Water Radical Cations in the Gas Phase: Methods and Mechanisms of Formation, Structure and Chemical Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Mechanisms of Formation of (H2O)n+•
2.1. Electron Bombardment
2.2. Corona Discharge at Atmospheric Pressure
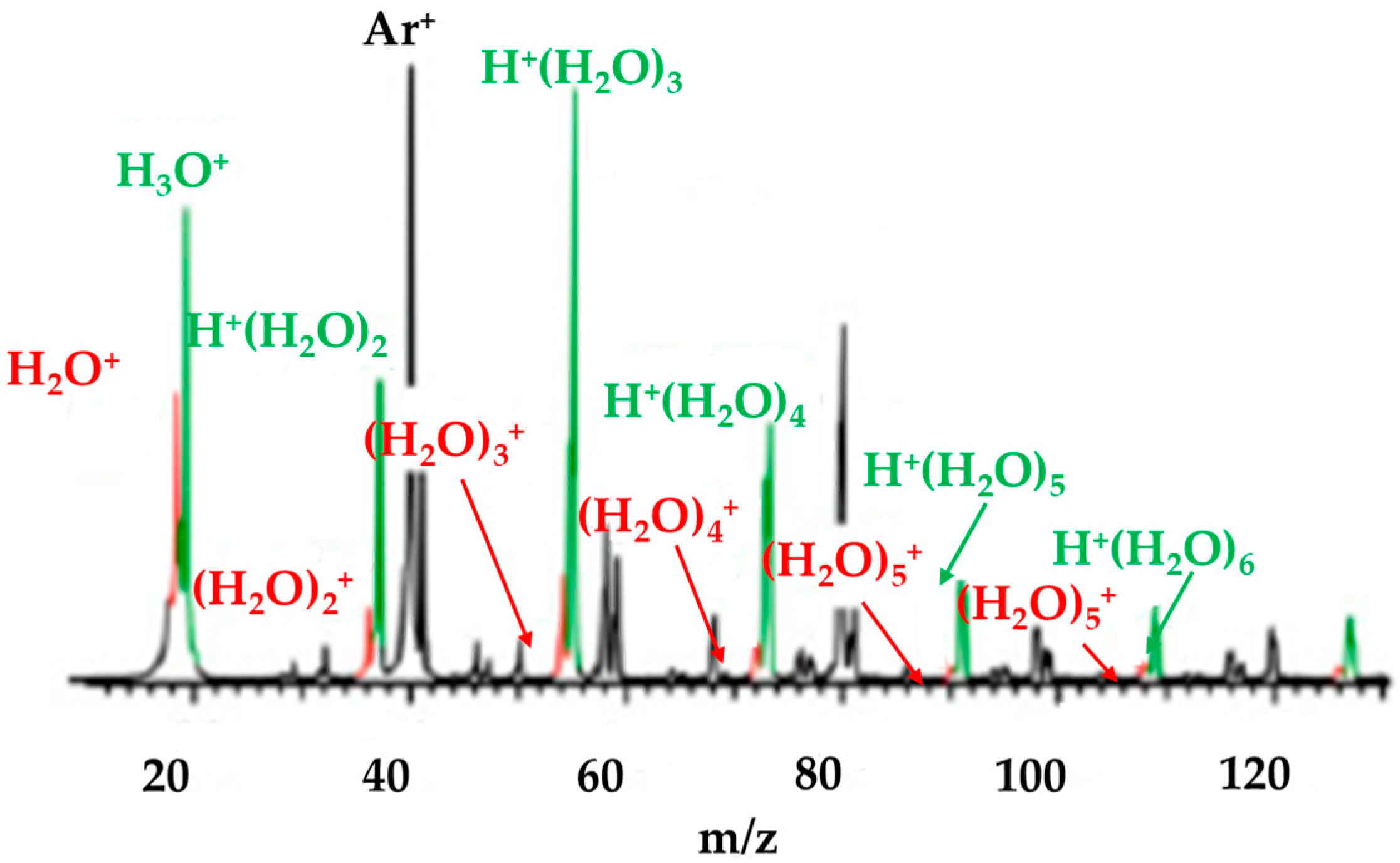
2.3. Photoionization of a Water Vapor Beam for the Formation of (H2O)n+• (n ≤ 3)
2.4. Photoionization of a Molecular Beam for the Formation of (H2O)n+• (n > 3)
2.5. Stabilization of (H2O)n+• Produced by Electron Impact in Helium Nanodroplets
2.6. Formation of (H2O)n+• Using High-Energy Photons or Particles
3. Structural Properties of (H2O)n+•: Simulations and Experimental Studies
3.1. Investigation on the Structure of the Water Dimer Radical Cation (H2O)2+•
3.2. Structural Trends for Water Radical Cations (H2O)n+• (3 ≤ n ≤ 10 and n > 10)
3.3. (H2O)n+• Isomers in a Global Potential Energy Surface
4. Chemical Properties of (H2O)n+•: Simulations and Experimental Studies
4.1. Proton Transfer to Form Hydroxyl Radicals
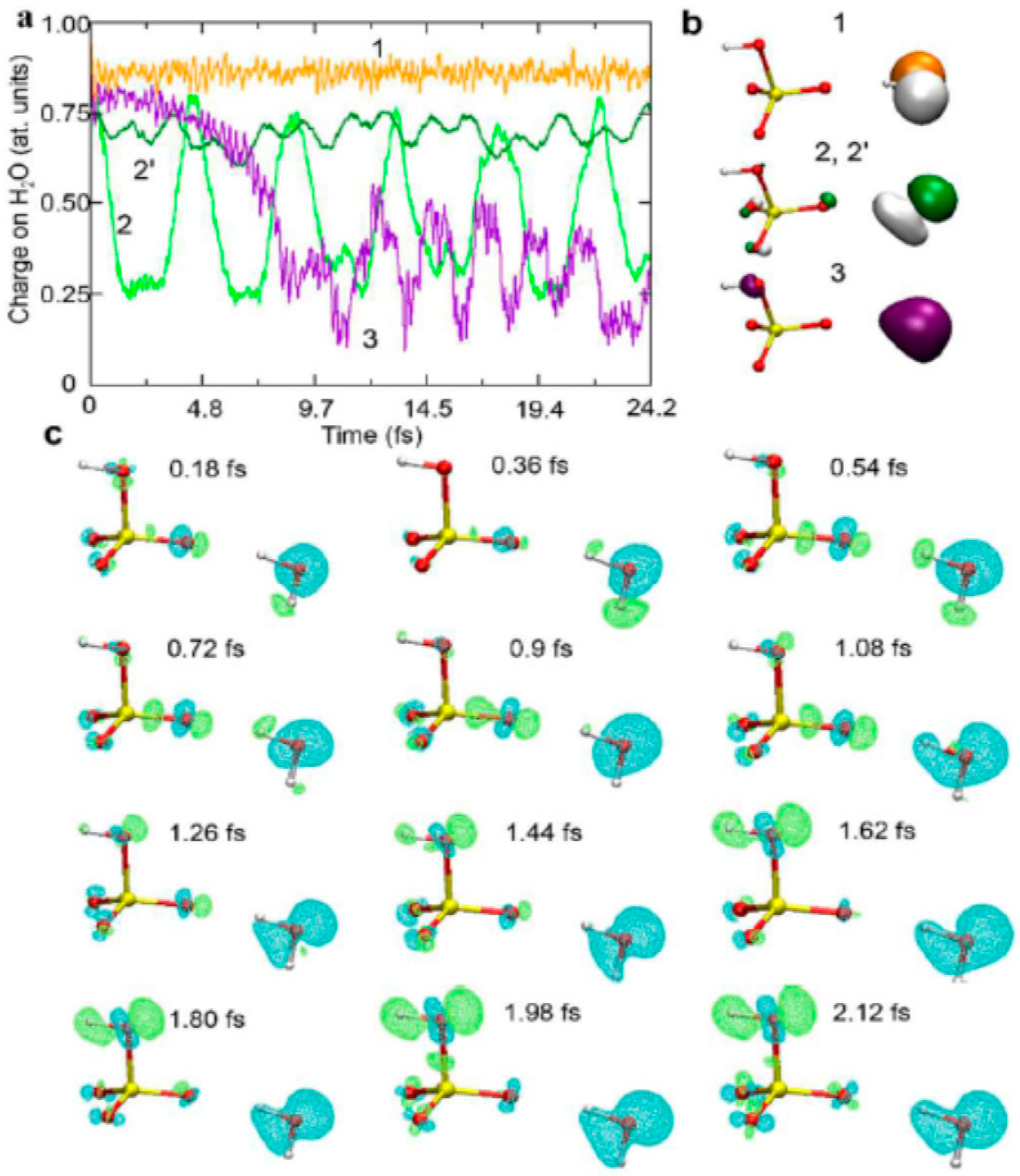
4.2. Initial Ultrarapid Charge Migration in the Chemistry of H2O+•
5. Summary and Outlook
5.1. Summary
- The currently known methods and mechanisms of formation of (H2O)n+• can be summarized as follows:
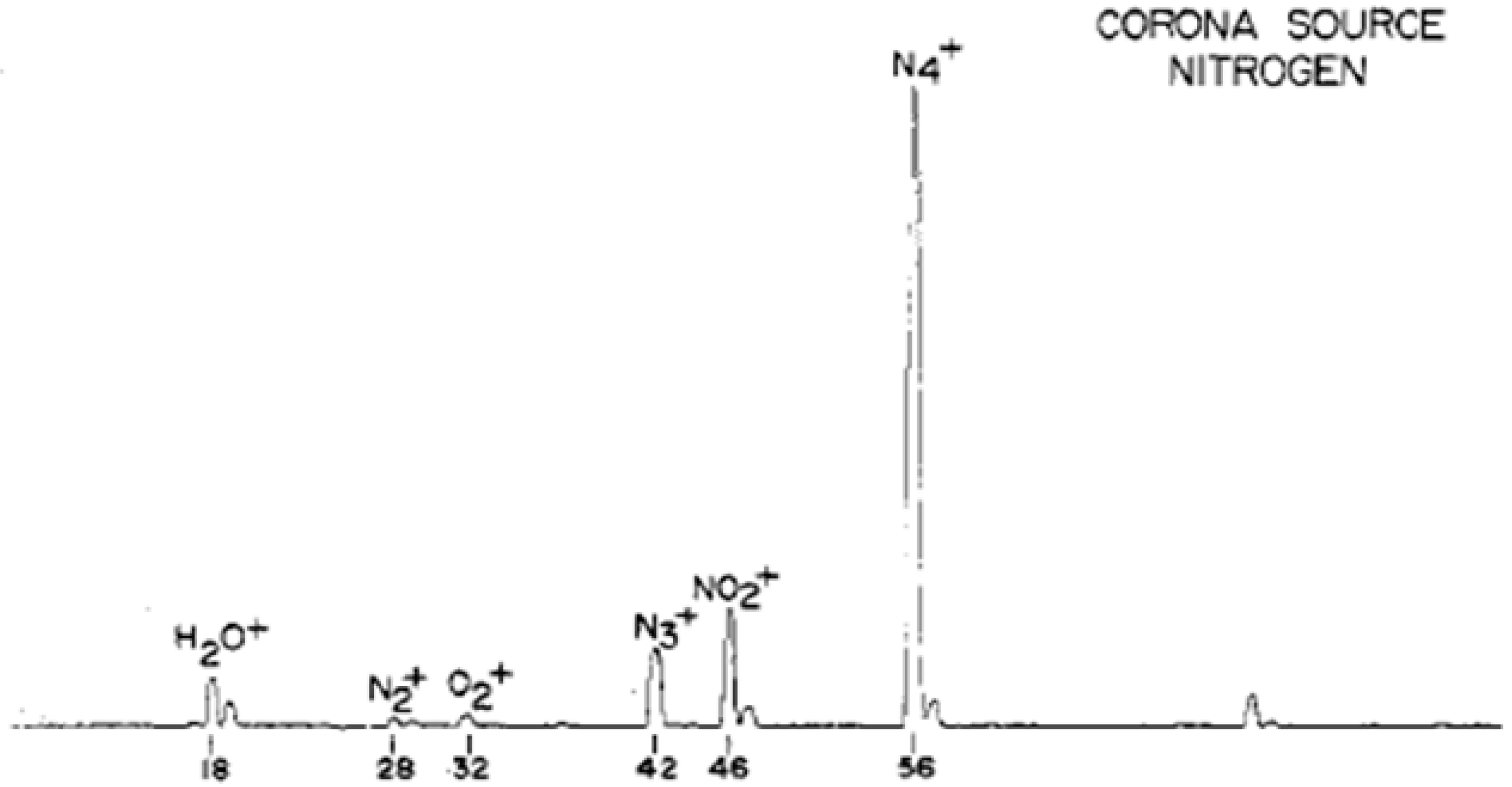
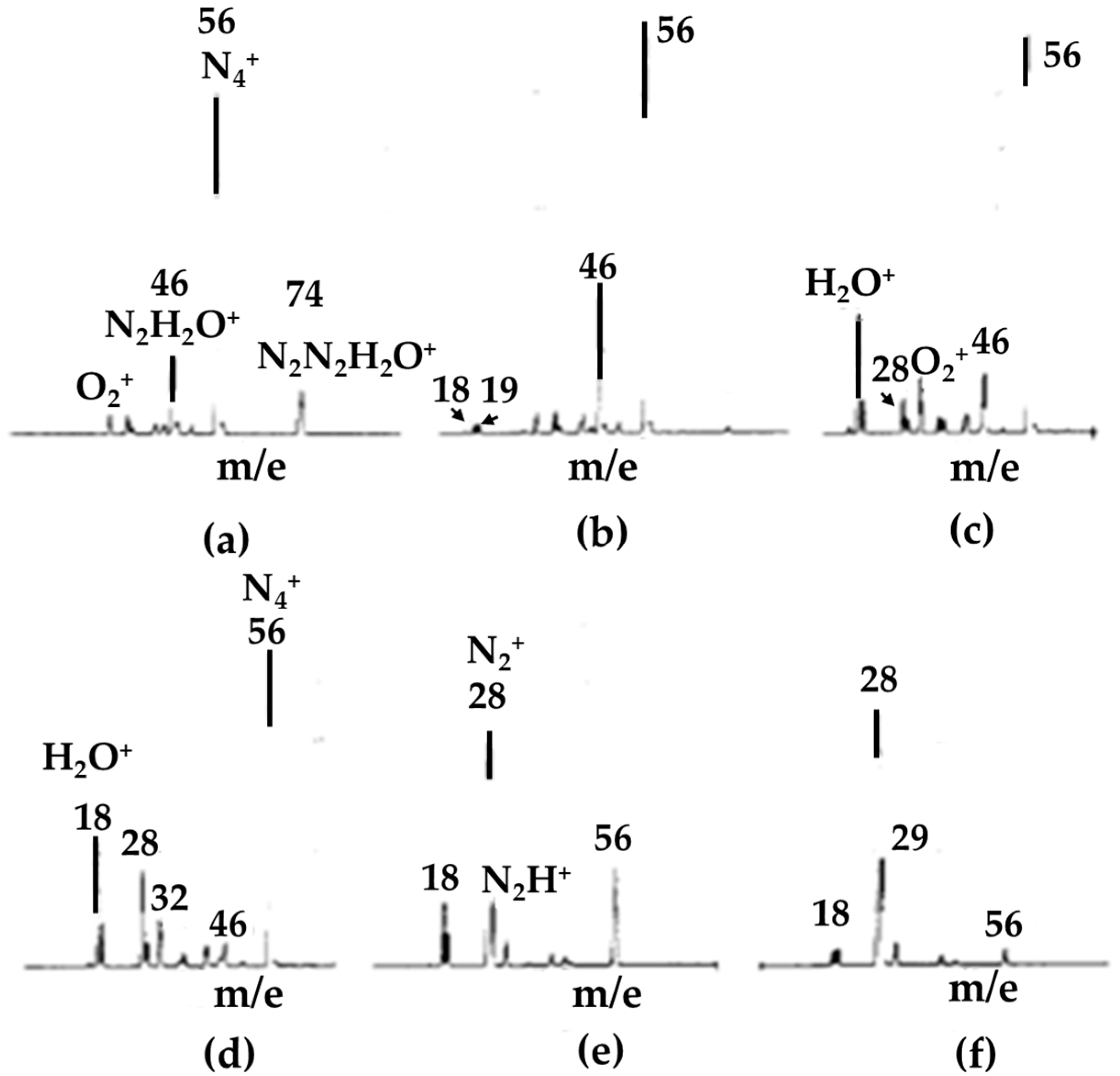
- Method: wet nitrogen ionized as a result of being subjected to electron bombardment or corona discharge.Mechanism: a series of charge transfer reactions started by the formation of N2+•.
