
Activated Carbons and Their Evaluation in Electric Double Layer Capacitors


Abstract
:1. Introduction
2. Carbonaceous Materials
2.1. Precursors for Activated Carbons
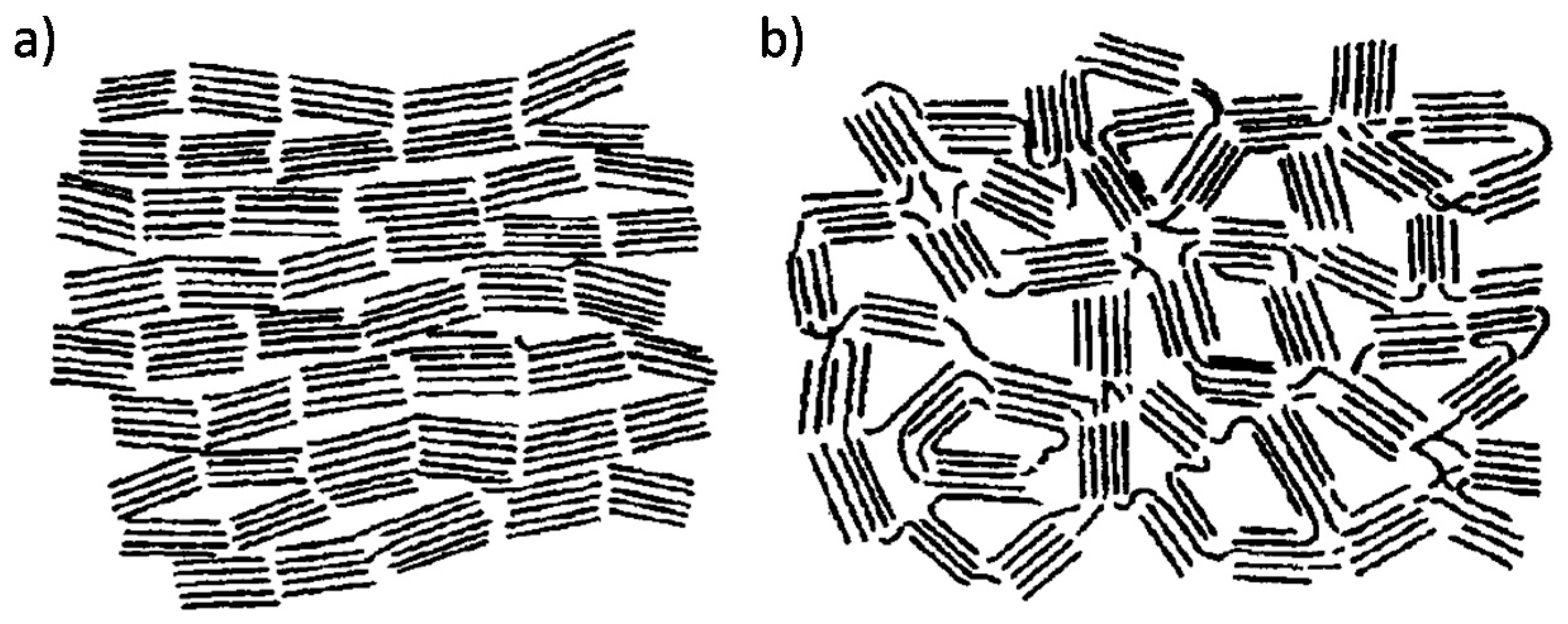
2.2. The Structure of Non-Graphitic Carbons
2.3. The Porous Texture in Carbon Materials
- SBET—the Brunauer-Emmett-Teller (BET) specific surface calculated from N2 adsorption isotherm data based on the BET model. BET theory has been verified using many mesoporous materials. However, recently, it has been strongly criticized when applied to the analysis of microporous carbons because it can significantly over-estimate the real surface area [33]. Despite this drawback, in the literature, this model is commonly used to differentiate between ACs in a series.
- SDFT—the SSA calculated from density functional theory (DFT) based on N2 and CO2 adsorption isotherm data. This parameter seems to be more reliable than SBET, and many reports have demonstrated the convergence of SDFT and the real surface areas of materials [34,35]. The DFT method is also commonly used to determine the pore size distribution precisely. Two implementations of DFT are described in the literature: Non-Local DFT (NLDFT) [36] and Quenched Solid DFT (QSDFT) [34]. These two analyses give slightly different values and are selected arbitrarily.
- V0 and S0 (or VDR and SDR)—the volume and surface area of micropores based on the Dubinin-Radushkevich (DR) model. These values can be determined from N2 and CO2 adsorption isotherm data. The volume of the micropores is directly calculated from the DR equation. In contrast, the surface is the derivative parameter estimated from V0 based on the assumption of a particular pore shape (slit or cylindrical).
- L0—the mean micropore size derived from the VDR based on the empirical equation proposed by Stoeckli [37]. For microporous carbons with a mean pore size smaller than 1.8 nm, the equation is as follows:For carbon with a wider pore size, the mean pore size is calculated according to the following equation:where E0 is the characteristic energy of adsorption.
- VT—the total pore volume calculated from N2 adsorption data. This parameter is directly related to the volume of liquefied nitrogen stored in the pores, assuming ideal capillary condensation. It is calculated for the last adsorption point of the isotherm or at a partial pressure of approximately 0.96 p p0−1, which corresponds to a pore size of 50 nm.
