Neuroprotective Effects of Cryptotanshinone in a Direct Reprogramming Model of Parkinson’s Disease

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. MG132-Induced In Vitro Model for PD Using hiNPCs from PD Patient
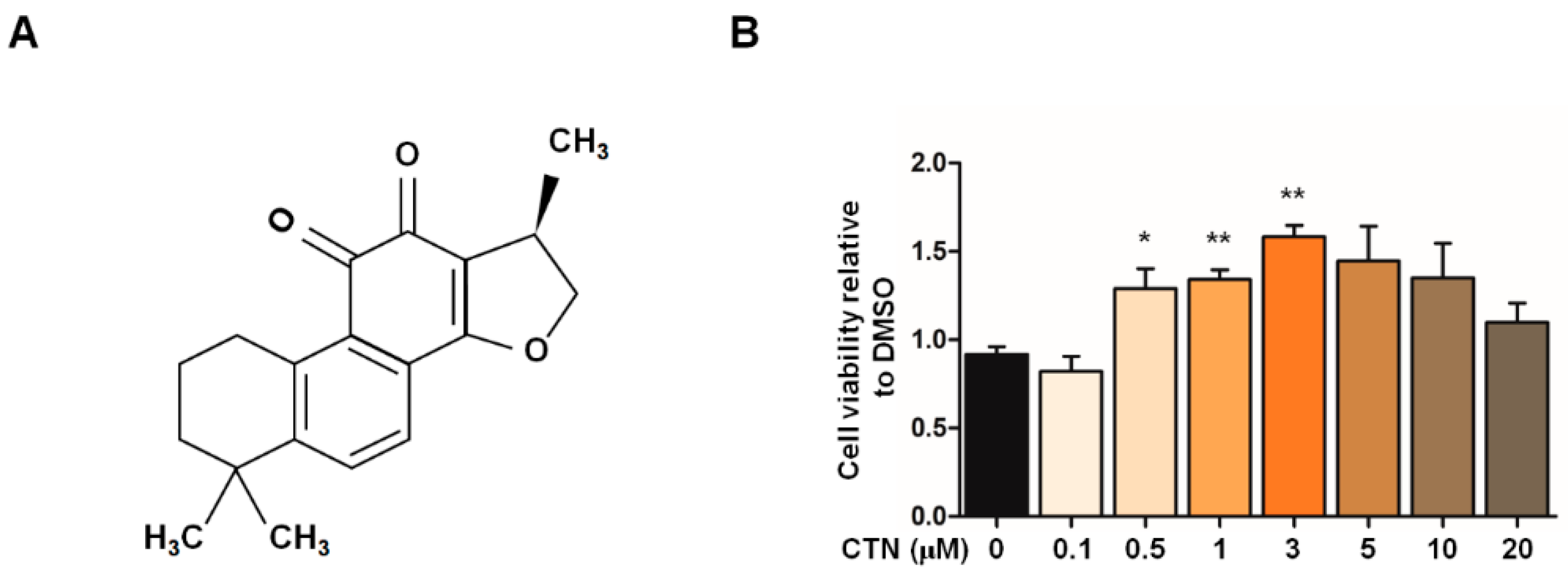
2.2. Cytoprotective Effects of CTN on the In Vitro PD Model
2.3. Cell Death Attenuation by CTN
2.4. Recovery of Mitochondrial Functions by CTN
2.5. Induction of NRF2-Mediated Oxidative Stress Response by CTN
2.6. Expression of Antioxidative and Mitochondrial Biogenesis Molecules by CTN
3. Materials and Methods
3.1. Cell Culture
3.2. Cell Viability Assay
3.3. Flow Cytometry Analysis
3.4. Western Blot Analysis
3.5. Real-Time PCR for mRNA Quantification
3.6. Immunofluorescence
3.7. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Singh, A.; Zhi, L.; Zhang, H. LRRK2 and mitochondria: Recent advances and current views. Brain Res. 2019, 1702, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Macdonald, R.; Payne, T.; Sassani, M.; Jenkins, T.; Bandmann, O. Translational approaches to restoring mitochondrial function in Parkinson’s disease. FEBS Lett. 2018, 592, 776–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raza, C.; Anjum, R. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Mitochondria, Oxidative Damage, and Inflammation in Parkinson’s Disease. Ann. N. Y. Acad. Sci. 2006, 991, 120–131. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Zeng, X.S.; Geng, W.S.; Jia, J.J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10. [Google Scholar] [CrossRef]
- Hattori, N.; Tanaka, M.; Ozawa, T.; Mizuno, Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson’s disease. Ann. Neurol. 1991, 30, 563–571. [Google Scholar] [CrossRef]
- Papkovskaia, T.D.; Chau, K.Y.; Inesta-vaquera, F.; Papkovsky, D.B.; Healy, D.G.; Nishio, K.; Staddon, J.; Duchen, M.R.; Hardy, J.; Schapira, A.H.V.; et al. G2019s leucine-rich repeat kinase 2 causes uncoupling protein-mediated mitochondrial depolarization. Hum. Mol. Genet. 2012, 21, 4201–4213. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial Complex I Deficiency in Parkinson’s Disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Janetzky, B.; Hauck, S.; Youdim, M.B.H.; Riederer, P.; Jellinger, K.; Pantucek, F.; Zöchling, R.; Boissl, K.W.; Reichmann, H. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci. Lett. 1994, 169, 126–128. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [Green Version]
- Sai, Y.; Zou, Z.; Peng, K.; Dong, Z. The Parkinson’s disease-related genes act in mitochondrial homeostasis. Neurosci. Biobehav. Rev. 2012, 36, 2034–2043. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef] [Green Version]
- Mendivil-Perez, M.; Velez-Pardo, C.; Jimenez-Del-Rio, M. Neuroprotective Effect of the LRRK2 Kinase Inhibitor PF-06447475 in Human Nerve-Like Differentiated Cells Exposed to Oxidative Stress Stimuli: Implications for Parkinson’s Disease. Neurochem. Res. 2016, 41, 2675–2692. [Google Scholar] [CrossRef]
- Albarracin, S.L.; Stab, B.; Casas, Z.; Sutachan, J.J.; Samudio, I.; Gonzalez, J.; Gonzalo, L.; Capani, F.; Morales, L.; Barreto, G.E. Effects of natural antioxidants in neurodegenerative disease. Nutr. Neurosci. 2012, 15, 1–9. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Olanow, C.W.; Greenamyre, J.T.; Bezard, E. Slowing of neurodegeneration in Parkinson’s disease and Huntington’s disease: Future therapeutic perspectives. Lancet 2014, 384, 545–555. [Google Scholar] [CrossRef]