- Method: photoionization of a water molecular beam or vapor, even including helium nanodroplets.Mechanism: (i) weakly bonded ions, e.g., (Ar)k(H2O)n+•, were cooled and then (H2O)n+• were formed after further evaporating the carrier gas; (ii) the Franck–Condon factor could be enlarged due to the presence of carrier gas atoms in complex ions, (Ar)k(H2O)n+•, preventing the contraction of the O-O bond upon ionization. (H2O)n+• were generated after the complex ions’ dissociation; (iii) rapid evaporation of the carrier gas could remove the excess energy in the (H2O)n+•; (iv) the excess energies remained in (H2O)n+• after exposure to high-energy photos or particles would be removed by the exiting electron.
- Method: photoionization by high-energy photons or 70 eV electrons impactions.Mechanism: the inner valence electrons could be excited and finally two H2O+• ion radicals could be formed through an ICD process.
- The current knowledge about the structure of (H2O)n+• can be summarized as follows:
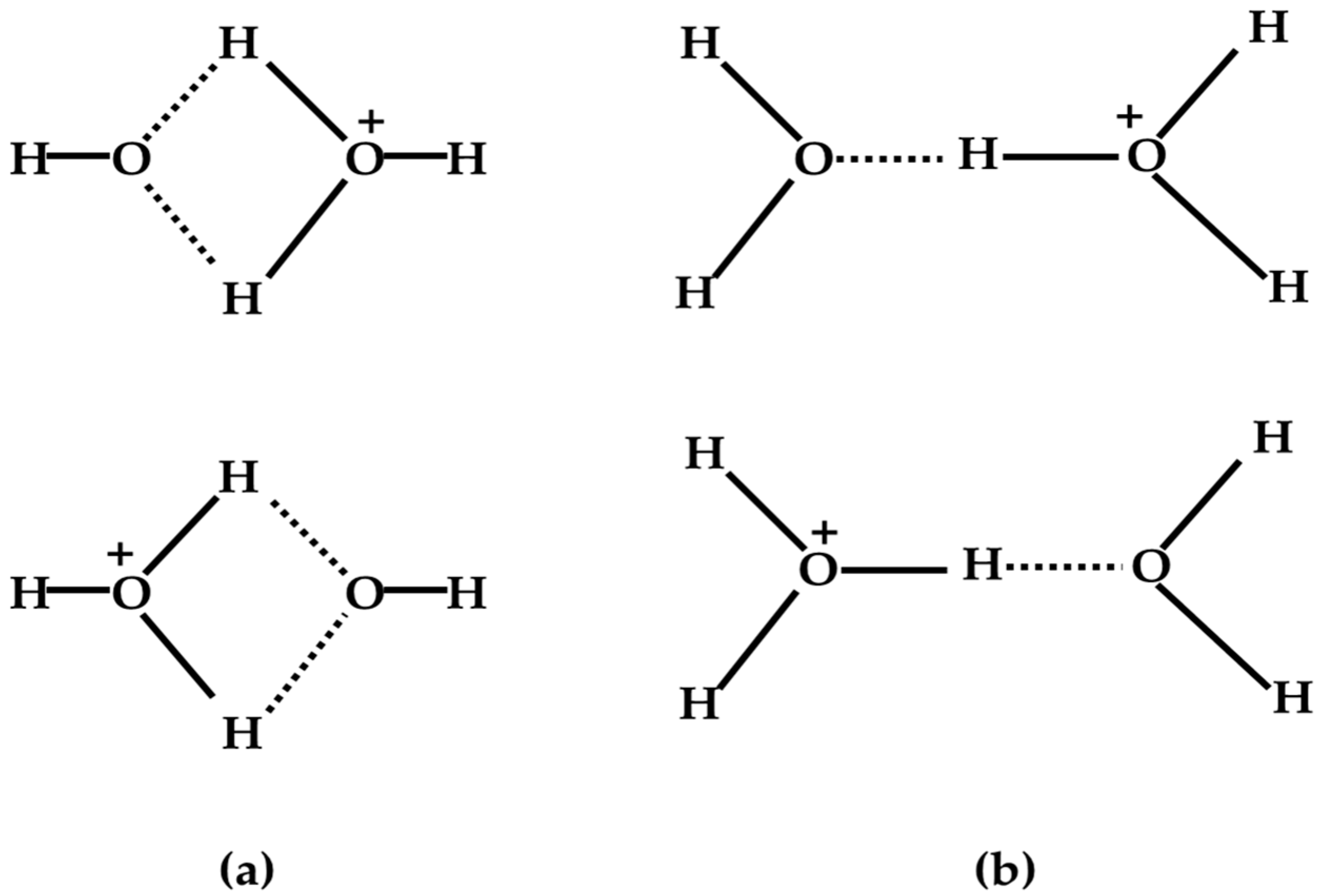
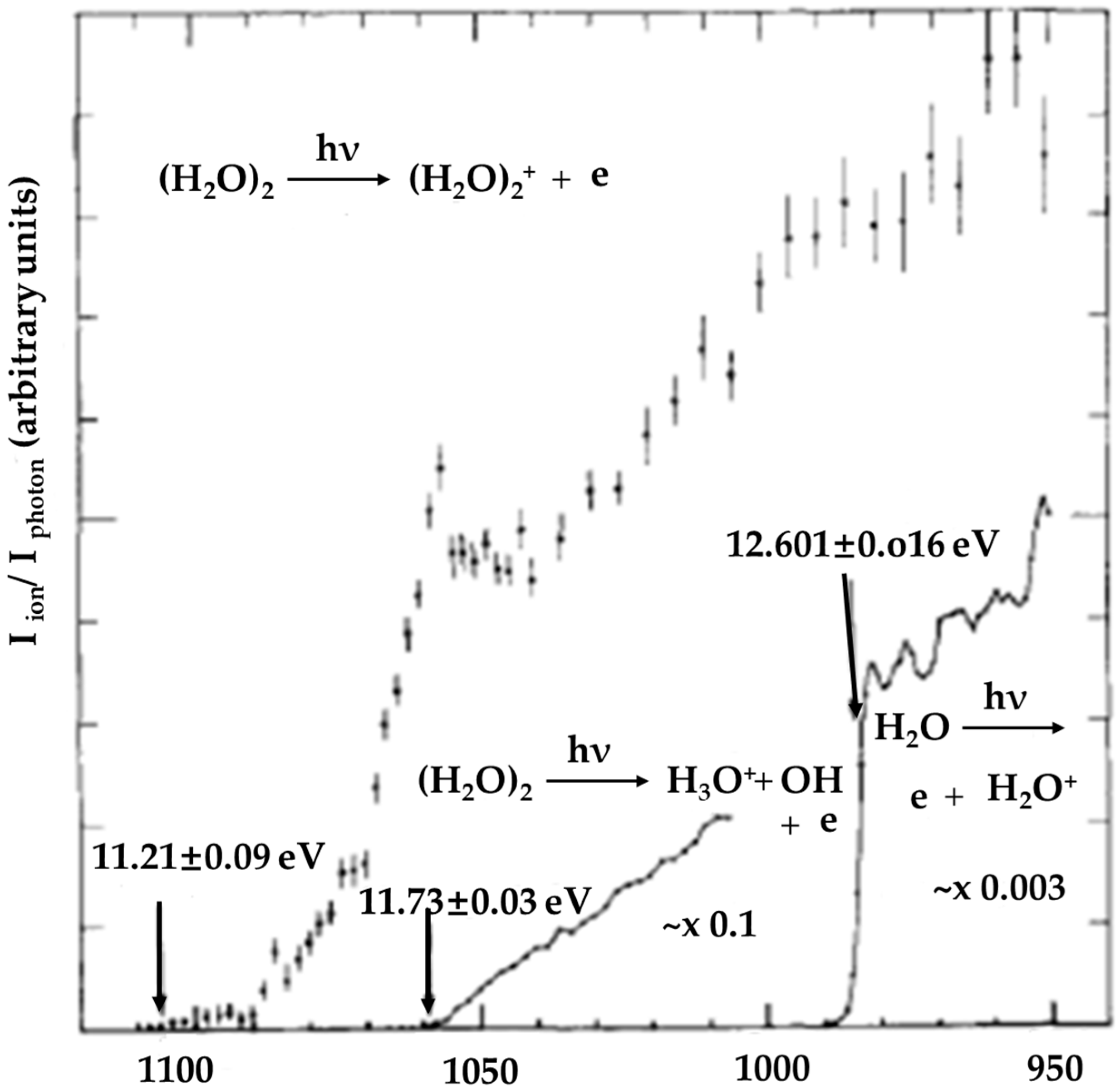
- For (H2O)2+•, a theoretical simulation led to minimum-energy arrangements of both the structure resulting from proton transfer (H3O+-•OH) and the dimer cation structure [H2O…OH2]+•, but with the former indicated to be more stable than the latter by 8.8 kcal/mol with an interconversion barrier of 15.1 kcal/mol. Experimental results based on infrared spectroscopy and collision-induced dissociation MS spectroscopy also tended to support the formation of structures resulting from proton transfer.
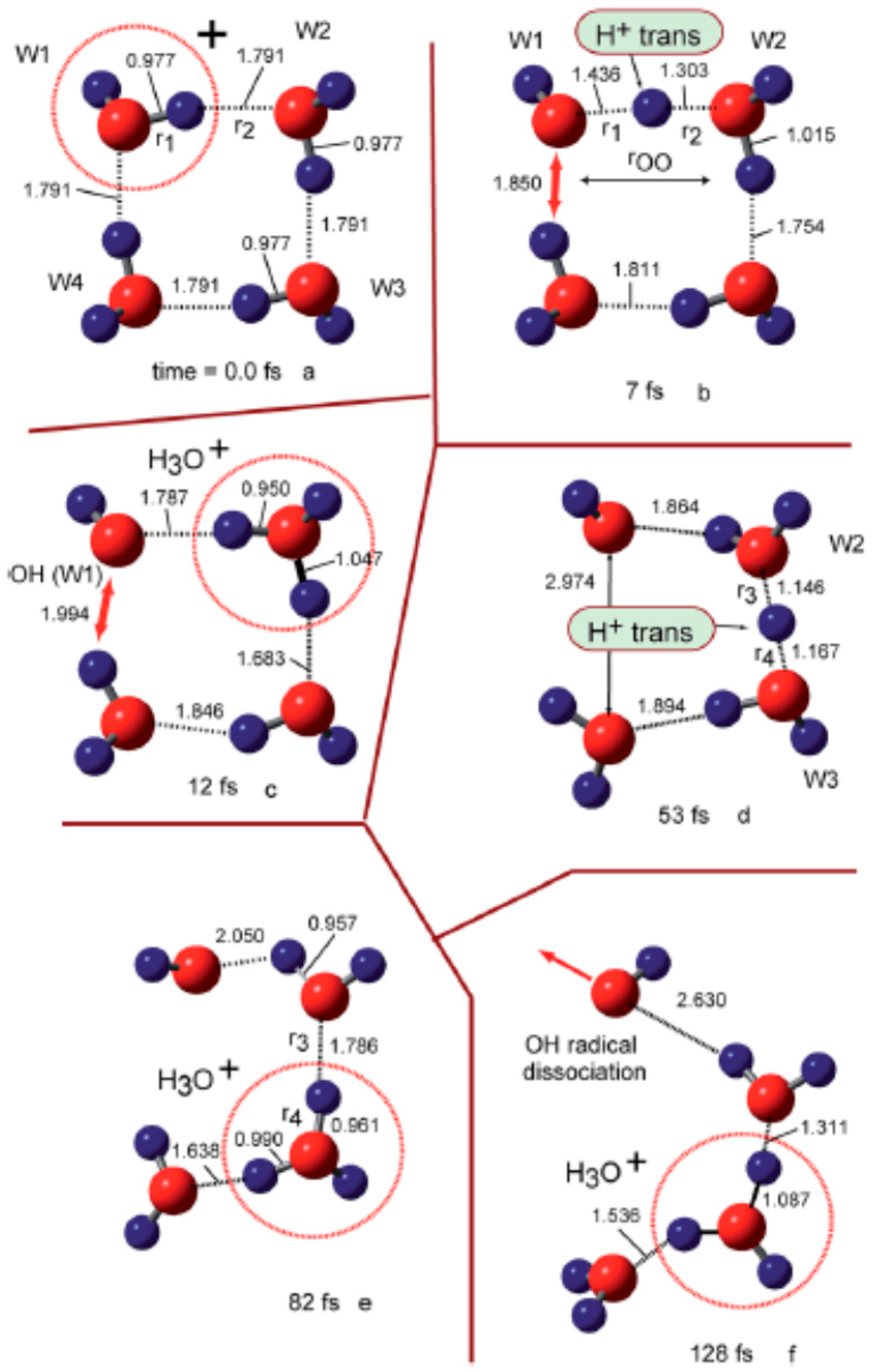
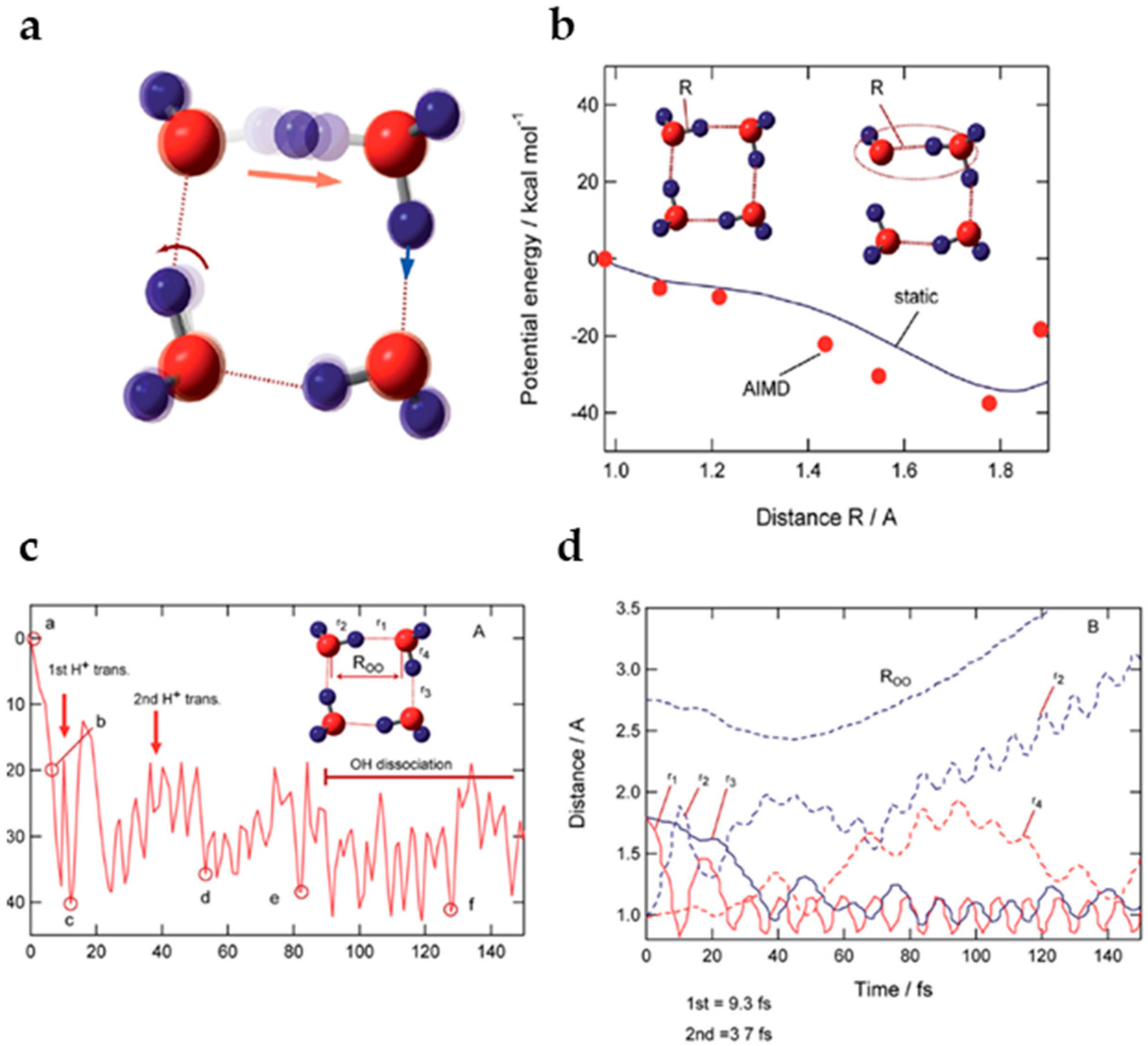
- A recently reported simulation of the dynamical process following water dimer ionization suggests that both the structure resulting from proton transfer and the dimer cation structures as well as other dissociated structures (H3O+ + •OH, H• + •OH + H2O+•) could be generated. The relative yields of these species are controlled by the populated electronic state of the radical cation. Proton transfer resulting from HOMO electron ionization is an ultrafast process, taking less than 100 fs; in the case of higher-energy ionization, the dynamical processes occur on longer timescales (200–300 fs).