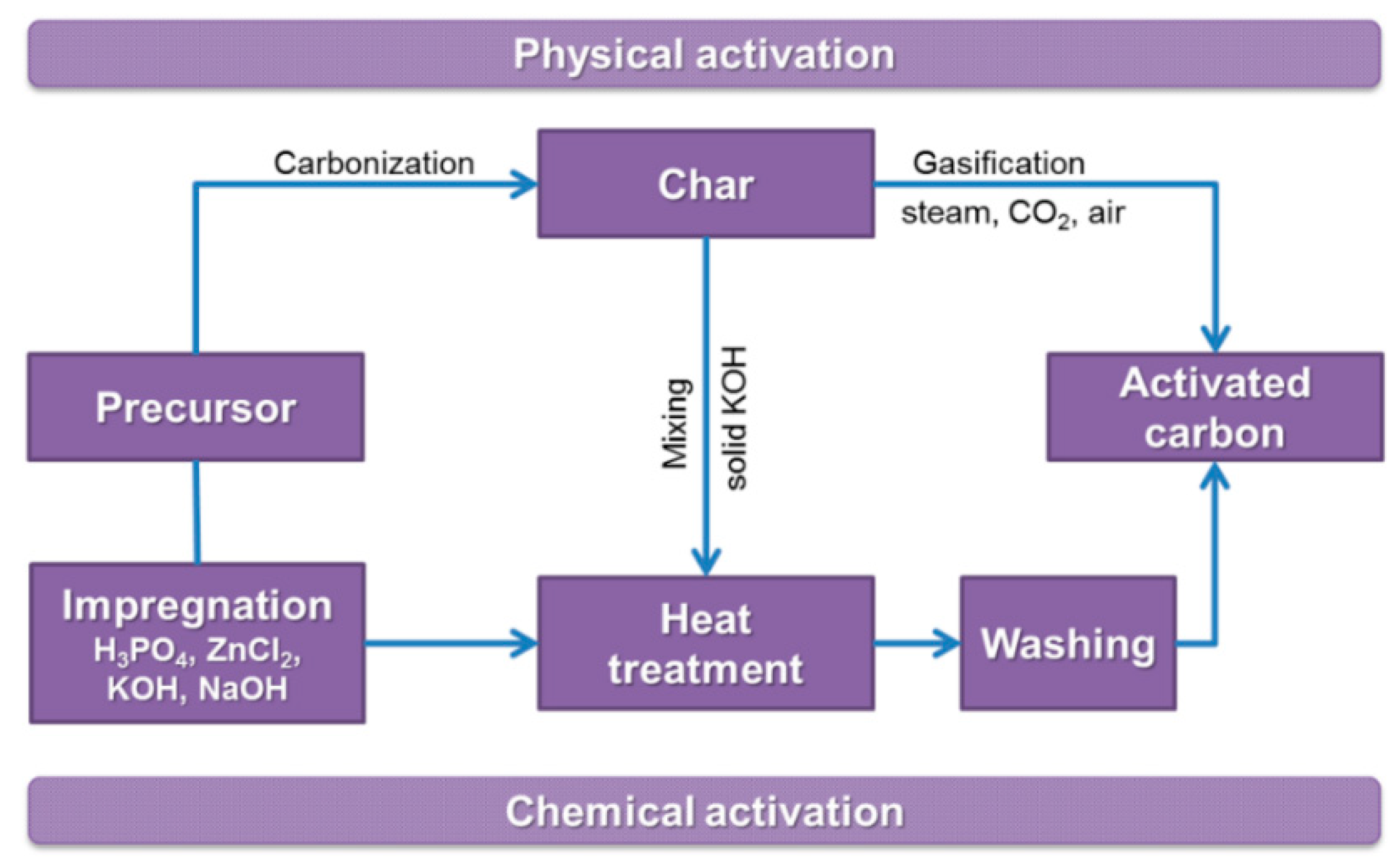
3. Activation Process
3.1. Physical Activation
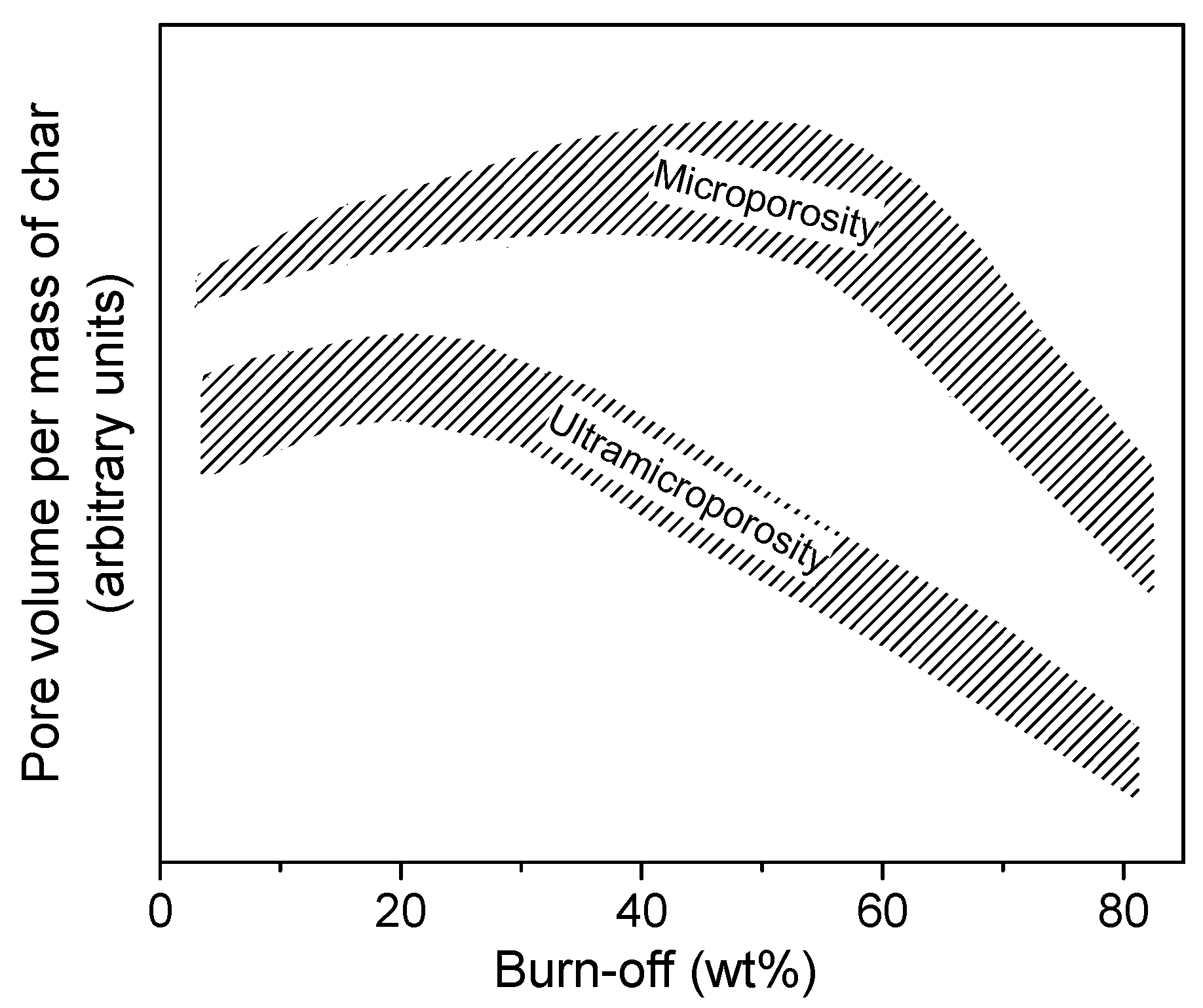
3.1.1. Effect of the Gasification Temperature
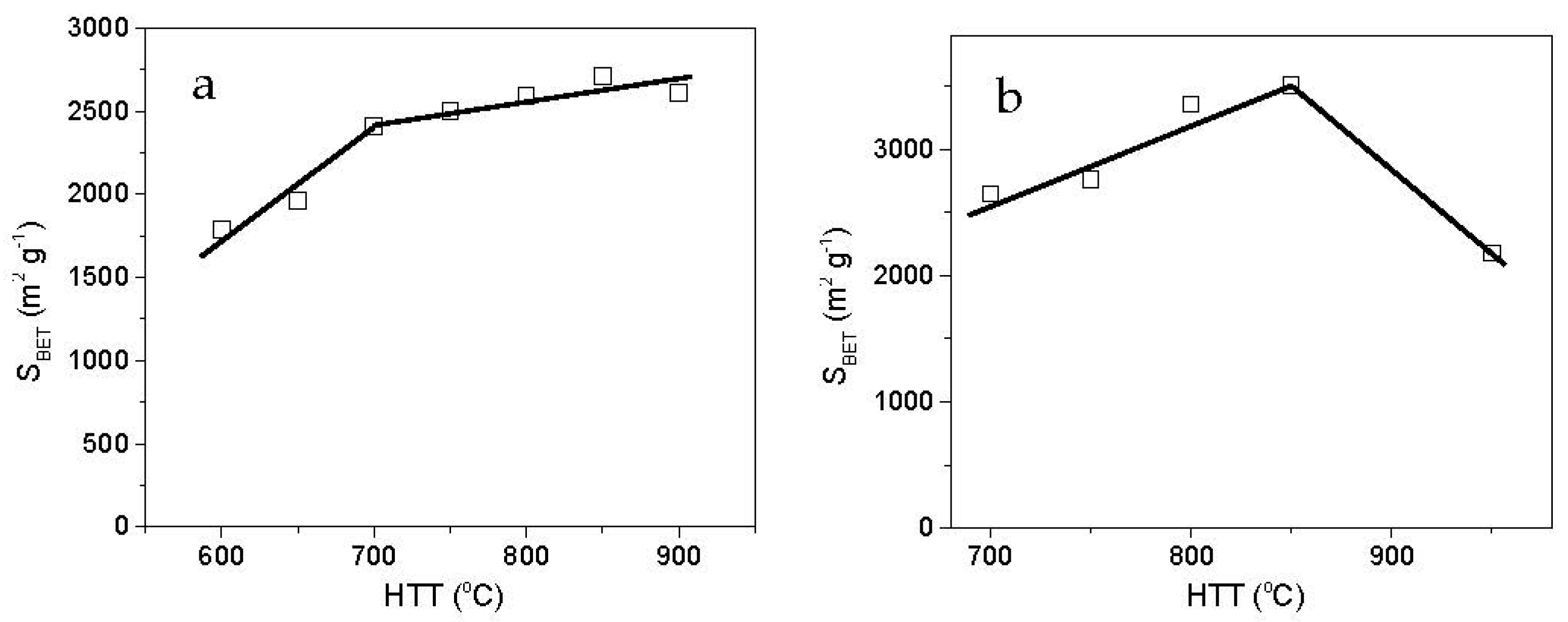
3.1.2. Effect of the Pre-Carbonization Temperature
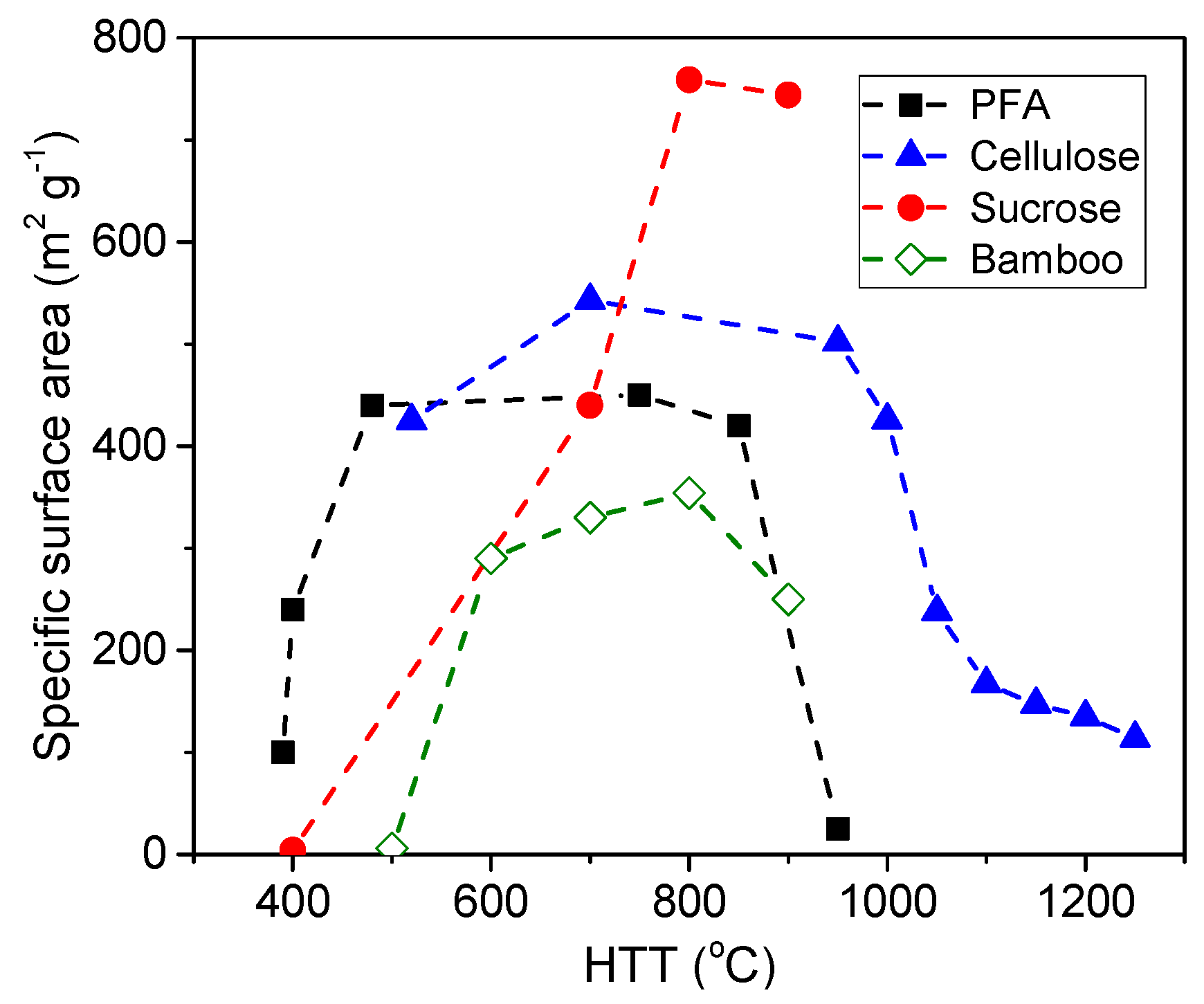
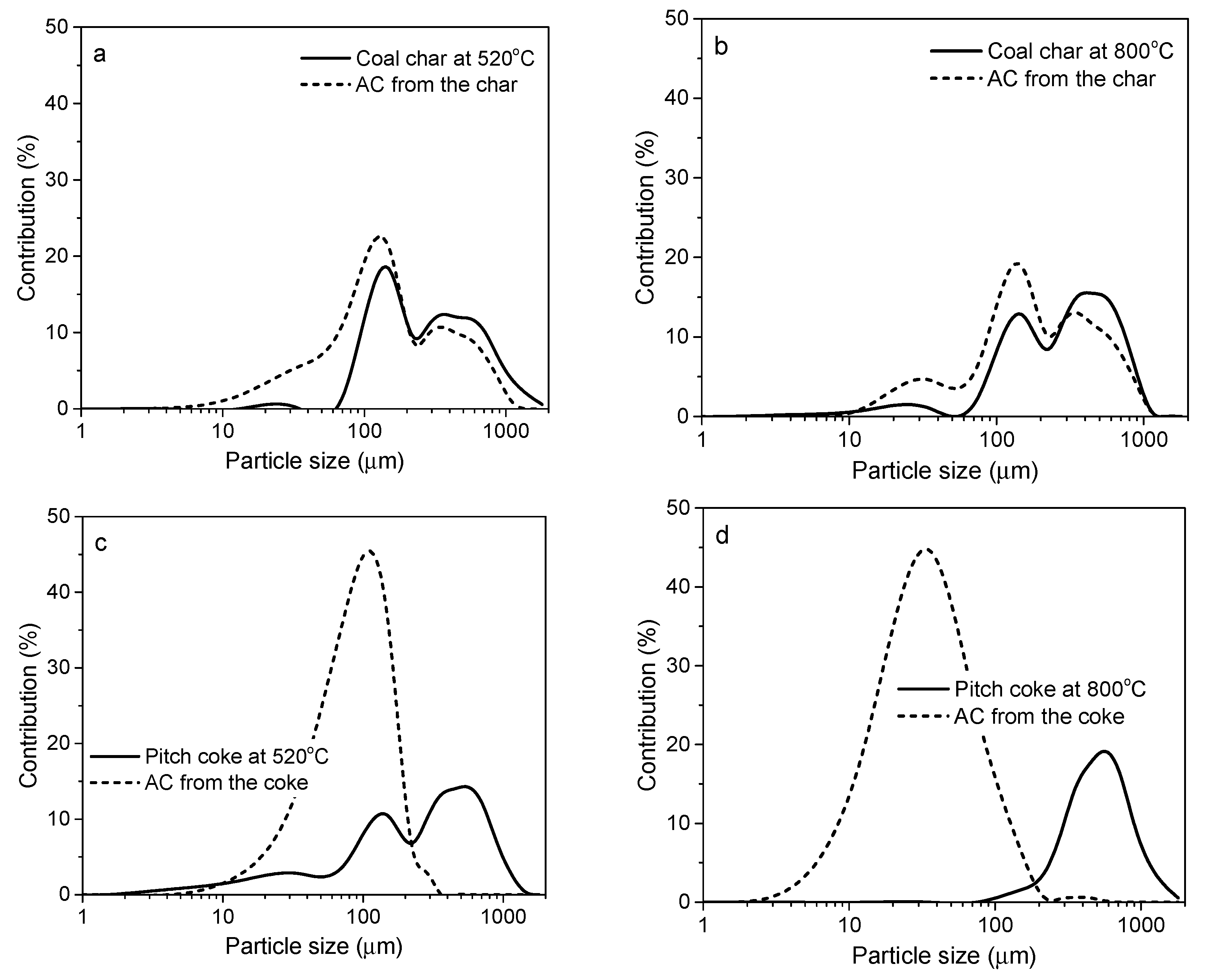
3.1.3. Effect of the Precursor Origin
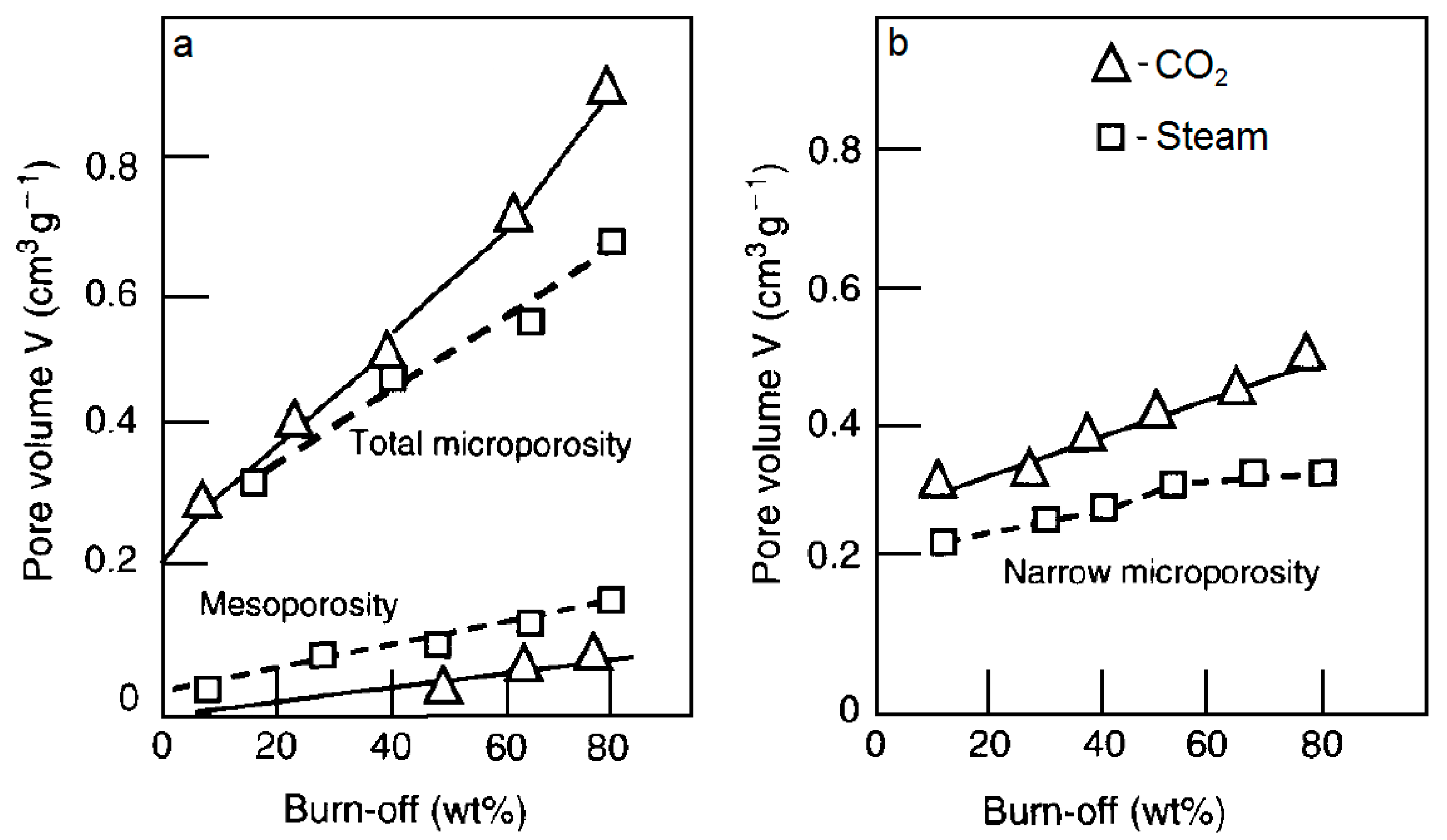
3.1.4. Carbon Dioxide Versus Steam
3.2. Chemical Activation
3.2.1. Activation with Alkali Metal Hydroxides
The Impact of the Reaction Temperature
The Impact of the KOH/Precursor Ratio
The Impact of the Precursor Nature
The Impact of the Reaction Time
The Impact of the Inert Gas Flow Rate
Other Factors
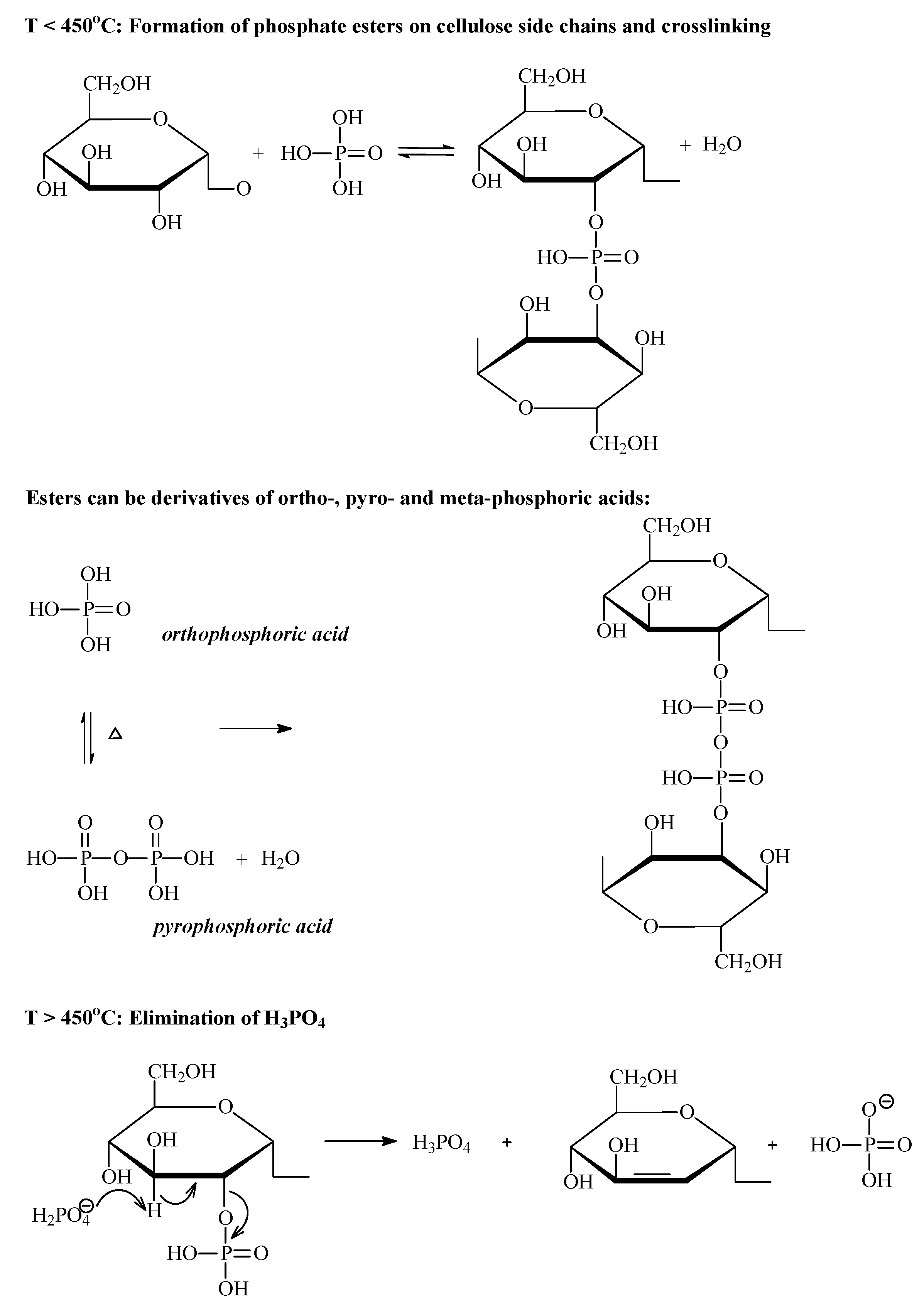
3.2.2. Phosphoric Acid Activation
3.2.3. Zinc Chloride Activation
