, B.T.; Liang, J.; McCarthy-Torrens, A.M.; Warren, C.M.; Heath, J.R. Protein-catalyzed capture agents. Chem. Rev. 2019, 119, 9950–9970. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, J.; Zhao, M.; Li, Q. Predicting linear B-cell epitopes by using sequence-derived structural and physicochemical features. Int. J. Data Min. Bioinform. 2012, 6, 557–569. [Google Scholar] [CrossRef]
- Kuang, L.; Yan, X.; Tan, X.; Li, S.; Yang, X. Predicting Taxi Demand Based on 3D Convolutional Neural Network and Multi-task Learning. Remote Sens. 2019, 11, 1265. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.P. Prediction of the subcellular location of prokaryotic proteins based on a new representation of the amino acid composition. Biopolymers 2015, 58, 491–499. [Google Scholar] [CrossRef]
- Garg, A.; Raghava, G.P. ESLpred2: Improved method for predicting subcellular localization of eukaryotic proteins. BMC Bioinform. 2008, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Zhao, Y.W.; Zou, P.; Zhang, C.M.; Chen, R.; Huang, P.; Lin, H. HBPred: A tool to identify growth hormone-binding proteins. Int. J. Biol. Sci. 2018, 14, 957–964. [Google Scholar] [CrossRef]
- Hao, L.; Qian-Zhong, L. Using pseudo amino acid composition to predict protein structural class: Approached by incorporating 400 dipeptide components. J. Comput. Chem. 2010, 28, 1463–1466. [Google Scholar]
- Ahmad, S.; Sarai, A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinform. 2005, 6, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST—A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Zhu, X.J.; Huang, J.; Ding, H.; Lin, H. A brief survey of machine learning methods in protein sub-Golgi localization. Curr. Bioinform. 2019, 14, 234–240. [Google Scholar] [CrossRef]
- Tang, H.; Chen, W.; Lin, H. Identification of immunoglobulins using Chou’s pseudo amino acid composition with feature selection technique. Mol. Biosyst. 2016, 12, 1269–1275. [Google Scholar] [CrossRef]
- Huang, H.L.; Lin, I.C.; Liou, Y.F.; Tsai, C.T.; Hsu, K.T.; Huang, W.L.; Ho, S.J.; Ho, S.Y. Predicting and analyzing DNA-binding domains using a systematic approach to identifying a set of informative physicochemical and biochemical properties. BMC Bioinform. 2011, 12, S47. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, S.; Kanehisa, M. AAindex: Amino Acid index database. Nucleic Acids Res. 1999, 27, 368–369. [Google Scholar] [CrossRef]
- Dong, Q.; Zhou, S.; Guan, J. A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics 2009, 25, 2655–2662. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Liu, B. A Review on the Recent Developments of Sequence-based Protein Feature Extraction Methods. Curr. Bioinform. 2019, 14, 190–199. [Google Scholar] [CrossRef]
- Hanchuan, P.; Fuhui, L.; Chris, D. Feature selection based on mutual information: Criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef]
- Wang, S.P.; Zhang, Q.; Lu, J.; Cai, Y.D. Analysis and Prediction of Nitrated Tyrosine Sites with the mRMR Method and Support Vector Machine Algorithm. Curr. Bioinform. 2018, 13, 3–13. [Google Scholar] [CrossRef]
- Friedman, J.H. Stochastic gradient boosting. Comput. Stat. Data Anal. 2002, 38, 367–378. [Google Scholar] [CrossRef]
- Hoque, M.T.; Chetty, M.; Lewis, A.; Sattar, A. Twin Removal in Genetic Algorithms for Protein Structure Prediction Using Low-Resolution Model. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 8, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Tang, Y.; Chao, F.; Chen, Z.; Lei, D. PredRBR: Accurate Prediction of RNA-Binding Residues in proteins using Gradient Tree Boosting. In Proceedings of the IEEE International Conference on Bioinformatics & Biomedicine, Shenzhen, China, 15–18 December 2016. [Google Scholar]
