Synthesis of α-Aminophosphonic Acid Derivatives Through the Addition of O- and S-Nucleophiles to 2H-Azirines and Their Antiproliferative Effect on A549 Human Lung Adenocarcinoma Cells




Abstract
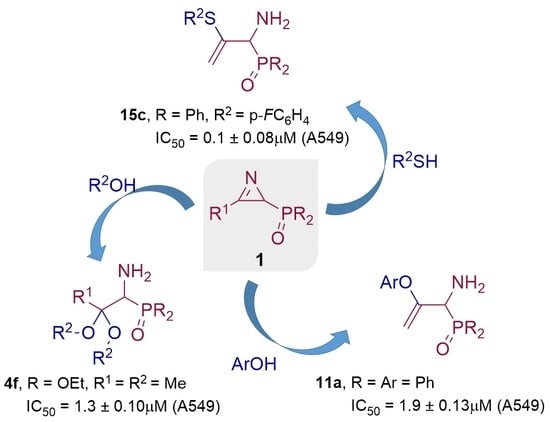
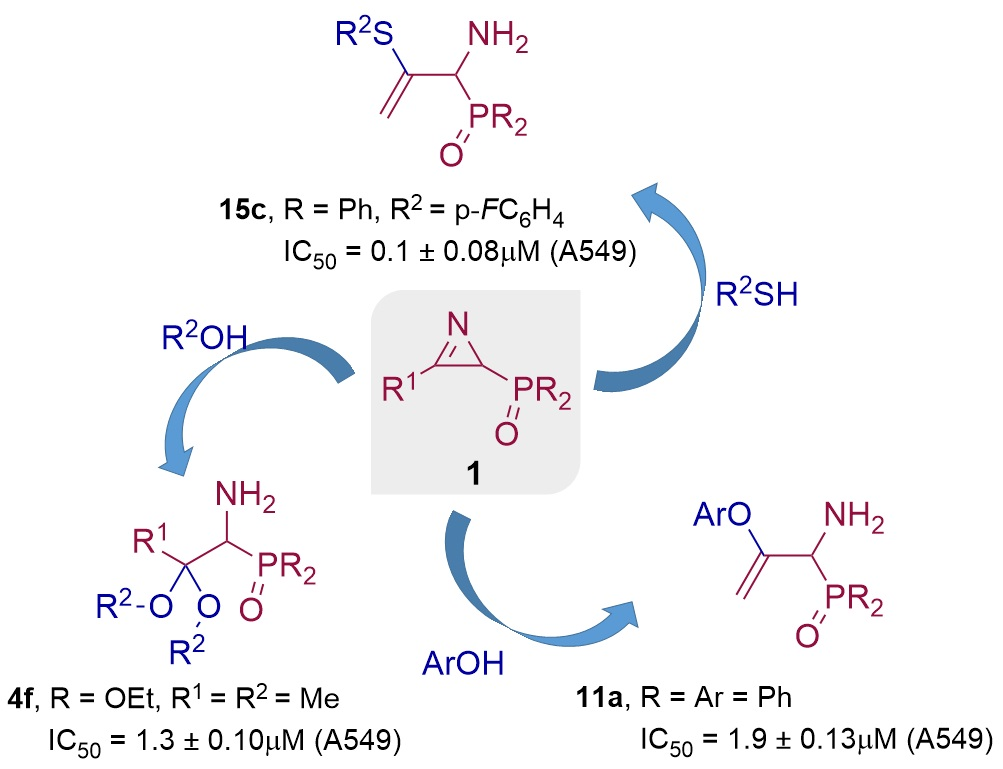
1. Introduction
2. Results
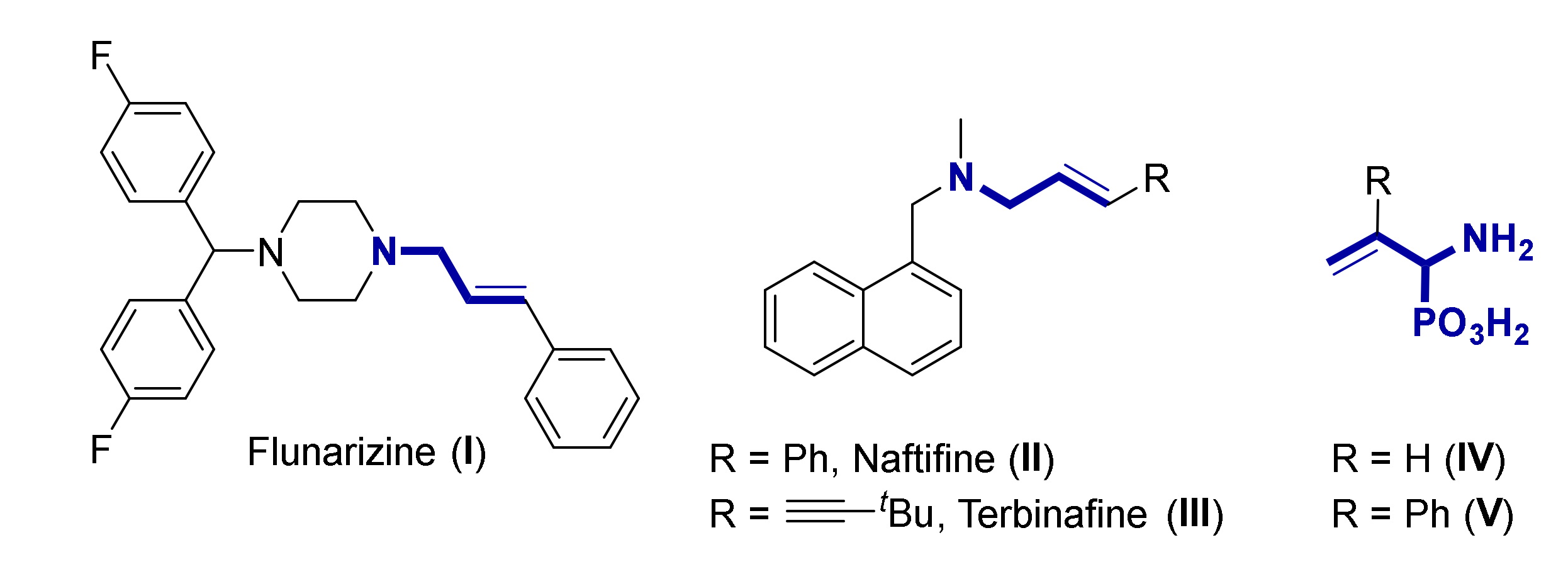
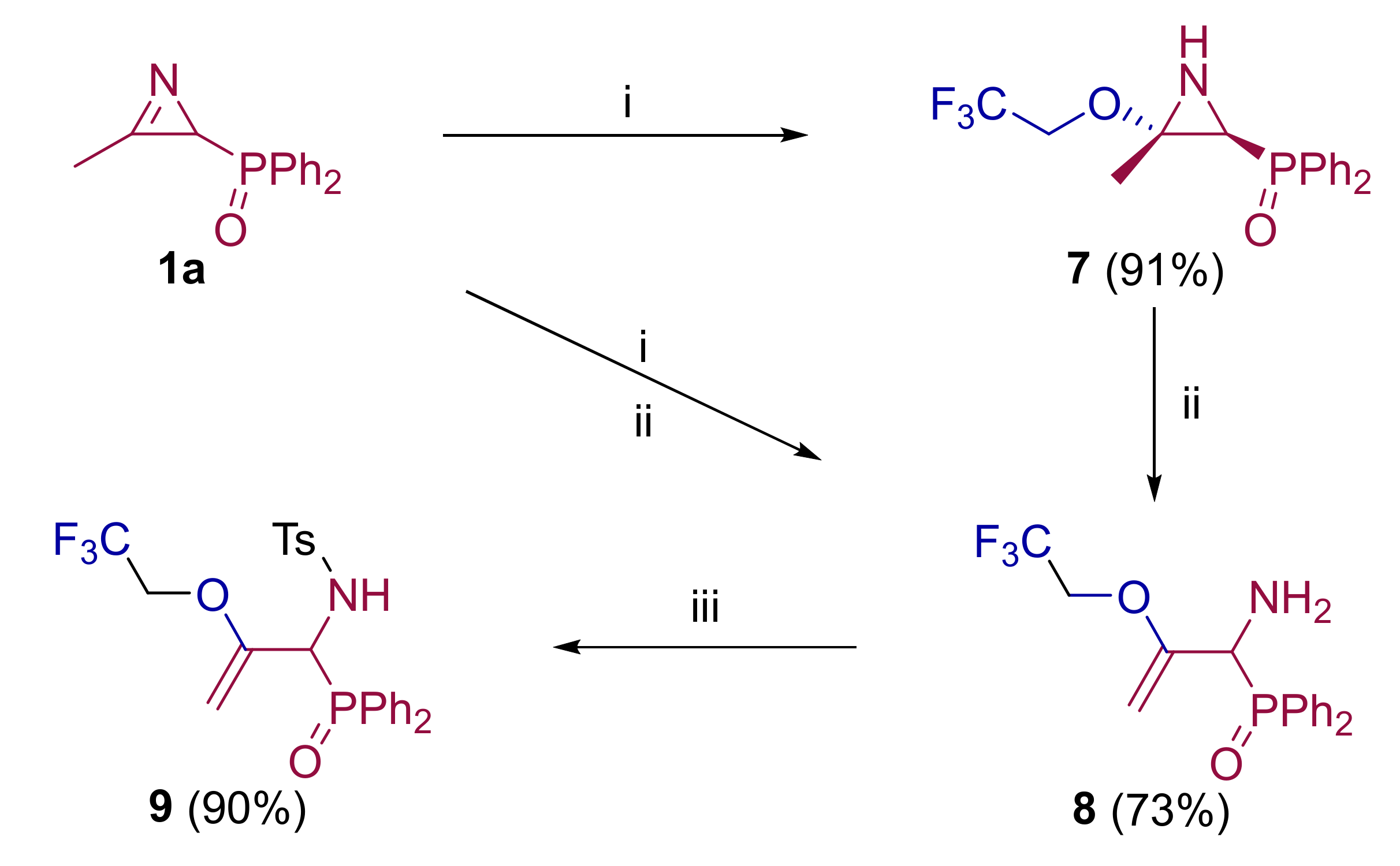
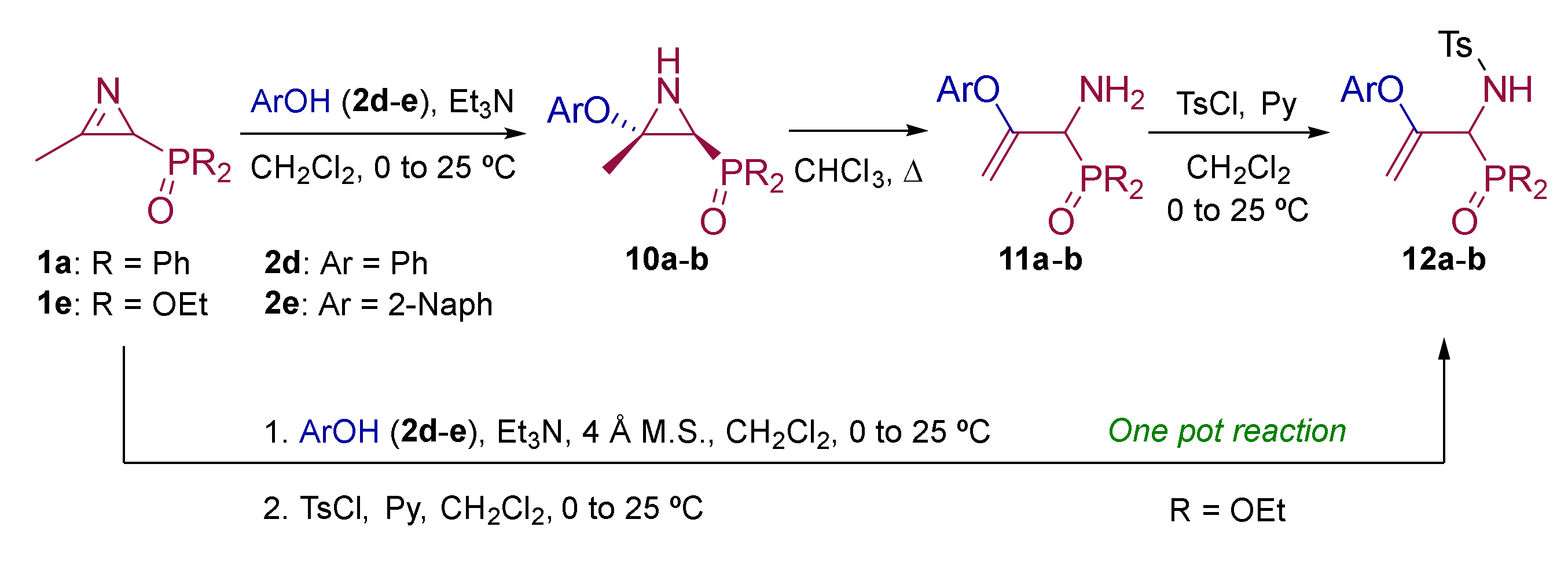
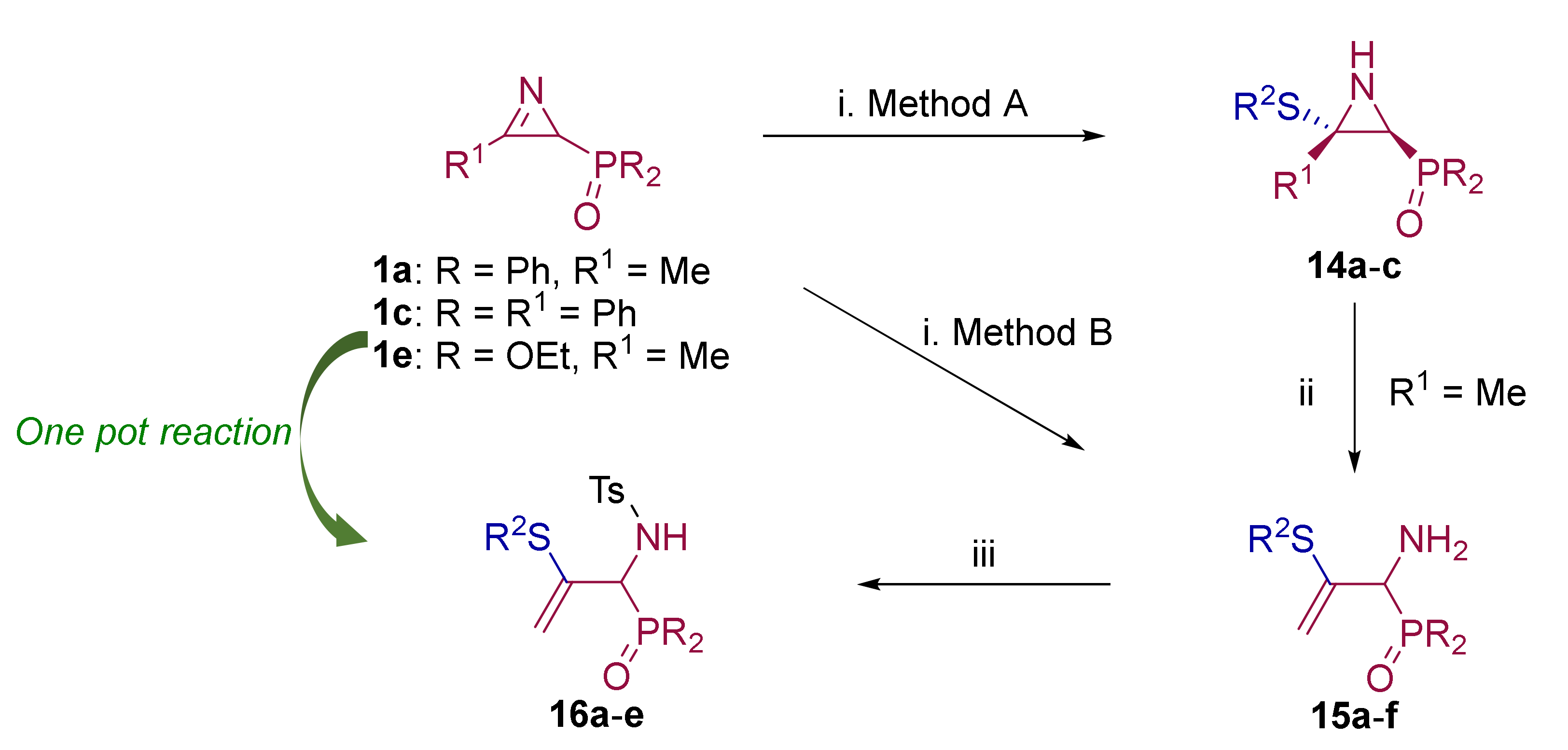
2.1. Chemistry
2.2. Biological Results
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Experimental Procedure and Characterization Data for Compounds 4, 5 and 6
General Procedure and Spectral Data for the Addition of Aliphatic Alcohols to Functionalized 2H-Azirines 1






General Procedure and Spectral Data for the N-Tosyl Functionalization of α-Aminophosphine Oxide and Phosphonate Acetals 4



One Pot Procedure for the Synthesis of N-tosyl-α-Aminophosphonate Acetal 5d

General Procedure and Spectral Data of β-Keto-α-Aminophosphine Oxide 6

3.1.3. Experimental Procedure and Characterization Data for Compounds 7, 8 and 9
General procedure and spectral data for the addition of 2,2,2-trifluoroethanol (2c) to functionalized 2H-azirines 1

General Procedure and Spectral Data of Allylic α-Aminophosphine Oxide 8

General Procedure and Spectral Data of Allylic N-Tosyl α-Aminophosphine Oxide 9

3.1.4. Experimental Procedure and Characterization Data for Compounds 10, 11 and 12
General Procedure and Spectral Data for the Addition of Phenols (2d–e) to Functionalized 2H-Azirines 1


General Procedure for the Preparation of Allylic α-Aminophosphine Oxides 11


One Pot Procedure for the Synthesis of N-Tosyl Allyl Amines 12 Derived From Phosphonate


3.1.5. Experimental Procedure and Characterization Data for Compounds 14, 15 and 16
General Procedure and Spectral Data for the Addition of Thiophenols and Thiols to 2H-Azirines 1









General Procedure and Spectral Data for the N-Tosyl Functionalization of Allylic α-Amino-phosphine Oxides and Phosphonates 15



One pot procedure for the synthesis of N-tosyl allylic α-aminophosphonates 16d–e


3.2. Biology
3.2.1. Materials
