3.5. Chemistry
All reagents and solvents were purchased from standard commercial sources and were of analytical grade. All synthetic compounds described in this study were checked with analytical TLC (Macherey−Nagel precoated F254 aluminum plates, Düren, Germany), visualized under UV light at 254 nm and purified by column chromatography (CC) on a Reveleris X2 (Grace, BÜCHI, Flawil, Switzerland) automated flash unit. All final compounds and some intermediates were measured with Varian Mercury 300/75 MHz (Palo Alto, CA, USA) or a Bruker AVANCE (Fällanden, Zürich, Switzerland) Neo
® 400/100 MHz spectrometer at 298.15 K using tetramethylsilane (TMS) as an internal standard. The analysis and confirmation of the final compounds were conducted with
1H,
13C, HSQC and HMBC NMR spectral data (
Supplementary Materials). High-resolution mass spectrometry was performed on a Waters LCT Premier XE
TM (Waters, Zellik, Belgium) time-of-flight (TOF) mass spectrometer equipped with a standard electrospray ionization (ESI) and modular LockSpray
TM interface (Waters, Zellik, Belgium). The purity of the tested compounds was determined by LC-MS analysis using a Waters AutoPurification system equipped with a Waters Cortecs C18 column (2.7 μm, 100 × 4.6 mm), as was a gradient system of formic acid in H
2O (0.2%,
v/v)/MeCN with a gradient of 95:5 to 0:100 in 6.5 min at a flow rate of 1.44 mL/min.
General procedure A: Synthesis of biphenyl ether aldehyde building blocks
. According to a literature report [
26], hydroxyphenylacetic ester derivatives (1.0 eq), phenylboronic acid derivatives (3.0 eq.), Cu(OAc)
2 (2.0 eq.), 4Å molecular sieves (0.18 g/mmol ester) and pyridine (3.0 eq.) in 1,2-dichloroethane (6.0 mL/mmol ester) afforded the biphenyletheracetic ester intermediates. To a solution of the biphenyletheracetic ester intermediates (1.0 eq.) in dry tetrahydrofuran (THF) (6.0 mL/mmol ester intermediate) was added LiAlH
4 (2.0 eq.) at 0 °C under N
2 atmosphere, and the resulting mixture was then stirred at room temperature for 1 h [
23]. After complete consumption of the starting material, the reaction mixture was quenched with aq. Na
+/K
+ tartrate solution (5.0 mL/mmol LiAlH
4), and the mixture was stirred at room temperature overnight and then filtered. The collected filtrate was dried and concentrated to afford a crude alcohol intermediate, which was oxidized by PCC (2.0 eq.) for 2 h in dichloromethane (DCM) (5.0 mL/mmol PCC) [
24,
25]. The reaction mixture was filtered through a short silica column. The collected filtrate was concentrated in vacuo and used in the next step without any additional purification.
General procedure B: Synthesis of final compounds
. To a solution of aldehyde intermediates (1.0 eq.) and
28 (1.0 eq.) in 1,2-dichloroethane (33 mL/mmol aldehyde) was added sodium triacetoxyborohydride (2.0 eq.). The resulting mixture was stirred at room temperature for overnight to afford the BOM-protected intermediate [
14], which was deprotected with TFA (17 mL/mmol aldehyde) in the presence of triethylsilane [
29] (17 mL/mmol aldehyde) at 73 °C for 4 h. After cooling down to room temperature, the reaction mixture was evaporated in vacuo to remove TFA, and the residue was adjusted to pH 6 with 1N aq. HCl. Purification of the resulting mixture by column chromatography gave the desired compounds.
2-([1,1’-Biphenyl]-4-yl)acetaldehyde (6). To a solution of methyl 2-([1,1’-biphenyl]-4-yl)acetate (0.3 g, 1.3 mmol) in dry THF (7.8 mL) was added LiAlH4 (0.10 g, 2.7 mmol) to give alcohol intermediate, which was oxidized with PCC (0.56 g, 2.6 mmol) in DCM (13.0 mL) to yield aldehyde 6 (C14H12O, 0.22 g, 1.1 mmol).
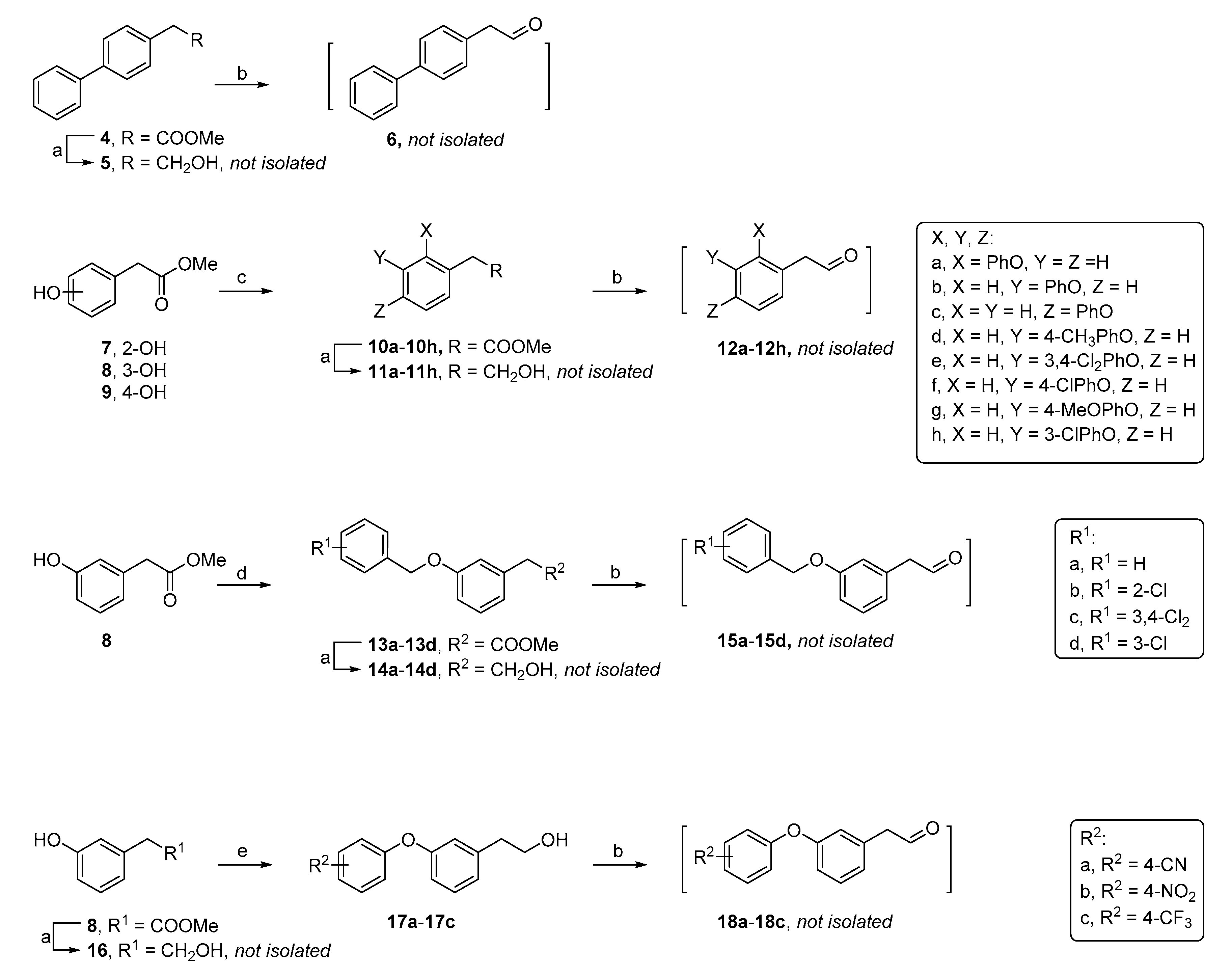
2-(2-Phenoxyphenyl)acetaldehyde (12a). Following the general procedure A, methyl 2-(2-hydroxyphenyl)acetate (1.1 g, 8.1 mmol), phenylboronic acid (2.9 g, 24 mmol), Cu(OAc)2 (2.9 g, 16 mmol), 4Å molecular sieves (1.5 g) and pyridine (1.9 mL, 24 mmol) in 1,2-dichloroethane (49 mL) afforded the ester intermediate methyl 2-(2-phenoxyphenyl)acetate 10a (eluent system: 10% ethylacetate in petroleum ether, C15H14O3, 0.40 g, 1.6 mmol, 21% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 3 H, OCH3), 3.72 (s, 2 H, CH2), 6.90 (dd, J = 8.1, 1.0 Hz, 1 H, Ph), 6.95–7.01 (m, 2 H, Ph), 7.06–7.15 (m, 2 H, Ph), 7.21–7.37 (m, 4 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 35.6 (1 C, CH2), 51.8 (1 C, OCH3), 118.3 (2 C, Ph), 118.8 (1 C, Ph), 123.0 (1 C, Ph), 123.6 (1 C, Ph), 125.8 (1 C, Ph), 128.6 (1 C, Ph), 129.6 (2 C, Ph), 131.4 (1 C, Ph), 155.0 (1 C, Ph), 157.2 (1 C, Ph), 171.7 (1 C, CO). Then, 10a (0.20 g, 0.83 mmol) was treated with LiAlH4 (63 mg, 1.7 mmol) in dry THF (5.0 mL) to give alcohol intermediate, which was oxidized with PCC (0.34 g, 1.6 mmol) in DCM (8.0 mL) to yield aldehyde 12a (C14H12O2, 0.15 g, 0.70 mmol).
2-(3-Phenoxyphenyl)acetaldehyde (12b). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (1.1 g, 8.1 mmol), phenylboronic acid (2.9 g, 24 mmol), Cu(OAc)2 (2.9 g, 16 mmol), 4Å molecular sieves (1.5 g) and pyridine (1.9 mL, 24 mmol) in 1,2-dichloroethane (49 mL) afforded the ester intermediate methyl 2-(3-phenoxyphenyl)acetate 10b (eluent system: 10% ethylacetate in petroleum ether, C15H14O3, 0.80 g, 3.3 mmol, 41% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.62 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.90 - 6.95 (m, 1 H, Ph), 6.98 (t, J = 2.1 Hz, 1 H, Ph), 7.01–7.07 (m, 3 H, Ph), 7.13 (tt, J = 7.4, 1.1 Hz, 1 H, Ph), 7.26–7.40 (m, 3 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.9 (1 C, CH2), 52.0 (1 C, OCH3), 117.3 (1 C, Ph), 119.0 (2 C, Ph), 119.7 (1 C, Ph), 123.3 (1 C, Ph), 124.0 (1 C, Ph), 129.7 (3 C, Ph), 135.7 (1 C, Ph), 156.9 (1 C, Ph), 157.4 (1 C, Ph), 171.6 (1 C, CO). Then, 10b (0.20 g, 0.83 mmol) was treated with LiAlH4 (63 mg, 1.7 mmol) in dry THF (5.0 mL) to give alcohol intermediate, which was oxidized with PCC (0.35 g, 1.6 mmol) in DCM (8.0 mL) to yield aldehyde 12b (C14H12O2, 0.15 g, 0.71 mmol).
