Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery

 ,
,  , , and
, , and 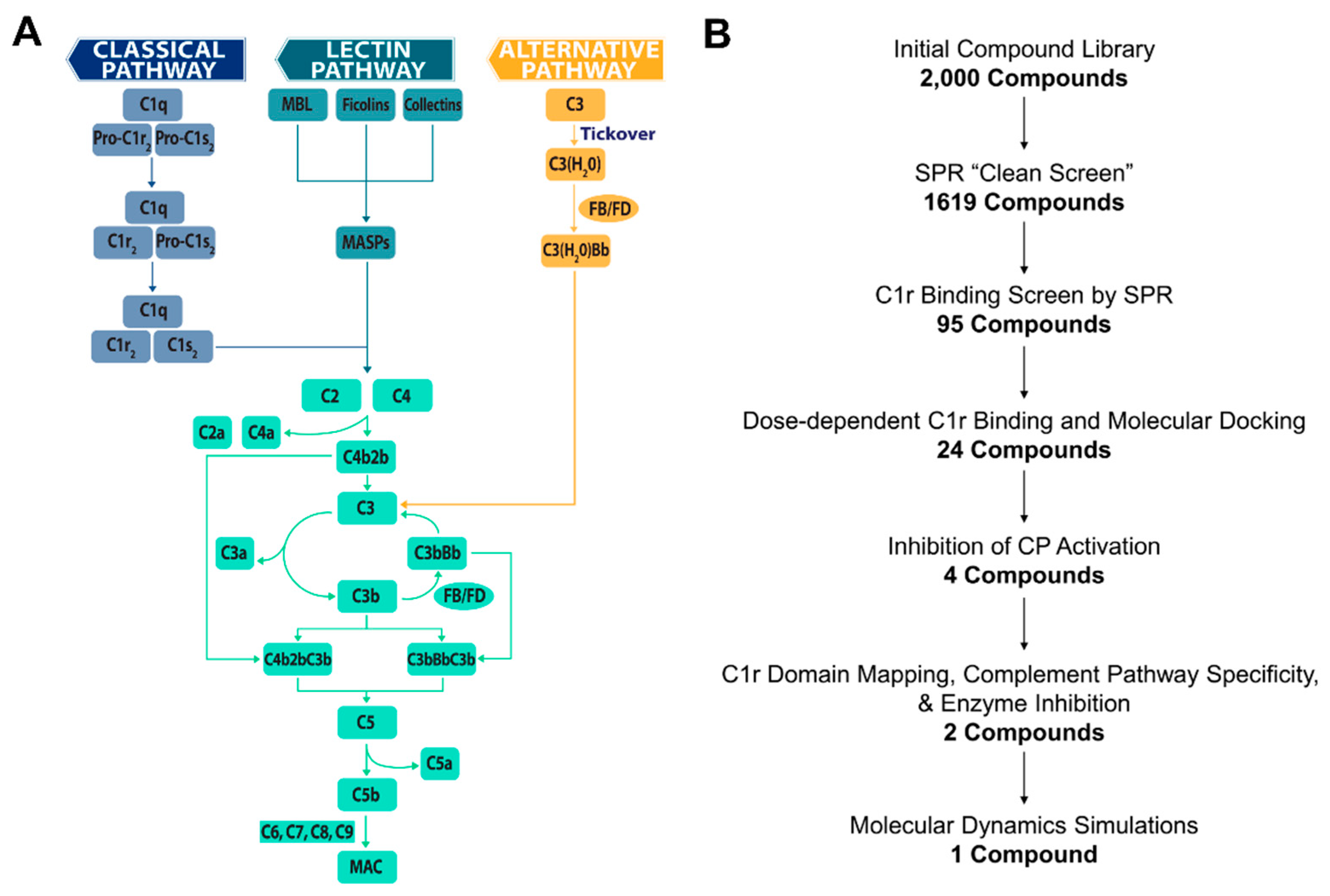{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Small-Molecule Fragment Library Design
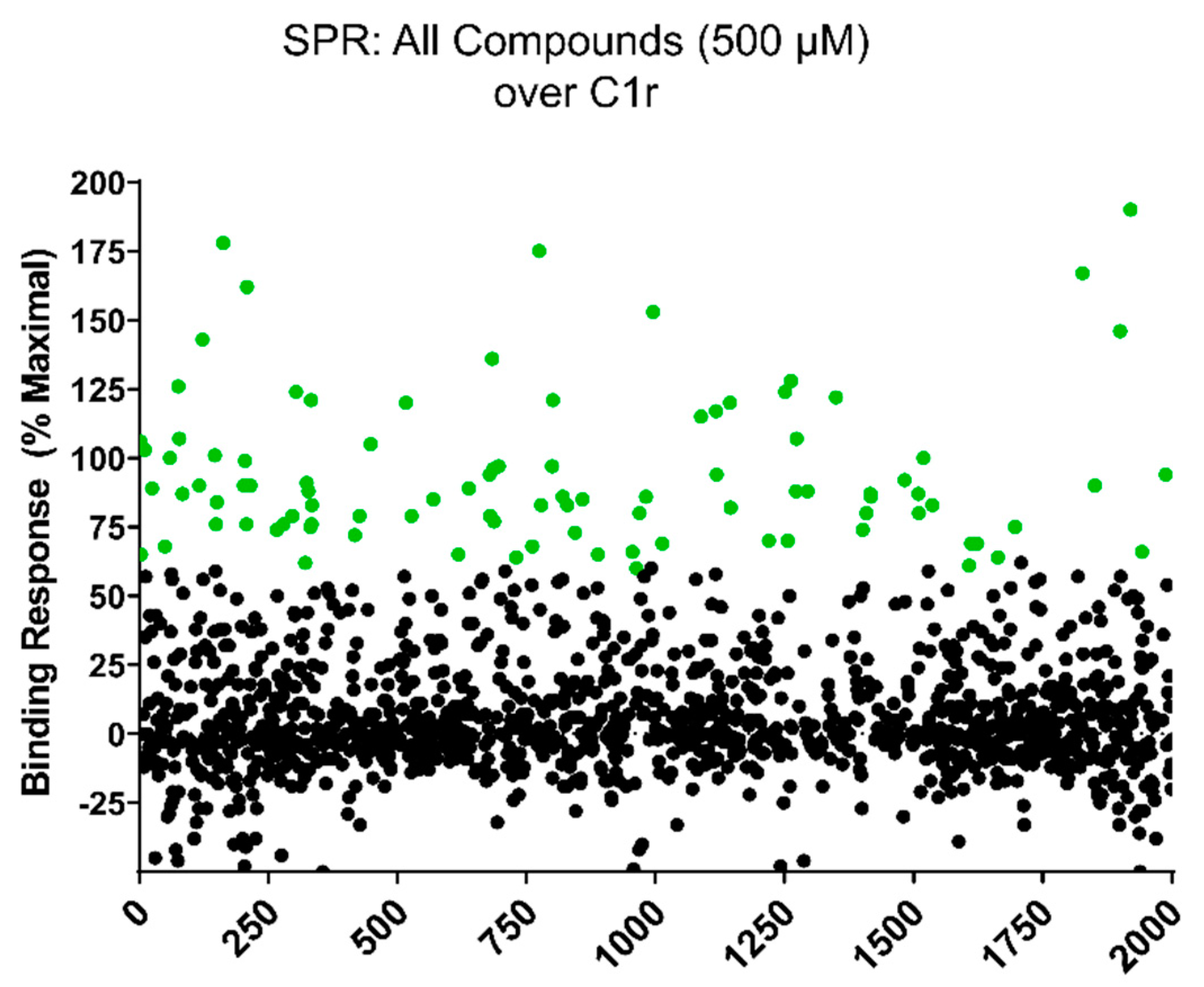
2.2. Initial Library Screening of C1r-Binding by Surface Plasmon Resonance
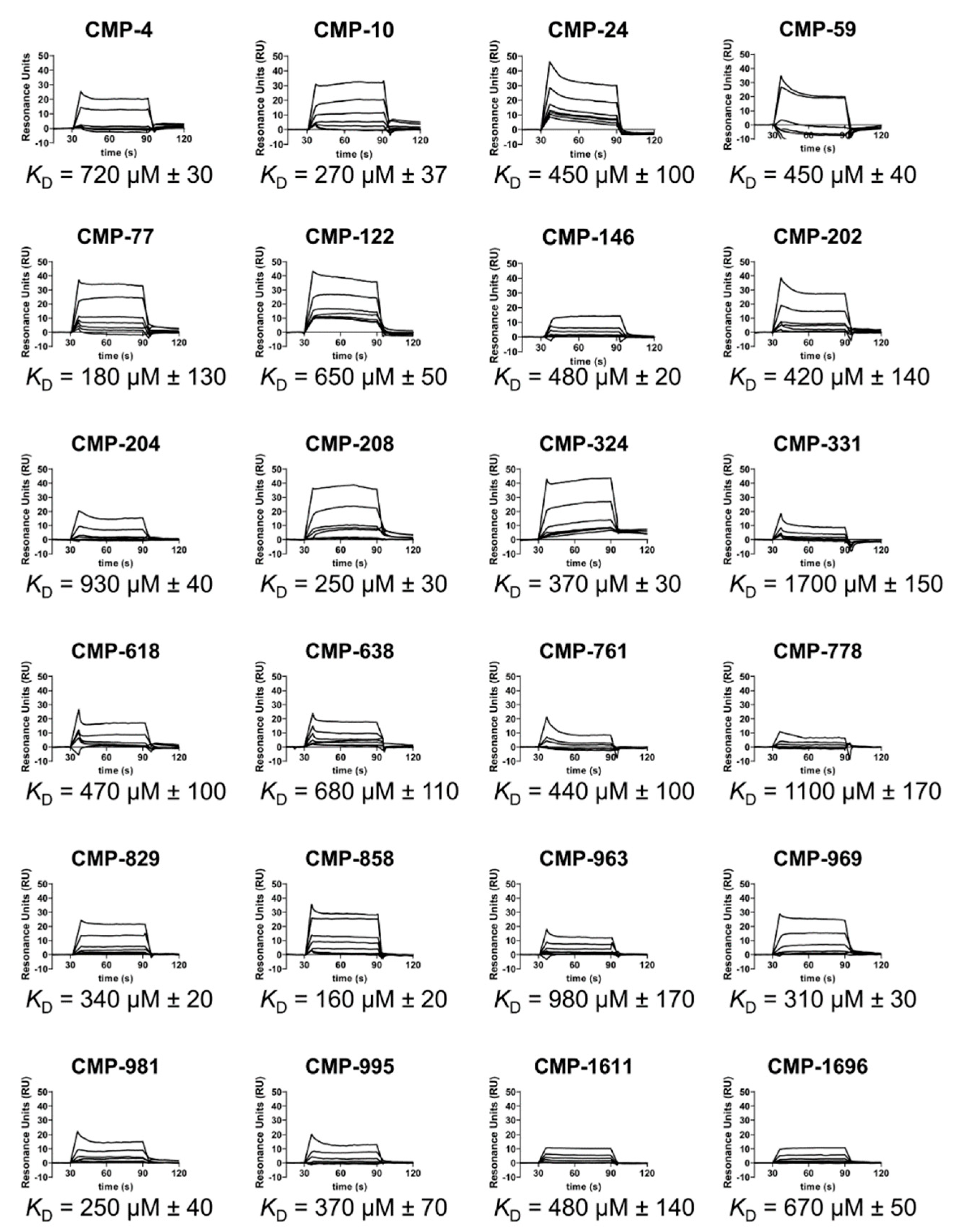
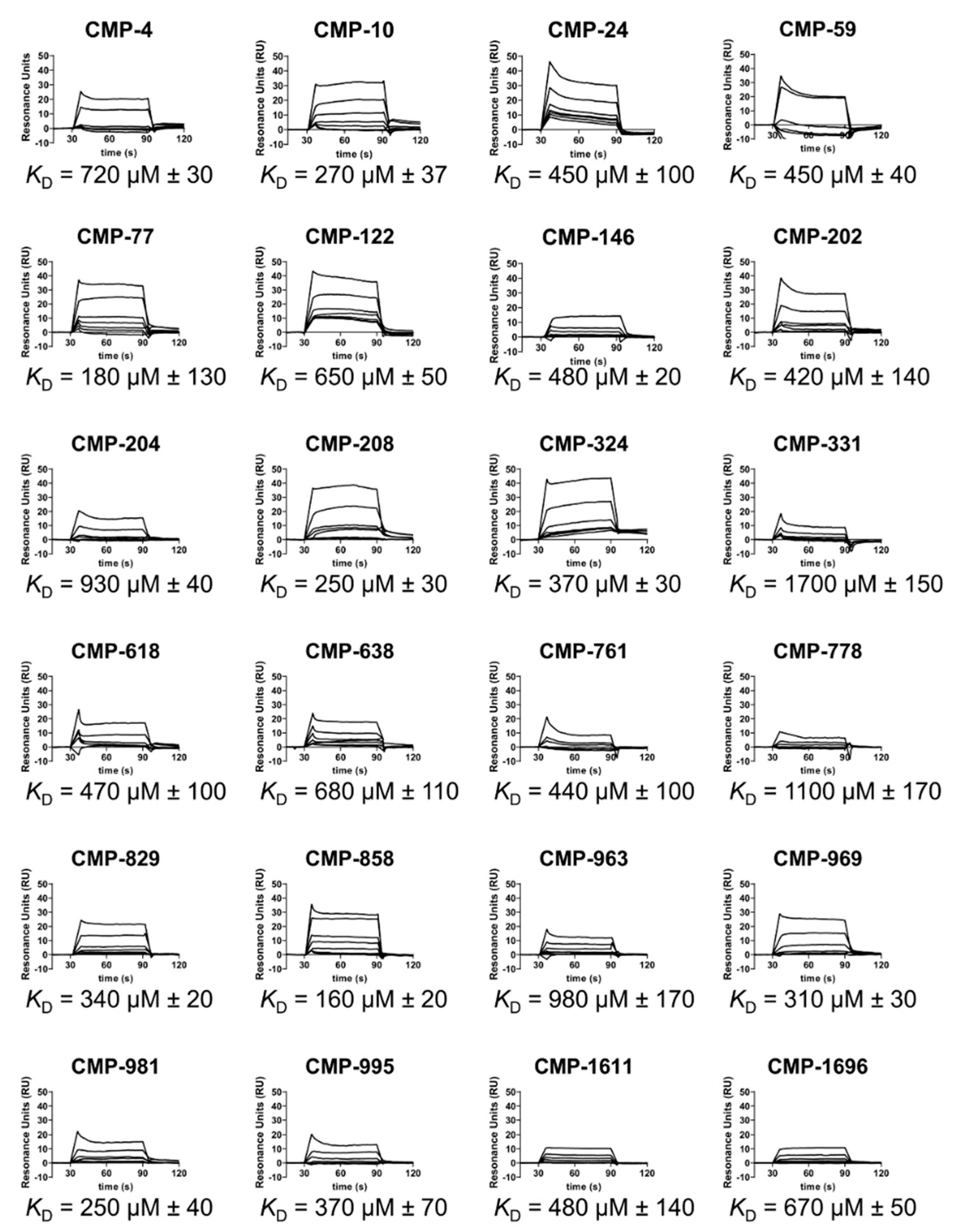
2.3. C1r-Binding Properties of Hit Compounds
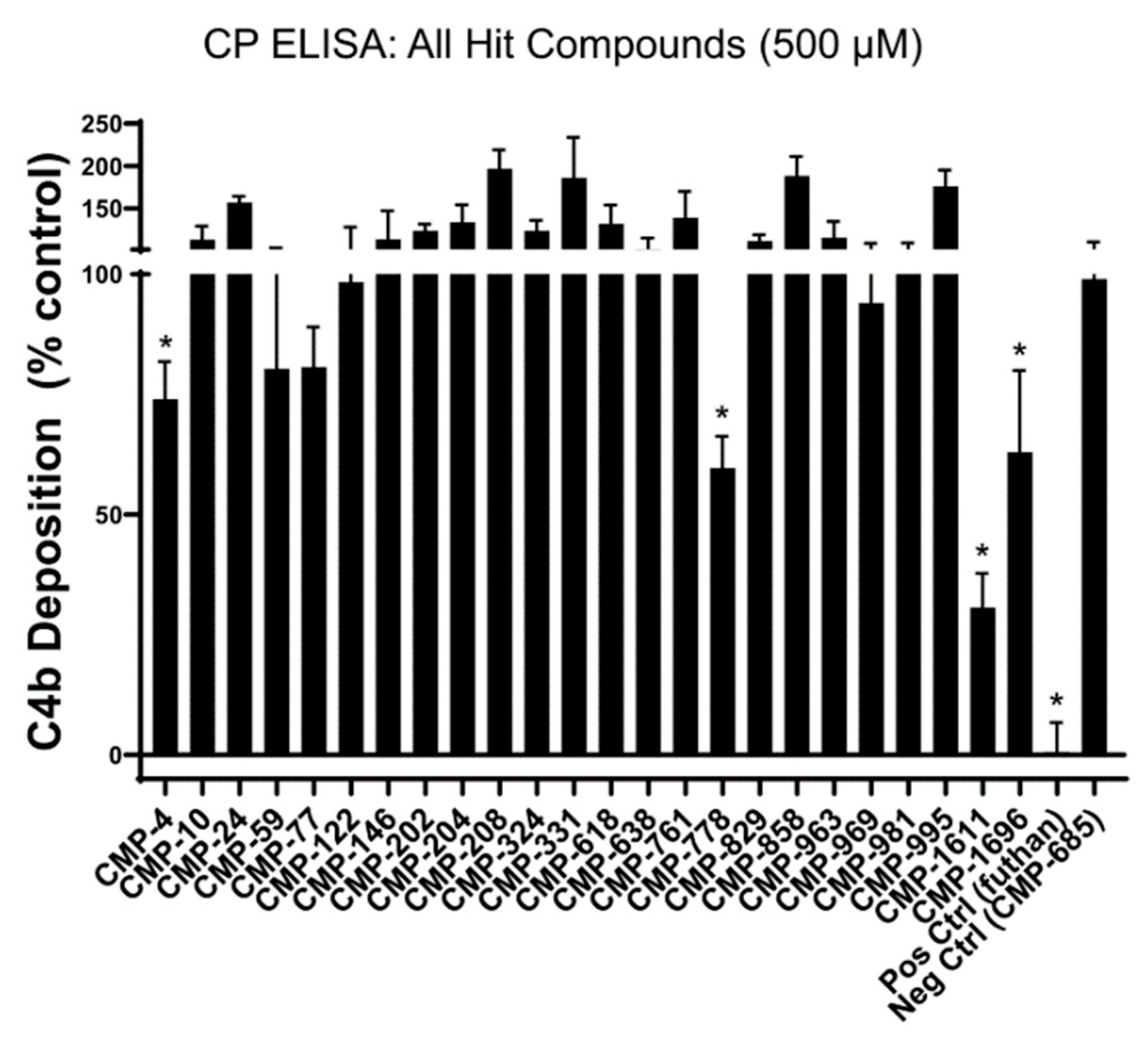
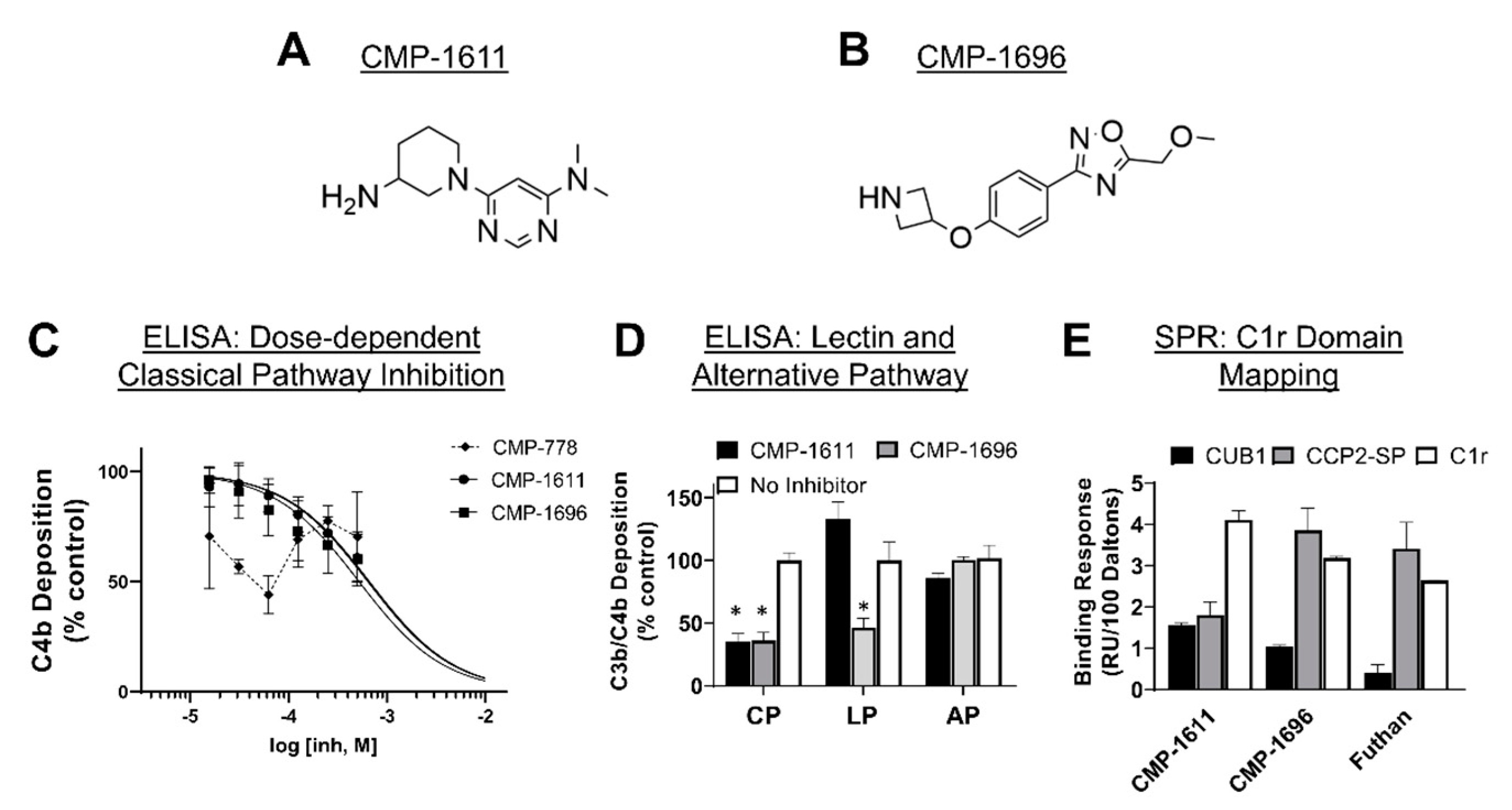
2.4. Identification of Two Structurally Distinct C1r-Binding and Complement Inhibitory Lead Fragments
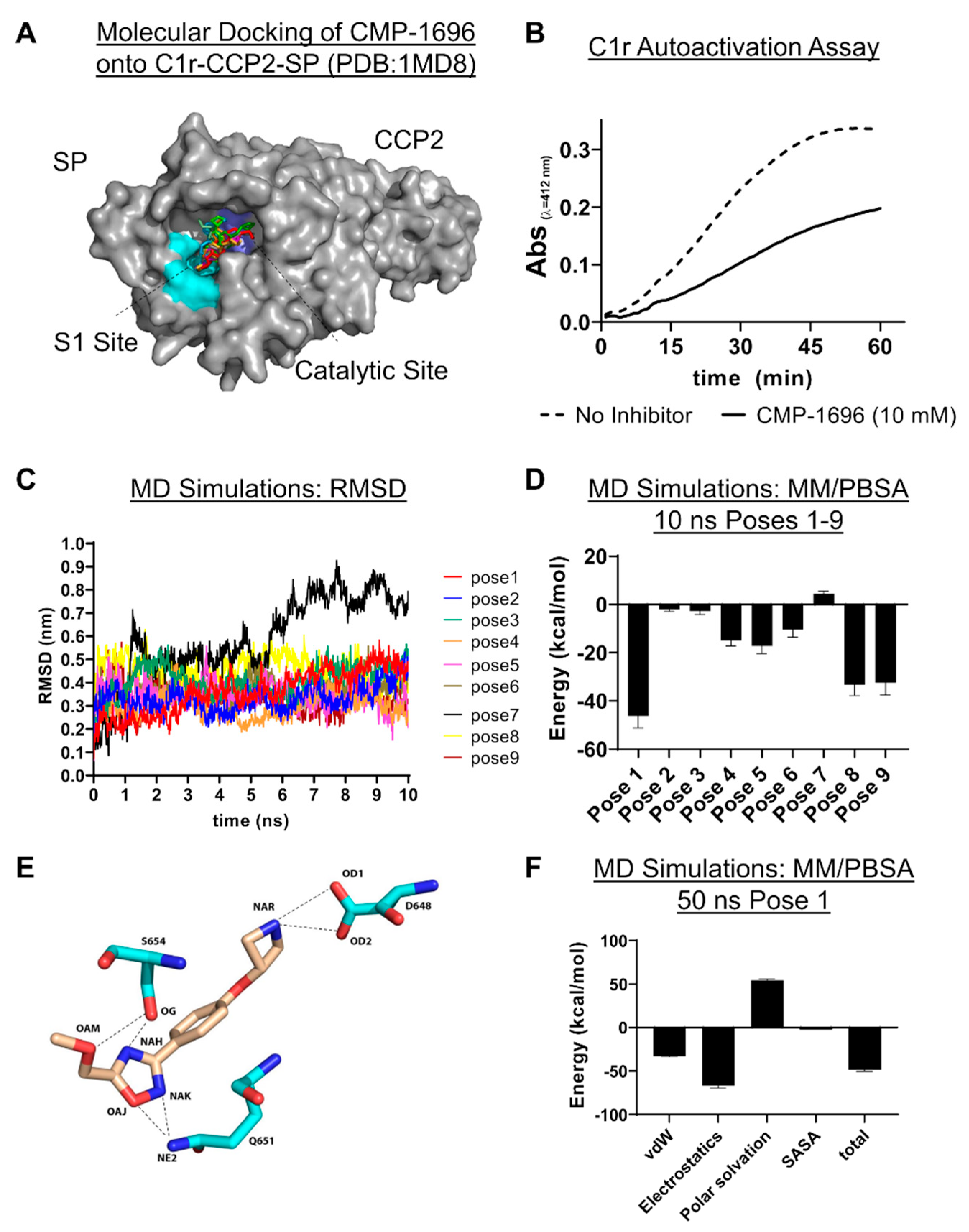
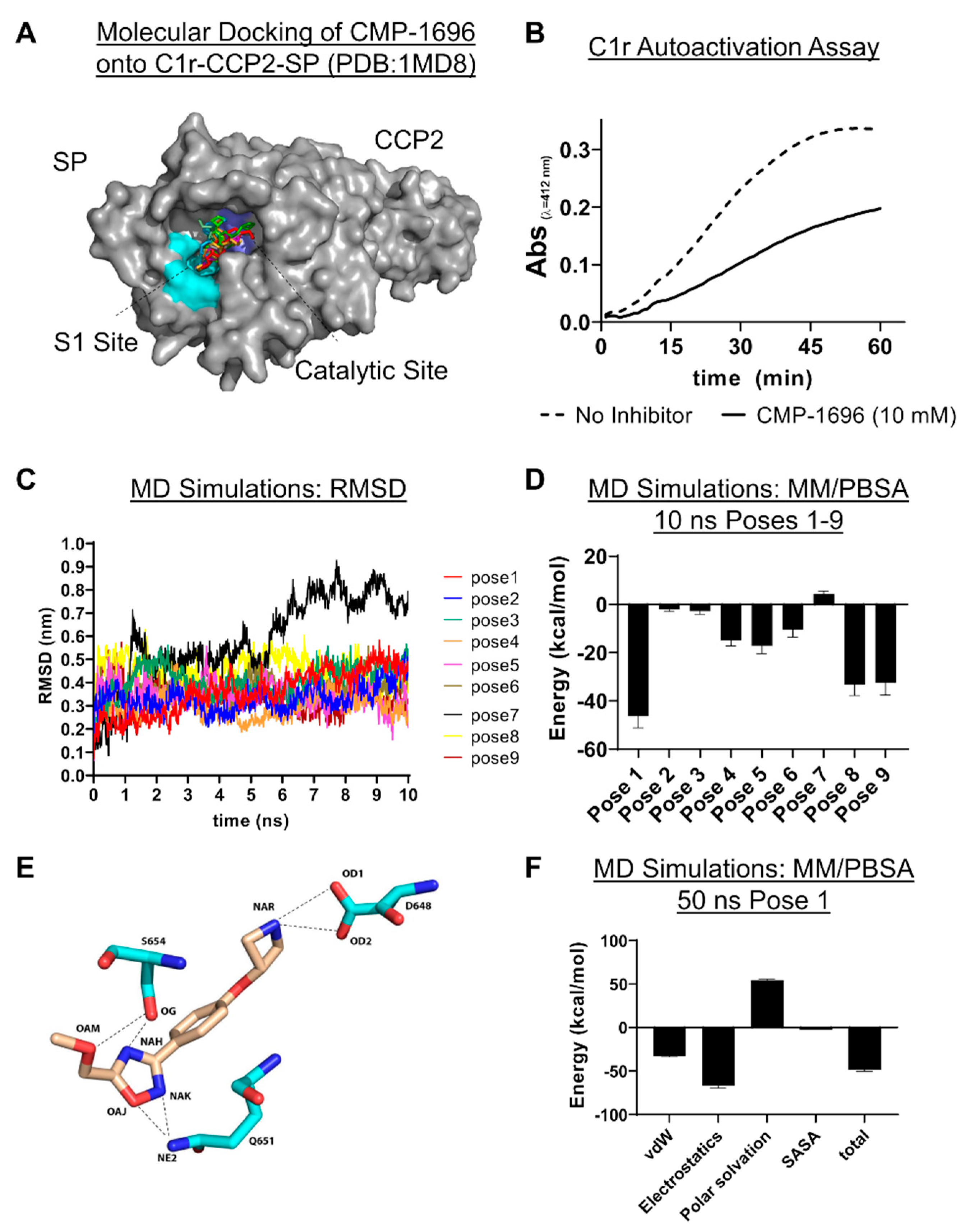
2.5. C1r-Binding Mode of CMP-1696
3. Discussion
4. Materials and Methods
4.1. Recombinant Expression, Purification, and Refolding of C1r-Domain Truncations
4.2. Compound Library
4.3. Surface Plasmon Resonance
4.4. Initial SPR Screening
4.5. Evaluation of Dose-Dependent Binding by SPR
4.6. Complement Inhibition Assay
4.7. Molecular Docking
4.8. C1r Proenzyme Activation Assay
4.9. Molecular Dynamics
4.10. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement: A key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part II: Role in Immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohlson, S.S.; Garred, P.; Kemper, C.; Tenner, A.J. Complement Nomenclature-Deconvoluted. Front. Immunol. 2019, 10, 1308. [Google Scholar] [CrossRef] [Green Version]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Therapeutic interventions. J. Immunol. 2013, 190, 3839–3847. [Google Scholar] [CrossRef]