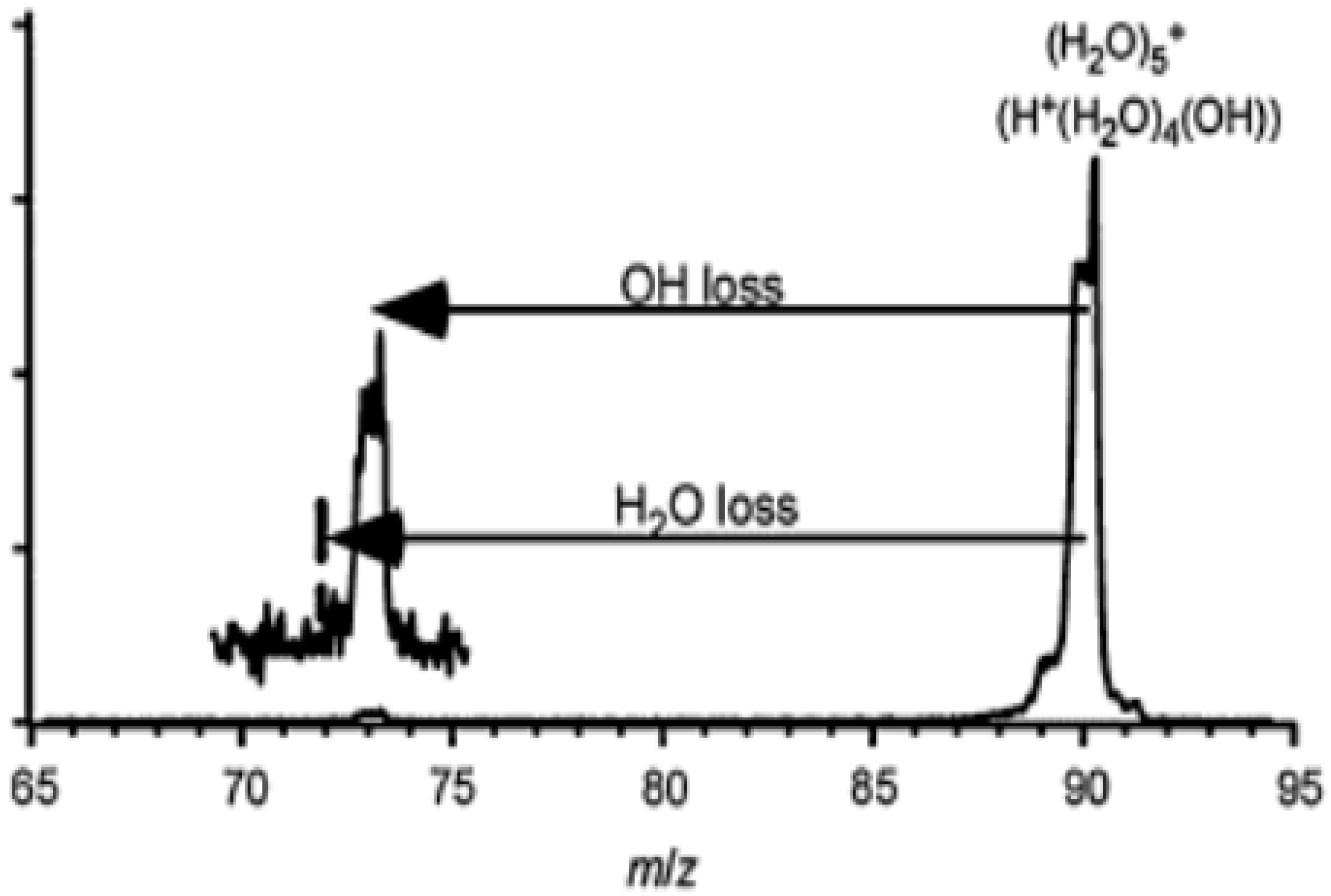
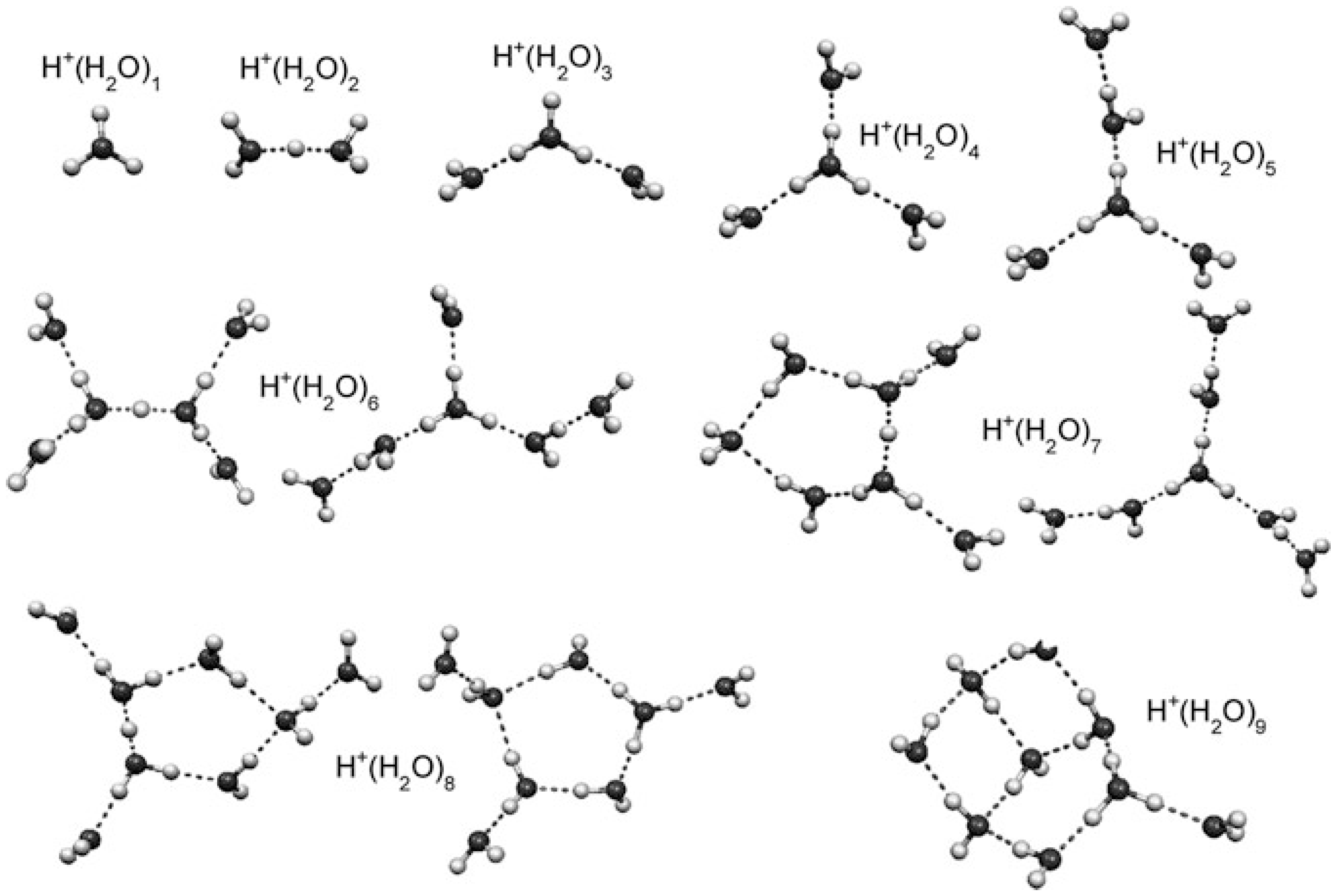
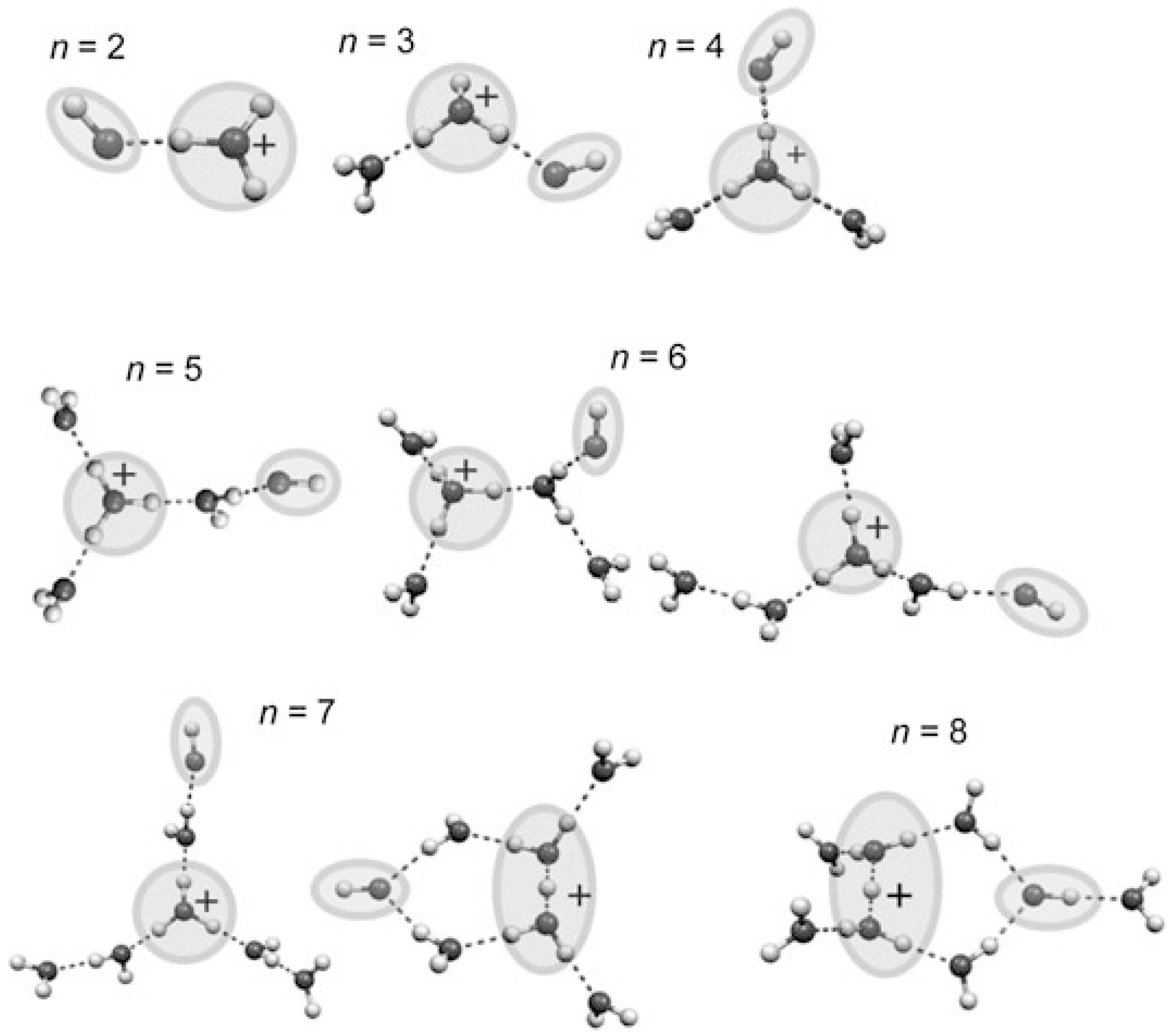
- For the larger (H2O)n+• clusters (i.e., n ≥ 3), most of the simulations and experimental results indicated the presence of the structures resulting from proton transfer. At first, researchers expected •OH to be bonded tightly with H3O+. Later, however, they proposed that the •OH group would be separated from the protonated site by one water molecule since an •OH is a weaker hydrogen bond acceptor than is a water molecule. Then, researchers found a preference for the •OH to stay on the terminal site of the H-bond network—with, in the case of the clusters becoming bigger (n > 10), the •OH groups beginning to serve as H-bond donors (instead of acceptors) and in this way form H-bonds with water molecules.