3.3. Self-Activation
3.4. Catalytic Activation
4. Activated Carbons for EDLC
4.1. The Role of the Porous Texture
4.2. The Role of Structure Ordering
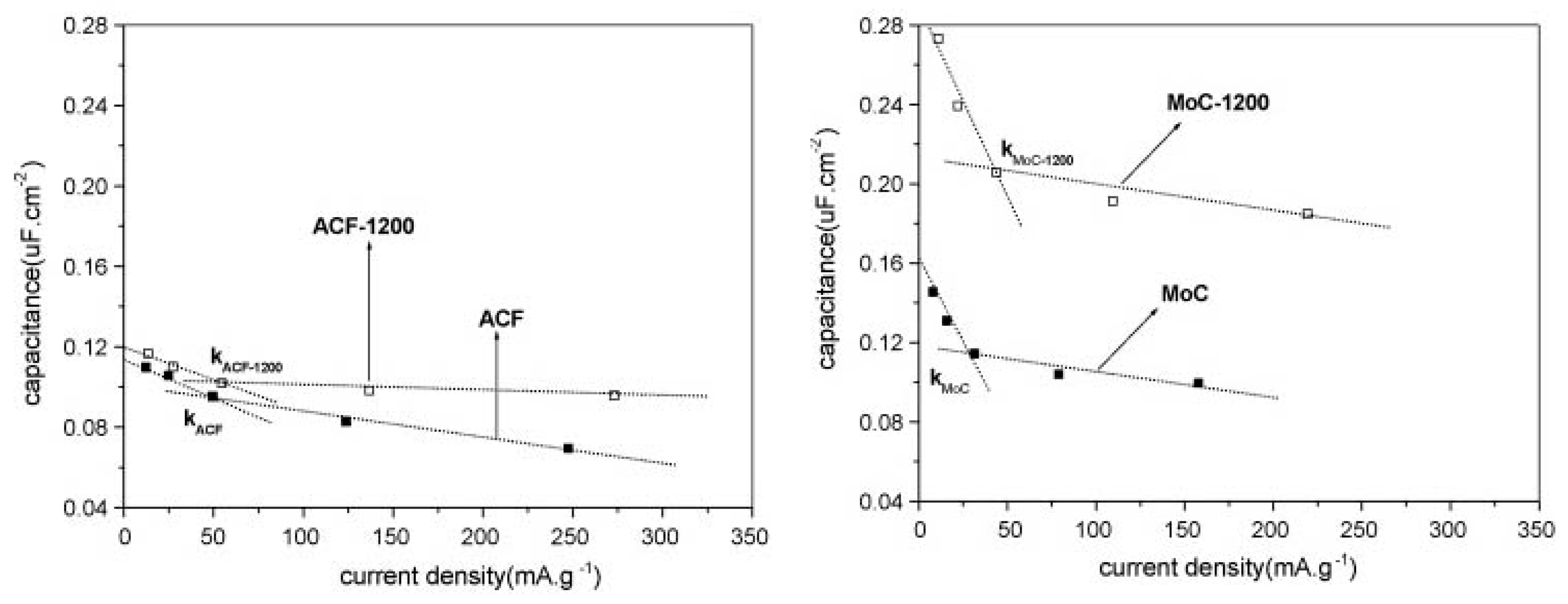
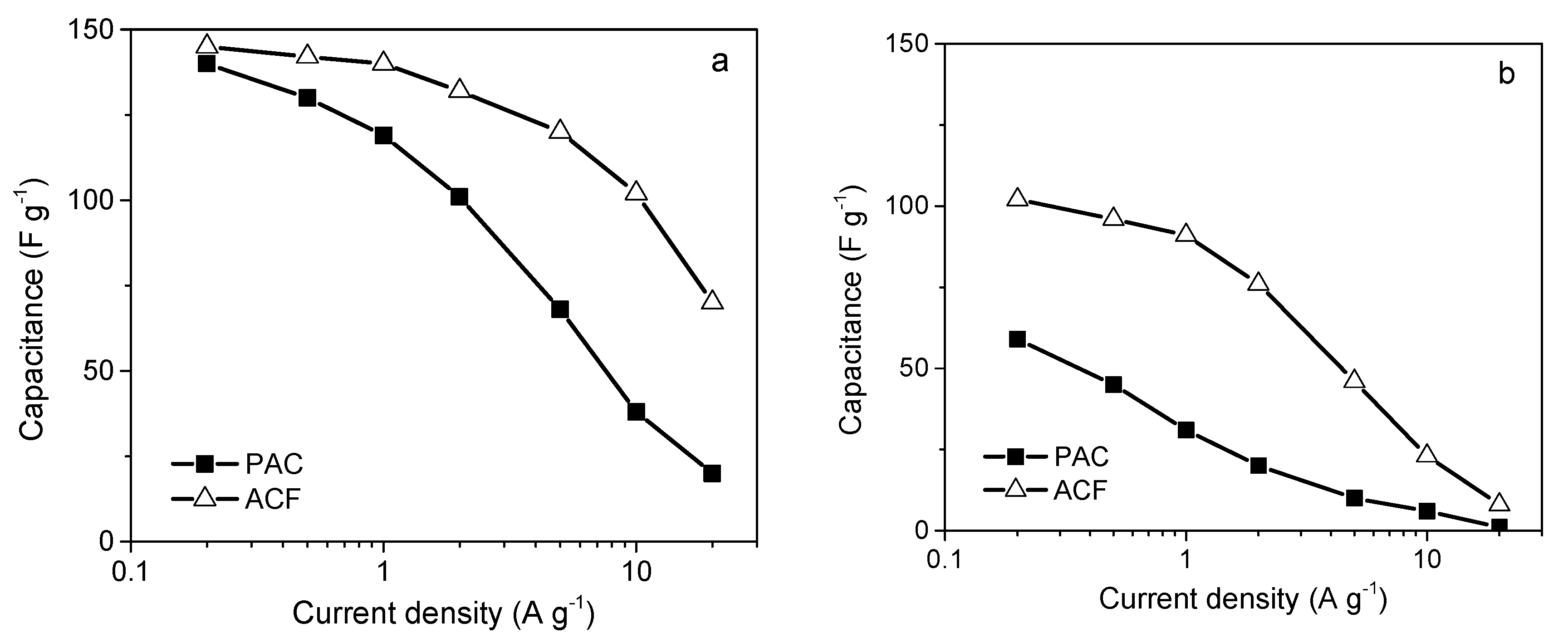
4.3. The Role of Particle Morphology
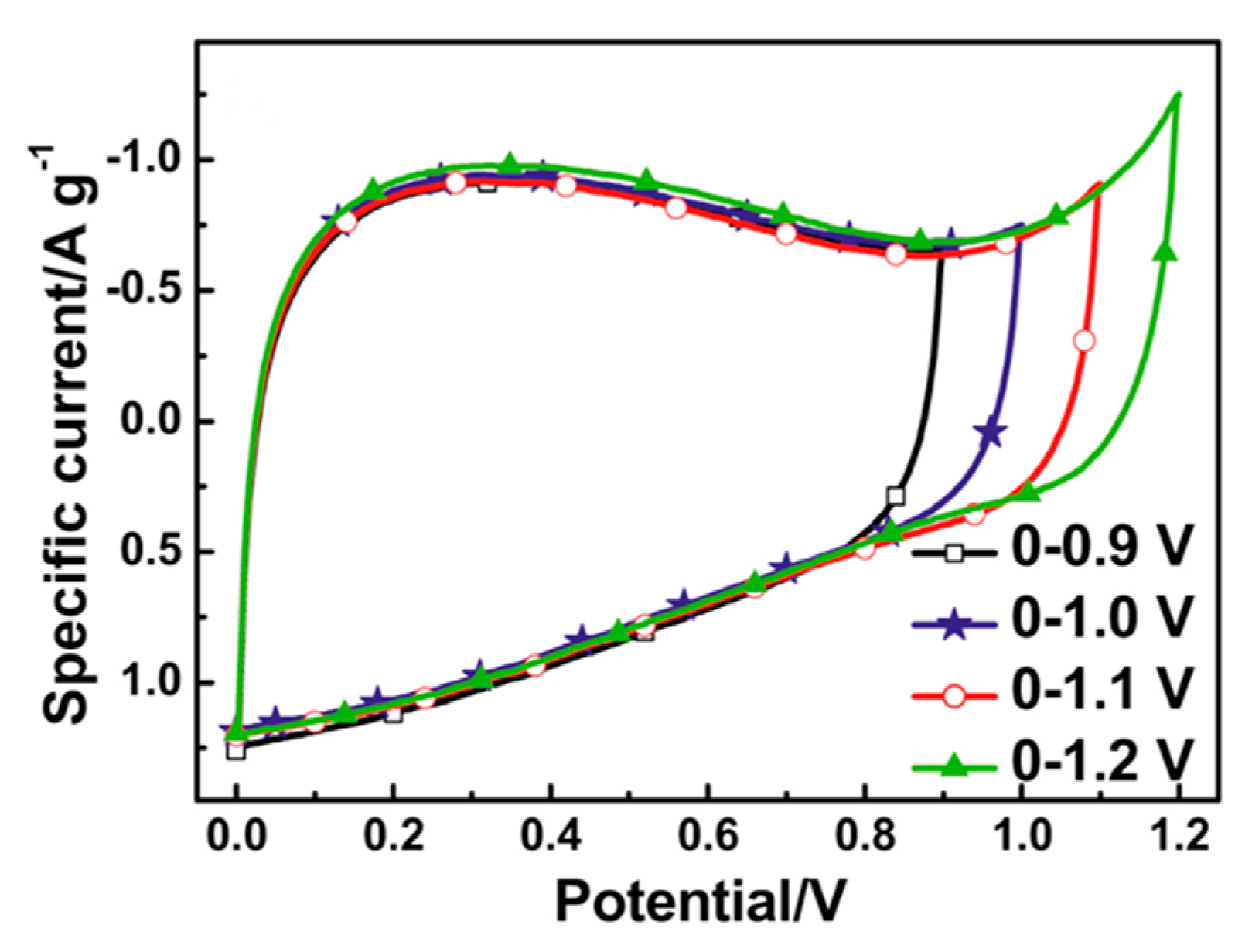
4.4. The Impurities in Activated Carbons
5. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Activated carbon |
| ACF | Activated carbon fiber |
| BET | Brunauer-Emmett-Teller |
| CEC | Cation exchange capacity |
| DFT | Density functional theory |
| DR | Dubinin-Radushkevich |
| EDL | Electric double layer |
| EDLC | Electric double layer capacitor |
| ESEM | Environmental scanning electron microscopy |
| FESEM | Field emission scanning electron microscopy |
| HRTEM | High-resolution transmission electron microscopy |