- An, L.K.; Bu, X.Z.; Wu, H.Q.; Guo, X.D.; Ma, L.; Gu, L.Q. Reaction of tanshinones with biogenic amine metabolites in vitro. Tetrahedron 2002, 58, 10315–10321. [Google Scholar] [CrossRef]
- Su, C.Y.; Ming, Q.L.; Rahman, K.; Han, T.; Qin, L.P. Salvia miltiorrhiza: Traditional medicinal uses, chemistry, and pharmacology. Chin. J. Nat. Med. 2015, 13, 163–182. [Google Scholar] [CrossRef]
- Ke, F.; Wang, Z.; Song, X.; Ma, Q.; Hu, Y.; Jiang, L.; Zhang, Y.; Liu, Y.; Zhang, Y.; Gong, W. Cryptotanshinone induces cell cycle arrest and apoptosis through the JAK2/STAT3 and PI3k/Akt/NfκB pathways in cholangiocarcinoma cells. Drug Des. Devel. Ther. 2017, 11, 1753–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, X.; Zhang, X.S.; Liang, C.Z. Cryptotanshinone Attenuates Oxidative Stress and Inflammation through the Regulation of Nrf-2 and NF-κB in Mice with Unilateral Ureteral Obstruction. Basic Clin. Pharmacol. Toxicol. 2018, 123, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Wang, X.; Ying, W.; Wu, D.; Zhong, P. Cryptotanshinone Attenuates Inflammatory Response of Microglial Cells via the Nrf2/HO-1 Pathway. Front. Neurosci. 2019, 13, 852. [Google Scholar] [CrossRef] [Green Version]
- Cao, G.Y.; Wang, X.H.; Li, K.K.; Zhao, A.H.; Shen, L.; Yu, D.N. Neuroprotective effects of cryptotanshinone and 1,2-dihydrotanshinone I against MPTP induced mouse model of Parkinson’s disease. Phytochem. Lett. 2018, 26, 68–73. [Google Scholar] [CrossRef]
- Wood-Kaczmar, A.; Gandhi, S.; Wood, N.W. Understanding the molecular causes of Parkinson’s disease. Trends Mol. Med. 2006, 12, 521–528. [Google Scholar] [CrossRef]
- Ke, M.; Chong, C.M.; Su, H. Using induced pluripotent stem cells for modeling Parkinson’s disease. World J. Stem Cells 2019, 11, 634–649. [Google Scholar] [CrossRef]
- Lee, M.; Sim, H.; Ahn, H.; Ha, J.; Baek, A.; Jeon, Y.J.; Son, M.Y.; Kim, J. Direct reprogramming to human induced neuronal progenitors from fibroblasts of familial and sporadic Parkinson’s disease patients. Int. J. Stem Cells 2019, 12, 474–483. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Marchetto, M.C.; Bardy, C.; Gage, F.H. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat. Rev. Neurosci. 2016, 17, 424–437. [Google Scholar] [CrossRef]
- Arbab, M.; Baars, S.; Geijsen, N. Modeling motor neuron disease: The matter of time. Trends Neurosci. 2014, 37, 642–652. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Suzuki, K.; Nivet, E.; Li, M.; Montserrat, N.; Yi, F.; Xu, X.; Ruiz, S.; Zhang, W.; et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 2012, 491, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentea, E.; Verbruggen, L.; Massie, A. The Proteasome Inhibition Model of Parkinson’s Disease. J. Parkinsons. Dis. 2017, 7, 31–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Han, E.S.; Park, E.S.; Bang, H. Inhibition of MG132-induced mitochondrial dysfunction and cell death in PC12 cells by 3-morpholinosydnonimine. Brain Res. 2005, 1036, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Zafar, K.S.; Inayat-Hussain, S.H.; Ross, D. A comparative study of proteasomal inhibition and apoptosis induced in N27 mesencephalic cells by dopamine and MG132. J. Neurochem. 2007, 102, 913–921. [Google Scholar] [CrossRef] [PubMed]