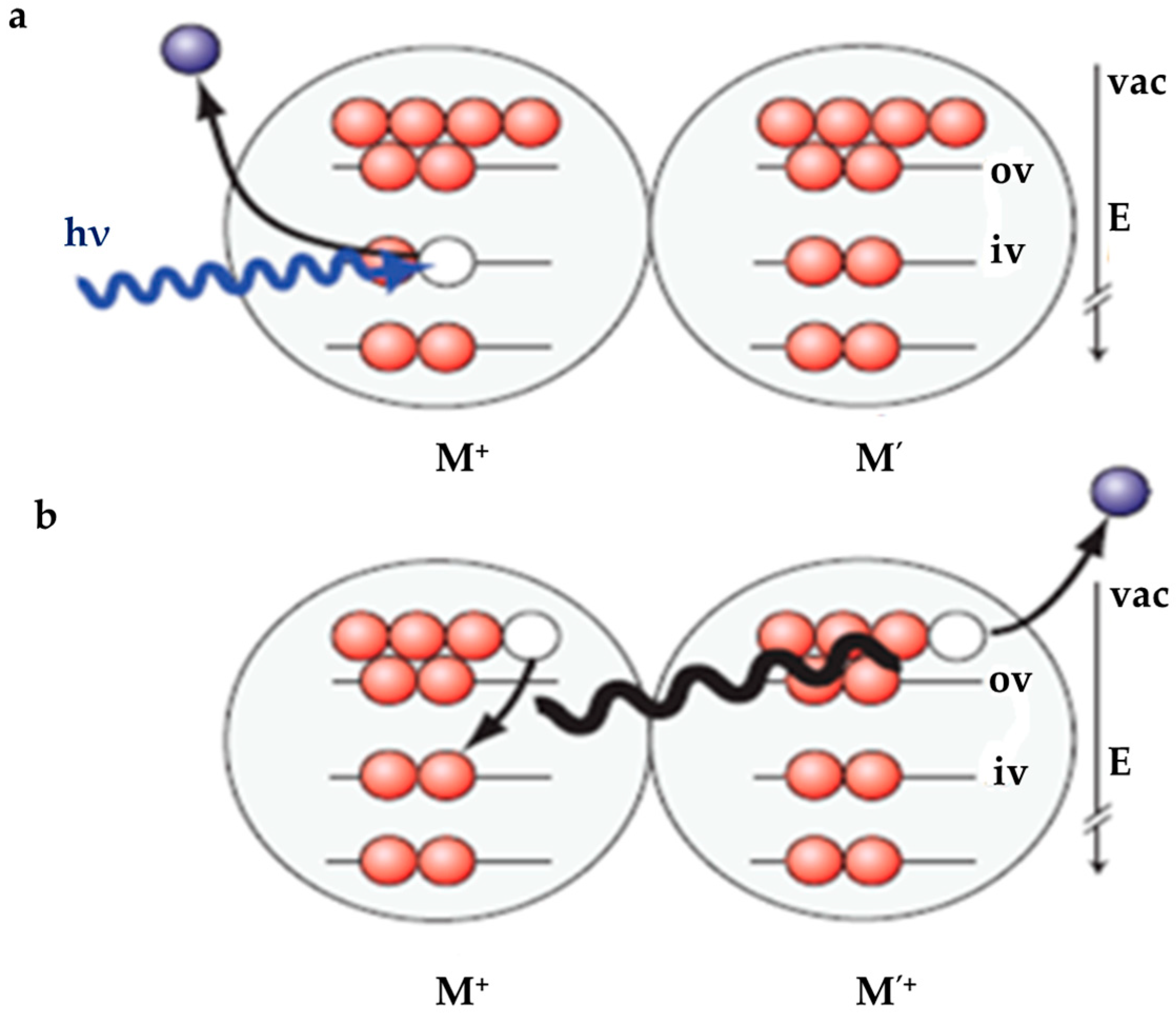
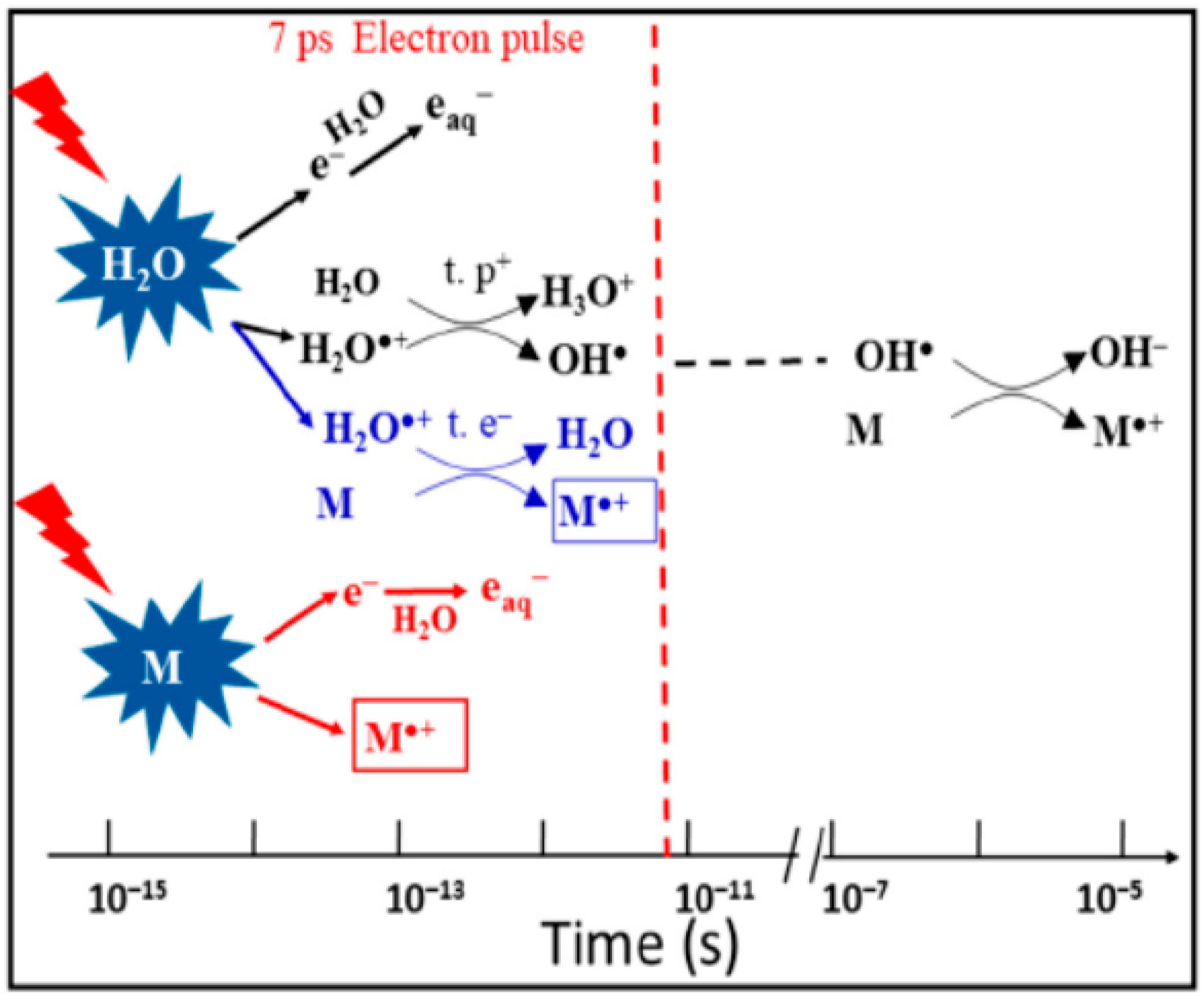
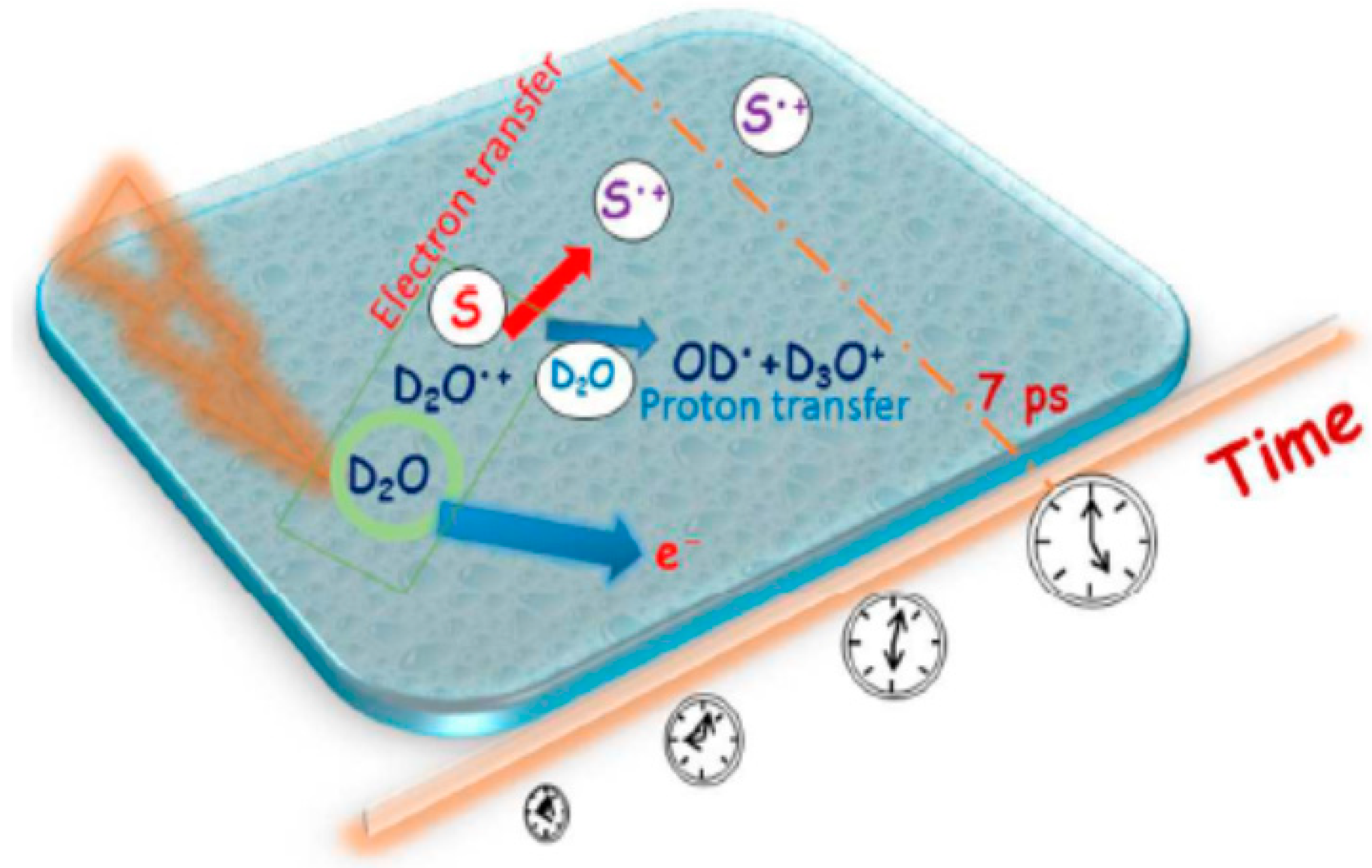
- It was recently found that, besides the ultra-fast proton transfer reaction between (H2O)n+• and its neighboring water molecules to generate protonated water clusters and highly oxidative hydroxyl radicals, (H2O)n+• should also be regarded as the strongest oxidant in the solution and that ultrafast charge migration could occur from a solute M to (H2O)n+• in highly concentrated solutions. The reaction rate of charge migration was indicated by both the simulation and experimental results to be competitive with the rate of proton transfer.
5.2. Outlook
- According to the electric field ionization theory, (H2O)n+• may be expected to be produced under ambient conditions by the exposure of water vapor to the low-energy corona discharge in open air.
- More accurate methods are needed to reliably predict the structures and chemical properties of (H2O)n+•.
- Experimental investigation of chemical properties of (H2O)n+• is greatly hindered due to the low amount of the generated (H2O)n+• in current methods. New methods are needed to allow a higher abundance of (H2O)n+•.
- Attosecond photoelectron spectroscopy based on high harmonic generation resolves H2O+• decay in pure water and therefore shows high potential for the studies of (H2O)n+•.
- Future studies of (H2O)n+• reactivity need to take into account the competition of alternative reactions such as H2O+• addition or H• abstraction with proton transfer and charge migration reactions.
- More research on the reactivity of (H2O)n+• in the condensed phase is essential in radio-biochemistry, nuclear industry and many other disciplines.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bellissent-Funel, M.C.; Hassanali, A.; Havenith, M.; Henchman, R.; Pohl, P.; Sterpone, F.; van der Spoel, D.; Xu, Y.; Garcia, A.E. Water Determines the Structure and Dynamics of Proteins. Chem. Rev. 2016, 116, 7673–7697. [Google Scholar] [CrossRef] [PubMed]
- Mizuse, K.; Mikami, N.; Fujii, A. Infrared Spectra and Hydrogen-Bonded Network Structures of Large Protonated Water Clusters H+(H2O)n (n = 20–200). Angew. Chem. Int. Ed. 2010, 49, 10119–10122. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, S. Biomolecular Action of Ionizing Radiation; Taylor & Francis Group: Oxfordshire, UK, 2008. [Google Scholar]
- Narcisi, R.S.; Bailey, A.D. Mass Spectrometric Measurements of Positive Ions at Altitudes from 64-112 kilometers. J. Geophys. Res. 1965, 70, 3687–3700. [Google Scholar] [CrossRef]
- Arnold, F.; Krankowsky, D.; Marien, K.H. First Mass Spectrometric Measurements of Positive Ions in the Stratosphere. Nature 1977, 267, 30–31. [Google Scholar] [CrossRef]
- Smith, D.; Spanel, P. Ions in the terrestrial atmosphere and in interstellar clouds. Mass Spectrom. Rev. 1995, 14, 255–278. [Google Scholar] [CrossRef]
- Draganic, I.G.; Draganic, Z.D. The Radiation Chemistry of Water; Academic Press: New York, NY, USA, 1971. [Google Scholar]
- Alizadeh, E.; Sanche, L. Precursors of Solvated Electrons in Radiobiological Physics and Chemistry. Chem. Rev. 2012, 112, 5578–5602. [Google Scholar] [CrossRef]
- Ceppatelli, M.; Bini, R.; Schettino, V. High-pressure photodissociation of water as a tool for hydrogen synthesis and fundamental chemistry. Proc. Natl. Acad. Sci. USA 2009, 106, 11454–11459. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, H.; Nishi, N.; Washida, N. Photoionization of water clusters at 11.83 eV: Observation of unprotonated cluster ions (H2O)+n (2 ≤ n ≤ 10). J. Chem. Phys. 1986, 84, 5561–5567. [Google Scholar] [CrossRef]
- Hatano, Y.; Mozumder, A. Charged Particle and Photo Interactions with Matter; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Garrett, B.C.; Dixon, D.A.; Camaioni, D.M.; Chipman, D.M.; Johnson, M.A.; Jonah, C.D.; Kimmel, G.A.; Miller, J.H.; Rescigno, T.N.; Rossky, P.J.; et al. Role of Water in Electron-Initiated Processes and Radical Chemistry Issues and Scientific Advances. Chem. Rev. 2005, 105, 355–390. [Google Scholar] [CrossRef]
- Buxton, G.V. Radiation Chemistry-From Basics to Applications in Material and Life Sciences; EDP Sciences: Les Ulis, France, 2008. [Google Scholar]
- Thurmer, H.; Oncak, M.; Ottosson, N.; Seidel, R.; Hergenhahn, U.; Bradforth, S.E.; Slavicek, P.; Winter, B. On the Nature and Origin of Dicationic, Charge-separated Species Formed in Liquid Water on X-ray Irradiation. Nat. Chem. 2013, 5, 590–596. [Google Scholar] [CrossRef]
- Mizuse, K.; Kuo, J.-L.; Fujii, A. Structural trends of ionized water networks: Infrared spectroscopy of watercluster radical cations (H2O)n+ (n = 3–11). Chem. Sci. 2011, 2, 868–876. [Google Scholar] [CrossRef]
- Jiang, J.C.; Wang, Y.S.; Chang, H.C.; Lin, S.H.; Lee, Y.T.; Niedner-Schatteburg, G.; Chang, H.C. Infrared Spectra of H+(H2O)5-8 Clusters Evidence for Symmetric Proton Hydration. J. Am. Chem. Soc. 2000, 122, 1398–1410. [Google Scholar] [CrossRef]
- Conlan, X.A.; Fletcher, J.S.; Lockyer, N.P.; Vickerman, J.C. A Comparative Study of Secondary of Secondary Ion Emission from Water Ice under Ion Bombardment by Au+, Au3+, and C60+. J. Phys. Chem. C 2010, 114, 5468–5479. [Google Scholar] [CrossRef]
- Good, A.; Durden, D.A.; Kebarle, P. Ion-molecule Reactions in Pure Nitrogen and Nitrogen Containing Traces of Water at Total Pressures 0.5-4 torr. Kinetics of Clustering Reaction Forming H+(H2O)n. J. Chem. Phys. 1970, 52, 212–221. [Google Scholar] [CrossRef]
- Good, A.; Durden, D.A.; Kebarle, P. Mechanism and Rate Constants of Ion-molecule Reactions Leading to Formation of H+(H2O)n in Moist Oxygen and Air. J. Chem. Phys. 1970, 52, 222–229. [Google Scholar] [CrossRef]
- Shahin, M.M. Mass-Spectrometric Studies of Corona Discharges in Air at Atmospheric Pressures. J. Chem. Phys. 1966, 45, 2600–2605. [Google Scholar] [CrossRef]
- Liu, Y.-Y.; Ying, Y.-L.; Hua, X.; Long, Y.-T. In-situ discrimination of the water cluster size distribution in aqueous solution by ToF-SIMS. Sci. China Chem. 2017, 61, 159–163. [Google Scholar] [CrossRef]
- Mozumder, A. Events in Radiation Chemistry An Introduction. Int. J. Radiat. Appl. Instrum. Part. C 1989, 34, 1–3. [Google Scholar] [CrossRef]
- Gauduel, Y.; Pommeret, S.; Migus, A.; Antonetti, A. Some Evidence of Ultrafast H2O+.-Water Molecule Reaction in Femosecond Photoionizaiton of Pure Liquid Water: Influence on Geminate Pair Recombination Dynamics. Chem. Phys. 1990, 149, 1–10. [Google Scholar] [CrossRef]
- Long, F.H.; Lu, H.; Eisenthal, K.B. Femtosecond studies of the presolvated electron: An excited state of the solvated electron? Phys. Rev. Lett. 1990, 64, 1469–1472. [Google Scholar] [CrossRef]
- Marsalek, O.; Elles, C.G.; Pieniazek, P.A.; Pluharova, E.; VandeVondele, J.; Bradforth, S.E.; Jungwirth, P. Chasing charge localization and chemical reactivity following photoionization in liquid water. J. Chem. Phys. 2011, 135, 224510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Nie, Z.; Zheng, Y.Y.; Dong, S.; Loh, Z.-H. Elementary Electron and Ion Dynamics in Ionized Liquid Water. J. Chem. Phys. Lett. 2013, 4, 3698–3703. [Google Scholar] [CrossRef]
- Kamarchik, E.; Kostko, O.; Bowman, J.M.; Ahmed, M.; Krylov, A.I. Spectroscopic signatures of proton transfer dynamics in the water dimer cation. J. Chem. Phys. 2010, 132, 194311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodupe, M.; Bertran, J.; Rodriguez-Santiago, L.; Baerends, E.J. Ground State of the (H2O)n+. Radical Cation DFT versus Post-Hartree-Fock Methods. J. Phys. Chem. A 1999, 103, 166–170. [Google Scholar] [CrossRef]
- Karpas, Z.; Huntress, W.T., Jr. Reactions of OH+ and H2O+. ions with some diatomic and simple polyatomic molecules. Chem. Phys. Lett. 1978, 59, 87–89. [Google Scholar] [CrossRef]
- Dotan, I.; Lindinger, W.; Rowe, B.; Fahey, D.W.; Fehsenfeld, F.C.; Albritton, D.L. Rate Constants for the Reaction of H2O+. with NO2, O2, NO, C2H4, CO, CH4, and H2 measured at relative kinetic energies 0.04-2 eV. Chem. Phys. Lett. 1980, 72, 67–70. [Google Scholar] [CrossRef]
- Asundi, R.K.; Schulz, G.J.; Chantry, P.J. Studies of N4+and N3+Ion Formation in Nitrogen Using High-Pressure Mass Spectrometry. J. Chem. Phys. 1967, 47, 1584–1591. [Google Scholar] [CrossRef]
- Thynne, J.C.; Harrison, A.G. Reactions of Thermal Energy Ions. Part 3.-Ion-Molecule Reactions in H2O and D2O. Trans. Faraday Soc. 1966, 62, 2468–2475. [Google Scholar] [CrossRef]
- Gupta, S.K.; Jones, E.G.; Harrison, A.G.; Myher, J.J. Reactions of Thermal Energy Ions. VI. Hydrogen-Transfer Ion-Molecule Reactions Involving Polar Molecules. Can. J. Chem. 1967, 45, 3107–3117. [Google Scholar] [CrossRef]
- Fehsenfeld, F.C.; Ferguson, E.E. Origin of water cluster ions in the Dregion. J. Geophys. Res. 1969, 74, 2217–2222. [Google Scholar] [CrossRef]
- Dzidic, I.; Carroll, D.I.; Stillwell, R.N.; Horning, E.C. Comparison of Positive Ions Formed in Nickel-63 and Corona Discharge Ion Source Using Nitrogen, Argon, Isobutane, Ammonia and Nitric Oxide as Reagents in Atmospheric Pressure Ionization Mass Spectrometry. Anal. Chem. 1976, 48, 1763–1768. [Google Scholar] [CrossRef]
- Kambara, H.; Mitsui, Y.; Kanomata, I. Identification of Clusters Produced in an Atmospheric Pressure Ionization Process by Collisional Dissociation Method. Anal. Chem. 1979, 51, 1447–1452. [Google Scholar] [CrossRef]
- Siegel, M.W.; Fite, W.L. Terminal Ions in Weak Atmospheric Pressure Plasmas. Applications of Atmospheric Pressure Ionization to Trace Impurity Analysis in Gases. J. Phys. Chem. 1976, 80, 2871–2881. [Google Scholar] [CrossRef]
- French, J.B.; Reid, N.M.; Poon, C.C. Proceedings of the 25th Annual Conference on Mass Spectroscopy and Allied Topics, Washington, DC, USA, 29 May–3 June 1977. Paper MD-4.