2-(4-Phenoxyphenyl)acetaldehyde (12c). Following the general procedure A, methyl 2-(4-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), phenylboronic acid (1.6 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.70 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(4-phenoxyphenyl)acetate 10c (eluent system: 10% ethylacetate in petroleum ether, C15H14O3, 0.66 g, 2.7 mmol, 62% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.72 (s, 3 H, OCH3), 6.95–7.06 (m, 4 H, Ph), 7.08–7.15 (m, 1 H, Ph), 7.23–7.29 (m, 2 H, Ph), 7.31–7.39 (m, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.3 (1 C, CH2), 52.0 (1 C, OCH3), 118.8 (4 C, Ph), 123.2 (1 C, Ph), 128.7 (1 C, Ph), 129.6 (2 C, Ph), 130.5 (2 C, Ph), 156.3 (1 C, Ph), 157.0 (1 C, Ph), 172.0 (1 C, CO). Then, 10c (0.20 g, 0.83 mmol) was treated with LiAlH4 (63 mg, 1.6 mmol) in dry THF (5.0 mL) to give alcohol intermediate, which was oxidized with PCC (0.3 g, 1.4 mmol) in DCM (7.0 mL) to yield aldehyde 12c (C14H12O2, 0.13 g, 0.62 mmol).
2-(3-(p-Tolyloxy)phenyl)acetaldehyde (12d). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60g, 4.4 mmol), p-tolylboronic acid (1.8 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(p-tolyloxy)phenyl)acetate 10d (eluent system: 10% ethylacetate in petroleum ether, C16H16O3, 0.34 g, 1.3 mmol, 30% yield). 1H NMR (300 MHz, CDCl3) δ ppm 2.37 (s, 3 H, CH3), 3.62 (s, 2 H, CH2), 3.72 (s, 3 H, OCH3), 6.89–6.99 (m, 4 H, Ph), 7.00–7.05 (m, 1 H, Ph), 7.14–7.21 (m, 2 H, Ph), 7.25–7.32 (m, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 20.6 (1 C, CH3), 40.9 (1 C, CH2), 51.9 (1 C, OCH3), 116.7 (1 C, Ph), 119.1 (3 C, Ph), 123.5 (1 C, Ph), 129.6 (1 C, Ph), 130.1 (2 C, Ph), 132.9 (1 C, Ph), 135.6 (1 C, Ph), 154.4 (1 C, Ph), 157.9 (1 C, Ph), 171.6 (1 C, CO). Then, 10d (0.20 g, 0.78 mmol) was treated with LiAlH4 (59 mg, 1.6 mmol) in dry THF (4.7 mL) to give alcohol intermediate, which was oxidized with PCC (0.28 g, 1.3 mmol) in DCM (6.5 mL) to yield aldehyde 12d (C15H14O2, 0.13 g, 0.58 mmol).
2-(3-(3,4-Dichlorophenoxy)phenyl)acetaldehyde (12e). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), (3,4-dichlorophenyl)boronic acid (2.5 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(3,4-dichlorophenoxy)phenyl)acetate 10e (eluent system: 10% ethylacetate in petroleum ether, C15H12Cl2O3, 0.73 g, 2.3 mmol, 53% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.86 (dd, J = 8.9, 2.8 Hz, 1 H, Ph), 6.90–6.99 (m, 2 H, Ph), 7.06–7.12 (m, 2 H, Ph), 7.31 (d, J = 7.9 Hz, 1 H, Ph), 7.37 (d, J = 8.8 Hz, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.8 (1 C, CH2), 52.1 (1 C, OCH3), 117.8 (1 C, Ph), 118.0 (1 C, Ph), 120.2 (1 C, Ph), 120.3 (1 C, Ph), 125.1 (2 C, Ph), 130.0 (1 C, Ph), 130.9 (1 C, Ph), 133.1 (1 C, Ph), 136.1 (1 C, Ph), 156.1 (1 C, Ph), 156.3 (1 C, Ph), 171.5 (1 C, CO). Then, 10e (0.20 g, 0.64 mmol) was treated with LiAlH4 (49 mg, 1.3 mmol) in dry THF (3.8 mL) to give alcohol intermediate, which was oxidized with PCC (0.23 g, 1.1 mmol) in DCM (5.5 mL) to yield aldehyde 12e (C14H10Cl2O2, 0.10 g, 0.36 mmol).
2-(3-(4-Chlorophenoxy)phenyl)acetaldehyde (12f). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), (4-chlorophenyl)boronic acid (2.1 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(4-chlorophenoxy)phenyl)acetate 10f (eluent system: 10% ethylacetate in petroleum ether, C15H13ClO3, 0.40 g, 1.4 mmol, 33% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.62 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.87–6.99 (m, 4 H, Ph), 7.05 (d, J = 7.3 Hz, 1 H, Ph), 7.25–7.34 (m, 3 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.9 (1 C, CH2), 52.1 (1 C, OCH3), 117.4 (1 C, Ph), 119.8 (1 C, Ph), 120.2 (2 C, Ph), 124.5 (1 C, Ph), 128.4 (1 C, Ph), 129.8 (2 C, Ph), 129.9 (1 C, Ph), 136.0 (1 C, Ph), 155.7 (1 C, Ph), 157.1 (1 C, Ph), 171.6 (1 C, CO). Then, 10f (0.20 g, 0.72 mmol) was treated with LiAlH4 (55 mg, 1.5 mmol) in dry THF (4.3 mL) to give alcohol intermediate, which was oxidized with PCC (0.31 g, 1.5 mmol) in DCM (7.5 mL) to yield aldehyde 12f (C14H11ClO2, 0.13 g, 0.53 mmol).
2-(3-(4-Methoxyphenoxy)phenyl)acetaldehyde (12g). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), (4-methoxyphenyl)boronic acid (2.0 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(4-methoxyphenoxy)phenyl)acetate 10g (eluent system: 10% ethylacetate in petroleum ether, C16H16O4, 0.33 g, 1.2 mmol, 28% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.60 (s, 2 H, CH2), 3.70 (s, 3 H, OCH3), 3.82 (s, 3 H, (Ph)OCH3), 6.82–6.87 (m, 1 H, Ph), 6.88–6.94 (m, 3 H, Ph), 6.95–7.03 (m, 3 H, Ph), 7.22–7.29 (m, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 41.0 (1 C, CH2), 52.0 (1 C, OCH3), 55.6 (1 C, (Ph)OCH3), 114.8 (2 C, Ph), 116.0 (1 C, Ph), 118.4 (1 C, Ph), 120.9 (2 C, Ph), 123.2 (1 C, Ph), 129.6 (1 C, Ph), 135.6 (1 C, Ph), 149.8 (1 C, Ph), 155.9 (1 C, Ph), 158.6 (1 C, Ph), 171.7 (1 C, CO). Then, 10g (0.20 g, 0.74 mmol) was treated with LiAlH4 (56 mg, 1.5 mmol) in dry THF (4.4 mL) to give alcohol intermediate, which was oxidized with PCC (0.32 g, 1.5 mmol) in DCM (7.5 mL) to yield aldehyde 12g (C15H14O3, 0.14 g, 0.58 mmol).
2-(3-(3-Chlorophenoxy)phenyl)acetaldehyde (12h). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), 3-chlorophenylboronic acid (1.4 g, 8.8 mmol), Cu(OAc)2 (1.1 g, 5.9 mmol), 4Å molecular sieves (0.50 g) and pyridine (0.71 mL, 8.8 mmol) in 1,2-dichloroethane (17 mL) afforded the ester intermediate methyl 2-(3-(3-chlorophenoxy)phenyl)acetate 10h (eluent system: 10% ethylacetate in petroleum ether, C15H13ClO3, 0.28 g, 1.0 mmol, 34% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.88–6.96 (m, 2 H, Ph), 6.98 (t, J = 1.9 Hz, 1 H, Ph), 7.00 (t, J = 2.2 Hz, 1 H, Ph), 7.04–7.14 (m, 2 H, Ph), 7.17–7.36 (m, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.9 (1 C, CH2), 52.1 (1 C, OCH3), 116.8 (1 C, Ph), 117.9 (1 C, Ph), 118.9 (1 C, Ph), 120.2 (1 C, Ph), 123.3 (1 C, Ph), 124.8 (1 C, Ph), 130.0 (1 C, Ph), 130.5 (1 C, Ph), 135.0 (1 C, Ph), 136.0 (1 C, Ph), 156.5 (1 C, Ph), 158.1 (1 C, Ph), 171.5 (1 C, CO). Then, 10h (0.20 g, 0.72 mmol) was treated with LiAlH4 (55 mg, 1.4 mmol) in dry THF (4.3 mL) to give alcohol intermediate, which was oxidized with PCC (0.19 g, 0.87 mmol) in DCM (4.3 mL) to yield aldehyde 12h (C14H11ClO2, 0.10 g, 0.41 mmol).
2-(3-(Benzyloxy)phenyl)acetaldehyde (
15a). According to a literature procedure [
27] with minor changes, methyl 2-(3-hydroxyphenyl)acetate (0.30 g, 2.2 mmol), benzyl bromide (0.26 mL, 2.2 mmol), K
2CO
3 (0.61 g, 4.4 mmol), sodium iodide (33 mg, 0.22 mmol) in dimethylformamide (DMF) (10 mL) at room temperature for overnight afforded the ester intermediate methyl 2-(3-(benzyloxy)phenyl)acetate
13a (eluent system: 10% ethylacetate in petroleum ether, C
16H
16O
3, 0.33 g, 1.3 mmol, 58% yield).
1H NMR (300 MHz, CDCl
3)
δ ppm 3.65 (s, 2 H, CH
2), 3.73 (s, 3 H, OCH
3), 5.10 (s, 2 H, (Ph)CH
2O), 6.83–7.05 (m, 3 H, Ph), 7.21–7.52 (m, 6 H, Ph).
13C NMR (75 MHz, CDCl
3)
δ ppm 41.1 (1 C, CH
2), 52.0 (1 C, OCH
3), 69.8 (1 C, (Ph)CH
2O), 113.4 (1 C, Ph), 115.8 (1 C, Ph), 121.8 (1 C, Ph), 127.5 (2 C, Ph), 127.9 (1 C, Ph), 128.5 (2 C, Ph), 129.5 (1 C, Ph), 135.4 (1 C, Ph), 136.9 (1 C, Ph), 158.9 (1 C, Ph), 171.8 (1 C, CO). Then,
13a (0.20 g, 0.78 mmol) was treated with LiAlH
4 (59 mg, 1.6 mmol) in dry THF (4.7 mL) to give alcohol intermediate, which was oxidized with PCC (0.34 g, 1.6 mmol) in DCM (8.0 mL) to yield aldehyde
15a (C
15H
14O
2, 0.14 g, 0.62 mmol).