3.2.2. Cytotoxicity Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, K.; Cheng, X.; Zhao, C.; Liu, C.-C.; Jia, C.; Feng, L.; Xiao, J.-M.; Zhou, L.-S.; Gao, H.-Z.; Yang, X.; et al. Synthesis and activity study of phosphonamidate dipeptides as potential inhibitors of VanX. Bioorg. Med. Chem. Lett. 2011, 21, 7224–7227. [Google Scholar] [CrossRef]
- Van Der Jeught, S.; Stevens, C.V. Direct Phosphonylation of Aromatic Azaheterocycles. Chem. Rev. 2009, 109, 2672–2702. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, S.; Xeilari, M.; Yiotakis, A.; Grembecka, J.; Pawelczak, M.; Kafarski, P.; Mucha, A. A synthetic method for diversification of the P1’ substituent in phosphinic dipeptides as a tool for exploration of the specificity of the S1’ binding pockets of leucine aminopeptidases. Bioorg. Med. Chem. Lett. 2007, 15, 3187–3200. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B. Aminophosphonic acids of potential medical importance. Curr. Med. Chem. Agents 2001, 1, 301–312. [Google Scholar] [CrossRef]
- Huang, R.-Z.; Wang, C.-Y.; Li, J.-F.; Yao, G.-Y.; Pan, Y.-M.; Ye, M.-Y.; Wang, H.-S.; Zhang, Y. Synthesis, antiproliferative and apoptosis-inducing effects of novel asiatic acid derivatives containing α-aminophosphonates. RSC Adv. 2016, 6, 62890–62906. [Google Scholar] [CrossRef]
- Yao, G.-y.; Ye, M.-y.; Huang, R.-z.; Li, Y.-j.; Pan, Y.-m.; Xu, Q.; Liao, Z.-X.; Wang, H.-s. Synthesis and antitumor activities of novel rhein α-minophosphonates conjugates. Bioorg. Med. Chem. Lett. 2014, 24, 501–507. [Google Scholar] [CrossRef]
- Lavielle, G.; Hautefaye, P.; Schaeffer, C.; Boutin, J.A.; Cudennec, C.A.; Pierre, A. New α-aminophosphonic acid derivatives of vinblastine: Chemistry and antitumor activity. J. Med. Chem. 1991, 34, 1998–2003. [Google Scholar] [CrossRef]
- Atherton, F.R.; Hassall, C.H.; Lambert, R.W. Synthesis and structure-activity relationships of antibacterial phosphonopeptides incorporating (1-aminoethyl)phosphonic acid and (aminomethyl) phosphonic acid. J. Med. Chem. 1986, 29, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Lejczak, B.; Kafarski, P.; Sztajer, H.; Mastalerz, P. Antibacterial activity of phosphono dipeptides related to alafosfalin. J. Med. Chem. 1986, 29, 2212–2217. [Google Scholar] [CrossRef] [PubMed]