| HTT | Heat treatment temperature |
| IUPAC | International Union of Pure and Applied Chemistry |
| L0 | Mean micropore size |
| La | Lateral size of crystallite |
| Lc | Height of crystallite |
| LiTFSI | Bis(trifluoromethane)sulfonamide lithium salt |
| MCMB | Mesocarbon microbeads |
| MWCNTs | Multi-wall carbon nanotubes |
| NLDFT | Non-local density functional theory |
| PAC | Powdered activated carbon |
| PAN | Polyacrylonitrile |
| PET | Poly(ethylene terephthalate) |
| PFA | Poly(furfuryl alcohol) |
| PVP | Polyvinylpyridine |
| SANS | Small-angle neutron scattering |
| SAXS | Small-angle X-ray scattering |
| SEM | Scanning electron microscopy |
| SQDFT | Quenched solid density functional theory |
| SSA | Specific surface area |
| TEABF4 | Tetraethylammonium tetrafluoroborate |
| TPD | Temperature-programmed desorption |
| XPS | X-ray photoelectron spectroscopy |
References
- Abioye, A.M.; Ani, F.N. Recent development in the production of activated carbon electrodes from agricultural waste biomass for supercapacitors: A review. Renew. Sustain. Energ. Rev. 2015, 52, 1282–1293. [Google Scholar] [CrossRef]
- Kalyani, P.; Anitha, A. Biomass carbon & its prospects in electrochemical energy systems. Int. J. Hydrog Energy 2013, 38, 4034–4045. [Google Scholar] [CrossRef]
- Dias, J.M.; Alvim-Ferraz, M.C.M.; Almeida, M.F.; Rivera-Utrilla, J.; Sánchez-Polo, M. Waste materials for activated carbon preparation and its use in aqueous-phase treatment: A review. J. Environ. Manag. 2007, 85, 833–846. [Google Scholar] [CrossRef]