2-(3-((2-Chlorobenzyl)oxy)phenyl)acetaldehyde (15b). Following the procedure as described for 15a, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), 1-(bromomethyl)-2-chlorobenzene (0.60 g, 2.9 mmol), K2CO3 (0.81 g, 5.9 mmol), sodium iodide (44 mg, 0.29 mmol) in DMF (13 mL) afforded the ester intermediate methyl 2-(3-((2-chlorobenzyl)oxy)phenyl)acetate 13b (eluent system: 10% ethylacetate in petroleum ether, C16H15ClO3, 0.26 g, 0.89 mmol, 30% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 5.17 (s, 2 H, (Ph)CH2O), 6.86–7.01 (m, 3 H, Ph), 7.23–7.36 (m, 3 H, Ph), 7.41 (dd, J = 7.0, 2.1 Hz, 1 H, Ph), 7.53–7.63 (m, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 41.2 (1 C, CH2), 52.1 (1 C, OCH3), 67.1 (1 C, (Ph)CH2O), 113.4 (1 C, Ph), 116.0 (1 C, Ph), 122.1 (1 C, Ph), 126.9 (1 C, Ph), 128.8 (1 C, Ph), 129.0 (1 C, Ph), 129.3 (1 C, Ph), 129.6 (1 C, Ph), 132.6 (1 C, Ph), 134.7 (1 C, Ph), 135.5 (1 C, Ph), 158.7 (1 C, Ph), 171.8 (1 C, CO). Then, 13b (0.20 g, 0.69 mmol) was treated with LiAlH4 (52 mg, 1.4 mmol) in dry THF (4.1 mL) to give alcohol intermediate, which was oxidized with PCC (0.25 g, 1.2 mmol) in DCM (6.0 mL) to yield aldehyde 15b (C15H13ClO2, 0.14 g, 0.53 mmol).
2-(3-((3,4-Dichlorobenzyl)oxy)phenyl)acetaldehyde (15c). Following the procedure as described for 15a, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), 3,4-dichlorobenzyl bromide (0.70 g, 2.9 mmol), K2CO3 (0.81 g, 5.9 mmol), sodium iodide (44 mg, 0.29 mmol) in DMF (13 mL) afforded the ester intermediate methyl 2-(3-((3,4-dichlorobenzyl)oxy)phenyl)acetate 13c (eluent system: 10% ethylacetate in petroleum ether, C16H14Cl2O3, 0.75 g, 2.3 mmol, 84% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.62 (s, 2 H, CH2), 3.70 (s, 3 H, OCH3), 5.01 (s, 2 H, (Ph)CH2O), 6.87 (d, J = 8.2 Hz, 1 H, Ph), 6.93 (br. s., 2 H, Ph), 7.21–7.30 (m, 2 H, Ph), 7.41–7.48 (m, 1 H, Ph), 7.54 (s, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 41.1 (1 C, CH2), 52.1 (1 C, OCH3), 68.4 (1 C, (Ph)CH2O), 113.4 (1 C, Ph), 115.8 (1 C, Ph), 122.3 (1 C, Ph), 126.5 (1 C, Ph), 129.2 (1 C, Ph), 129.7 (1 C, Ph), 130.5 (1 C, Ph), 131.8 (1 C, Ph), 132.6 (1 C, Ph), 135.6 (1 C, Ph), 137.3 (1 C, Ph), 158.4 (1 C, Ph), 171.7 (1 C, CO). Then, 13c (0.20 g, 0.62 mmol) was treated with LiAlH4 (47 mg, 1.2 mmol) in dry THF (3.7 mL) to give alcohol intermediate, which was oxidized with PCC (0.26 g, 1.2 mmol) in DCM (6.0 mL) to yield aldehyde 15c (C15H12Cl2O2, 0.18 g, 0.60 mmol).
2-(3-((3-Chlorobenzyl)oxy)phenyl)acetaldehyde (15d). Following the procedure as described for 15a, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), m-chlorobenzyl bromide (0.60 g, 2.9 mmol), K2CO3 (0.81 g, 5.9 mmol), sodium iodide (44 mg, 0.29 mmol) in DMF (13 mL) afforded the ester intermediate methyl 2-(3-((3-chlorobenzyl)oxy)phenyl)acetate 13d (eluent system: 10% ethylacetate in petroleum ether, C16H15ClO3, 0.62 g, 2.1 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ ppm 3.61 (s, 2 H, CH2), 3.70 (s, 3 H, OCH3), 5.03 (s, 2 H, (Ph)CH2O), 6.84–6.94 (m, 3 H, Ph), 7.23–7.28 (m, 1 H, Ph), 7.29–7.32 (m, 3 H, Ph), 7.35–7.48 (m, 1 H, Ph). 13C NMR (101 MHz, CDCl3) δ ppm 41.2 (1 C, CH2), 52.1 (1 C, OCH3), 69.1 (1 C, (Ph)CH2O), 113.4 (1 C, Ph), 115.8 (1 C, Ph), 122.1 (1 C, Ph), 125.3 (1 C, Ph), 127.4 (1 C, Ph), 128.0 (1 C, Ph), 129.6 (1 C, Ph), 129.8 (1 C, Ph), 134.5 (1 C, Ph), 135.5 (1 C, Ph), 139.0 (1 C, Ph), 158.6 (1 C, Ph), 171.9 (1 C, CO). Then, 13d (0.20 g, 0.69 mmol) was treated with LiAlH4 (52 mg, 1.4 mmol) in dry THF (4.1 mL) to give alcohol intermediate, which was oxidized with PCC (0.26 g, 1.2 mmol) in DCM (6.0 mL) to yield aldehyde 15d (C15H13ClO2, 0.14 g, 0.52 mmol).
2-(3-(4-Cyanophenoxy)phenyl)acetaldehyde (
18a). To a solution of methyl 2-(3-hydroxyphenyl)acetate (0.35 g, 2.1 mmol) in THF (13 mL) was added LiAlH
4 (0.16 g, 4.2 mmol) afforded 3-(2-hydroxyethyl)phenol (0.24 g, 1.4 mmol), which was reacted with 4-fluorobenzonitrile (0.21 g, 1.7 mmol), K
2CO
3 (0.40 g, 2.9 mmol) in DMF (10 mL) at 90 °C gave 4-(3-(2-hydroxyethyl)phenoxy)benzonitrile [
28]
17a (eluent system: 25% ethylacetate in petroleum ether, C
15H
13NO
2, 0.14 g, 0.59 mmol, 40% yield).
1H NMR (300 MHz, CDCl
3)
δ ppm 2.89 (t,
J = 6.6 Hz, 2 H, CH
2), 3.89 (t,
J = 6.4 Hz, 2 H, CH
2(OH)), 6.89–7.06 (m, 4 H, Ph), 7.11 (dd,
J = 7.6, 0.59 Hz, 1 H, Ph), 7.36 (t,
J = 7.4 Hz, 1 H, Ph), 7.49–7.69 (m, 2 H, Ph).
13C NMR (75 MHz, CDCl
3)
δ ppm 38.9 (1 C, CH
2), 63.3 (1 C, CH
2(OH)), 105.8 (1 C, Ph), 110.0 (1 C, Ph), 118.0 (2 C, Ph), 118.3 (1 C, CN), 120.9 (1 C, Ph), 125.7 (1 C, Ph), 130.2 (1 C, Ph), 134.1 (2 C, Ph), 141.4 (1 C, Ph), 154.9 (1 C, Ph), 161.5 (1 C, Ph). Then,
17a was oxidized by PCC (0.25 g, 1.2 mmol) in DCM (6.0 mL) to give aldehyde
18a (C
15H
11NO
2, 0.11 g, 0.44 mmol).
2-(3-(4-Nitrophenoxy)phenyl)acetaldehyde (18b). Following the procedure described for 18a, 3-(2-hydroxyethyl)phenol (0.30 g, 2.2 mmol), 1-fluoro-4-nitrobenzene (0.37 g, 2.6 mmol) and K2CO3 (0.60 g, 4.3 mmol) in DMF (16 mL) afforded 2-(3-(4-nitrophenoxy)phenyl)ethan-1-ol 17b (eluent system: 25% ethylacetate in petroleum ether, C14H13NO4, 0.52 g, 2.0 mmol, 92% yield). 1H NMR (300 MHz, CDCl3) δ ppm 2.90 (t, J = 6.4 Hz, 2 H, CH2), 3.89 (t, J = 6.4 Hz, 2 H, CH2(OH)), 6.93–7.07 (m, 4 H, Ph), 7.13 (d, J = 7.6 Hz, 1 H, Ph), 7.37 (t, J = 7.7 Hz, 1 H, Ph), 8.13–8.24 (m, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 38.8 (1 C, CH2), 63.2 (1 C, CH2(OH)), 117.1 (2 C, Ph), 118.4 (1 C, Ph), 121.0 (1 C, Ph), 125.9 (2 C, Ph), 126.0 (1 C, Ph), 130.3 (1 C, Ph), 136.5 (1 C, Ph), 141.5 (1 C, Ph), 154.9 (1 C, Ph), 163.2 (1 C, Ph). Then, 17b was oxidized by PCC (0.86 g, 4.0 mmol) in DCM (20 mL) to give aldehyde 18b (C14H11NO4, 0.36 g, 1.4 mmol).
2-(3-(4-(Trifluoromethyl)phenoxy)phenyl)acetaldehyde (18c). Following the procedure described for 18a, 3-(2-hydroxyethyl)phenol (0.30 g, 2.2 mmol), 1-fluoro-4-(trifluoromethyl)benzene (0.43 g, 2.6 mmol) and CsCO3 (0.85 g, 2.6 mmol) in DMF (16 mL) at 90 °C gave 2-(3-(4-(trifluoromethyl)phenoxy)phenyl)ethan-1-ol 17c (eluent system: 25% ethylacetate in petroleum ether, C15H13F3O2, 0.13 g, 0.46 mmol, 21% yield). 1H NMR (300 MHz, CDCl3) δ ppm 2.87 (t, J = 6.6 Hz, 2 H, CH2), 3.86 (t, J = 6.6 Hz, 2 H, CH2(OH)), 6.89–6.98 (m, 2 H, Ph), 7.01–7.12 (m, 3 H, Ph), 7.33 (t, J = 6.6 Hz, 1 H, Ph), 7.58 (d, J = 8.8 Hz, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 38.9 (1 C, CH2), 63.3 (1 C, CH2(OH)), 117.7 (1 C, Ph), 117.8 (2 C, Ph), 120.4 (2 C, Ph), 124.2 (q, J = 271.5 Hz, 1 C, CF3), 125.1 (1 C, Ph), 127.0 (2 C, Ph), 130.0 (1 C, Ph), 141.1 (1 C, Ph), 155.9 (1 C, Ph), 160.3 (1 C, Ph). Then, 17c was oxidized by PCC (0.20 g, 0.92 mmol) in DCM (4.6 mL) to give aldehyde 18c (C15H11F3O2, 0.12 g, 0.43 mmol).