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The Most Potent Organophosphorus Inhibitors of Leucine Aminopeptidase. Structure-Based Design, Chemistry, and Activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Bird, J.; De Mello, R.C.; Harper, G.P.; Hunter, D.J.; Karran, E.H.; Markwell, R.E.; Miles-Williams, A.J.; Rahman, S.S.; Ward, R.W. Synthesis of novel N-phosphonoalkyl dipeptide inhibitors of human collagenase. J. Med. Chem. 1994, 37, 158–169. [Google Scholar] [CrossRef]
- Lan, X.; Xie, D.; Yin, L.; Wang, Z.; Chen, J.; Zhang, A.; Song, B.; Hu, D. Novel α,β-unsaturated amide derivatives bearing α-amino phosphonate moiety as potential antiviral agents. Bioorganic Med. Chem. Lett. 2017, 27, 4270–4273. [Google Scholar] [CrossRef] [PubMed]
- Hirschmann, R.; Smith, A.; Taylor, C.M.; Benkovic, P.; Taylor, S.; Yager, K.; Sprengeler, P.; Benkovic, S. Peptide synthesis catalyzed by an antibody containing a binding site for variable amino acids. Science 1994, 265, 234–237. [Google Scholar] [CrossRef]
- Allen, M.C.; Fuhrer, W.; Tuck, B.; Wade, R.; Wood, J.M. Renin inhibitors. Synthesis of transition-state analog inhibitors containing phosphorus acid derivatives at the scissile bond. J. Med. Chem. 1989, 32, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Logusch, E.W.; Walker, D.M.; McDonald, J.F.; Leo, G.C.; Franz, J.E. ChemInform Abstract: Synthesis of α- and γ-Alkyl Substituted Phosphinothricins: Potent New Inhibitors of Glutamine Synthetase. ChemInform 1989, 20, 4069–4074. [Google Scholar] [CrossRef]
- Patel, D.V.; Rielly-Gauvin, K.; Ryono, D.E. Preparation of peptidic α-hydroxy phosphonates a new class of transition state analog renin inhibitors. Tetrahedron Lett. 1990, 31, 5587–5590. [Google Scholar] [CrossRef]
- Deng, S.-L.; Baglin, I.; Nour, M.; Flekhter, O.; Vita, C.; Cavé, C. Synthesis of Ursolic Phosphonate Derivatives as Potential Anti-HIV Agents. Phosphorus Sulfur Silicon Relat. Elements 2007, 182, 951–967. [Google Scholar] [CrossRef]
- Stowasser, B.; Budt, K.-H.; Jian-Qi, L.; Peyman, A.; Ruppert, D. New hybrid transition state analog inhibitors of HIV protease with peripheric C2-symmetry. Tetrahedron Lett. 1992, 33, 6625–6628. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Skoda, E.M.; Davis, G.C.; Wipf, P. Allylic Amines as Key Building Blocks in the Synthesis of (E)-Alkene Peptide Isosteres. Org. Process. Res. Dev. 2012, 16, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Crawley, M.L. Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. ChemInform 2003, 34, 2921–2943. [Google Scholar] [CrossRef]