- Thurman, J.M.; Yapa, R. Complement Therapeutics in Autoimmune Disease. Front. Immunol. 2019, 10, 672. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.L.; Pouw, R.B.; Kavanagh, D.; Sun, R.; Ricklin, D. Developments in anti-complement therapy; from disease to clinical trial. Mol. Immunol. 2018, 102, 89–119. [Google Scholar] [CrossRef]
- Ricklin, D.; Mastellos, D.C.; Reis, E.S.; Lambris, J.D. The renaissance of complement therapeutics. Nat. Rev. Nephrol. 2018, 14, 26–47. [Google Scholar] [CrossRef] [Green Version]
- Veerhuis, R.; Nielsen, J.M.; Tenner, A.J. Complement in the brain. Mol. Immunol. 2011, 48, 1592–1603. [Google Scholar] [CrossRef]
- Morgan, B.P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopath 2018, 40, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Ricklin, D.; Lambris, J.D. Complement in immune and inflammatory disorders: Pathophysiological mechanisms. J. Immunol. (Baltimore, Md. 1950) 2013, 190, 3831–3838. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, W.-S.; Verghese, P.B.; Chakraborty, C.; Joung, J.; Hyman, B.T.; Ulrich, J.D.; Holtzman, D.M.; Barres, B.A. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 10186–10191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Ackermann, S.; Ma, Z.; Mohanta, S.K.; Zhang, C.; Li, Y.; Nietzsche, S.; Westermann, M.; Peng, L.; Hu, D.; et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med. 2019, 25, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Arepally, G.M. Heparin-induced thrombocytopenia. Blood 2017, 129, 2864–2872. [Google Scholar] [CrossRef]
- Khandelwal, S.; Ravi, J.; Rauova, L.; Johnson, A.; Lee, G.M.; Gilner, J.B.; Gunti, S.; Notkins, A.L.; Kuchibhatla, M.; Frank, M.; et al. Polyreactive IgM initiates complement activation by PF4/heparin complexes through the classical pathway. Blood 2018, 132, 2431–2440. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.L. Expanding horizons in complement drug discovery: Challenges and emerging strategies. Semin. Immunopathol. 