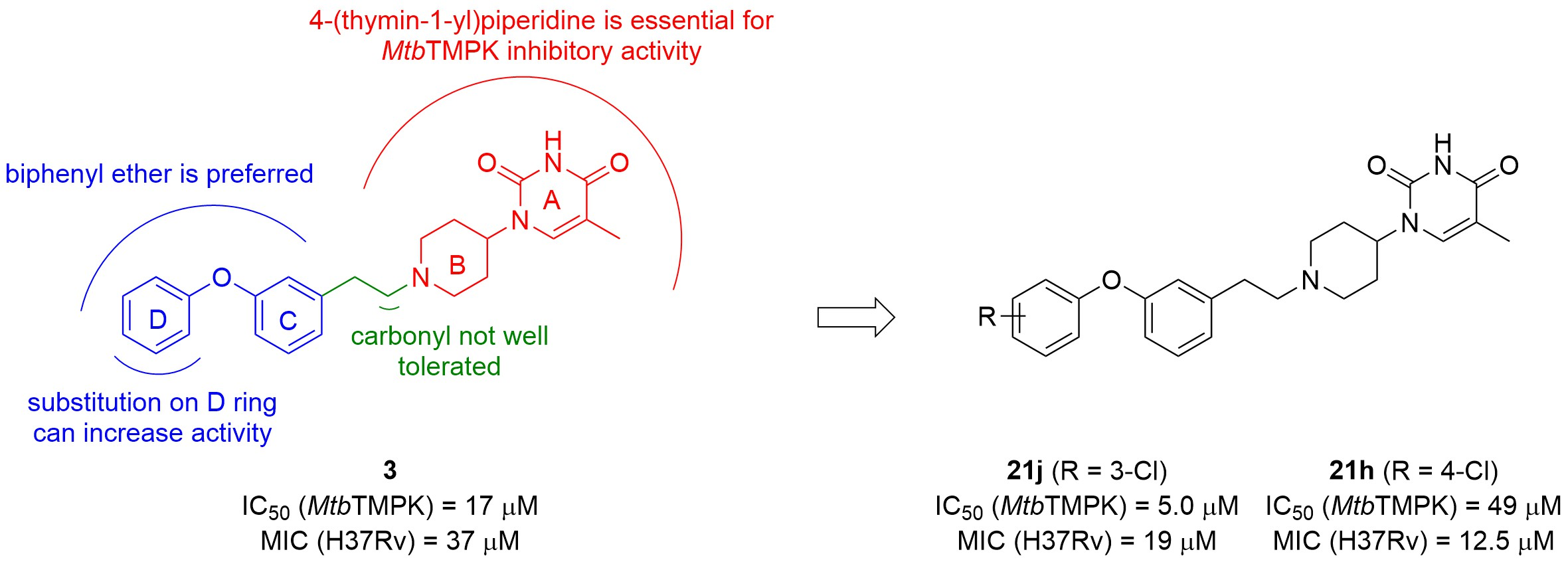
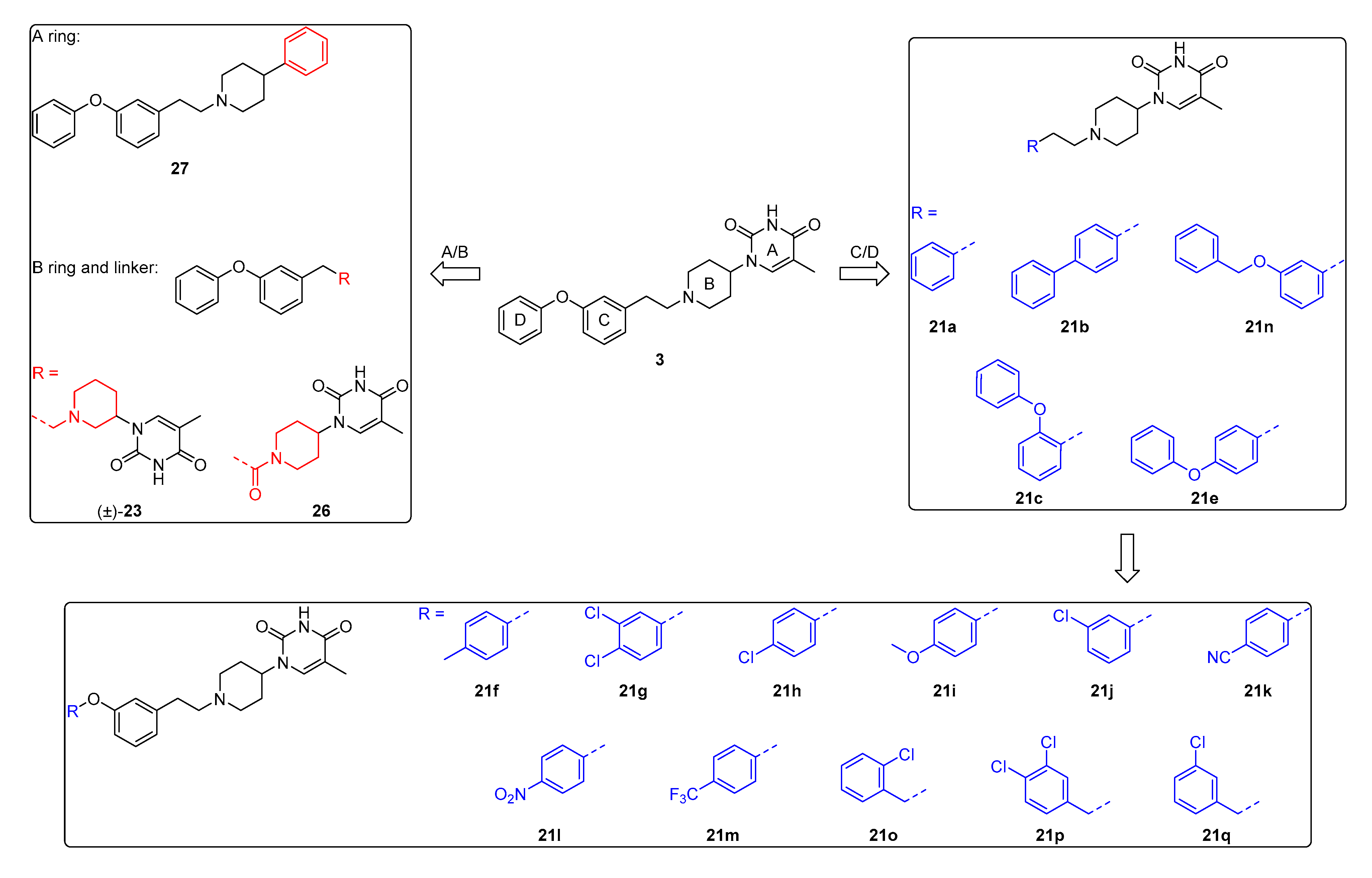
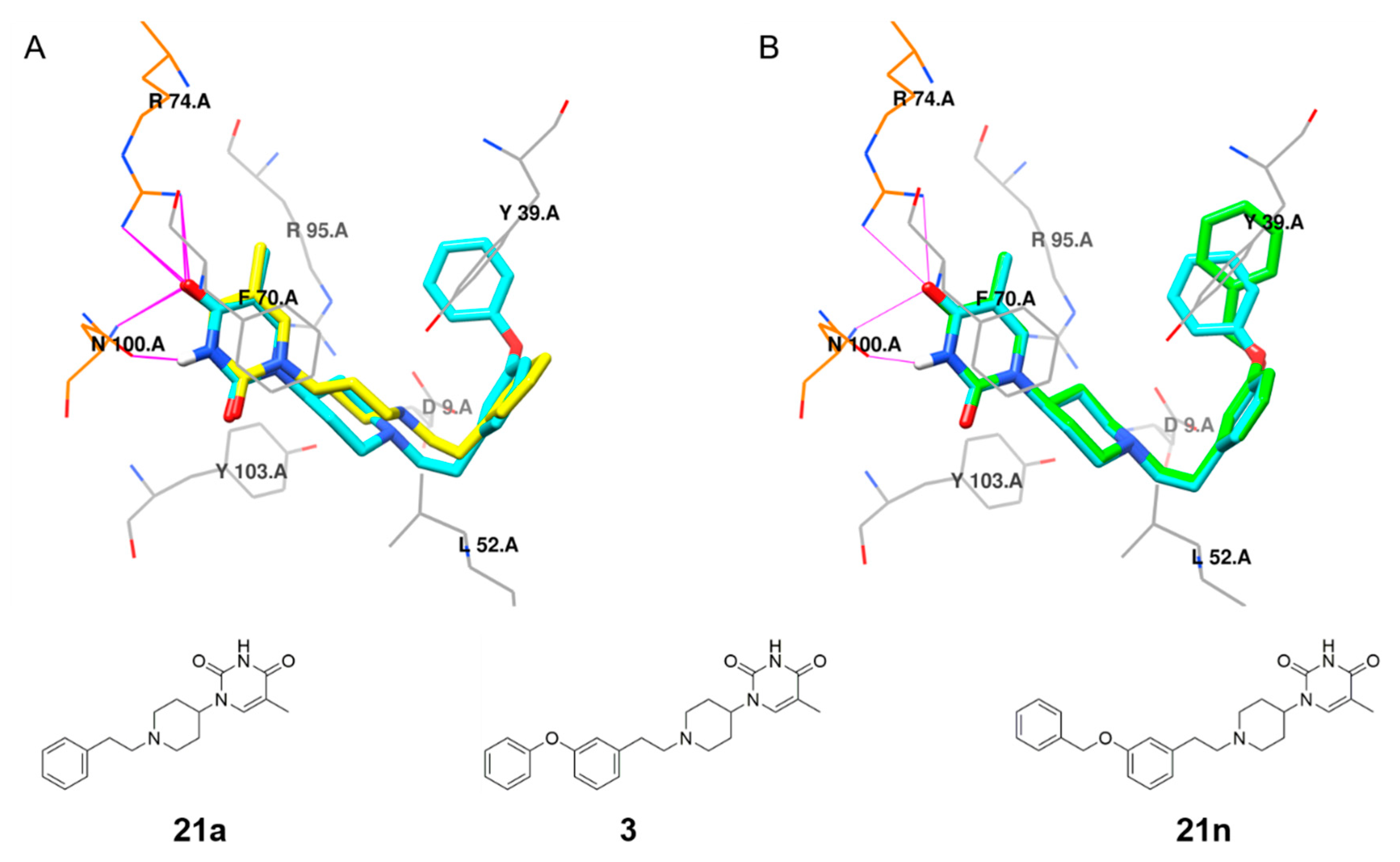
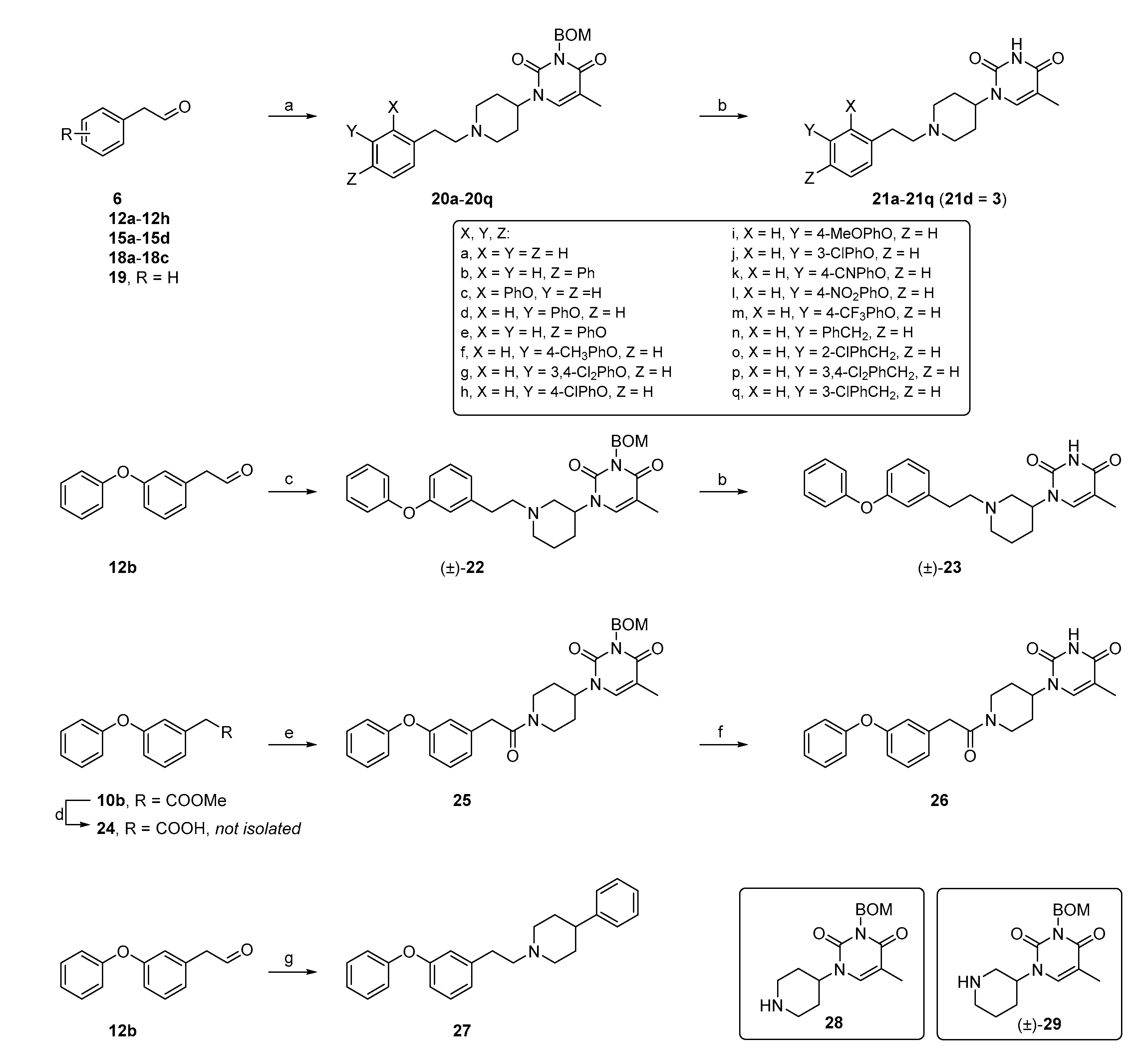
5-Methyl-1-(1-phenethylpiperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21a). Following the general procedure B, phenylacetaldehyde (36 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21a (eluent system: 5% MeOH in DCM, 53 mg, 0.17 mmol, 56% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.53–1.93 (m, 7 H, 5-CH3, piperdyl-3-yl, piperidyl-5-yl), 2.00–2.16 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.57 (d, J = 8.2 Hz, 2 H, CH2N), 2.66–2.82 (m, 2 H, PhCH2), 3.06 (d, J = 10.8 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.16–4.36 (m, 1 H, piperidyl-4-yl), 7.10–7.37 (m, 5 H, Ph), 7.63 (s, 1 H, H-6), 11.19 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 29.9 (2 C, piperdyl-3-yl, piperidyl-5-yl), 33.0 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.6 (1 C, piperidyl-4-yl), 59.3 (1 C, CH2N), 108.9 (1 C, C-5), 125.8 (1 C, Ph), 128.2 (2 C, Ph), 128.6 (2 C, Ph), 137.7 (1 C, C-6), 140.4 (1 C, Ph), 150.8 (1 C, C-2), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C18H23N3O2 + H]+ 314.1863, found 314.1855.
1-(1-(2-([1,1’-Biphenyl]-4-yl)ethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21b). Following the general procedure B, 6 (60 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21b (eluent system: 5% MeOH in DCM, 39 mg, 0.10 mmol, 33% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.72–1.93 (m, 5 H, piperdyl-3a-yl, piperidyl-5a-yl, 5-CH3), 1.98–2.20 (m, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.89–3.04 (m, 2 H, PhCH2), 3.26–3.61 (m, 4 H, piperidyl-2-yl, piperidyl-6-yl), 4.39–4.53 (m, 1 H, piperidyl-4-yl), 7.33–7.40 (m, 3 H, Ph), 7.46 (t, J = 7.6 Hz, 2 H, Ph), 7.59–7.71 (m, 5 H, H-6, Ph), 11.30 (s, 1 H, NH), 2 H (CH2N) could not be observed. 