- Fehsenfeld, F.C.; Mosesman, M.; Ferguson, E.E. Ion-Molecule Reactions in an O2+-H2O System. J. Chem. Phys. 1971, 55, 2115–2120. [Google Scholar] [CrossRef]
- Ng, C.Y.; Trevor, D.J.; Tiedemann, P.W.; Ceyer, S.T.; Kronebusch, P.L.; Mahan, B.H.; Lee, Y.T. Photoionization of dimeric polyatomic molecules: Proton affinities of H2O and HF. J. Chem. Phys. 1977, 67, 4235–4237. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, L.; Mattsson, L.; Jadrny, R.; Albridge, R.G.; Pinchas, S.; Bergmark, T.; Siegbahn, K. Isotopic and vibronic coupling effects in the valence electron spectra of H216O, H218O, and D216O. J. Chem. Phys. 1975, 62, 4745–4752. [Google Scholar] [CrossRef]
- Belau, L.; Wilson, K.R.; Leone, S.R.; Ahmed, M. Vacuum Ultraviolet (VUV) Photoionization of Small Water Clusters. J. Phys. Chem. A 2007, 111, 10075–10083. [Google Scholar] [CrossRef] [PubMed]
- Tomoda, S.; Achiba, Y.; Kimura, K. Photoelectron Spectrum of the Water Dimer. Chem. Phys. Lett. 1982, 87, 197–200. [Google Scholar] [CrossRef]
- Tomoda, S.; Kimura, K. Proton-Transfer Potential-Energy Surfaces of the Water Dimer Cation (H2O)2+. in the 12A″, and 12A′ States. Chem. Phys. 1983, 82, 215–227. [Google Scholar] [CrossRef]
- Tiee, J.J.; Ferris, M.J.; Loge, G.W.; Wampler, F.B. Two-Photon Laser-induced Fluorescence Studies of HS Radicals, DS Radicals, and I Atoms. Chem. Phys. Lett. 1983, 96, 422–425. [Google Scholar] [CrossRef]
- Ozaki, Y.; Murano, K.; Izumi, K.; Fukuyama, T. Unprotonated (D2O)n+. ions by electron impact ionization of (CO2)m•(H2O)n clusters. In 50th Spring Annual Meeting; Japan Chemical Society: Tokyo, Japan, 1985. [Google Scholar]
- Shiromaru, H.; Shinohara, H.; Washida, N.; Woo, H.-S.; Kimura, K. Synchrotron Radiation Measurements of Appearance Potentials for (H2O)2+., (H2O)3+., (H2O)2H+, and (H2O)3H+ in Supersonic Jets. Chem. Phys. Lett. 1987, 141, 7–11. [Google Scholar] [CrossRef]
- Jongma, R.T.; Huang, Y.; Shi, S.; Wodtke, A.M. Rapid Evaporative Cooling Suppresses Fragmentation in Mass Spectrometry: Synthesis of “unprotonated” Water Cluster Ions. J. Phys. Chem. A 1998, 102, 8847–8854. [Google Scholar] [CrossRef]
- Radi, P.P.; Beaud, P.; Franzke, D.; Frey, H.-M.; Gerber, T.; Mischler, B.; Tzannis, A.-P. Femtosecond Photoionization of (H2O)n and (D2O)n Clusters. J. Chem. Phys. 1999, 111, 512–518. [Google Scholar] [CrossRef]
- Mizuse, K. Spectroscopic Investigations of Hydrogen Bond Network Structures in Water Clusters. Ph.D. Thesis, Tohoku University, Tokyo, Japan, 2013. [Google Scholar]
- Even, U.; Jortner, J.; Noy, D.; Lavie, N.; Cossart-Magos, C. Cooling of large molecules below 1 K and He clusters formation. J. Chem. Phys. 2000, 112, 8068–8071. [Google Scholar] [CrossRef] [Green Version]
- Toennies, J.P.; Vilesov, A.F. Superfluid helium droplets: A uniquely cold nanomatrix for molecules and molecular complexes. Angew. Chem. 2004, 43, 2622–2648. [Google Scholar] [CrossRef]
- Lewerenz, M.; Schilling, B.; Toennies, J.P. Successive Capture and Coagulation of Atoms and Molecules to Small Clusters in Large Liquid Helium Clusters. J. Chem. Phys. 1995, 102, 8191–8207. [Google Scholar] [CrossRef]
- Fröchtenicht, R.; Kaloudis, M.; Koch, M.; Huisken, F. Vibrational spectroscopy of small water complexes embedded in large liquid helium clusters. J. Chem. Phys. 1996, 105, 6128–6140. [Google Scholar] [CrossRef]
- Yang, S.; Brereton, S.M.; Nandhra, S.; Ellis, A.M. Electron Impact Ionization of Water-doped Superfluid Helium Nanodroplets: Observation of He•(H2O)n+. Clusters. J. Chem. Phys. 2007, 127, 134303. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Heinbuch, S.; Rocca, J.J.; Bernstein, E.R. Dynamics and Fragmentation of Van der Waals Clusters: (H2O)n, (CH3OH)n, and (NH3)n upon ionization by a 26.5 eV Soft X-ray Laser. J. Chem. Phys. 2006, 124, 224319. [Google Scholar] [CrossRef] [Green Version]
- Mucke, M.; Braune, M.; Barth, S.; Förstel, M.; Lischke, T.; Ulrich, V.; Arion, T.; Becker, U.; Bradshaw, A.; Hergenhahn, U. A hitherto unrecognized source of low-energy electrons in water. Nat. Phys. 2010, 6, 143–146. [Google Scholar] [CrossRef]
- Jahnke, T.; Sann, H.; Havermeier, T.; Kreidi, K.; Stuck, C.; Meckel, M.; Schöffler, M.; Neumann, N.; Wallauer, R.; Voss, S.; et al. Ultrafast energy transfer between water molecules. Nat. Phys. 2010, 6, 139–142. [Google Scholar] [CrossRef]
- Svoboda, O.; Hollas, D.; Ončák, M.; Slavíček, P. Reaction selectivity in an ionized water dimer: Nonadiabatic ab initio dynamics simulations. Phys. Chem. Chem. Phys. 2013, 15, 11531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, U.; Winter, M. Electron Bombardment Induced Fragmentation of Size Selected Neutral (D2O)n Clusters. Z. Phys. D 1994, 31, 291–297. [Google Scholar] [CrossRef]
- Sato, K.; Tomoda, S.; Kimura, K.; Iwata, S. Electronic and Molecular Structure of the Water Dimer Cation. A Theoretical Study. Chem. Phys. Lett. 1983, 95, 579–583. [Google Scholar] [CrossRef]
- De Visser, S.P.; De Koning, L.J.; Nibbeering, N.N.M. Reactivity and Thermochemical Properties of the Water Dimer Radical Cation in the Gas Phase. J. Phys. Chem. 1995, 99, 15444–15447. [Google Scholar] [CrossRef]
- Yang, X.; Castleman, A.W. Large Protonated Water Clusters H+(H2O)n ( 1. Ltoreq. N > 60): The Production and Reactivity of Clathrate-like Structures Under Thermal Conditions. J. Am. Chem. Soc. 1989, 111, 6845–6846. [Google Scholar] [CrossRef]
- Wei, S.; Shi, Z.; Castleman, J.A.W. Mixed Cluster Ions as a Structure Probe: Experimental Evidence for Clathrate Structure of (H2O)20H+ and (H2O)21H+. J. Chem. Phys. 1991, 94, 3268–3270. [Google Scholar] [CrossRef]