2018, 40, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Morgan, B.P.; Harris, C.L. Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug Discov. 2015, 14, 857–877. [Google Scholar] [CrossRef]
- Drag, M.; Salvesen, G.S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 2010, 9, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Dobó, J.; Kocsis, A.; Gál, P. Be on Target: Strategies of Targeting Alternative and Lectin Pathway Components in Complement-Mediated Diseases. Front. Immunol. 2018, 9, 1851. [Google Scholar] [CrossRef]
- Phuan, P.-W.; Zhang, H.; Asavapanumas, N.; Leviten, M.; Rosenthal, A.; Tradtrantip, L.; Verkman, A.S. C1q-targeted monoclonal antibody prevents complement-dependent cytotoxicity and neuropathology in in vitro and mouse models of neuromyelitis optica. Acta Neuropathol. 2013, 125, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ujiie, H.; Shibaki, A.; Wang, G.; Moriuchi, R.; Qiao, H.; Morioka, H.; Shinkuma, S.; Natsuga, K.; Long, H.A.; et al. Human IgG1 monoclonal antibody against human collagen 17 noncollagenous 16A domain induces blisters via complement activation in experimental bullous pemphigoid model. J. Immunol. 2010, 185, 7746–7755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, G.; Diercks, G.F.H.; Seelen, M.A.J.; Horvath, B.; van Doorn, M.B.A.; Damman, J. Complement Activation in Autoimmune Bullous Dermatoses: A Comprehensive Review. Front. Immunol. 2019, 10, 1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berentsen, S. Complement Activation and Inhibition in Autoimmune Hemolytic Anemia: Focus on Cold Agglutinin Disease. Semin. Hematol. 2018, 55, 141–149. [Google Scholar] [CrossRef]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177. [Google Scholar] [CrossRef] [Green Version]
