1. Introduction
Coumarins are plant-derived secondary metabolites, with the parent compound being coumarin, chromen-2-one. As with other naturally derived compounds such as flavones, chalcones, etc., there are a large number and wide variety of structures due to potential substitution of hydrogen atoms with other functional groups. The biological activities of these compounds have been well studied and include competing with Vitamin K to produce the anticoagulant activity of coumarins such as warfarin [
1].
Citrus plants produce coumarins and furanocoumarins and different citrus species show diversity in coumarin and furanocoumarin content. Interestingly, a 2015 study shows that pomelos, citrons and papedas synthesize high amounts of these compounds, whereas mandarins produce less [
2]. The furanocoumarin, bergamottin (5-Geranoxypsoralen),
Scheme 1, is a mechanism-based inhibitor of the Cytochrome P450 CYP3A4 enzyme, the major liver and intestinal P450 in humans. It is mainly responsible for the “grapefruit effect”, whereby the bioavailability of some pharmaceutical compounds is increased. CYP3A4 is the predominant member of the P450 family, a group of mixed-function oxidases that account for the metabolism of a majority of xenobiotic compounds as well as endogenous biochemicals such as cholesterol [
3]. Bergamottin also inhibits other members of the P450 family such as CYP1A1, and to a lesser extent, CYP1A2.
A recent review highlights the structural features of cytochrome P450 enzymes which catalyze a large variety of oxidation reactions and which show an extraordinary flexibility in the dimensions of the P450 active site that depends on the chemical features of their substrates [
4]. The coordinates of a large number of cytochrome P450 enzyme structures that have been studied are available from the Protein Data Bank (PDB) [
5]. Observing those structures in the PDB with bergamottin or its derivatives at the active site, we note a crystal structure of CYP1A1 (PDB 6DWM) that shows the bergamottin geranoxy tail bent over the heme, while its psoralen ring system displays π–π interactions with Phe-224 of the F-helix [
6]. A recent report of a crystal structure of CYP3A4 with 6′,7′-dihydroxybergamottin, DHB, a metabolite of bergamottin, bound at the active site, also exists (PDB 6OOB) [
7]. The DHB psoralen ring lies above the heme and parallel to the I-helix, with the carbonyl oxygen H-bonded to the S119 hydroxyl group. Very recently, docking calculations on bergamottin, and a synthesized bergamottin derivative containing a nitroxide moiety and having antitumor effects, with CYP3A4 (PDB 2V0M), were performed [
8].
We also investigated a C-prenylated coumarin-derivative, osthole (7-methoxy-8-(3-methyl-2-butenyl)-2
H-1-benzopyran-2-one),
Scheme 1, that shows neuroprotective, osteogenic, anticancer, hepatoprotective and potential antioxidant properties. Osthole is found in citrus peels [
2] and in
Cnidium monnieri, a flowering plant commonly used in traditional Chinese medicine.
C. monnieri has been traditionally used to treat itching, inflammation, and other skin ailments [
9].
The third coumarin studied is fraxidin (6,7-dimethoxy-8-hydroxycoumarin),
Scheme 1. It can be found, among other natural sources, in the inner shell of the Japanese chestnut,
Castanea crenata. It is of interest as a potential antidiabetic compound because of its insulin mimetic ability [
10].
In this work, we describe the experimental results of single crystal X-ray structure determinations of three coumarin compounds as well as computational studies describing the docking of these coumarins into the active site of a CYP3A4 protein (PDB 4D78) after removal of ritonavir [
11]. Single crystal studies are ideal for studying the packing between molecules which is dependent on the overall effect of all the intra- and intermolecular interactions in the crystal. The π–π stacking and hydrogen bonding are two important and significant forces in the interaction of molecules, regardless of whether they are a protein or a small molecule. The former interaction is not as simple to understand as hydrogen bonding and has been the subject of a recent review [
12]. Janiak performed an early systematic and statistical investigation on stacking in metal–π–ligands by analyzing quantitative experimental crystal data found in the Cambridge Structural Database, CSD [
13] and concluded that most interactions were parallel displaced and that atom–atom contact distances are an important measure to suggest the strength of π–π interactions with values around 3.3 Å (strong) > 3.6 Å (weaker) > 3.8 Å (weakest distance for which π–π interactions are acknowledged) [
14]. Although weaker than H-bonding, the stabilization energy in these widely found interactions increases when there are heteroatoms in the rings. For example, the stacking energy in nucleic acid bases is 10–17 kcal/mol [
15].