- Perez-Alvarez, S.; Solesio, M.E.; Manzanares, J.; Jordán, J.; Galindo, M.F. Lactacystin requires reactive oxygen species and Bax redistribution to induce mitochondria-mediated cell death. Br. J. Pharmacol. 2009, 158, 1121–1130. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Xie, X.; Liu, C.; Lim, K.L.; Zhang, C.W.; Li, L. The Sources of Reactive Oxygen Species and Its Possible Role in the Pathogenesis of Parkinson’s Disease. Parkinsons. Dis. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Puspita, L.; Chung, S.Y.; Shim, J.W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Heo, H.Y.; Park, J.M.; Kim, C.H.; Han, B.S.; Kim, K.S.; Seol, W. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp. Cell Res. 2010, 316, 649–656. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Perelman, A.; Wachtel, C.; Cohen, M.; Haupt, S.; Shapiro, H.; Tzur, A. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 2012, 3, e430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vomund, S.; Schäfer, A.; Parnham, M.J.; Brüne, B.; Von Knethen, A. Nrf2, the master regulator of anti-oxidative responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Yuan, L.; Wang, W.; Zhang, M.; Wang, Q.; Li, S.; Zhang, L.; Hu, K. Tricetin protects against 6-OHDA-induced neurotoxicity in Parkinson’s disease model by activating Nrf2/HO-1 signaling pathway and preventing mitochondria-dependent apoptosis pathway. Toxicol. Appl. Pharmacol. 2019, 378, 114617. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.C.; Lee-Chen, G.J.; Chen, C.M.; Wu, Y.R.; Chen, Y.J.; Lin, J.L.; Lo, Y.S.; Yao, C.F.; Chang, K.H. Neuroprotection of Indole-Derivative Compound NC001-8 by the Regulation of the NRF2 Pathway in Parkinson’s Disease Cell Models. Oxid. Med. Cell. Longev. 2019, 2019, 5074367. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.; Zhang, R.; Hong, B.H.; Yang, E.J.; Kang, K.A.; Choi, M.; Kim, K.C.; Noh, S.J.; Kim, H.S.; Lee, N.H.; et al. Phloroglucinol Attenuates Motor Functional Deficits in an Animal Model of Parkinson’s Disease by Enhancing Nrf2 Activity. PLoS ONE 2013, 8, e71178. [Google Scholar] [CrossRef]
- Tsou, Y.H.; Shih, C.T.; Ching, C.H.; Huang, J.Y.; Jen, C.J.; Yu, L.; Kuo, Y.M.; Wu, F.S.; Chuang, J.I. Treadmill exercise activates Nrf2 antioxidant system to protect the nigrostriatal dopaminergic neurons from MPP+ toxicity. Exp. Neurol. 2015, 263, 50–62. [Google Scholar] [CrossRef]
- Tufekci, K.U.; Civi Bayin, E.; Genc, S.; Genc, K. The Nrf2/ARE pathway: A promising target to counteract mitochondrial dysfunction in Parkinson’s disease. Parkinsons. Dis. 2011, 2011, 314082. [Google Scholar] [CrossRef] [Green Version]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef] [Green Version]
- Son, H.J.; Choi, J.H.; Lee, J.A.; Kim, D.J.; Shin, K.J.; Hwang, O. Induction of NQO1 and Neuroprotection by a Novel Compound KMS04014 in Parkinson’s Disease Models. J. Mol. Neurosci. 2015, 56, 263–272. [Google Scholar] [CrossRef]
- Ye, Q.; Chen, C.; Si, E.; Cai, Y.; Wang, J.; Huang, W.; Li, D.; Wang, Y.; Chen, X. Mitochondrial effects of PGC-1alpha silencing in MPP+ treated human SH-SY5Y neuroblastoma cells. Front. Mol. Neurosci. 2017, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinsons disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Ferretta, A.; Gaballo, A.; Tanzarella, P.; Piccoli, C.; Capitanio, N.; Nico, B.; Annese, T.; Di Paola, M.; Dell’Aquila, C.; De Mari, M.; et al. Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 902–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkelä, J.; Tselykh, T.V.; Kukkonen, J.P.; Eriksson, O.; Korhonen, L.T.; Lindholm, D. Peroxisome proliferator-activated receptor-γ (PPARγ) agonist is neuroprotective and stimulates PGC-1α expression and CREB phosphorylation in human dopaminergic neurons. Neuropharmacology 2016, 102, 266–275. [Google Scholar] [CrossRef]