13C NMR (101 MHz, DMSO-d6) δ ppm 12.1 (1 C, 5-CH3), 27.7 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.9 (1 C, PhCH2), 51.4 (3 C, piperidyl-2-yl, piperidyl-6-yl, piperidyl-4-yl), 109.1 (1 C, C-5), 126.6 (2 C, Ph), 126.8 (2 C, Ph), 127.4 (1 C, Ph), 128.9 (2 C, Ph), 129.3 (2 C, Ph), 137.5 (3 C, C-6, Ph), 139.9 (1 C, Ph), 150.8 (1 C, C-2), 163.7 (1 C, C-4), C (CH2N) could not be observed. HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O2 + H]+ 390.2176, found 390.2186.
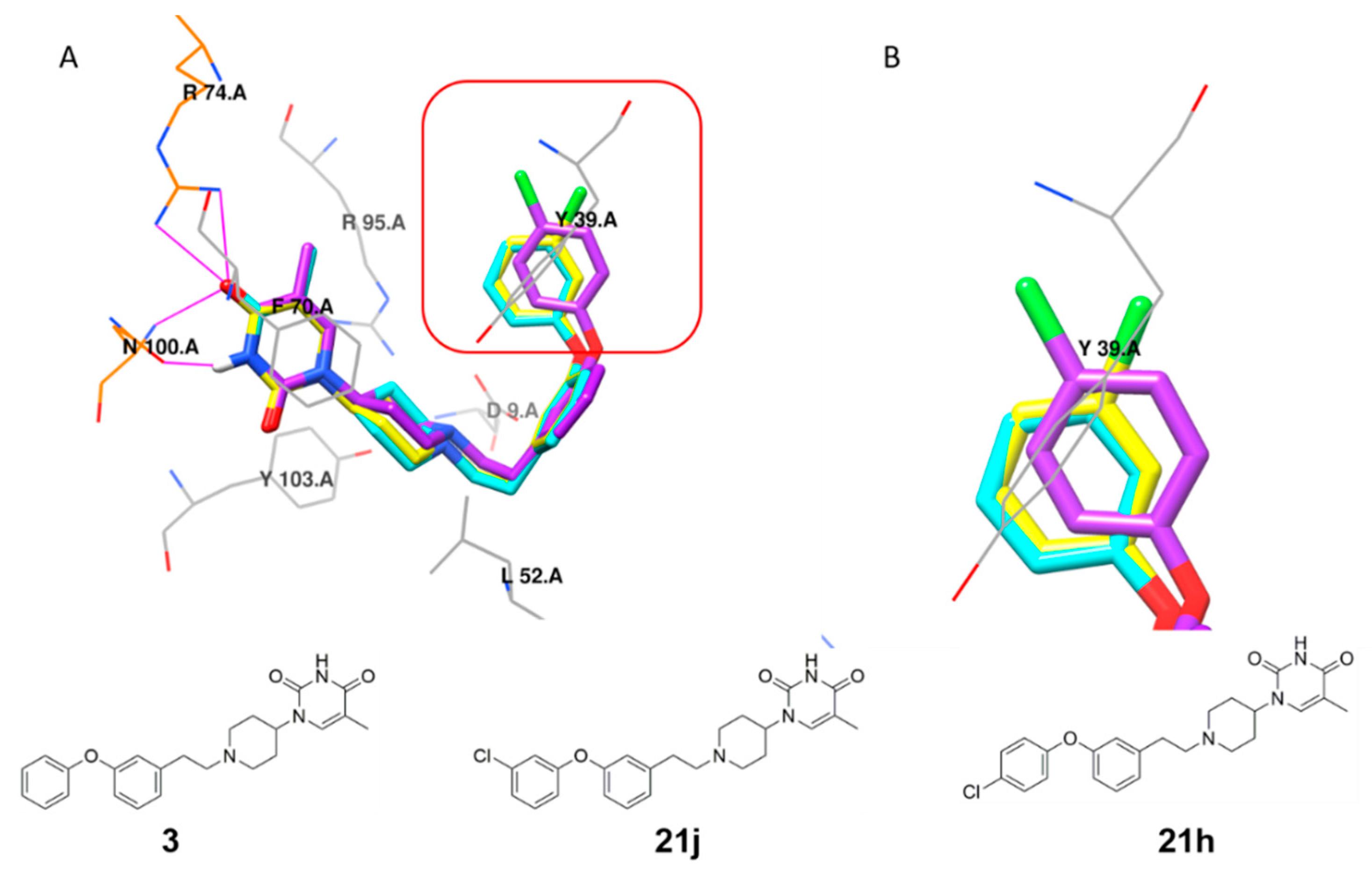
5-Methyl-1-(1-(2-phenoxyphenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21c). Following the general procedure B, 12a (64 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21c (eluent system: 5% MeOH in DCM, 40 mg, 0.098 mmol, 32% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.52–1.62 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.68–1.84 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 1.92–2.04 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.50–2.55 (m, 2 H, CH2N), 2.64–2.75 (m, 2 H, PhCH2), 2.92 (d, J = 11.1 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.12–4.26 (m, 1 H, piperidyl-4-yl), 6.85–6.92 (m, 3 H, Ph), 7.02–7.15 (m, 2 H, Ph), 7.17–7.26 (m, 1 H, Ph), 7.29–7.38 (m, 3 H, Ph), 7.57 (d, J = 1.2 Hz, 1 H, H-6), 11.16 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 27.3 (1 C, PhCH2), 29.9 (2 C, piperdyl-3-yl, piperidyl-5-yl), 52.3 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.3 (1 C, piperidyl-4-yl), 57.9 (1 C, CH2N), 108.9 (1 C, C-5), 117.1 (2 C, Ph), 119.8 (1 C, Ph), 122.6 (1 C, Ph), 124.2 (1 C, Ph), 127.7 (1 C, Ph), 129.9 (2 C, Ph), 131.1 (1 C, Ph), 131.8 (1 C, Ph), 137.6 (1 C, C-6), 150.8 (1 C, C-2), 153.8 (1 C, Ph), 157.6 (1 C, Ph), 163.6 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O3 + H]+ 406.2125, found 406.2133.