- Deniz, U.; Stuart, C.H.; de Kreuk, Bart-Jan; Roman, I.K.; de Jong Rob, N.; Frank, J.B.; Janine, S.; Abraham, J.K.; Thomas, H.S.; Parren Paul, W.H.I.; et al. Structures of C1-IgG1 provide insights into how danger pattern recognition activates complement. Science (80-) 2018, 359, 794–797. [Google Scholar] [CrossRef] [Green Version]
- Diebolder, C.A.; Beurskens, F.J.; de Jong, R.N.; Koning, R.I.; Strumane, K.; Lindorfer, M.A.; Voorhorst, M.; Ugurlar, D.; Rosati, S.; Heck, A.J.R.; et al. Complement is activated by IgG hexamers assembled at the cell surface. Science 2014, 343, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Sharp, T.H.; Boyle, A.L.; Diebolder, C.A.; Kros, A.; Koster, A.J.; Gros, P. Insights into IgM-mediated complement activation based on in situ structures of IgM-C1-C4b. Proc. Natl. Acad. Sci. USA 2019, 116, 11900–11905. [Google Scholar] [CrossRef] [Green Version]
- Girija, U.V.; Gingras, A.R.; Marshall, J.E.; Panchal, R.; Sheikh, M.A.; Harper, J.A.J.; Gál, P.; Schwaeble, W.J.; Mitchell, D.A.; Moody, P.C.E.; et al. Structural basis of the C1q/C1s interaction and its central role in assembly of the C1 complex of complement activation. Proc. Natl. Acad. Sci. USA 2013, 110, 13916–13920. [Google Scholar] [CrossRef] [Green Version]
- Phillips, A.E.; Toth, J.; Dodds, A.W.; Girija, U.V.; Furze, C.M.; Pala, E.; Sim, R.B.; Reid, K.B.M.; Schwaeble, W.J.; Schmid, R.; et al. Analogous Interactions in Initiating Complexes of the Classical and Lectin Pathways of Complement. J. Immunol. 2009, 182, 7708–7717. [Google Scholar] [CrossRef] [Green Version]