- Johannsen, M.; Jørgensen, K.A. Allylic Amination. Chem. Rev. 1998, 98, 1689–1708. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L.; Majláth, Z.; Szok, D.; Csáti, A.; Tajti, J. Drug safety and tolerability in prophylactic migraine treatment. Expert Opin. Drug Saf. 2015, 14, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Petranyi, G.; Ryder, N.S.; Stutz, A. Allylamine derivatives: New class of synthetic antifungal agents inhibiting fungal squalene epoxidase. Science 1984, 224, 1239–1241. [Google Scholar] [CrossRef]
- Georgopoulos, A.; Petranyi, G.; Mieth, H.; Drews, J. In vitro activity of naftifine, a new antifungal agent. Antimicrob. Agents Chemother. 1981, 19, 386–389. [Google Scholar] [CrossRef]
- Stuetz, A.; Petranyi, G. Synthesis and antifungal activity of (E)-N-(6,6-dimethyl-2-hepten-4-ynyl)-N-methyl-1-naphthalenemethanamine (SF 86-327) and related allylamine derivatives with enhanced oral activity. J. Med. Chem. 1984, 27, 1539–1543. [Google Scholar] [CrossRef]
- Mensch, A.C.; Hernandez, R.T.; Kuether, J.E.; Torelli, M.D.; Feng, Z.V.; Hamers, R.J.; Pedersen, J.A. Natural Organic Matter Concentration Impacts the Interaction of Functionalized Diamond Nanoparticles with Model and Actual Bacterial Membranes. Environ. Sci. Technol. 2017, 51, 11075–11084. [Google Scholar] [CrossRef]
- Anusionwu, C.G.; Aderibigbe, B.; Mbianda, X.Y. Hybrid Molecules Development: A Versatile Landscape for the Control of Antifungal Drug Resistance: A Review. Mini-Reviews Med. Chem. 2019, 19, 450–464. [Google Scholar] [CrossRef]
- Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef]
- Lacoste, A.M.; Darriet, M.; Neuzil, E.; Le Goffic, F. Inhibition of alanine racemase by vinylglycine and its phosphonic analog: A proton nuclear magnetic resonance spectroscopy study. Biochem. Soc. Trans. 1988, 16, 606–608. [Google Scholar] [CrossRef]
- Yen, V.Q.; Carniato, D.; Quang, L.V.; Lacoste, A.M.; Neuzil, E.; Le Goffic, F. (1-Amino-2-propenyl)phosphonic acid, an inhibitor of alanine racemase and D-alanine:D-alanine ligase. J. Med. Chem. 1986, 29, 579–581. [Google Scholar] [CrossRef]