5-Methyl-1-(1-(4-phenoxyphenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21e). Following the general procedure B, 12c (64 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21e (eluent system: 5% MeOH in DCM, 65 mg, 0.16 mmol, 53% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.57–1.93 (m, 7 H, 5-CH3, piperdyl-3-yl, piperidyl-5-yl), 1.97–2.16 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.54 (d, J = 7.3 Hz, 2 H, CH2N), 2.64–2.77 (m, 2 H, PhCH2), 3.04 (d, J = 11.1 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.19–4.29 (m, 1 H, piperidyl-4-yl), 6.87–6.98 (m, 4 H, Ph), 7.04–7.13 (m, 1 H, Ph), 7.18–7.26 (m, 2 H, Ph), 7.29–7.41 (m, 2 H, Ph), 7.60 (d, J = 0.9 Hz, 1 H, H-6), 11.17 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 29.9 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.2 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.6 (1 C, piperidyl-4-yl), 59.3 (1 C, CH2N), 108.9 (1 C, C-5), 118.2 (2 C, Ph), 118.7 (2 C, Ph), 123.1 (1 C, Ph), 130.0 (2 C, Ph), 130.1 (2 C, Ph), 135.6 (1 C, Ph), 137.7 (1 C, C-6), 150.8 (1 C, C-2), 154.6 (1 C, Ph), 157.0 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O3 + H]+ 406.2125, found 406.2124.
5-Methyl-1-(1-(3-(p-tolyloxy)phenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21f). Following the general procedure B, 12d (69 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21f (eluent system: 5% MeOH in DCM, 40 mg, 0.095 mmol, 31% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.64 (d, J = 10.6 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.73 - 1.87 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 2.05 (t, J = 11.1 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.28 (s, 3 H, PhCH3), 2.54 (d, J = 8.4 Hz, 2 H, CH2N), 2.72 (t, J = 7.5 Hz, 2 H, PhCH2), 3.02 (d, J = 11.5 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.19–4.29 (m, 1 H, piperidyl-4-yl), 6.77 (d, J = 8.1 Hz, 1 H, Ph), 6.87 (s, 1 H, Ph), 6.90 (d, J = 8.3 Hz, 2 H, Ph), 6.98 (d, J = 7.6 Hz, 1 H, Ph), 7.19 (d, J = 8.4 Hz, 2 H, Ph), 7.26 (t, J = 7.8 Hz, 1 H, Ph), 7.61 (s, 1 H, H-6), 11.20 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 20.2 (1 C, PhCH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.8 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.5 (1 C, piperidyl-4-yl), 59.0 (1 C, CH2N), 108.9 (1 C, C-5), 115.5 (1 C, Ph), 118.4 (1 C, Ph), 118.8 (2 C, Ph), 123.4 (1 C, Ph), 129.7 (1 C, Ph), 130.3 (2 C, Ph), 132.5 (1 C, Ph), 137.7 (1 C, C-6), 142.7 (1 C, Ph), 150.8 (1 C, C-2), 154.2 (1 C, Ph), 157.1 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H29N3O3 + H]+ 420.2282, found 420.2286.
1-(1-(3-(3,4-Dichlorophenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21g). Following the general procedure B, 12e (85 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21g (eluent system: 5% MeOH in DCM, 56 mg, 0.12 mmol, 39% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 1.91–2.06 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.10–2.19 (m, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.96–3.08 (m, 2 H, PhCH2), 3.09–3.22 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.25–3.37 (m, 2 H, CH2N), 3.56–3.73 (m, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.46–4.59 (m, 1 H, piperidyl-4-yl), 7.02 (dd, J = 8.9, 2.9 Hz, 2 H, Ph), 7.06 (br. s., 1 H, Ph), 7.15 (d, J = 7.3 Hz, 1 H, Ph), 7.30 (d, J = 2.8 Hz, 1 H, Ph), 7.34–7.46 (m, 2 H, H-6, Ph), 7.65 (d, J = 8.9 Hz, 1 H, Ph), 11.34 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.1 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.4 (1 C, PhCH2), 50.5 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.3 (1 C, CH2N), 109.3 (1 C, C-5), 117.8 (1 C, Ph), 118.7 (1 C, Ph), 119.5 (1 C, Ph), 120.2 (1 C, Ph), 125.16 (d, J = 43.5 Hz, 1 C, Ph), 130.6 (1 C, Ph), 131.6 (2 C, Ph), 132.0 (1 C, Ph), 137.4 (1 C, C-6), 139.6 (1 C, Ph), 150.7 (1 C, C-2), 155.8 (1 C, Ph), 156.4 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H25Cl2N3O3 + H]+ 474.1346, found 474.1333.
1-(1-(3-(4-Chlorophenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21h). Following the general procedure B, 12f (75 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21h (eluent system: 5% MeOH in DCM, 40 mg, 0.091 mmol, 30% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.57–1.67 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.68–1.87 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 2.03 (t, J = 12.3 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.54 (d, J = 8.5 Hz, 2 H, CH2N), 2.72 (t, J = 7.4 Hz, 2 H, PhCH2), 3.00 (d, J = 12.0 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.22 (tt, J = 12.1, 4.5 Hz, 1 H, piperidyl-4-yl), 6.83 (dd, J = 7.8, 1.9 Hz, 1 H, Ph), 6.92 (s, 1 H, Ph), 6.96–7.06 (m, 3 H, Ph), 7.24–7.33 (m, 1 H, Ph), 7.38–7.43 (m, 2 H, Ph), 7.59 (s, 1 H, H-6), 11.17 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.7 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.5 (1 C, piperidyl-4-yl), 59.0 (1 C, CH2N), 108.9 (1 C, C-5), 116.3 (1 C, Ph), 119.2 (1 C, Ph), 120.0 (2 C, Ph), 124.3 (1 C, Ph), 126.9 (1 C, Ph), 129.8 (2 C, Ph), 129.9 (1 C, Ph), 137.6 (1 C, C-6), 143.0 (1 C, Ph), 150.8 (1 C, C-2), 155.8 (1 C, Ph), 156.1 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H26ClN3O3 + H]+ 440.1736, found 440.1750.
1-(1-(3-(4-Methoxyphenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21i). Following the general procedure B, 12g (74 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21i (eluent system: 5% MeOH in DCM, 40 mg, 0.092 mmol, 30% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.65 (d, J = 10.6 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.74–1.87 (m, 5 H, piperdyl-3b-yl, piperidyl-5b-yl, 5-CH3), 2.05 (t, J = 11.3 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.52–2.56 (m, 2 H, CH2N), 2.70 (t, J = 8.3 Hz, 2 H, PhCH2), 3.02 (d, J = 11.4 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 3.74 (s, 3 H, OCH3), 4.19–4.29 (m, 1 H, piperidyl-4-yl), 6.72 (d, J = 8.1 Hz, 1 H, Ph), 6.83 (s, 1 H, Ph), 6.92–7.01 (m, 5 H, Ph), 7.24 (t, J = 7.9 Hz, 1 H, Ph), 7.61 (s, 1 H, H-6), 11.20 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.8 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.5 (1 C, piperidyl-4-yl), 55.4 (1 C, OCH3), 59.0 (1 C, CH2N), 108.9 (1 C, C-5), 114.7 (1 C, Ph), 115.0 (2 C, Ph), 117.6 (1 C, Ph), 120.6 (2 C, Ph), 123.0 (1 C, Ph), 129.6 (1 C, Ph), 137.7 (1 C, C-6), 142.6 (1 C, Ph), 149.4 (1 C, Ph), 150.8 (1 C, C-2), 155.5 (1 C, Ph), 157.9 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H29N3O4 + H]+ 436.2231, found 436.2234.
1-(1-(3-(3-Chlorophenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21j). Following the general procedure B, 12h (75 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21j (eluent system: 5% MeOH in DCM, 63 mg, 0.14 mmol, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.65 (d, J = 9.8 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.71–1.89 (m, 5 H, piperdyl-3b-yl, piperidyl-5b-yl, 5-CH3), 2.06 (t, J = 11.0 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.53–2.62 (m, 2 H, CH2N), 2.69–2.79 (m, 2 H, PhCH2), 3.03 (d, J = 11.6 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.17–4.31 (m, 1 H, piperidyl-4-yl), 6.88 (dd, J = 8.1, 1.8 Hz, 1 H, Ph), 6.92–7.00 (m, 2 H, Ph), 7.02 (t, J = 2.2 Hz, 1 H, Ph), 7.08 (d, J = 7.8 Hz, 1 H, Ph), 7.15–7.21 (m, 1 H, Ph), 7.33 (t, J = 7.8 Hz, 1 H, Ph), 7.37–7.43 (m, 1 H, Ph), 7.60 (d, J = 0.9 Hz, 1 H, H-6), 11.20 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.7 (1 C, PhCH2), 52.4 (3 C, piperidyl-2-yl, piperidyl-6-yl, piperidyl-4-yl), 58.9 (1 C, CH2N), 108.9 (1 C, C-5), 116.7 (1 C, Ph), 116.8 (1 C, Ph), 118.0 (1 C, Ph), 119.6 (1 C, Ph), 123.0 (1 C, Ph), 124.7 (1 C, Ph), 130.0 (1 C, Ph), 131.4 (1 C, Ph), 133.9 (1 C, Ph), 137.6 (1 C, C-6), 143.1 (1 C, Ph), 150.8 (1 C, C-2), 155.5 (1 C, Ph), 158.1 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H26ClN3O3 + H]+ 440.1736, found 440.1740.