- Dalleska, N.F.; Honma, K.; Armentrout, P.B. Stepwise Solvation Enthalpies of Protonated Water Clusters: Collision-Induced Dissociation as an Alternative to Equilibrium Studies. J. Am. Chem. Soc. 1993, 115, 12125–12131. [Google Scholar] [CrossRef]
- Hansen, K.; Andersson, P.U.; Uggerud, E. Activation Energies for Evaporation From Protonated and Deprotonated Water Clusters From Mass Spectra. J. Chem. Phys. 2009, 131, 124303. [Google Scholar] [CrossRef]
- Hulthe, G.; Stenhagen, G.; Wennerstrom, O.; Ottosson, C.-H. Water Clusters Studied by Electrospray Mass Spectrometry. J. Chromatogr. A 1997, 777, 155–156. [Google Scholar] [CrossRef]
- Haberland, H.; Ludewigt, C.; Schindler, H.G.; Worsnop, D.R. Experimental observation of the negatively charged water dimer and other small (H2O)−n clusters. J. Chem. Phys. 1984, 81, 3742–3744. [Google Scholar] [CrossRef]
- Lin, C.K.; Wu, C.C.; Wang, Y.S.; Lee, Y.T.; Chang, H.C.; Kuo, O.J.; Klein, M.L. Vibrational predissociation spectra and hydrogen-bond topologies of H+(H2O)9–11. Phys. Chem. Chem. Phys. 2005, 7, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Lisy, J.M. Infrared studies of ionic clusters: The influence of Yuan T. Lee. J. Chem. Phys. 2006, 125, 132302. [Google Scholar] [CrossRef] [PubMed]
- Okumura, M.; Yeh, L.I.; Myers, J.D.; Lee, Y.T. Infrared Spectra of the Solvated Hydronium Ion: Vibrational Predissociation Spectroscopy of Mass-Selected H3O+•(H2O)n•(H2)m. J. Phys. Chem. 1990, 94, 3146–3427. [Google Scholar] [CrossRef]
- Mizuse, K.; Fujii, A. Infrared Photodissociation Spectroscopy of H+(H2O)6•Mm (M = Ne, Ar, Kr, Xe, H2, N2, and CH4): Messenger-Dependent Balance Between H3O+ and H5O2+ Core Isomers. Phys. Chem. Chem. Phys. 2011, 13, 7129–7135. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, V.F.; Khusnatdinov, N.N. On the nature of photo charge carriers in ice. J. Chem. Phys. 1994, 100, 9096–9105. [Google Scholar] [CrossRef]
- Narten, A.H.; Levy, H.A. Liquid Water: Molecular Correlation Functions from X-Ray Diffraction. J. Chem. Phys. 1971, 55, 2263–2269. [Google Scholar] [CrossRef]
- Castleman, A.W.; Bowen, K.H. Clusters: Structure, Energetics, and Dynamics of Intermediate States of Matter. J. Phys. Chem. 1996, 100, 12911–12944. [Google Scholar] [CrossRef]
- Niedner-Schatteburg, G.; Bondybey, V.E. FT-ICR Studies of Solvation Effects in Ionic Water Cluster Reactions. Chem. Rev. 2000, 100, 4059–4086. [Google Scholar] [CrossRef]
- Bondybey, V.E.; Beyer, M.K. How many molecules make a solution? Int. Rev. Phys. Chem. 2010, 21, 277–306. [Google Scholar] [CrossRef]
- Mohammed, O.F.; Pines, D.; Dreyer, J.; Pines, E.; Nibbering, E.T. Sequential proton transfer through water bridges in acid-base reactions. Science 2005, 310, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Headrick, J.M.; Diken, E.G.; Walters, R.S.; Hammer, N.I.; Christie, R.A.; Cui, J.; Myshakin, E.M.; Duncan, M.A.; Johnson, M.A.; Jordan, K.D. Spectral signatures of hydrated proton vibrations in water clusters. Science 2005, 308, 1765–1769. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; de Maeyer, L. Self-dissociation and protonic charge transport in water and. Proc. Math. Phys. Eng. Sci. 1958, 247, 505–533. [Google Scholar] [CrossRef]
- Eigen, M. Proton Transfer, Acid-Base Catalysis, and Enzymatic Hydrolysis. Angew. Chem. Int. Ed. 1964, 3, 1–19. [Google Scholar] [CrossRef]
- Marx, D.; Tuckerman, M.E.; Hutter, J.; Parrinello, M. The Nature of the Hydrated Excess Proton. Nature 1999, 397, 601–604. [Google Scholar] [CrossRef]
- De Grotthuss, C.J.T. Memoir on the Decomposition of Water and of the Bodies That It Holds in Solution by Means of Galvanic Electricity. Biochim. Biophys. Acta 2006, 1957, 871–875. [Google Scholar] [CrossRef] [Green Version]
- Pimblott, S.M.; LaVerne, J.A. Stochastic Simulation of the Electron Radiolysis of Water and Aqueous Solutions. J. Phys. Chem. A 1997, 101, 5828–5838. [Google Scholar] [CrossRef]
- Pimblott, S.M.; LaVerne, J.A. Cooperative Effects of Scavengers on the Scavenged Yield of the Hydrated Electron. J. Phys. Chem. 1992, 96, 8904–8909. [Google Scholar] [CrossRef]
- Angel, L.; Stace, A.J. Reappraisal of the Contribution From [O2•(H2O)n]+ Cluster Ions to the Chemistry of the Ionosphere. J. Phys. Chem. A 1999, 103, 2999–3005. [Google Scholar] [CrossRef]
- Tomoda, S.; Kimura, K. Ionization Energies and Hydrogen-bond Strength of the Water Clusters. Chem. Phys. Lett. 1983, 102, 560–564. [Google Scholar] [CrossRef]
- Novakovskaya, Y.V.; Stepanov, N.F. Small Charged Water Clusters Cations. J. Phys. Chem. A 1999, 103, 3285–3288. [Google Scholar] [CrossRef]
- Barnett, R.N.; Landman, U. Structure and Energetics of Ionized Water Clusters (H2O)n+., n = 2–5. J. Phys. Chem. A 1997, 101, 164–169. [Google Scholar] [CrossRef]
- Gill, P.M.W.; Radom, L. Structures and Stabilities of Singly Charged Three-Electron Hemi-bonded Systems and Their Hydrogen-Bonded Isomers. J. Am. Chem. Soc. 1988, 110, 4931–4941. [Google Scholar] [CrossRef]
- Sodupe, M.; Oliva, A.; Bertran, J. Theoretical Study of the Ionization of the H2O-H2O, NH3-H2O, and FH-H2O Hydrogen-Bonded Molecules. J. Am. Chem. Soc. 1994, 116, 8249–8258. [Google Scholar] [CrossRef]
- Cheng, Q.; Evangelista, F.A.; Simmonett, A.C.; Yamaguchi, Y.; Schaefer III, H.F. Water Dimer Radical Cation Structures, Vibrational Frequencies, and Energetics. J. Phys. Chem. A 2009, 113, 13779–13789. [Google Scholar] [CrossRef]
- Xantheas, S.S. Theoretical Study of Hydroxide Ion-Water Clusters. J. Am. Chem. Soc. 1995, 117, 10373–10380. [Google Scholar] [CrossRef]