- Ye, Q.; Huang, W.; Li, D.; Si, E.; Wang, J.; Wang, Y.; Chen, C.; Chen, X. Overexpression of PGC-1α Influences Mitochondrial Signal Transduction of Dopaminergic Neurons. Mol. Neurobiol. 2016, 53, 3756–3770. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The SH-SY5Y cell line in Parkinson’s disease research: A systematic review. Mol. Neurodegener. 2017, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Jang, W.; Kim, H.J.; Li, H.; Jo, K.D.; Lee, M.K.; Yang, H.O. The Neuroprotective Effect of Erythropoietin on Rotenone-Induced Neurotoxicity in SH-SY5Y Cells Through the Induction of Autophagy. Mol. Neurobiol. 2016, 53, 3812–3821. [Google Scholar] [CrossRef]
- Torrent, R.; De Angelis Rigotti, F.; Dell’Era, P.; Memo, M.; Raya, A.; Consiglio, A. Using iPS Cells toward the Understanding of Parkinson’s Disease. J. Clin. Med. 2015, 4, 548–566. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.; Paquola, A.C.M.; Ku, M.; Hatch, E.; Böhnke, L.; Ladjevardi, S.; McGrath, S.; Campbell, B.; Lee, H.; Herdy, J.R.; et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 2015, 17, 705–718. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.M.; Blithikioti, C.; Ku, M.; et al. Mitochondrial Aging Defects Emerge in Directly Reprogrammed Human Neurons due to Their Metabolic Profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef] [Green Version]
- Huh, C.J.; Zhang, B.; Victor, M.B.; Dahiya, S.; Batista, L.F.Z.; Horvath, S.; Yoo, A.S. Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. eLife 2016, 5, e18648. [Google Scholar] [CrossRef] [PubMed]
- Böhnke, L.; Traxler, L.; Herdy, J.R.; Mertens, J. Human neurons to model aging: A dish best served old. Drug Discov Today Dis Model. 2018, 27, 43–49. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound are available from the authors. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.-E.; Sim, H.; Yoo, H.M.; Lee, M.; Baek, A.; Jeon, Y.-J.; Seo, K.-S.; Son, M.-Y.; Yoon, J.S.; Kim, J. Neuroprotective Effects of Cryptotanshinone in a Direct Reprogramming Model of Parkinson’s Disease. Molecules 2020, 25, 3602. https://doi.org/10.3390/molecules25163602
Lee J-E, Sim H, Yoo HM, Lee M, Baek A, Jeon Y-J, Seo K-S, Son M-Y, Yoon JS, Kim J. Neuroprotective Effects of Cryptotanshinone in a Direct Reprogramming Model of Parkinson’s Disease. Molecules. 2020; 25(16):3602. https://doi.org/10.3390/molecules25163602
Chicago/Turabian StyleLee, Joo-Eun, Hyuna Sim, Hee Min Yoo, Minhyung Lee, Aruem Baek, Young-Joo Jeon, Kang-Sik Seo, Mi-Young Son, Joo Seog Yoon, and Janghwan Kim. 2020. "Neuroprotective Effects of Cryptotanshinone in a Direct Reprogramming Model of Parkinson’s Disease" Molecules 25, no. 16: 3602. https://doi.org/10.3390/molecules25163602
APA StyleLee, J.-E., Sim, H., Yoo, H. M., Lee, M., Baek, A., Jeon, Y.-J., Seo, K.-S., Son, M.-Y., Yoon, J. S., & Kim, J. (2020). Neuroprotective Effects of Cryptotanshinone in a Direct Reprogramming Model of Parkinson’s Disease. Molecules, 25(16), 3602. https://doi.org/10.3390/molecules25163602