4-(3-(2-(4-(5-Methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)piperidin-1- yl)ethyl)phenoxy)benzonitrile (21k). Following the general procedure B, 18a (72 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21k (eluent system: 5% MeOH in DCM, 50 mg, 0.12 mmol, 43% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 2.00 (d, J = 10.8 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.21 (d, J = 11.6 Hz, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 3.05 (br. s., 2 H, PhCH2), 3.15 (br. s., 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.32 (s, 2 H, CH2N), 3.66 (br. s., 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.44–4.63 (m, 1 H, piperidyl-4-yl), 7.03–7.16 (m, 4 H, Ph), 7.21 (d, J = 6.6 Hz, 1 H, Ph), 7.35–7.53 (m, 2 H, Ph, H-6), 7.86 (d, J = 8.4 Hz, 2 H, Ph), 11.32 (br. s., 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.1 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.3 (1 C, PhCH2), 50.3 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.2 (1 C, CH2N), 105.2 (1 C, Ph), 109.3 (1 C, C-5), 118.1 (2 C, Ph), 118.7 (2 C, Ph, CN), 120.5 (1 C, Ph), 125.6 (1 C, Ph), 130.7 (1 C, Ph), 134.7 (2 C, Ph), 137.3 (1 C, C-6), 139.7 (1 C, Ph), 150.7 (1 C, C-2), 154.7 (1 C, Ph), 161.0 (1 C, Ph), 163.6 (1 C. C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H26N4O3 + H]+ 431.2078, found 431.2085.
5-Methyl-1-(1-(3-(4-nitrophenoxy)phenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21l). Following the general procedure B, 18b (78 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21l (eluent system: 5% MeOH in DCM, 67 mg, 0.15 mmol, 40% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 1.93–2.06 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.20 (d, J = 11.6 Hz, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.98–3.08 (m, 2 H, PhCH2), 3.09–3.24 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.41–3.55 (m, 2 H, CH2N), 3.67 (d, J = 10.6 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.44–4.62 (m, 1 H, piperidyl-4-yl), 7.09–7.17 (m, 4 H, Ph), 7.24 (d, J = 7.5 Hz, 1 H, Ph), 7.38 (s, 1 H, H-6), 7.49 (t, J = 7.8 Hz, 1 H, Ph), 8.27 (d, J = 9.1 Hz, 2 H, Ph), 11.32 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.3 (1 C, PhCH2), 50.5 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.2 (1 C, CH2N), 109.3 (1 C, C-5), 117.5 (2 C, Ph), 118.9 (1 C, Ph), 120.7 (1 C, Ph), 125.9 (1 C, Ph), 126.2 (2 C, Ph), 130.8 (1 C, Ph), 137.3 (1 C, C-6), 139.4 (1 C, Ph), 142.3 (1 C, Ph), 150.7 (1 C, C-2), 154.5 (1 C, Ph), 162.7 (1 C, Ph), 163.6 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H26N4O5 + H]+ 451.1976, found 451.1971.
5-Methyl-1-(1-(3-(4-(trifluoromethyl)phenoxy)phenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21m). Following the general procedure B, 18c (85 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21m (eluent system: 5% MeOH in DCM, 64 mg, 0.14 mmol, 44% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 2.00 (d, J = 12.0 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.20 (d, J = 11.4 Hz, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.99 - 3.08 (m, 2 H, PhCH2), 3.16 (d, J = 9.5 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.39–3.55 (m, 2 H, CH2N), 3.67 (d, J = 10.6 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.47–4.59 (m, 1 H, piperidyl-4-yl), 7.05 (d, J = 7.9 Hz, 1 H, Ph), 7.10–7.20 (m, 4 H, Ph), 7.39 (br. s., 1 H, H-6), 7.44 (t, J = 7.8 Hz, 1 H, Ph), 7.75 (d, J = 8.5 Hz, 2 H, Ph), 11.32 (s, 1 H, NH). 19F NMR (377 MHz, DMSO-d6) δ ppm -60.16 (s, 3 F). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.1 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.3 (1 C, PhCH2), 50.4 (1 C, piperidyl-4-yl), 51.1 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.3 (1 C, CH2N), 109.3 (1 C, C-5), 118.0 (2 C, Ph), 118.4 (1 C, Ph), 120.2 (1 C, Ph), 123.20 (q, J = 32.6 Hz, 1 C, Ph), 124.27 (q, J = 271.3 Hz, 1 C, CF3), 125.2 (1 C, Ph), 127.5 (2 C, Ph), 130.6 (1 C, Ph), 137.3 (1 C, C-6), 139.6 (1 C, Ph), 150.7 (1 C, C-2), 155.2 (1 C, Ph), 160.3 (1 C, Ph), 163.6 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H26F3N3O3 + H]+ 474.1999, found 474.1989.
1-(1-(3-(Benzyloxy)phenethyl)piperidin-4-yl)-5-methylpyridine-2,4(1H,3H)-dione (21n). Following the general procedure B, 15a (69 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21n (eluent system: 5% MeOH in DCM, 58 mg, 0.14 mmol, 46% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.66 (d, J = 10.8 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.74–1.90 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 2.07 (t, J = 11.3 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.55 (t, J = 9.0 Hz, 2 H, CH2N), 2.70 (t, J = 7.4 Hz, 2 H, PhCH2), 3.04 (d, J = 11.3 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.20–4.31 (m, 1 H, piperidyl-4-yl), 5.08 (s, 2 H, (Ph)CH2O), 6.82 (t, J = 7.8 Hz, 2 H, Ph), 6.90 (s, 1 H, Ph), 7.19 (t, J = 7.9 Hz, 1 H, Ph), 7.30–7.35 (m, 1 H, Ph), 7.39 (t, J = 7.4 Hz, 2 H, Ph), 7.43–7.48 (m, 2 H, Ph), 7.63 (s, 1 H, H-6), 11.20 (s, 1 H, NH).13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 33.0 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.4 (1 C, piperidyl-4-yl), 59.2 (1 C, CH2N), 69.0 (1 C, (Ph)CH2O), 108.9 (1 C, C-5), 112.1 (1 C, Ph), 115.2 (1 C, Ph), 121.1 (1 C, Ph), 127.7 (2 C, Ph), 127.8 (1 C, Ph), 128.4 (2 C, Ph), 129.2 (1 C, Ph), 137.2 (1 C, Ph), 137.7 (1 C, C-6), 142.0 (1 C, Ph), 150.8 (1 C, C-2), 158.4 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H29N3O3 + H]+ 420.2282, found 420.2277.
1-(1-(3-((2-Chlorobenzyl)oxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21o). Following the general procedure B, 15b (79 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21o (eluent system: 5% MeOH in DCM, 90 mg, 0.20 mmol, 65% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.73–2.23 (m, 7 H, piperdyl-3-yl, piperidyl-5-yl, 5-CH3), 2.92 (br. s., 2 H, PhCH2), 3.21–3.67(m, 4 H, piperidyl-2-yl, piperidyl-6-yl), 4.35–4.68 (m, 1 H, piperidyl-4-yl), 5.12 (s, 2 H, (Ph)CH2O), 6.85–6.93 (m, 2 H, Ph), 6.97 (br. s., 1 H, Ph), 7.26 (t, J = 7.8 Hz, 1 H, Ph), 7.36–7.49 (m, 4 H, Ph), 7.52 (s, 1 H, H-6), 11.31 (br. s., 1 H, NH), 2 H (CH2N) could not be observed. 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 30.2 (1 C, PhCH2), 50.4 (1 C, piperidyl-4-yl), 51.5 (2 C, piperidyl-2-yl, piperidyl-6-yl), 68.2 (1 C, (Ph)CH2O), 109.2 (1 C, C-5), 112.7 (1 C, Ph), 115.6 (1 C, Ph), 121.4 (1 C, Ph), 126.2 (2 C, Ph), 127.3 (1 C, Ph), 127.8 (1 C, Ph), 129.7 (1 C, Ph), 130.4 (1 C, Ph), 133.1 (1 C, Ph), 137.5 (1 C, C-6), 139.7 (1 C, Ph), 150,8 (1 C, C-2), 158.3 (1 C, Ph), 163.7 (1 C, C-4), C (CH2N) could not be observed. HRMS (ESI): m/z [M + H]+ Calcd. for [C25H28ClN3O3 + H]+ 454.1892, found 454.1902.
1-(1-(3-((3,4-Dichlorobenzyl)oxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21p). Following the general procedure B, 15c (90 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21p (eluent system: 5% MeOH in DCM, 55 mg, 0.11 mmol, 37% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 1.92–2.06 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.08–2.24 (m, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.85–3.02 (m, 2 H, PhCH2), 3.06–3.22 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.24–3.34 (m, 2 H, CH2N), 3.65 (br. s., 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.45–4.59 (m, 1 H, piperidyl-4-yl), 5.13 (s, 2 H, (Ph)CH2O), 6.85–6.95 (m, 2 H, Ph), 6.98 (br. s., 1 H, Ph), 7.28 (t, J = 7.9 Hz, 1 H, Ph), 7.38–7.48 (m, 2 H, H-6, Ph), 7.68 (d, J = 8.3 Hz, 1 H, Ph), 7.73 (d, J = 1.5 Hz, 1 H, Ph), 11.33 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.2 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.7 (1 C, PhCH2), 50.6 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.5 (1 C, CH2N), 67.5 (1 C, (Ph)CH2O), 109.3 (1 C, C-5), 112.8 (1 C, Ph), 115.5 (1 C, Ph), 121.5 (1 C, Ph), 127.8 (1 C, Ph), 129.4 (1 C, Ph), 129.8 (1 C, Ph), 130.4 (1 C, Ph), 130.7 (1 C, Ph), 131.1 (1 C, Ph), 137.4 (1 C, C-6), 138.3 (2 C, Ph), 150.7 (1 C, C-2), 158.2 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H27Cl2N3O3 + H]+ 488.1502, found 488.1518.