- Bata, Z.; Qian, R.; Roller, A.; Horak, J.; Bencze, L.C.; Paizs, C.; Hammerschmidt, F.; Vértessy, B.G.; Poppe, L. A Methylidene Group in the Phosphonic Acid Analogue of Phenylalanine Reverses the Enantiopreference of Binding to Phenylalanine Ammonia-Lyases. Adv. Synth. Catal. 2017, 359, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Potashman, M.H.; Duggan, M.E. Covalent modifiers: Anorthogonal approach to drug design. J. Med. Chem. 2009, 52, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Vaidergorn, M.M.; Carneiro, Z.A.; Lopes, C.D.; De Albuquerque, S.; Reis, F.C.; Nikolaou, S.; E Mello, J.F.; Genesi, G.L.; Trossini, G.; Ganesan, A.; et al. β-amino alcohols and their respective 2-phenyl-N-alkyl aziridines as potential DNA minor groove binders. Eur. J. Med. Chem. 2018, 157, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Del Burgo, A.V.; De Retana, A.M.O.; Santos, J.M.D.L.; Palacios, F. ChemInform Abstract: Reaction of 2H-Azirine-Phosphine Oxides and -Phosphonates with Enolates Derived from β-Keto Esters. Cheminform 2016, 47, 100–108. [Google Scholar] [CrossRef]
- Palacios, F.; De Retana, A.M.O.; Alonso, J.M. Regioselective synthesis of fluoroalkylated β-aminophosphorus derivatives and aziridines from phosphorylated oximes and nucleophilic reagents. J. Org. Chem. 2006, 71, 6141–6148. [Google Scholar] [CrossRef]
- Palacios, F.; De Retana, A.M.O.; I Gil, J. Easy and efficient synthesis of enantiomerically enriched 2H-azirines derived from phosphonates. Tetrahedron Lett. 2000, 41, 5363–5366. [Google Scholar] [CrossRef]
- Palacios, F.; De Retana, A.M.O.; Gil, J.I.; Ezpeleta, J.M. Simple Asymmetric Synthesis of 2H-Azirines Derived from Phosphine Oxides†. J. Org. Chem. 2000, 65, 3213–3217. [Google Scholar] [CrossRef]
- Palacios, F.; De Retana, A.M.O.; Del Burgo, A.V. Selective Synthesis of Substituted Pyrrole-2-phosphine Oxides and -phosphonates from 2H-Azirines and Enolates from Acetyl Acetates and Malonates. J. Org. Chem. 2011, 76, 9472–9477. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; Aparicio, D.; De Retana, A.M.O.; Santos, J.M.D.L.; Gil, J.I.; Alonso, J.M. Asymmetric Synthesis of 2H-Azirines Derived from Phosphine Oxides Using Solid-Supported Amines. Ring Opening of Azirines with Carboxylic Acids. J. Org. Chem. 2002, 67, 7283–7288. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; De Retana, A.M.O.; Alonso, J.M. Reaction of 2H-Azirine Phosphine Oxide and -Phosphonates with Nucleophiles. Stereoselective Synthesis of Functionalized Aziridines and α- and β-Aminophosphorus Derivatives†. J. Org. Chem. 2005, 70, 8895–8901. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; Aparicio, D.; Ochoa de Retana, A.M.; de los Santos, J.M.; Gil, J.I.; López de Munain, R. Asymmetric synthesis of 2H-aziridine phosphonates, and α- or β-aminophosphonates from enantiomerically enriched 2H-azirines. Tetrahedron Asymmetry 2003, 14, 689–700. [Google Scholar] [CrossRef]
- Carramiñana, V.; De Retana, A.M.O.; Del Burgo, A.V.; Santos, J.M.D.L.; Palacios, F. Synthesis and biological evaluation of cyanoaziridine phosphine oxides and phosphonates with antiproliferative activity. Eur. J. Med. Chem. 2019, 163, 736–746. [Google Scholar] [CrossRef]
- Carramiñana, V.; De Retana, A.M.O.; Santos, J.M.D.L.; Palacios, F. First synthesis of merged hybrids phosphorylated azirino[2,1-b]benzo[e][1,3]oxazine derivatives as anticancer agents. Eur. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Fotsing, J.R.; Banert, K. New Way to Methylene-2H-azirines and Their Use as Powerful Intermediates for the Stereo- and Regioselective Synthesis of Compounds with Vinylamine Substructure. Eur. J. Org. Chem. 2006, 2006, 3617–3625. [Google Scholar] [CrossRef]
- Sakharov, P.A.; Rostovskii, N.V.; Khlebnikov, A.F.; Novikov, M.S. Annulation of five-membered cyclic enols with 3-aryl-2 H -azirines: Catalytic versus non-catalytic cycloaddition. Tetrahedron 2017, 73, 4663–4670. [Google Scholar] [CrossRef]
- Recio, R.; Vengut-Climent, E.; Mouillac, B.; Orcel, H.; López-Lázaro, M.; Calderón-Montaño, J.M.; Álvarez, E.; Khiar, N.; Fernández, I. Design, synthesis and biological studies of a library of NK1-Receptor Ligands Based on a 5-arylthiosubstituted 2-amino-4,6-diaryl-3-cyano-4 H -pyran core: Switch from antagonist to agonist effect by chemical modification. Eur. J. Med. Chem. 2017, 138, 644–660. [Google Scholar] [CrossRef]
- Gondru, R.; Saini, R.; Vaarla, K.; Singh, S.; Sirassu, N.; Bavantula, R.; Saxena, A.K. Synthesis and Characterization of Chalcone-Pyridinium Hybrids as Potential Anti-Cancer and Anti-Microbial Agents. ChemistrySelect 2018, 3, 1424–1431. [Google Scholar] [CrossRef]
- George, R.F. Facile synthesis of simple 2-oxindole-based compounds with promising antiproliferative activity. Futur. Med. Chem. 2018, 10, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Palacios, F.; De Retana, A.M.O.; Gil, J.I.; De Munain, R.L. Synthesis of Pyrazine-phosphonates and -Phosphine Oxides from 2H-Azirines or Oximes. Org. Lett. 2002, 4, 2405–2408. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | R | R1 | R2 | Yield (%) 1 |