- Klijanienko, A.; Lorenc-Grabowska, E.; Gryglewicz, G. Development of mesoporosity during phosphoric acid activation of wood in steam atmosphere. Bioresour. Technol. 2007, 99, 7208–7214. [Google Scholar] [CrossRef]
- Vassilev, S.V.; Baxter, D.; Andersen, L.K.; Vassileva, C.G.; Morgan, T.J. An overview of the organic and inorganic phase composition of biomass. Fuel 2012, 94, 1–33. [Google Scholar] [CrossRef]
- Raveendran, K.; Ganesh, A.; Khilar, K.C. Influence of mineral matter on biomass pyrolysis characteristics. Fuel 1995, 74, 1812–1822. [Google Scholar] [CrossRef]
- Rodríguez, G.; Lama, A.; Rodríguez, R.; Jiménez, A.; Guillén, R.; Fernández-Bolaños, J. Olive stone an attractive source of bioactive and valuable compounds. Bioresour. Technol. 2008, 99, 5261–5269. [Google Scholar] [CrossRef]
- Uysal, T.; Duman, G.; Onal, Y.; Yasa, I.; Yanik, J. Production of activated carbon and fungicidal oil from peach stone by two-stage process. J. Anal. Appl. Pyrolysis 2014, 108, 47–55. [Google Scholar] [CrossRef]