- He, T.; Heidemeyer, M.; Ban, F.; Cherkasov, A.; Ester, M. SimBoost: A read-across approach for predicting drug–target binding affinities using gradient boosting machines. J. Cheminform. 2017, 9, 24. [Google Scholar] [CrossRef]
- Li, Y.; Niu, M.; Zou, Q. ELM-MHC: An improved MHC Identification method with Extreme Learning Machine Algorithm. J. Proteome Res. 2019, 18, 1392–1401. [Google Scholar] [CrossRef]
- Dou, K.; Guo, B.; Kuang, L. A privacy-preserving multimedia recommendation in the context of social network based on weighted noise injection. Multimed. Tools Appl. 2019, 78, 26907–26926. [Google Scholar] [CrossRef]
- Fan, C.; Liu, D.; Huang, R.; Chen, Z.; Deng, L. PredRSA: A gradient boosted regression trees approach for predicting protein solvent accessibility. BMC Bioinform. 2016, 17 (Suppl. 1), 8. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Wang, Z.; Zhan, W.; Deng, L. Computational identification of binding energy hot spots in protein-RNA complexes using an ensemble approach. Bioinformatics 2018, 34, 1473–1480. [Google Scholar] [CrossRef]
- Wen, Z.; Hua, Z.; Luo, L.; Liu, Q.; Wu, W.; Xiao, W. Predicting potential side effects of drugs by recommender methods and ensemble learning. Neurocomputing 2016, 173, 979–987. [Google Scholar]
- Deng, L.; Li, W.; Zhang, J. LDAH2V: Exploring meta-paths across multiple networks for lncRNA-disease association prediction. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Features | Accuracy | SN | SP | AUC | MCC | F1 |
|---|---|---|---|---|---|---|
| PSSM | 0.913 | 0.814 | 0.968 | 0.968 | 0.809 | 0.870 |
| AAindex | 0.759 | 0.514 | 0.899 | 0.810 | 0.457 | 0.604 |
| OAAC | 0.778 | 0.572 | 0.895 | 0.843 | 0.503 | 0.649 |
| NetSurfP | 0.874 | 0.780 | 0.927 | 0.938 | 0.723 | 0.817 |
| LSE | 0.668 | 0.430 | 0.782 | 0.646 | 0.219 | 0.456 |
| Dipeptide | 0.838 | 0.645 | 0.948 | 0.912 | 0.644 | 0.741 |
| DisEMBL | 0.682 | 0.421 | 0.818 | 0.670 | 0.258 | 0.477 |
| Features | Accuracy | SN | SP | AUC | MCC | F1 |
|---|---|---|---|---|---|---|
| PredPSD | 0.912 | 0.784 | 0.975 | 0.956 | 0.799 | 0.854 |
| Wang 2017_SVM | 0.855 | 0.585 | 0.972 | 0.927 | 0.643 | 0.709 |
| Wang 2017_RF | 0.784 | 0.578 | 0.902 | 0.827 | 0.518 | 0.661 |
| Features | Accuracy | SN | SP | AUC | MCC | F1 |
|---|---|---|---|---|---|---|
| PredPSD | 0.770 | 0.512 | 0.855 | 0.708 | 0.373 | 0.525 |
| Wang 2017_SVM | 0.703 | 0.366 | 0.814 | 0.692 | 0.185 | 0.380 |
| Wang 2017_RF | 0.721 | 0.341 | 0.847 | 0.620 | 0.203 | 0.378 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, C.; Wang, T.; Yang, W.; Deng, L. PredPSD: A Gradient Tree Boosting Approach for Single-Stranded and Double-Stranded DNA Binding Protein Prediction. Molecules 2020, 25, 98. https://doi.org/10.3390/molecules25010098
Tan C, Wang T, Yang W, Deng L. PredPSD: A Gradient Tree Boosting Approach for Single-Stranded and Double-Stranded DNA Binding Protein Prediction. Molecules. 2020; 25(1):98. https://doi.org/10.3390/molecules25010098
Chicago/Turabian StyleTan, Changgeng, Tong Wang, Wenyi Yang, and Lei Deng. 2020. "PredPSD: A Gradient Tree Boosting Approach for Single-Stranded and Double-Stranded DNA Binding Protein Prediction" Molecules 25, no. 1: 98. https://doi.org/10.3390/molecules25010098