|---|---|---|---|---|---|
| 1 | 4a | Ph | Me | Me | 74 |
| 2 | 4b | Ph | Et | Me | 92 |
| 3 | 4c | Ph | Ph | Me | 81 |
| 4 | 4d | Ph | Me | Et | 56 |
| 5 | 4e | OiPr | Me | Me | 69 |
| 6 | 4f | OEt | Me | Me | 61 |
| 7 | 5a | Ph | Me | Me | 80 |
| 8 | 5b | OiPr | Me | Me | 62 |
| 9 | 5c | OEt | Me | Me | 64 |
| 10 | 5d | OEt | Me | Et | 70 2 |
| 11 | 6 | OiPr | Me | 68 |
| Entry | Compound 1 | R | Ar | Yield (%) 2 |
|---|---|---|---|---|
| 1 | 10a | Ph | Ph | 70 |
| 2 | 10b | Ph | 2-Naph | 3 |
| 3 | 11a | Ph | Ph | 93 |
| 4 | 11b | Ph | 2-Naph | 74 |
| 5 | 12a | OEt | Ph | 88 4 |
| 6 | 12b | OEt | 2-Naph | 67 4 |
| Entry | Compound | R | R1 | R2 | Yield (%) 1 |
|---|---|---|---|---|---|
| 1 | 14a | Ph | Me | Ph | 92 2 |
| 2 | 14b | Ph | Me | p-MeC6H4 | 3,4 |
| 3 | 14c | Ph | Ph | Ph | 60 2 |
| 4 | 15a | Ph | Ph | 91 2 | |
| 5 | 15b | Ph | p-MeC6H4 | 89 4 | |
| 6 | 15c | Ph | p-FC6H4 | 76 4 | |
| 7 | 15d | Ph | p-MeOC6H4 | 70 4 | |
| 8 | 15e | OEt | Ph | 41 4 | |
| 9 | 15f | Ph | Et | 64 2 | |
| 10 | 16a | Ph | Ph | 87 | |
| 11 | 16b | Ph | p-MeC6H4 | 84 | |
| 12 | 16c | OEt | Ph | 85 | |
| 13 | 16d | OEt | p-FC6H4 | 85 5 | |
| 14 | 16e | OEt | p-MeC6H4 | 73 5 |
| Entry | Comp. | R | R1 | R2 | Cytotoxicity IC50 (µM) 1 | |
|---|---|---|---|---|---|---|
| Lung A549 | MRC-5 | |||||
| 1 |  | 0.48 ± 0.017 [50] | >50 [51] | |||
 | ||||||
| 2 2 | 4a | Ph | Me | Me | 4.4 ± 0.72 | >50 |
| 3 | 4b | Ph | Et | Me | 21.3 ± 0.22 | >50 |
| 4 | 4c | Ph | Ph | Me | 16.1 ± 2.03 | >50 |
| 5 | 4d | Ph | Me | Et | 9.6 ± 1.13 | >50 |
| 6 | 4e | OiPr | Me | Me | 4.6 ± 0.31 | >50 |
| 7 | 4f | OEt | Me | Me | 1.3 ± 0.10 | >50 |
 | ||||||
| 8 | 5a | Ph | Me | Me | 8.2 ± 0.23 | >50 |
| 9 | 5b | OiPr | Me | Me | 1.7 ± 0.30 | >50 |
| 10 | 5c | OEt | Me | Me | 4.5 ± 0.45 | >50 |
| 11 | 5d | OEt | Me | Et | 3.7 ± 0.49 | >50 |
 | ||||||
| 12 | 6 | OiPr | Me | >50 | 3 | |
 | ||||||
| 13 | 7 | Ph | CH2CF3 | 3.6 ± 0.70 | >50 | |
| 14 | 10a | Ph | Ph | 13.3 ± 1.69 | >50 | |
 | ||||||
| 15 | 8 | Ph | CH2CF3 | >50 | >50 | |
| 16 2 | 11a | Ph | Ph | 1.9 ± 0.13 | >50 | |
| 17 | 11b | Ph | 2-Naph | 2.7 ± 0.44 | 33.6 ± 3.73 | |