1-(1-(3-((3-Chlorobenzyl)oxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21q). Following the general procedure B, 15d (79 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21q (eluent system: 5% MeOH in DCM, 38 mg, 0.084 mmol, 28% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.78 (s, 5 H, 5-CH3, piperdyl-3a-yl, piperidyl-5a-yl), 2.00 (br. s., 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.84 (br. s., 2 H, PhCH2), 4.31–4.46 (m, 1 H, piperidyl-4-yl), 5.12 (s, 2 H, (Ph)CH2O), 6.87 (t, J = 8.3 Hz, 2 H, Ph), 6.94 (br.s., 1 H, Ph), 7.23 (t, J = 7.8 Hz, 1 H, Ph), 7.37–7.47 (m, 3 H, Ph), 7.50–7.59 (m, 2 H, Ph, H-6), 11.26 (s, 1 H, NH), 2 H (CH2N) and 4 H (piperidyl-2-yl, piperidyl-6-yl) could not be observed. 13C NMR (101 MHz, DMSO-d6) δ ppm 12.1 (1 C, 5-CH3), 28.6 (2 C, piperdyl-3-yl, piperidyl-5-yl), 31.3 (1 C, PhCH2), 51.5 (1 C, piperidyl-4-yl), 51.7 (2 C, piperidyl-2-yl, piperidyl-6-yl), 68.1 (1 C, (Ph)CH2O), 109.0 (1 C, C-5), 112.5 (1 C, Ph), 115.4 (1 C, Ph), 121.3 (1 C, Ph), 126.1 (1 C, Ph), 127.3 (1 C, Ph), 127.7 (1 C, Ph), 129.5 (1 C, Ph), 130.4 (1 C, Ph), 133.1 (1 C, Ph), 137.5 (1 C, C-6), 139.7 (1 C, Ph), 150.8 (1 C, C-2), 158.2 (1 C, Ph), 163.7 (1 C, C-4), C (CH2N) and C (Ph) could not be observed. HRMS (ESI): m/z [M + H]+ Calcd. for [C25H28ClN3O3 + H]+ 454.1892, found 454.1899.
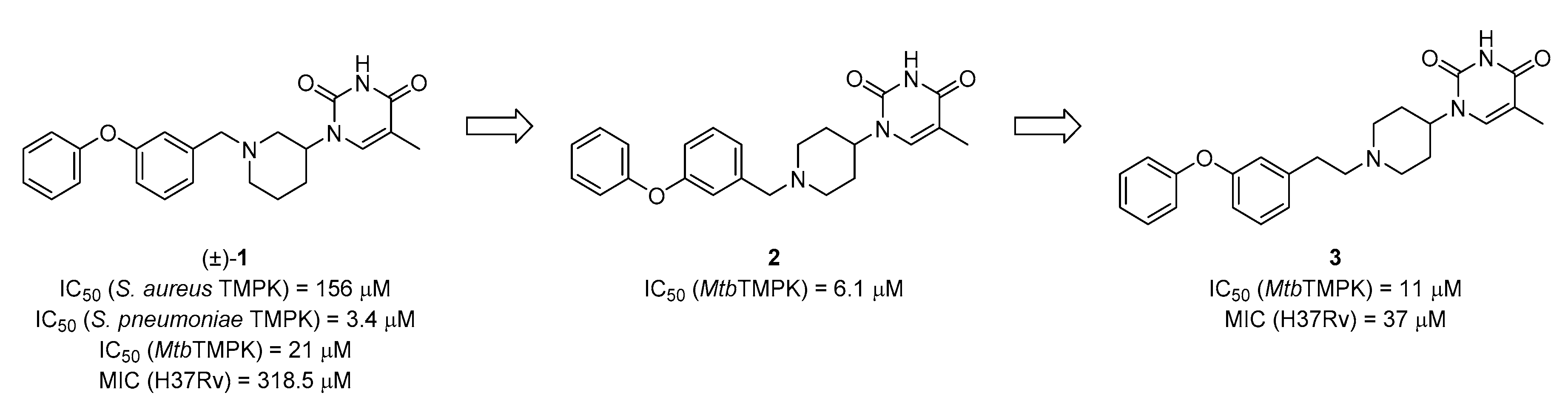
5-Methyl-1-(1-(3-phenoxyphenethyl)piperidin-3-yl)pyrimidine-2,4(1H,3H)-dione (23). Following the general procedure B, 12b (65 mg, 0.30 mmol), 29 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 23 (eluent system: 5% MeOH in DCM, 57 mg, 0.14 mmol, 46% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.43–1.56 (m, 1 H, piperdyl-5a-yl), 1.59–1.80 (m, 6 H, 5-CH3, piperdyl-4-yl, piperdyl-5b-yl), 2.07 (t, J = 12.9 Hz, 1 H, piperdyl-6a-yl), 2.22 (t, J = 10.3 Hz, 1 H, piperdyl-2a-yl), 2.56 (t, J = 4.6 Hz, 2 H, CH2N), 2.68–2.81 (m, 3 H, piperdyl-6b-yl, PhCH2), 2.82–2.90 (m, 1 H, piperdyl-2b-yl), 4.31–4.43 (m, 1 H, piperdyl-3-yl), 6.81 (dd, J = 8.1, 1.8 Hz, 1 H, Ph), 6.90 (s, 1 H, Ph), 6.95–7.04 (m, 3 H, Ph), 7.12 (t, J = 7.6 Hz, 1 H, Ph), 7.28 (t, J = 7.8 Hz, 1 H, Ph), 7.38 (t, J = 7.9 Hz, 2 H, Ph), 7.68 (s, 1 H, H-6), 11.23 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 23.9 (1 C, piperdyl-5-yl), 28.1 (1 C, piperdyl-4-yl), 32.3 (1 C, PhCH2), 51.0 (1 C, piperdyl-3-yl), 52.2 (1 C, piperdyl-6-yl), 56.5 (1 C, piperdyl-2-yl), 59.2 (1 C, CH2N), 108.6 (1 C, C-5), 116.1 (1 C, Ph), 118.5 (2 C, Ph), 119.0 (1 C, Ph), 123.3 (1 C, Ph), 123.9 (1 C, Ph), 129.7 (1 C, Ph), 130.0 (2 C, Ph), 138.0 (1 C, C-6), 142.7 (1 C, Ph), 150.8 (1 C, C-2), 156.5 (1 C, Ph), 156.7 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O3 + H]+ 406.2125, found 406.2125.
5-Methyl-1-(1-(2-(3-phenoxyphenyl)acetyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (
26). To a solution of
10b (0.2 g, 0.82 mmol) in MeOH (5.0 mL) was added 1M NaOH (5.0 mL), the resulting reaction mixture was stirred at 50 °C for 2 h. After cooling to room temperature, the reaction mixture was treated with 1N aq. HCl to pH 2–3. The generated white precipitate was collected through filtration and dried
in vacuo. The intermediate (0.17 g, 0.74 mmol),
28 (0.26 g, 0.79 mmol), EDC HCl (0.29 g, 1.5 mmol) and 4-dimethylaminopyridine (DMAP) (9.1 mg, 7.4 μmol) were dissolved in DCM (30 mL), and the reaction mixture was stirred at room temperature for overnight to afford BOM protected intermediate, which was deprotected with Pd/C (10%), H
2 and HCOOH (0.5%) in i-propanol/H
2O (10/1 mL) [
30] to give
26 (eluent system: 5% MeOH in DCM, 45 mg, 0.11 mmol, 13% yield).
1H NMR (300 MHz, DMSO-d
6)
δ ppm 1.54–1.83 (m, 7 H, 5-CH
3, piperdyl-3-yl, piperidyl-5-yl), 2.53–2.67 (m, 1 H, piperidyl-2/6-yl), 3.07 (t,
J = 10.8 Hz, 1 H, piperidyl-2/6-yl), 3.59–3.85 (m, 2 H, COCH
2), 4.05 (d,
J = 13.5 Hz, 1 H, piperidyl-2/6-yl), 4.43–4.56 (m, 2 H, piperidyl-4-yl, piperidyl-2/6-yl), 6.82–6.91 (m, 2 H, Ph), 6.95–7.03 (m, 3 H, Ph), 7.08–7.15 (m, 1 H, Ph), 7.31 (t,
J = 7.8 Hz, 1 H, Ph), 7.34–7.41 (m, 2 H, Ph), 7.53 (d,
J = 0.9 Hz, 1 H, H-6), 11.20 (s, 1 H, NH).
13C NMR (101 MHz, DMSO-d
6)
δ ppm 12.1 (1 C, 5-CH
3), 29.7 (1 C, piperidyl-3/5-yl), 30.5 (1 C, piperidyl-3/5-yl), 38.9 (1 C, (CO)CH
2), 40.8 (1 C, piperidyl-2/6-yl), 44.7 (1 C, piperidyl-2/6-yl), 52.1 (1 C, piperidyl-4-yl), 109.1 (1 C, C-5), 116.7 (1 C, Ph), 118.7 (2 C, Ph), 119.5 (1 C, Ph), 123.5 (1 C, Ph), 124.4 (1 C, Ph), 129.9 (1 C, Ph), 130.1 (2 C, Ph), 137.7 (1 C, Ph), 138.2 (1 C, C-6), 150.8 (1 C, C-2), 156.6 (1 C, Ph), 156.7 (1 C, Ph), 163.8 (1 C, C-4), 168.5 (1 C, CO(CH
2)). HRMS (ESI):
m/z [M + H]
+ Calcd. for [C
24H
25N
3O
4 + H]
+ 420.1918, found 420.1925.
1-(3-Phenoxyphenethyl)-4-phenylpiperidine (27). To a solution of 12b (0.12 g, 0.56 mmol) and 4-phenylpiperidine (0.14 g, 0.87 mmol) in dichloroethane (10 mL) was added sodium triacetoxyborohydride (0.24 g, 1.1 mmol), the resulting mixture was stirred at room temperature for overnight. The reaction mixture was diluted with DCM, and extracted with sat. NaHCO3 and brine. The collected organic layer was dried, concentrated and purified to give 27 (58 mg, 0.16 mmol, 29% yield). 1H NMR (300 MHz, CDCl3) δ ppm 1.91 (br. s., 4 H, piperdyl-3-yl, piperidyl-5-yl), 2.12–2.31 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.46–2.62 (m, 1 H, piperidyl-4-yl), 2.65–2.77 (m, 2 H, CH2N), 2.83–2.97 (m, 2 H, PhCH2), 3.20 (d, J = 10.8 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 6.82 - 6.95 (m, 2 H, Ph), 6.96–7.07 (m, 3 H, Ph), 7.12 (t, J = 7.3 Hz, 1 H, Ph), 7.17–7.46 (m, 8 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 33.0 (2 C, piperdyl-3-yl, piperidyl-5-l), 33.2 (1 C, PhCH2), 42.4 (1 C, piperidyl-4-yl), 54.1 (2 C, piperidyl-2-yl, piperidyl-6-yl), 60.3 (1 C, CH2N), 116.5 (1 C, Ph), 118.8 (1 C, Ph), 119.1 (1 C, Ph), 123.2 (1 C, Ph), 123.6 (1 C, Ph), 126.2 (1 C, Ph), 126.8 (3 C, Ph), 128.4 (2 C, Ph), 129.6 (1 C, Ph), 129.7 (2 C, Ph), 142.1 (1 C, Ph), 145.9 (1 C, Ph), 157.1 (1 C, Ph), 157.8 (1 C, Ph). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H27NO + H]+ 358.2166, found 358.2164.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}