- Weis, P.; Kemper, P.R.; Xantheas, S.S. A New Determination of the Fluoride Ion-Water Bond Energy. J. Am. Chem. Soc. 1999, 121, 3531–3532. [Google Scholar] [CrossRef]
- Roscioli, J.R.; Diken, E.G.; Johnson, M.A.; Horvath, S.; McCoy, A.B. Prying Apart a Water Molecule with Anionic H-Bonding: A Comparative Spectroscopic Study of the X-•H2O (X = OH, O, F, Cl, and Br) Binary Complexes in the 600–3800 cm-1 Region. J. Phys. Chem. A 2006, 110, 4943–4952. [Google Scholar] [CrossRef]
- Horvath, S.; McCoy, A.B.; Roscioli, J.R.; Johnson, M.A. Vibrationally Induced Proton Transfer in F-(H2O) and F-(D2O). J. Phys. Chem. A 2008, 112, 12337–12344. [Google Scholar] [CrossRef]
- Kurnig, I.J.; Scheiner, S. Additivity of the Effects of External Ions and Dipoles upon the Energetics of Proton Transfe. Int. J. Quantum Chem. 1986, 13, 71–79. [Google Scholar]
- Hunt, S.W.; Higgins, K.J.; Craddock, M.B.; Brauer, C.S.; Leopold, K.R. Influence of a Polar Near-Neighbor on Incipient Proton Transfer in a Strongly Hydrogen Bonded Complex. J. Am. Chem. Soc. 2003, 125, 13850–13860. [Google Scholar] [CrossRef] [PubMed]
- Gardenier, G.H.; Johnson, M.A. Spectroscopic Study of the Ion-Radical H-Bond in H4O2+. J. Phys. Chem. A 2009, 113, 4772–4779. [Google Scholar] [CrossRef] [PubMed]
- Angel, L.; Stace, A.J. Dissociation Patterns of (H2O)n+. Cluster Ions, for n = 2–6. Chem. Phys. Lett. 2001, 101, 164–169. [Google Scholar] [CrossRef]
- Pieniazek, P.A.; Vondele, J.V.; Jungwirth, P.; Krylov, A.I.; Bradforth, S.E. Electronic Structure of the Water Dimer Cation. J. Phys. Chem. A 2008, 112, 6159–6170. [Google Scholar] [CrossRef]
- Lu, E.-P.; Pan, P.-R.; Li, Y.-C.; Tsai, M.-K.; Kuo, J.-L. Structural evolution and solvation of the OH radical in ionized water radical cations (H2O)n+, n = 5–8. Phys. Chem. Chem. Phys. 2014, 16, 18888–18895. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Kudoh, S.; Kawai, Y.; Okada, Y.; Orii, T.; Takeuchi, K. Collisional Reaction of Water Cluster Cations (H2O)n+. (n = 2 and 3) with D2O. Chem. Phys. Lett. 2003, 377, 37–42. [Google Scholar] [CrossRef]
- Wang, Y.S.; Jiang, J.C.; Cheng, C.L.; Lin, S.H.; Lee, Y.T.; Chang, H.C. Identifying 2- and 3-coordinated H2O in Protonated Ion-Water Clusters by Vibrational Pre-Dissociation Spectroscopy and ab initio Calculations. J. Chem. Phys. 1997, 107, 9695–9698. [Google Scholar] [CrossRef]
- Wang, Y.S.; Tsai, C.H.; Lee, Y.T.; Chang, H.C.; Jiang, J.C.; Asvany, O.; Schlemmer, S.; Gerlich, D. Investigation of Protonated and Deprotonated Water Clusters Using a Low-Temperature 22-Pole Ion Trap. J. Phys. Chem. A 2003, 107, 4217–4225. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, M.; Fujii, A.; Ebata, T.; Mikami, N. Infrared spectroscopic evidence for protonated water clusters forming nanoscale cages. Science 2004, 304, 1134–1137. [Google Scholar] [CrossRef]
- Shin, J.W.; Hammer, N.I.; Diken, E.G.; Johnson, M.A.; Walters, R.S.; Jaeger, T.D.; Duncan, M.A.; Christie, R.A.; Jordan, K.D. Infrared signature of structures associated with the H+(H2O)n (n = 6 to 27) clusters. Science 2004, 304, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Momose, T.; Fushitani§, M.; Hoshina¶, H. Chemical reactions in quantum crystals. Int. Rev. Phys. Chem. 2005, 24, 533–552. [Google Scholar] [CrossRef]
- Douberly, G.E.; Rick, A.M.; Duncan, M.A. Infrared Spectroscopy of Perdeuterated Protonated Water Clusters in the Vicinity of the Clathrate Cage. J. Phys. Chem. A 2009, 113, 8449–8453. [Google Scholar] [CrossRef] [PubMed]
- Mizuse, K.; Fujii, A. Characterization of a Solvent-Separated Ion-Radical Pair in Cationized Water Networks: Infrared Photodissociation and Ar-Attachment Experiments for Water Cluster Radical Cations (H2O)n+. (n= 3–8). J. Phys. Chem. A 2013, 117, 929–938. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K. Structures of Water Octamers (H2O)8: Exploration of Ab initio Potential Energy Surfaces by the Scaled Hypersphere Search Method. J. Phys. Chem. A 2007, 111, 4527–4534. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Maeda, S.; Ohno, K. Quantum Chemistry Study of H+(H2O)8: A Global Search for Its Isomers by the Scaled Hypersphere Search Method, and Its Thermal Behavior. J. Phys. Chem. A 2007, 111, 10732–10737. [Google Scholar] [CrossRef]
- Luo, Y.; Maeda, S.; Ohno, K. Automated Exploration of Stable Isomers of H+(H2O)n (n = 5–7) via ab initio Calculations: An Application of the Anharmonic Downward Distortion Following Algorithm. J. Comput. Chem. 2008, 30, 952–961. [Google Scholar] [CrossRef]
- Ohno, K.; Maeda, S. A Scaled Hypersphere Search Method for the Topography of Reaction Pathways on the Potential Energy Surface. Chem. Phys. Lett. 2004, 109, 5742–5753. [Google Scholar] [CrossRef]
- Maeda, S.; Ohno, K. Global Mapping of Equilibrium and Transition Structures on Potential Energy Surfaces by the Scaled Hypersphere Search Method: Applications to ab Initio Surfaces of Formaldehyde and Propyne Molecules. J. Phys. Chem. A 2005, 109, 5742–5753. [Google Scholar] [CrossRef]
- Ohno, K.; Maeda, S. Global Reaction Route Mapping on Potential Energy Surfaces of Formaldehyde, Formic Acid, and Their Metal-Substituted Analogues. J. Phys. Chem. A 2006, 110, 8933–8941. [Google Scholar] [CrossRef]
- Tachikawa, H. A Direct ab-Initio Trajectory Study on the Ionization Dynamics of the Water Dimer. J. Phys. Chem. A 2002, 106, 6915–6921. [Google Scholar] [CrossRef]
- Tachikawa, H.; Takada, T. Proton transfer rates in ionized water clusters (H2O)n (n = 2–4). RSC Adv. 2015, 5, 6945–6953. [Google Scholar] [CrossRef]