The purpose of the current study was to study how the crystal structure results of three different coumarin structures can assist us in probing CYP3A4 active site flexibility. We evaluate the relationship of the hydrogen bonding and interactions between conjugated systems in these three coumarin structures relative to the positions of these small molecules as they are docked into the CYP3A4 active site. In particular, we consider how the molecular structure of bergamottin can influence the active site molecular interactions and might induce the adverse physiological effect seen in many individuals upon eating grapefruit.
4. Theoretical Computational Studies
In order to study the interactions at the CYP3A4 active site, we perform docking studies using the crystal structure coordinates of our three coumarin structures placed into the active site of a CYP3A4 protein whose crystal structure coordinates are available from the Protein Data Bank. We chose the protein (PDB 4D78) because it was co-crystallized with a large drug molecule ritonavir at the active site. Ritonavir is a strong CYP3A4 inactivator [
11]. Therefore, after the protein was “prepared” following the standard protocol, which includes H generation, the sulfur (ritonavir) atom was selected to establish the center of a sphere whose radius was 8 Å. Then, ritonavir was removed and bergamottin was docked using the program CDocker, which applies the molecular mechanic system CHARMm. Since the P450 protein contains the heme prosthetic group with an Fe metal iron, we employed CHARMm36 instead of using the default CHARMm setting. The resulting 10 configurations show two well distinct groups of interaction with the P450 heme. Poses 1–5 have the coumarin carbonyl coordinated to iron, in contrast with poses 6, 8–10 that show π–π co-planarity between the flat bergamottin aromatic rings and protein heme.
Figure 5 shows the protein amino acid residues interacting with bergamottin pose 6; a H(Ser−119) has a contact with furan ring O of bergamottin. This is described as a van der Waals type by the docking procedure which keeps the protein fixed, yet it is indeed a H-bond after free rotation by the corresponding serine moiety. Thus, after a serine torsion rotation from H-C-O-Hc 65.7° to 55°, [d[Hc-O(Berg6)]] becomes 2.59 Å. This may be an interesting specific interaction for bergamottin compared with other coumarins, see below. It should be noted that both terminal NH
2 groups of Arg−105 are engaged with both P450 heme carboxylates through H-bonds (
Figure 6 and
Figure 7). However, bergamottin carbonyl in pose 6 also interacts with Arg−105 through the adjacent NH group. The role of Ser−119 and Arg−105 will also be explored using a quantum-mechanical approach.