- Garcia, B.L.; Zwarthoff, S.A.; Rooijakkers, S.H.M.; Geisbrecht, B. V Novel Evasion Mechanisms of the Classical Complement Pathway. J. Immunol. 2016, 197, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Fragment-based lead discovery grows up. Nat. Rev. Drug Discov. 2013, 12, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of three” for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.J.; Erlanson, D.A. Learning from our mistakes: The “unknown knowns” in fragment screening. Bioorg. Med. Chem. Lett. 2013, 23, 2844–2852. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Reynolds, C.H. Performance of similarity measures in 2D fragment-based similarity searching: Comparison of structural descriptors and similarity coefficients. J. Chem. Inf. Comput. Sci. 2002, 42, 1407–1414. [Google Scholar] [CrossRef]
- Cao, Y.; Jiang, T.; Girke, T. A maximum common substructure-based algorithm for searching and predicting drug-like compounds. Bioinformatics 2008. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Gal, P.; Szilagyi, L.; Thielens, N.M.; Szilagyi, K.; Lorincz, Z.; Kulcsar, P.; Graf, L.; Arlaud, G.J.; Zavodszky, P. The Role of the Individual Domains in the Structure and Function of the Catalytic Region of a Modular Serine Protease, C1r. J. Immunol. 2001, 167, 5202–5208. [Google Scholar] [CrossRef] [Green Version]
- Budayova-Spano, M.; Lacroix, M.; Thielens, N.M.; Arlaud, G.J.; Fontecilla-Camps, J.C.; Gaboriaud, C. The crystal structure of the zymogen catalytic domain of complement protease C1r reveals that a disruptive mechanical stress is required to trigger activation of the C1 complex. EMBO J. 2002, 21, 231–239. [Google Scholar] [CrossRef]