 | ||||||
| 18 | 9 | Ph | CH2CF3 | 3.5 ± 0.77 | >50 | |
| 19 | 12a | OEt | Ph | 4.8 ± 0.90 | >50 | |
| 20 | 12b | OEt | 2-Naph | 2.1 ± 0.22 | 17.5 ± 1.47 | |
| Entry | Comp. | R | R1 | R2 | Cytotoxicity IC50 (µM) 1 | |
|---|---|---|---|---|---|---|
| Lung A549 | MRC-5 | |||||
| 1 | DOX | 0.48 ± 0.017 [27] | >50 [28] | |||
 | ||||||
| 2 2 | 14c | Ph | Ph | Ph | 1.1 ± 0.32 | 4.9 ± 0.49 |
 | ||||||
| 3 3 | 15a | Ph | Ph | 2.6 ± 0.68 | 15.9 ± 2.79 | |
| 4 | 15b | Ph | p-MeC6H4 | 5.1 ± 0.77 | 14.9 ± 1.61 | |
| 5 3 | 15c | Ph | p-FC6H4 | 0.1 ± 0.08 | >50 | |
| 6 | 15d | Ph | p-MeOC6H4 | 2.6 ± 0.42 | >50 | |
| 7 | 15e | OEt | Ph | 7.2 ± 0.49 | >50 | |
 | ||||||
| 8 | 16a | Ph | Ph | 1.2 ± 0.09 | >50 | |
| 9 | 16b | Ph | p-MeC6H4 | 2.1 ± 0.15 | >50 | |
| 10 3 | 16c | OEt | Ph | 0.2 ± 0.07 | 24.1 ± 3.55 | |
| 11 | 16d | OEt | p-FC6H4 | 3.0 ± 0.98 | >50 | |
| 12 | 16e | OEt | p-MeC6H4 | 3.9 ± 0.63 | >50 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carramiñana, V.; Ochoa de Retana, A.M.; Palacios, F.; de los Santos, J.M. Synthesis of α-Aminophosphonic Acid Derivatives Through the Addition of O- and S-Nucleophiles to 2H-Azirines and Their Antiproliferative Effect on A549 Human Lung Adenocarcinoma Cells. Molecules 2020, 25, 3332. https://doi.org/10.3390/molecules25153332
Carramiñana V, Ochoa de Retana AM, Palacios F, de los Santos JM. Synthesis of α-Aminophosphonic Acid Derivatives Through the Addition of O- and S-Nucleophiles to 2H-Azirines and Their Antiproliferative Effect on A549 Human Lung Adenocarcinoma Cells. Molecules. 2020; 25(15):3332. https://doi.org/10.3390/molecules25153332
Chicago/Turabian StyleCarramiñana, Victor, Ana M. Ochoa de Retana, Francisco Palacios, and Jesús M. de los Santos. 2020. "Synthesis of α-Aminophosphonic Acid Derivatives Through the Addition of O- and S-Nucleophiles to 2H-Azirines and Their Antiproliferative Effect on A549 Human Lung Adenocarcinoma Cells" Molecules 25, no. 15: 3332. https://doi.org/10.3390/molecules25153332
APA StyleCarramiñana, V., Ochoa de Retana, A. M., Palacios, F., & de los Santos, J. M. (2020). Synthesis of α-Aminophosphonic Acid Derivatives Through the Addition of O- and S-Nucleophiles to 2H-Azirines and Their Antiproliferative Effect on A549 Human Lung Adenocarcinoma Cells. Molecules, 25(15), 3332. https://doi.org/10.3390/molecules25153332