- Khorana, S.; Hamill, W.H. Electronic Processes in the Pulse Radiolysis of Aqueous Solutions of Halide Ions. J. Phys. Chem. 1971, 75, 3081–3088. [Google Scholar] [CrossRef]
- Kim, K.J.; Hamill, W.H. Direct and Indirect Effects in Pulse Irradiated Concentrated Aqueous Solutions of Chloride and Sulfate Ions. J. Phys. Chem. 1976, 80, 2320–2325. [Google Scholar] [CrossRef]
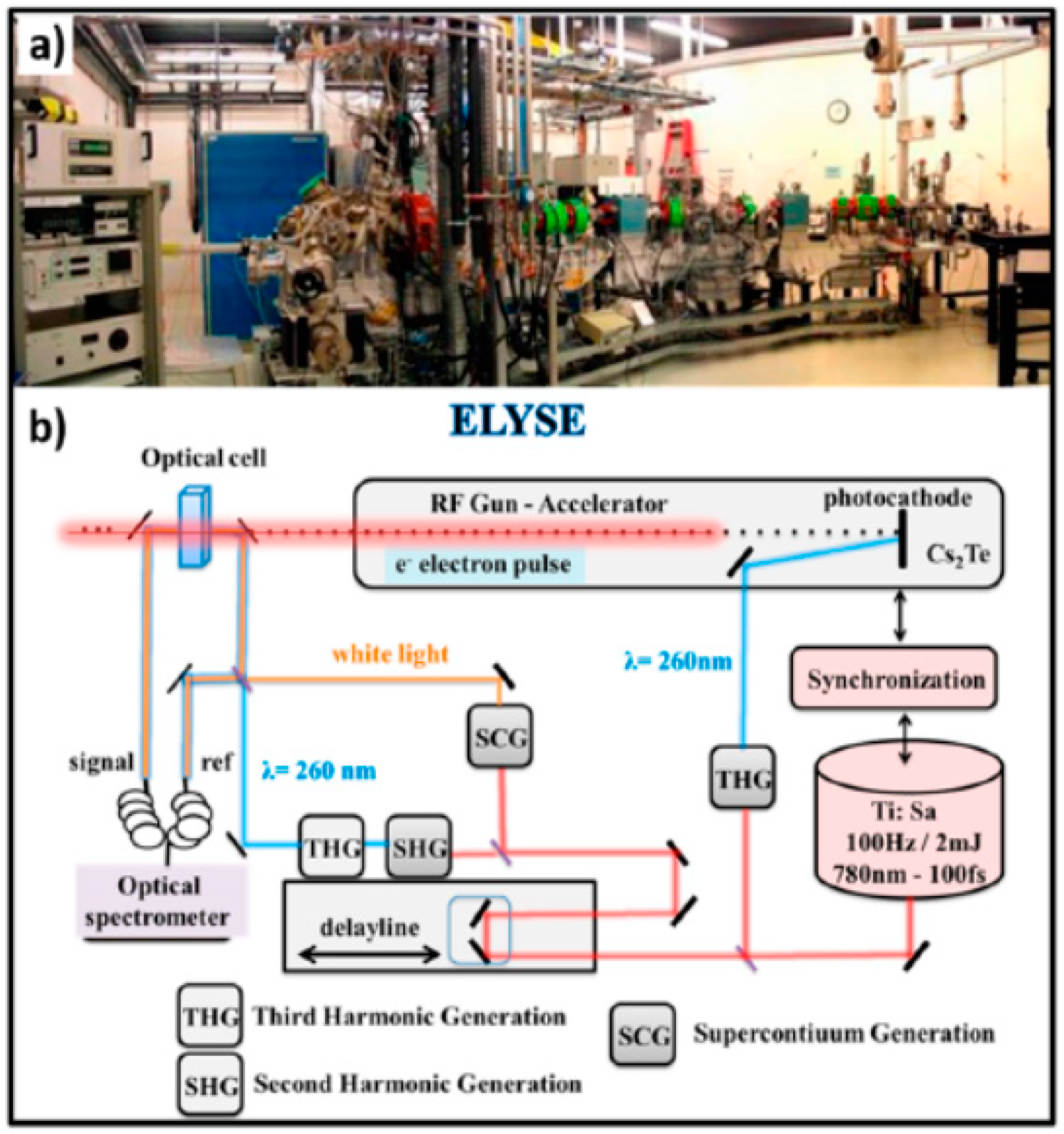
- Belloni, J.; Monard, H.; Gobert, F.; Larbre, J.P.; Demarque, A.; De Waele, V.; Lampre, I.; Marignier, J.L.; Mostafavi, M.; Bourdon, J.C.; et al. ELYSE—A picosecond electron accelerator for pulse radiolysis research. Nucl. Instrum. Methods Phys. Res. Sec. A 2005, 539, 527–539. [Google Scholar] [CrossRef]
- Schmidhammer, U.; Jeunesse, P.; Stresing, G.; Mostafavi, M. A Broadband Ultrafast Transient Absorption Spectrometer Covering the Range from Near-Infrared (NIR) Down to Green. Appl. Spectrosc. 2014, 68, 1137–1147. [Google Scholar] [CrossRef]
- Ma, J.; Wang, F.; Mostafavi, M. Ultrafast Chemistry of Water Radical Cation, H(2)O(*+), in Aqueous Solutions. Molecules 2018, 232, 244. [Google Scholar] [CrossRef] [Green Version]
- Balcerzyk, A.; LaVerne, J.; Mostafavi, M. Direct and indirect radiolytic effects in highly concentrated aqueous solutions of bromide. J. Phys. Chem. A 2011, 115, 4326–4333. [Google Scholar] [CrossRef]
- EI Omar, A.K.; Schmidhammer, U.; Rousseau, B.; Laverne, J.; Mostafavi, M. Competition Reaction of H2O+. Radical in Concentrated Cl- Aqueous Solutions: Picosecond Pulse Radiolysis Study. J. Phys. Chem. A 2012, 116, 11509–11518. [Google Scholar] [CrossRef]
- Balcerzyk, A.; Schmidhammer, U.; El Omar, A.K.; Jeunesse, P.; Larbre, J.P.; Mostafavi, M. Picosecond Pulse Radiolysis of Direct and Indirect Radiolytic Effects in Highly Concentrated Halide Aqueous Solutions. J. Phys. Chem. A 2011, 115, 9151–9159. [Google Scholar] [CrossRef]
- El Omar, A.K.; Schmidhammer, U.; Balcerzyk, A.; La Verne, J. Spur Reactions Observed by Picosecond Pulse Radiolysis in Highly Concentrated Bromide Aqueous Solutions. J. Phys. Chem. A 2013, 117, 2287–2293. [Google Scholar] [CrossRef]
- Ghalei, M.; Ma, J.; Schmidhammer, U.; Vandenborre, J.; Fattahi, M.; Mostafavi, M. Picosecond Pulse Radiolysis of Highly Concentrated Carbonate Solutions. J. Phys. Chem. B 2016, 120, 2434–2439. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Schmidhammer, U.; Mostafavi, M. Picosecond Pulse Radiolysis of Highly Concentrated Sulfuric Acid Solutions: Evidence for the Oxidation Reactivity of Radical Cation H2O+. J. Phys. Chem. A 2014, 118, 4030–4037. [Google Scholar] [CrossRef] [PubMed]
- Choe, Y.K.; Tsuchida, E.; Ikeshoji, T. First-principles molecular dynamics study on aqueous sulfuric acid solutions. J. Chem. Phys. 2007, 126, 154510. [Google Scholar] [CrossRef]
- Kameda, Y.; Hosoya, K.; Sakamoto, S.; Suzuki, H.; Usuki, T.; Uemura, O. Hydrogen-Bonded Structure in Aqueous Sulfuric Acid Solutions. J. Mol. Liq. 1995, 65, 305–308. [Google Scholar] [CrossRef]
- Ma, J.; Schmidhammer, U.; Pernot, P.; Mostafavi, M. Reactivity of the Strongest Oxidizing Species in Aqueous Solutions: The Short-Lived Radical Cation H2O+. J. Phys. Chem. Lett. 2014, 5, 258–261. [Google Scholar] [CrossRef]
- Tang, Y.; Thorn, R.P.; Mauldin, R.L.; Wine, P.H. Kinetics and Spectroscopy of SO4- Radical in Aqueous Solution. J. Photochem. Photobiol. A 1988, 44, 243–258. [Google Scholar] [CrossRef]
- Hayon, E.; Treinin, A.; Wilf, J. Electronic Spectra, Photochemistry, and Autoxidation Mechanism of the Sulfite-Bisulfite-Pyrosulfite Systems. SO2-, SO3-, SO4-, and SO5- Radicals. J. Am. Chem. Soc. 1972, 94, 47–57. [Google Scholar] [CrossRef]
- Ma, J.; Schmidhammer, U.; Mostafavi, M. icosecond Pulse Radiolysis of Highly Concentrated Phosphoric Acid Solutions: Mechanism of Phosphate Radical Formation. J. Phys. Chem. B 2015, 119, 7180–7185. [Google Scholar] [CrossRef]
- Wang, F.; Schmidhammer, U.; Lande, A.; Mostafavi, M. Ultra-Fast Charge Migration Competes with Proton Transfer in the Early Chemistry of H2O+. Phys. Chem. Chem. Phys. 2017, 19, 2894–2899. [Google Scholar] [CrossRef]
- Lépine, F.; Ivanov, M.Y.; Vrakking, M.J.J. Attosecond molecular dynamics: Fact or fiction? Nat. Photonics 2014, 8, 195–204. [Google Scholar] [CrossRef]



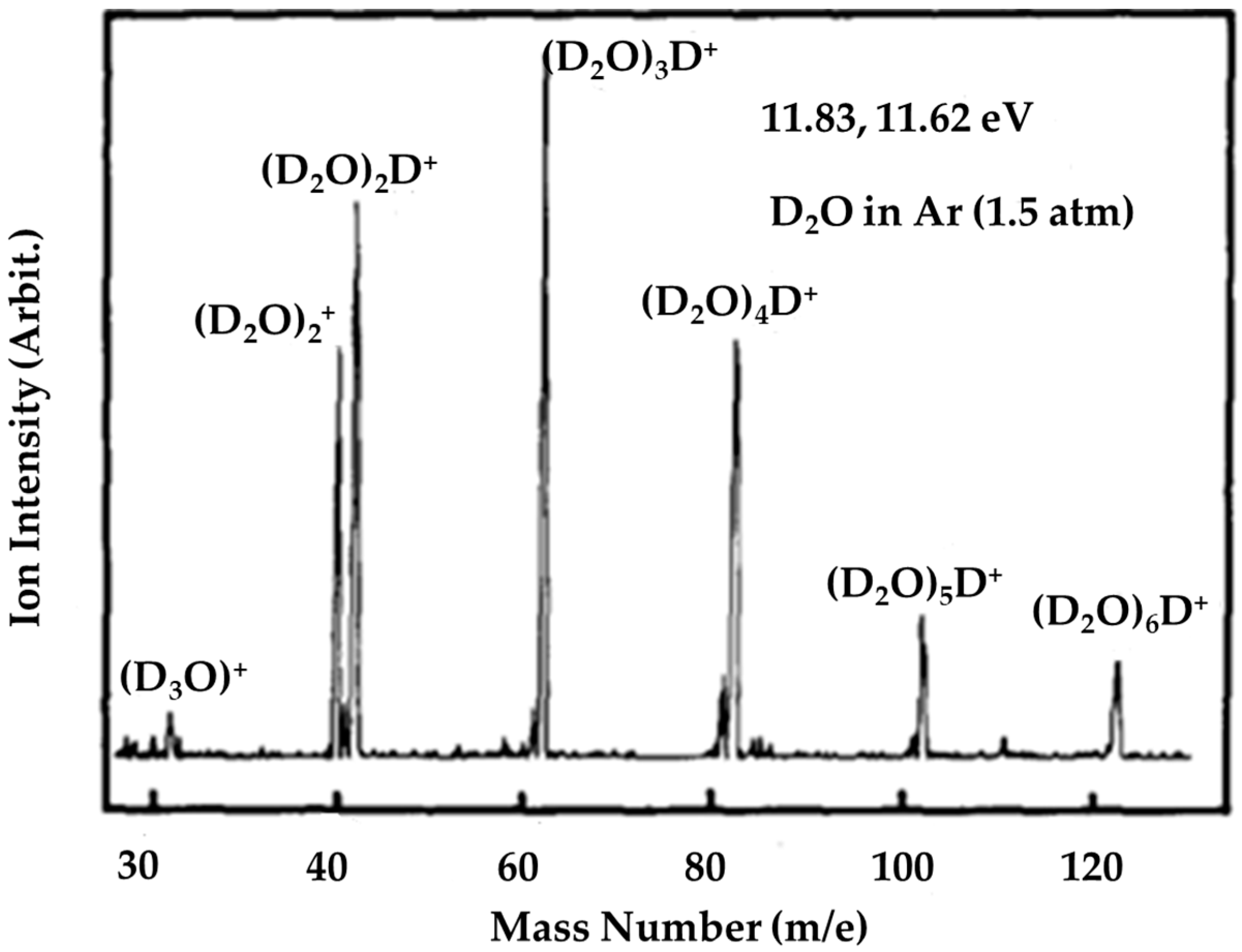

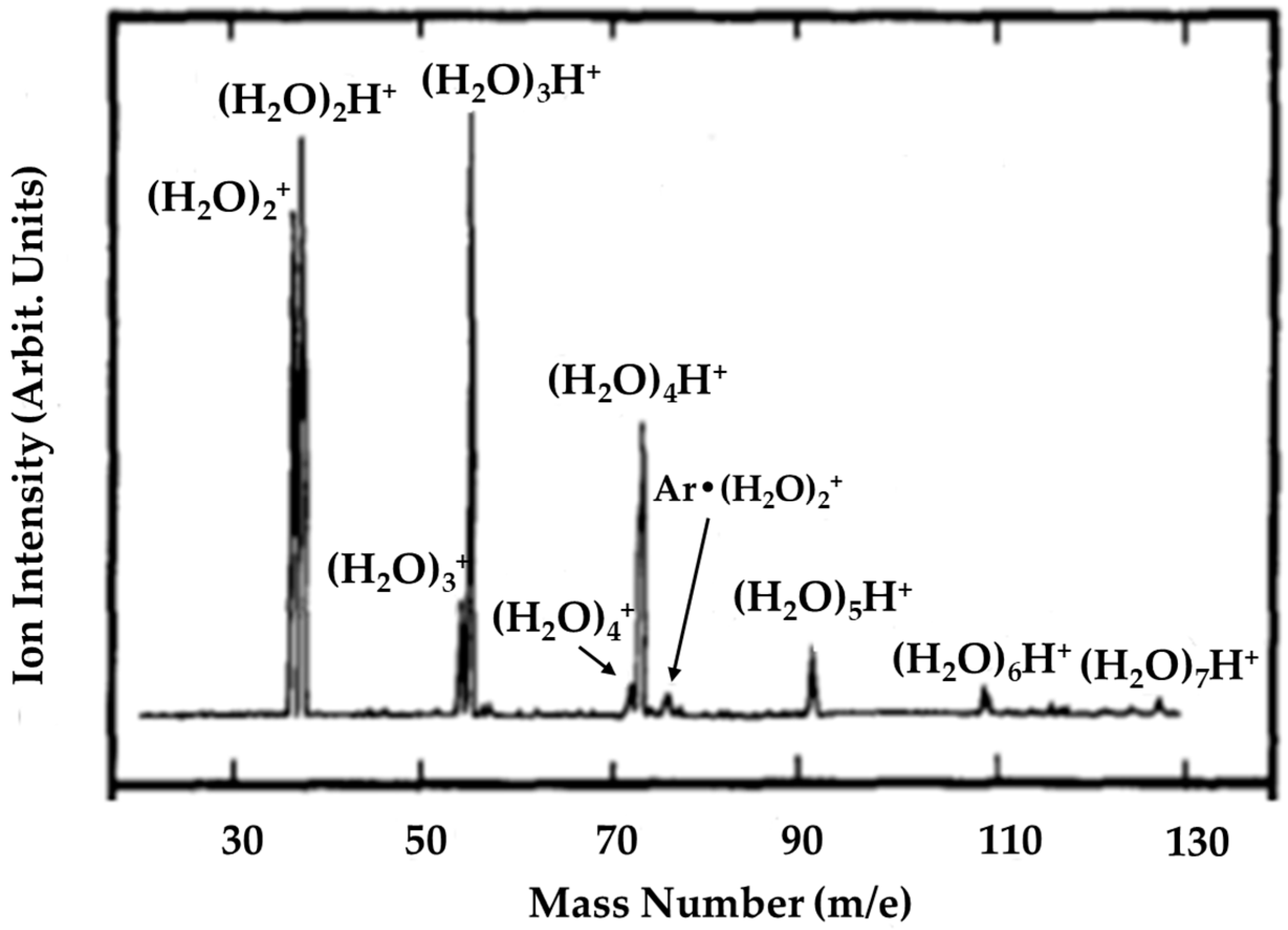
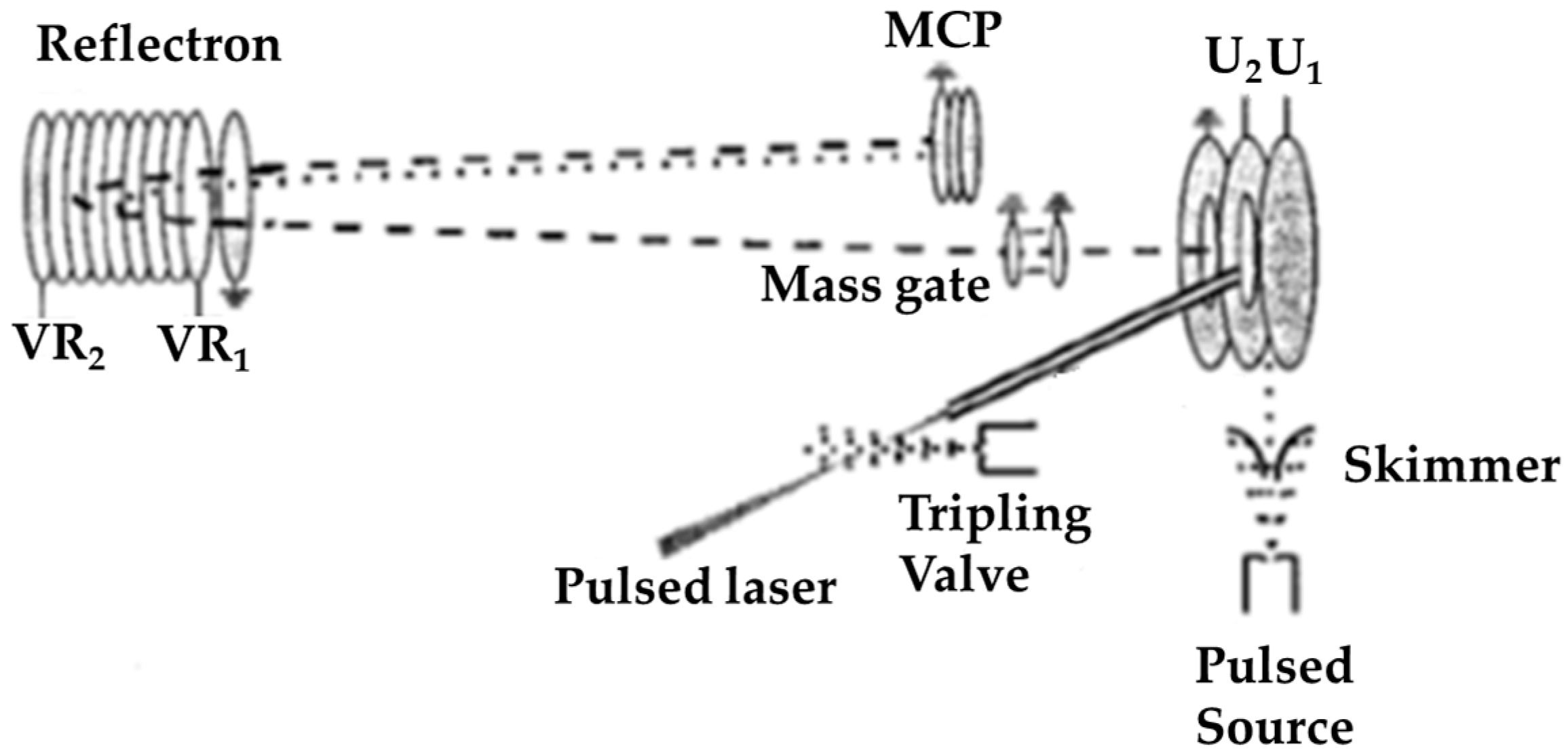
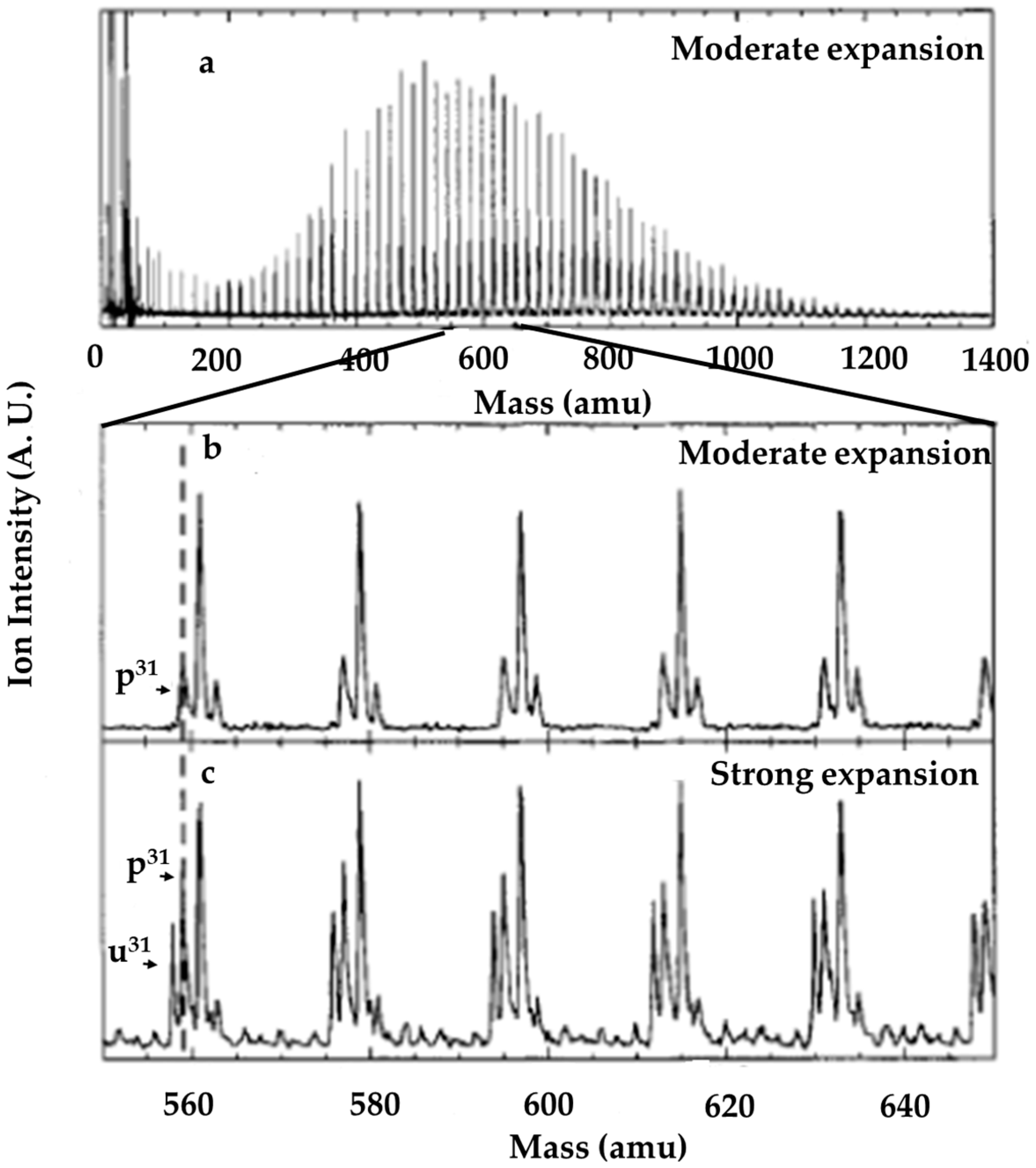







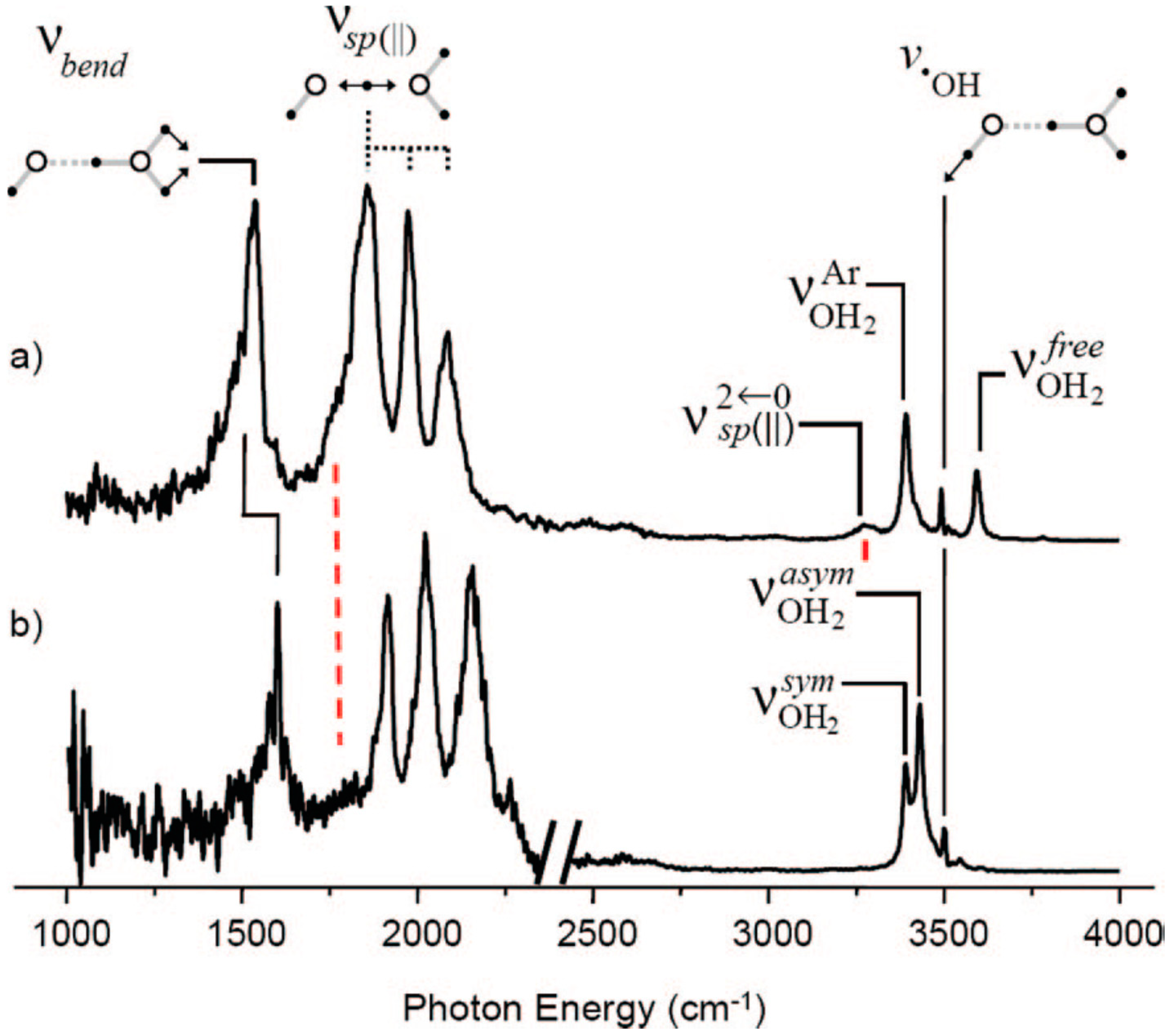
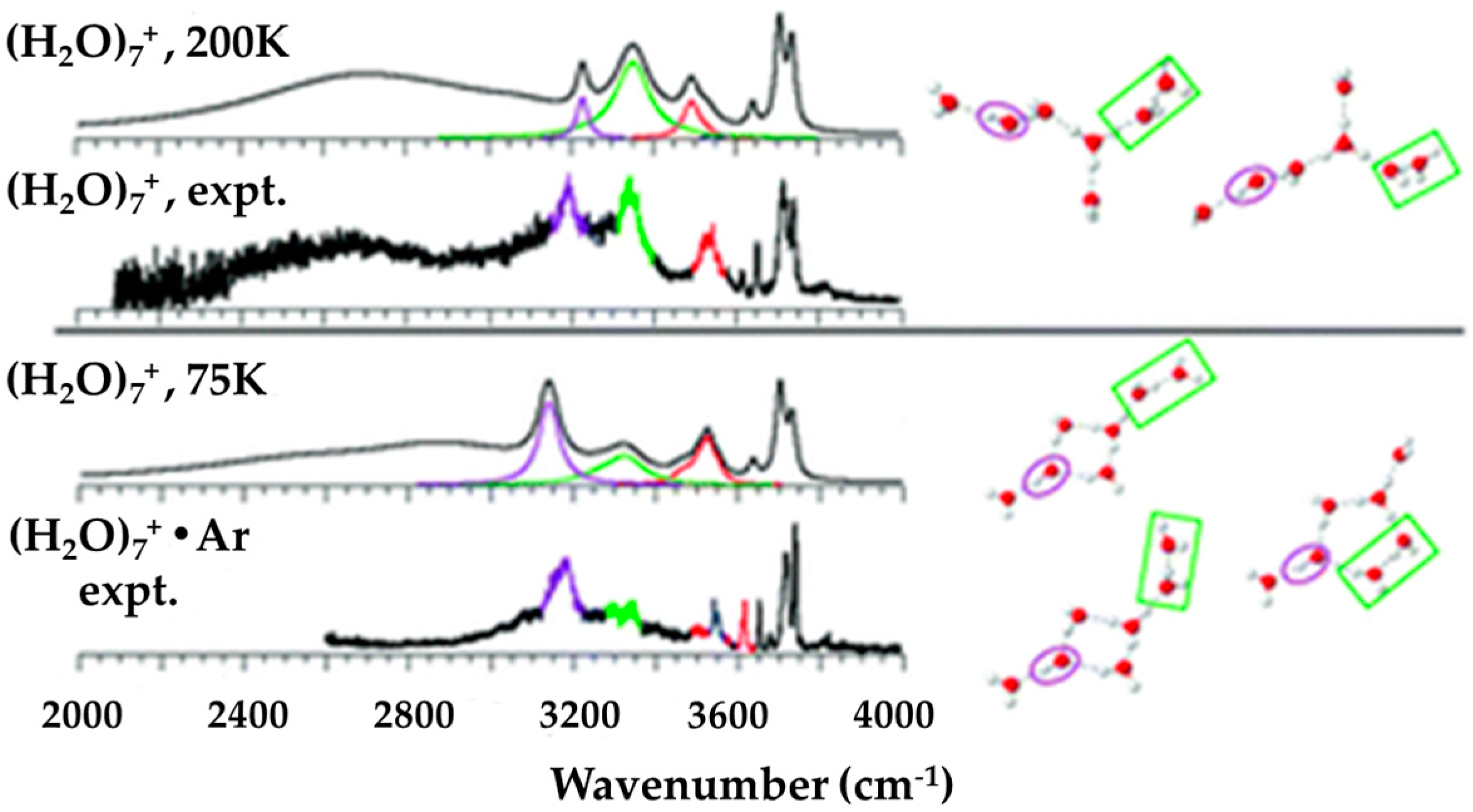
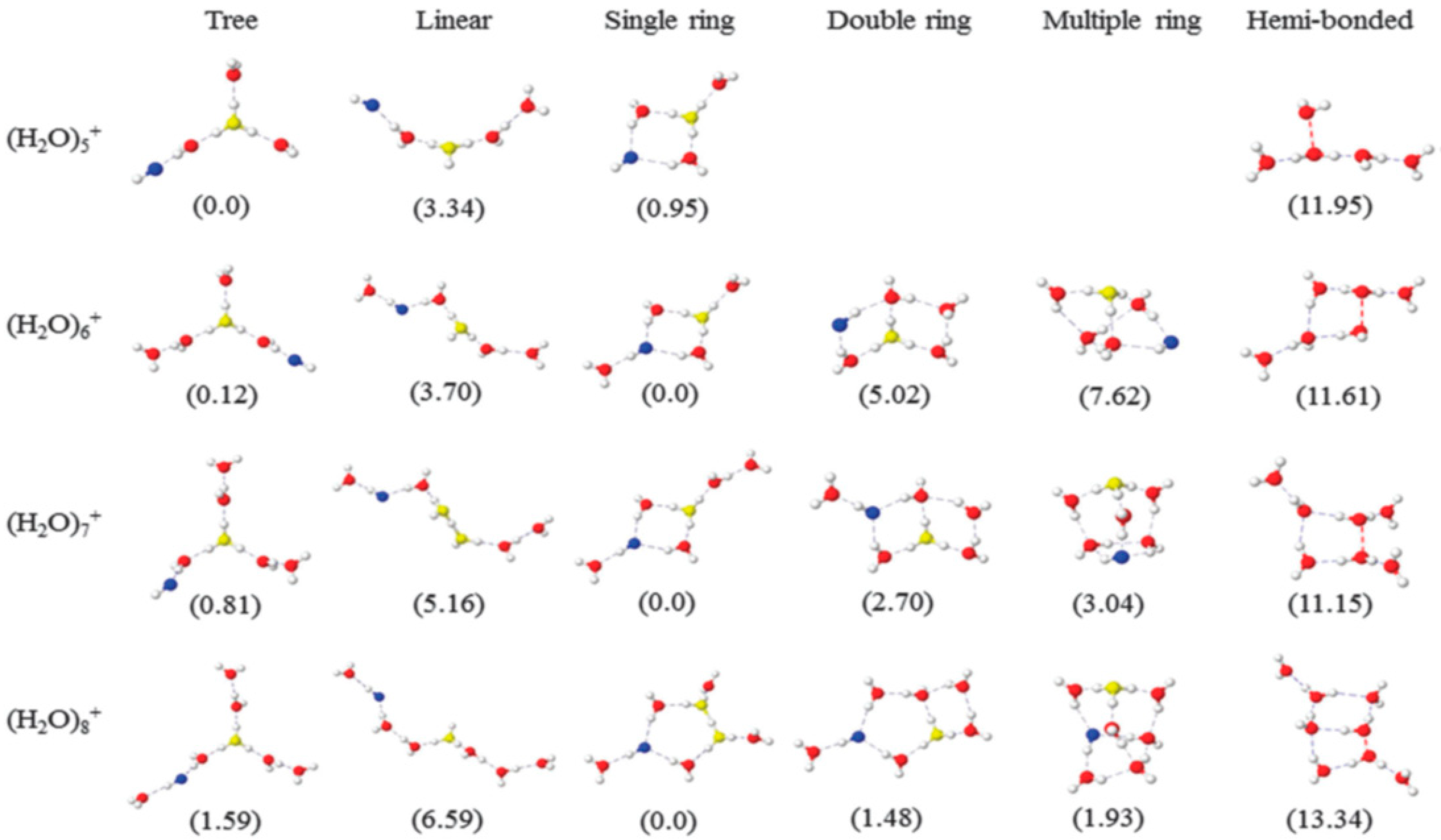

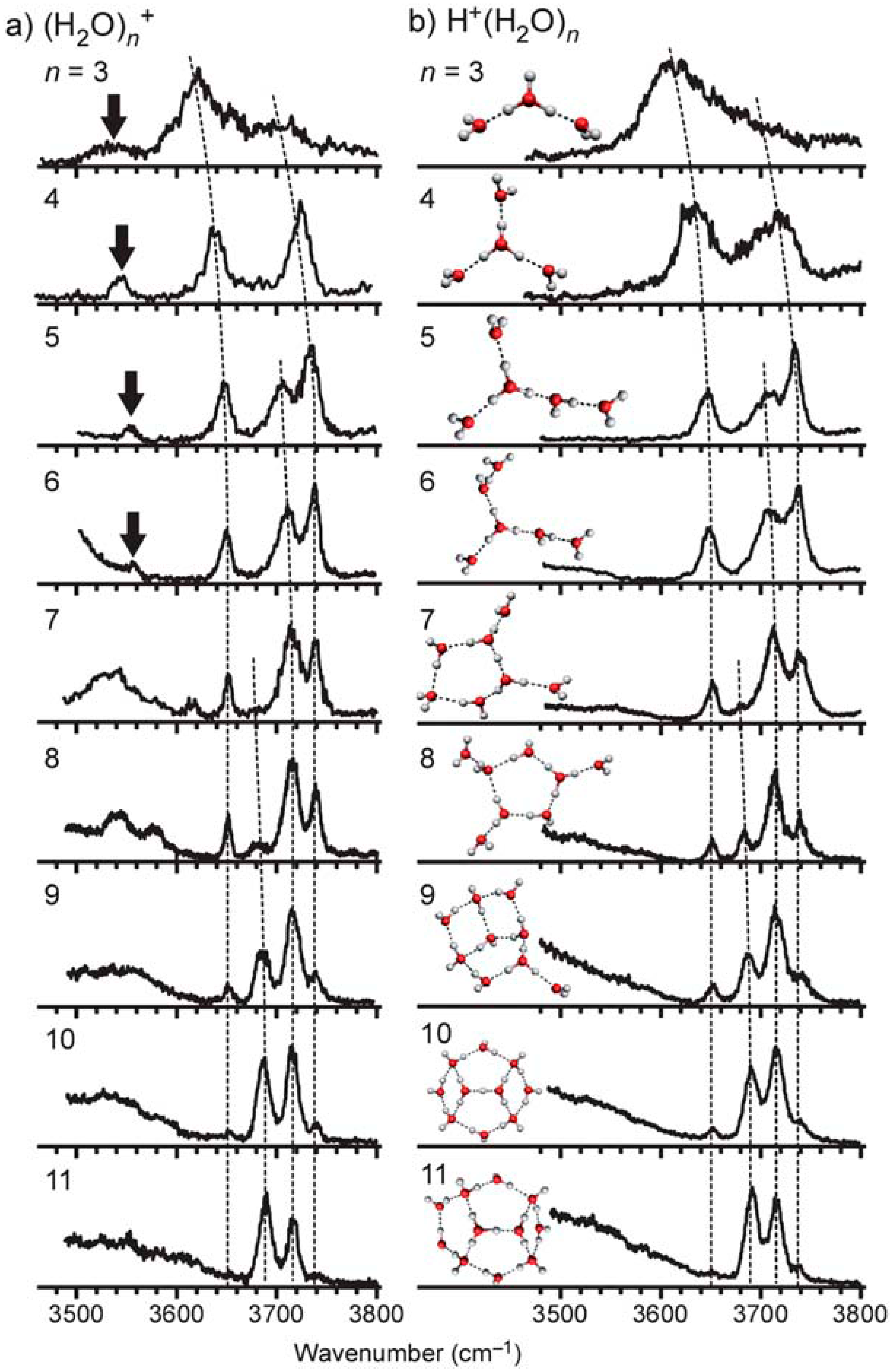
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mi, D.; Chingin, K. Water Radical Cations in the Gas Phase: Methods and Mechanisms of Formation, Structure and Chemical Properties. Molecules 2020, 25, 3490. https://doi.org/10.3390/molecules25153490
Mi D, Chingin K. Water Radical Cations in the Gas Phase: Methods and Mechanisms of Formation, Structure and Chemical Properties. Molecules. 2020; 25(15):3490. https://doi.org/10.3390/molecules25153490
Chicago/Turabian StyleMi, Dongbo, and Konstantin Chingin. 2020. "Water Radical Cations in the Gas Phase: Methods and Mechanisms of Formation, Structure and Chemical Properties" Molecules 25, no. 15: 3490. https://doi.org/10.3390/molecules25153490
APA StyleMi, D., & Chingin, K. (2020). Water Radical Cations in the Gas Phase: Methods and Mechanisms of Formation, Structure and Chemical Properties. Molecules, 25(15), 3490. https://doi.org/10.3390/molecules25153490