- Budayova-Spano, M.; Grabarse, W.; Thielens, N.M.; Hillen, H.; Lacroix, M.; Schmidt, M.; Fontecilla-Camps, J.C.; Arlaud, G.J.; Gaboriaud, C. Monomeric structures of the zymogen and active catalytic domain of complement protease c1r: Further insights into the c1 activation mechanism. Structures 2002, 10, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Kardos, J.; Harmat, V.; Palló, A.; Barabás, O.; Szilágyi, K.; Gráf, L.; Náray-Szabó, G.; Goto, Y.; Závodszky, P.; Gál, P. Revisiting the mechanism of the autoactivation of the complement protease C1r in the C1 complex: Structure of the active catalytic region of C1r. Mol. Immunol. 2008, 45, 1752–1760. [Google Scholar] [CrossRef] [PubMed]
- Almitairi, J.O.M.; Girija, U.V.; Furze, C.M.; Simpson-Gray, X.; Badakshi, F.; Marshall, J.E.; Schwaeble, W.J.; Mitchell, D.A.; Moody, P.C.E.; Wallis, R. Structure of the C1r-C1s interaction of the C1 complex of complement activation. Proc. Natl. Acad. Sci. USA 2018, 115, 768–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plummer, J.S.; Cai, C.; Hays, S.J.; Gilmore, J.L.; Emmerling, M.R.; Michael, W.; Narasimhan, L.S.; Watson, M.D.; Wang, K.; Nath, R.; et al. Benzenesulfonamide derivatives of 2-substituted 4H-3,1-benzoxazin-4-ones and benzthiazin-4-ones as inhibitors of complement C1r protease. Bioorg. Med. Chem. Lett. 1999, 9, 815–820. [Google Scholar] [CrossRef]
- Thompson, D.C.; Humblet, C.; Joseph-McCarthy, D. Investigation of MM-PBSA rescoring of docking poses. J. Chem. Inf. Model 2008, 48, 1081–1091. [Google Scholar] [CrossRef]
- Greenidge, P.A.; Kramer, C.; Mozziconacci, J.C.; Sherman, W. Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J. Chem. Inf. Model 2014, 54, 2697–2717. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Clinical promise of next-generation complement therapeutics. Nat. Rev. Drug Discov. 2019, 18, 707–729. [Google Scholar] [CrossRef]
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Sweeney, A.M.; Liao, S.-M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef] [Green Version]