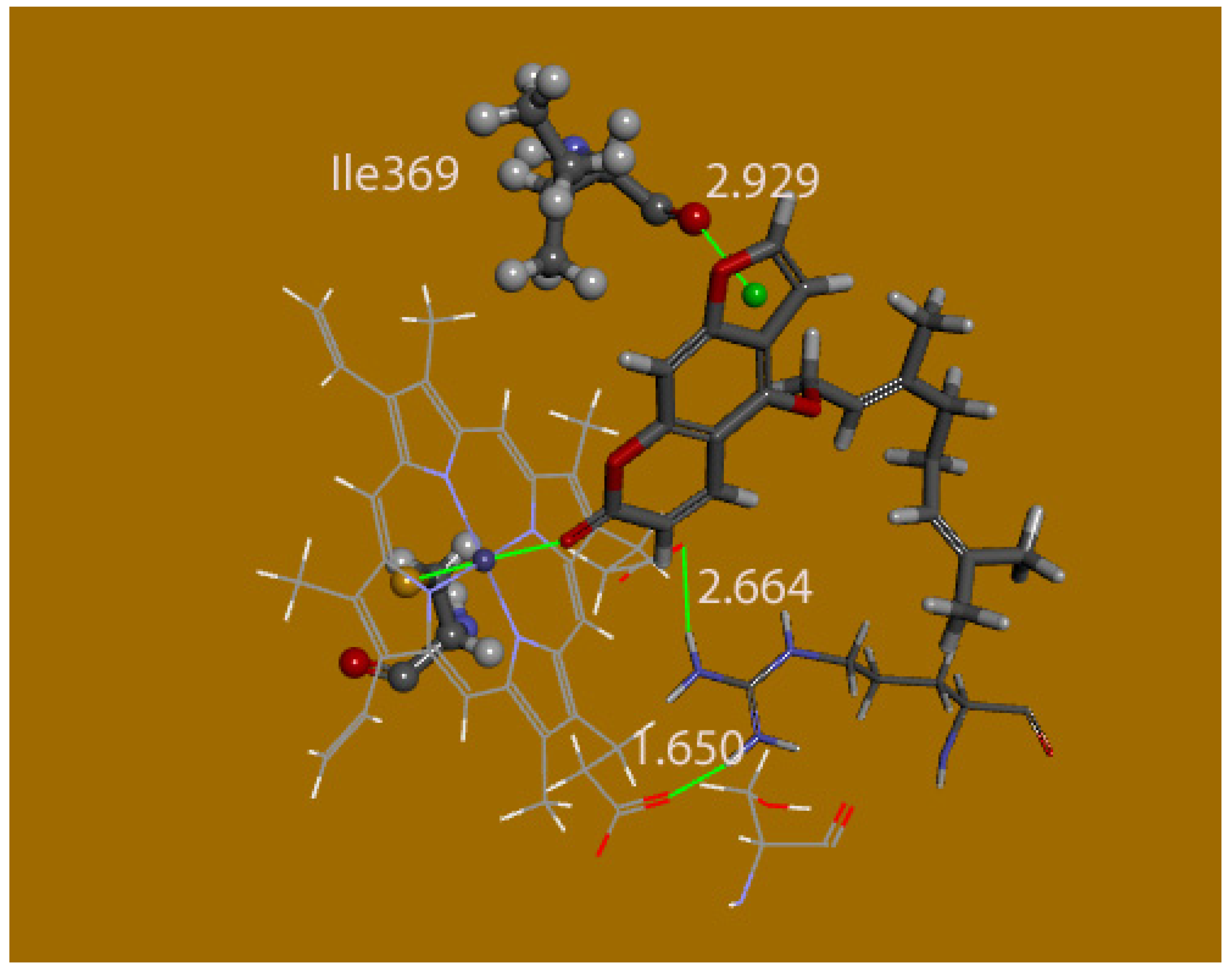
The protein amino acid residues interacting with bergamottin pose 5 are shown in
Figure 8. Bergamottin pose 5 (
Figure 9) shows the iron heme center displaying an octahedral configuration. Most of residual interactions are of the van der Waals type. In addition, bergamottin pose 4 shows the heme coordination sphere with the coumarin carbonyl pointing to Fe, and, in addition, a lone pair–π interaction between Ile−369 and the furan ring, 2.292 Å. This is interesting because it can clarify a specific role for bergamottin, compared with other coumarins missing the furan moiety.
The protein amino acid residues interacting with bergamottin pose 4 are shown in
Figure S5. Bergamottin pose 4 (
Figure 10) shows heme coordination sphere and a lone pair–π interaction between Ile−369 and the furan ring at 2.292 Å. This is noteworthy because it can clarify a specific role for bergamottin interaction.
Cdocker energy for poses 4, 5 and 6 of bergamottin are positive at 12.0, 12.1, and 12.4 kcal/mol, respectively. The CDocker interaction energies are all negative at −44.3, −41.8 and −38.4 kcal/mol respectively. A standard dynamic cascade was performed for the Berg6 conformer, without atomic restrictions, and the π–π arrangement was kept. For instance, the distance between the furan ring centroid and one C(heme) adjacent to one N(heme) is 3.815 Å. In contrast, the iron atom became displaced out of the N4 heme plane. This may be expected when a molecular mechanics program is used to describe a metal coordination sphere, and so a quantum-mechanical approach was employed, as shown below.
4.1. Quantum-Mechanical Calculations for Poses 6,5,4
Berg6: The input file was taken from the DS procedure, that is, the docked molecule of Berg6 was transferred into the Dmol3 program to perform geometry optimization. To conserve the protein environment, the following amino acid residues, Phe−302_Ile−303_Phe−304_Ala−305_Gly−306, were included in the DFT calculation, and their atomic coordinates were fixed. We also included some other residues, engaged in important H-bonds; these were less restrained. Thus, Ser−119, engaged in H-bonds with Berg6 and Arg−105 had the maximum freedom, that is, its connected residues Ile−118 and Ile−120 were replaced by H atoms, which were kept fixed. Arg−105 and Arg−106 were included and partially refined, that is, only the hydrocarbon chain of Arg−105 was not restricted, while their associated peptides Asn−104 and Pro−107 were substituted by H atoms, kept fixed. In addition, heme and Berg6 were not restricted at all.
Figure S6 shows this initial situation. After minimization, the obtained structure confirms the π–π arrangement (
Figure 11). However, some important changes are observed, for instance, the potential interaction between bergamottin ether oxygen with iron seen in
Figure 7 was not confirmed, resulting in the lengthening of the corresponding Fe-O distance from 3.652 to 5.368 Å. Neither is the bergamottin carbonyl interaction with NH(Arg−105) seen after docking, as both atoms appeared completely apart at 4.782 Å. Cys−442 remains basically at the same distance from iron as in the docked molecule. The most important feature is shown by Ser−119 strongly bound to the O(furan) bergamottin, which may be important to explain bergamottin biological inhibitory action.
Berg5: The DFT geometry minimization of Berg pose 5 (
Figure 12) also shows interesting features. The bergamottin O(carbonyl) moves markedly apart from the metal center at 3.132 Å; its initial separation after docking was much closer at 2.311 Å. After such shifting, Berg5 does not establish any other new interaction. Arg−105 and Ser−119 were treated the same way as was done with Berg6. This resulted in confirmation of important H-bond interactions. Thus, the Ser−119 H-bond to the NH
2(Arg−105), which was 2.664 Å in the 4D78 protein, became strengthened to 1.998 Å. In addition, H-bonds seen between Arg−105 NH
2 and NH groups to both heme carboxylates were confirmed at distances of 1.802 and 1.857 Å. Therefore, an important result of applying the DFT procedure to the docked molecular arrangement is the displacement of bergamottin from the heme coordination sphere, that is, the loss of the Fe-O(carbonyl) bond.
Berg4: The minimization of Berg4 shows the π–π interaction between the furan ring and the Ile−369 O(carbonyl) not confirmed, as the original distance of 2.292 Å becomes 5.59 Å. In addition, bergamottin O(carbonyl) bonding to the iron center is also lost, becoming d[O(carbonyl)-Fe] = 3.616 Å. We conclude that the active site interaction based on Berg4 docked configuration is not realistic.
The comparison of energies between Berg6 and Berg5 after Dmol application is of interest. Since both processes involved exactly the same molecules, including the same fixed atoms, this parameter may describe which configuration is preferred. The difference is 6.2 kcal/mol favoring Berg6, and so, the π–π configuration is energetically more stable. Moreover, as already described, the interaction between the furan oxygen with Ser−119 in Berg6 seems to provide another important element of consideration, as this is a specific structural feature of bergamottin, perhaps related to its characteristic biological activity, and so, we conclude that this π–π configuration for bergamottin suggests its inhibitory role of on CYP3A4, which may explain why bergamottin disturbs the metabolism of drugs, that is, it makes P450 be less available to perform its expected task [
3].
4.1.1. Comparison of Bergamottin with Other Coumarins
Fraxidin
Fraxidin was docked using the same procedure described earlier for bergamottin. The 10 conformers always show an O(carbonyl) interaction with Fe. The most interesting conformer is pose 8, as seen by its 2D interactions below (
Figure S7). This differs from the others by also having an H-bond with Ser−119. Therefore, its coordinates were input into the Dmol3 program to run a geometry minimization. This resulted in confirmation of the heme Fe-O(carbonyl) bonding (
Figure 13).
4.1.2. Osthole
Osthole was docked using the same procedure described earlier for bergamottin and fraxidin; of the 10 poses, pose 1, Ost1, and pose 3, Ost3, were selected for further study. The 2D interactions of pose 1 show O(carbonyl) bound to iron (
Figure S8). The same diagram for Pose 3 has osthole O(carbonyl) engaged in 2 H-bonds and does not have a π-π interaction (
Figure S9).
The two configurations were geometry optimized with DFT methods and Ost3 was energetically more favored by 27.3 kcal/mol. The converged structures for pose 1 are shown in
Figure 14 and
Figure S10. The converged structures for the more stable pose 3, Ost3, are seen in
Figure 15;
Figure 16.
5. Results and Discussion
The crystal structures of three coumarin derivatives reveal their propensity for parallel-displaced π–π stacking, which is to be expected since coumarin contains an extended π-system. Short π–π distances imply strong charge transfer possibilities and makes them important contributors to the overall stability of the crystals. Additionally, in all three crystal structures, when proton donors are present, specific and directional hydrogen bonds are seen. Intermolecular interactions regulated by chemical functionality and molecular geometry thus, help manage molecular organization. These contacts help to identify potential molecular specificity or recognition and many chemical and biological pathways rely on their presence.