- Karki, R.G.; Powers, J.; Mainolfi, N.; Anderson, K.; Belanger, D.B.; Liu, D.; Ji, N.; Jendza, K.; Gelin, C.F.; Sweeney, A.M.; et al. Design, Synthesis, and Preclinical Characterization of Selective Factor D Inhibitors Targeting the Alternative Complement Pathway. J. Med. Chem. 2019, 62, 4656–4668. [Google Scholar] [CrossRef]
- Maibaum, J.; Liao, S.-M.; Vulpetti, A.; Ostermann, N.; Randl, S.; Rüdisser, S.; Lorthiois, E.; Erbel, P.; Kinzel, B.; Kolb, F.A.; et al. Small-molecule factor D inhibitors targeting the alternative complement pathway. Nat. Chem. Biol. 2016, 12, 1105–1110. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx J. Am. Soc. Exp. Neurother 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Drug transport across the blood-brain barrier. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Lamoree, B.; Hubbard, R.E. Current perspectives in fragment-based lead discovery (FBLD). Essays Biochem. 2017, 61, 453–464. [Google Scholar] [CrossRef]
- Geisbrecht, B.V.; Bouyain, S.; Pop, M. An optimized system for expression and purification of secreted bacterial proteins. Protein Expr. Purif. 2006, 46, 23–32. [Google Scholar] [CrossRef]
- Xie, J.; Zhi, H.; Garrigues, R.J.; Keightley, A.; Garcia, B.L.; Skare, J.T. Structural determination of the complement inhibitory domain of Borrelia burgdorferi BBK32 provides insight into classical pathway complement evasion by Lyme disease spirochetes. PLoS Pathog. 2019, 15, e1007659. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Backman, T.W.H.; Cao, Y.; Girke, T. ChemMine tools: An online service for analyzing and clustering small molecules. Nucleic Acids Res. 2011, 39, 486–491. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Giannetti, A.M. From Experimental Design to Validated Hits. In Methods in Enzymology; Kuo, L.C., Ed.; Academic Press: San Diego, CA, USA, 2011; Volume 493, pp. 169–218. ISBN 0123812747. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DA, G.J.S.W. Big Data Analysis for Bioinformatics and Biomedical Discoveries; Chapman and Hall/CRC: Boca Raton, FL, USA, 2016. [Google Scholar]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Aalten, D.M.F. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided. Mol. Des. 1996, 10, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8875. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa -A gromacs tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
Sample Availability: Samples of CMP-1611 and CMP-1696 may be requested from the authors and will be made available if supply allows. |




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rushing, B.R.; Rohlik, D.L.; Roy, S.; Skaff, D.A.; Garcia, B.L. Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery. Molecules 2020, 25, 4016. https://doi.org/10.3390/molecules25174016
Rushing BR, Rohlik DL, Roy S, Skaff DA, Garcia BL. Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery. Molecules. 2020; 25(17):4016. https://doi.org/10.3390/molecules25174016
Chicago/Turabian StyleRushing, Blake R., Denise L. Rohlik, Sourav Roy, D. Andrew Skaff, and Brandon L. Garcia. 2020. "Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery" Molecules 25, no. 17: 4016. https://doi.org/10.3390/molecules25174016
APA StyleRushing, B. R., Rohlik, D. L., Roy, S., Skaff, D. A., & Garcia, B. L. (2020). Targeting the Initiator Protease of the Classical Pathway of Complement Using Fragment-Based Drug Discovery. Molecules, 25(17), 4016. https://doi.org/10.3390/molecules25174016