Taking advantage of the crystal structures of bergamottin, osthole and fraxidin, we inserted their atomic coordinates into a molecular mechanics docking procedure (Discovery Studio program) using CHARMm36 forcefield, specially designed to deal with metals such as iron, in the heme group at the enzyme active site. The selected receptor for this process was PDB 4D78, which contains coordinates for the CYP3A4 enzyme plus ritonavir, a potent enzyme inactivator [
11]. We proceeded by docking bergamottin and found two distinct configurations. In one, there is an O(carbonyl)-Fe bond establishing an octahedral metal coordination, corresponding to bergamottin acting as a sixth ligand (pose 5). The other arrangement is described as π–π configuration involving the heme moiety (pose 6). Our work tried to differentiate between the two configurations.
Although energy is a discriminatory parameter, and the – system has a lower energy, suggesting it to be more stable as indicated by the CDocker interaction energy ∆E = 3.3 kcal/mol, we must proceed cautiously when comparing docking output results. Therefore, a standard dynamic cascade procedure was applied to both configurations, and some atoms at the active site showed substantial changes. For instance, the Fe atom became displaced out of the heme plane of pose 5 and the O(carbonyl)-Fe bond was lost, while in the π–π configuration (pose 6), there was only some shifting in the bergamottin position. In both configurations, a square-pyramidal coordination was shown by the iron center. Hence, a quantum-mechanical approach was used to discern the two docked configurations—one having the π–π arrangement, Berg6, and the other displaying bergamottin carbonyl coordinated to the Fe center, Berg5. Thus, we performed geometry optimizations for both configurations using the DFT program Dmol3. This program uses fewer parameters than those dealt with in a molecular mechanics program, and so, we had to restrain the protein environment. To save the active site conditions imposed on bergamottin, some amino acids were introduced in our calculation; the location of most of them implied van der Waals interactions and so, they were kept fixed to decrease calculation times. However, associated amino acids showing interactions such as H-bonds were allowed to vary in our calculation since these are very important. For instance, in both cases, the two heme carboxylate moieties are linked through H-bonds to Arg−105, and a substantial part of this amino acid was not fixed.
After operating with the two configurations, we reached the following conclusions:
In the pose 5 configuration, Berg5, the iron bonding to the coumarin carbonyl was lost after geometry optimization, and so, Fe-O(carbonyl) distance, which was initially 2.311 after docking, became 3.132 Å. It has to be noted that the whole protein system was kept in place, for instance, Arg−105 had H-bonds with heme carboxylates and also Ser−119 with Arg−104 amino groups were conserved. In addition, Berg5, after removal from the iron coordination sphere, did not establish alternative interactions.
On the other hand, Berg6, pose 6 conserved the π–π arrangement, even though a slight shift was generated by the geometry optimization. More importantly, the initial H-bond established between Ser−119 and O(furan) bergamottin was conserved. This may be important to explain bergamottin biological inhibitory action, i.e., this feature seems to be specific for bergamottin.
An additional arrangement, pose 4, was also analyzed quantum mechanically, but resulted not feasible. The difference in energy between poses 5 and 6 was marked, 6.2 kcal/mol favoring pose 6, and so, the π–π configuration is energetically more stable. It is interesting to compare our pose 6 configuration with a docking study of bergamottin [
8], which used a different receptor (PDB 2V0M) derived from the crystal structure of CYP3Y4 together with inhibitor ketoconazole. After docking, a stacking configuration was also concluded. However, in contrast with our study, H-bonds between the hydrophilic amino acid (Thr309) and the furan oxygen were described. There was no Ser119 H-bonding with bergamottin. Our docking study used the default setting of 10 poses and since we were intrigued by these different outcomes, we performed an additional docking inquiry increasing to 40 poses; this confirmed no interaction involving Thr309. The location of Thr309 is essentially opposite to Ser119, as both amino acids are just across from the Fe atom. Thus, the distance between O(Thr309) (potentially involved in H bonds) and Fe is 6.18 Å, slightly shorter than that between Fe and O(Ser119) at 8.44 Å. We conclude that the different display for the stacking configuration obtained in both studies is the result of different spatial availability for bergamottin in the active site, after eliminating ketoconazole [
8] or ritonavir [
11] in our study.
Another interesting comparison can be made with the substrate 6′,7′-dihydroxybergamottin, DHB, which has been described recently in a CYP3A4 crystal structure, PDB 6OOB [
7]. Here, a marked difference is a molecule of water coordinated to the Fe atom and the aromatic system of DHB positioned 3.6–5.2 Å above the heme (~15° tilt). Interestingly, Ser119 forms an H-bond with the coumarin carbonyl. It is obvious that the polar part of DHB plays an important role in situating the guest in the crystal. That bergamottin and DHB would interact differently at the active site is to be expected as this study presented some experimental binding differences between bergamottin and DHB at the active site based on the shape of the titration curves—sigmoidal for bergamottin and hyperbolic for DHB [
7].
Only one other P450 crystal structure exists with bergamottin at the active site, CYP1A1 (PDB 6DWM) [
6]. In that situation, bergamottin’s aromatic ring system is involved in π–π interactions with Phe−224 of the F-helix (and not with the heme group), while its geranoxy tail is bent over the heme with the terminal 6′,7′ double bond lying offset from the heme iron at distances of 4.8 and 5.1 Å. From the placement and interactions of bergamottin at the active site, the authors suggest that CYP1A1 is likely to metabolize bergamottin into the 6′,7′-dihydroxy metabolite, DHB.
Therefore, the docking and DFT treatment of bergamottin, according to our study, seems to be consistent with the biologically important property of P450 inhibition. Bergamottin is found in the active site with the planar psoralen ring system stacked above the heme and with the furan oxygen atom about 4 Å distance from the heme iron, in contrast to the bergamottin location seen in two crystal structures described earlier. That is, the π–π display has lower energy than the Fe-carbonyl coordinated configuration. In addition, our π–π arrangement is closely related to the one established by the strong inactivator ritonavir [
11], as in both cases, no Fe-O(carbonyl) bond is seen. Furthermore, the Fe-O pose within bonding distance, pose 5, was not confirmed by DFT, suggesting it is nonexistent. To our knowledge, there are no other structural studies of P450 protein crystal structures with other coumarins, except those described earlier. None show any Fe-O(carbonyl) bonding.
When reviewing the literature for fraxidin, we found no studies describing its interaction with the P450 enzymes. Our computational studies of 10 docked configurations always show an O(carbonyl) interaction with Fe, as was seen for Berg6. The most interesting conformer is Fraxi8, as it differs from the others by also incorporating an H-bond between Ser−119 and [O(7-methoxy)]. The quantum-mechanical approach confirmed this H-bond as well as the Fe-O(carbonyl) bond. Osthole docking studies confirm Fe-O(carbonyl) coordination plus hydrogen bonding as the lowest energy configuration.
These results led us to infer that the distinguishing feature of the bergamottin molecular structure that permits such a strong inhibition reaction with CYP3A4 was the very strong stacking interaction. We searched the Cambridge Crystallographic Database (CSD) [
13] for other furanocoumarin natural products and saw that presence of the O(ether) functional group played a major role in their stacking distance. In fact, furanocoumarin (CSD-FURCOU), with no substituent at all, had a stacking distance of 3.5 Å. Furanocoumarins with 8-O(ether) tail substituents (across from bergamottin’s) showed weaker stacking 3.7–3.8 Å (CSD-TOPXIH, CSD-YALZES). On the other hand, furanocoumarins with an 5-O(ether) tail in the same location as bergamottin, oxypeucedanin (CSD-PEXXAS), isoimperatorin (CSD-NUVHER), imperatorin (CSD-YAGHOE), and pranferol (CSD-KAHGEI), all had strong stacking between the furanocoumarin ring structure, around 3.4 Å. In particular, a natural furanocoumarin whose molecular structure is strikingly similar to bergamottin, notopterol (CSD-SOLXUN), was catalogued [
23]. This compound is a widely used component of Chinese herbal medicine and its molecular structure is the same as bergamottin, except for a hydroxyl substitution at 5′. The comparison of the crystal packing for bergamottin and notopterol is shown in
Figure 17.
It is clear that the extra polar 5′-OH group in notopterol causes a different type of packing (head–tail) than that seen in bergamottin (head–head). However, in both compounds, the furanocoumarin three-ring system has a very short stacking distance at 3.34 Å. With this similitude, we would expect notopterol to also be a strong inactivator of cytochrome P450 enzymes, and, indeed, the literature studies show it to be a strong inhibitor of CYP3A4 [
24]. Additionally, a very recent article [
25] demonstrated that notopterol inhibits around 94% of CYP2D6 activity, followed by CYP2B6 (77%) and CYP3A4 (50%).


 ,
, 




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}