- Lupul, I.; Yperman, J.; Carleer, R.; Gryglewicz, G. Tailoring of porous texture of hemp stem-based activated carbon produced by phosphoric acid activation in steam atmosphere. J. Porous Mater. 2015, 22, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Saidur, R.; Abdelaziz, E.A.; Demirbas, A.; Hossain, M.S.; Mekhilef, S. A review on biomass as a fuel for boilers. Renew. Sustain. Energ. Rev. 2011, 15, 2262–2289. [Google Scholar] [CrossRef]
- McKendry, P. Energy production from biomass (Part 1): Overview of biomass. Bioresour. Technol. 2002, 83, 37–46. [Google Scholar] [CrossRef]
- Śliwak, A.; Díez, N.; Miniach, E.; Gryglewicz, G. Nitrogen-containing chitosan-based carbon as an electrode material for high-performance supercapacitors. J. Appl. Electrochem. 2016, 46, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Lee, J.G.; Yun, H.; Deng, S.; Kim, Y.J.; Lee, J.E.; Kwak, S.K.; Lee, K.B. Solving two environmental issues simultaneously: Waste polyethylene terephthalate plastic bottle-derived microporous carbons for capturing CO2. Chem. Eng. J. 2020, 397, 125350. [Google Scholar] [CrossRef]
- Castro, C.S.D.; Viau, L.N.; Andrade, J.T.; Mendonça, T.A.P.; Gonçalves, M. Mesoporous activated carbon from polyethyleneterephthalate (PET) waste: Pollutant adsorption in aqueous solution. New J. Chem. 2018, 42, 14612–14619. [Google Scholar] [CrossRef]
- Bratek, W.; Świątkowski, A.; Pakuła, M.; Biniak, S.; Bystrzejewski, M.; Szmigielski, R. Characteristics of activated carbon prepared from waste PET by carbon dioxide activation. J. Anal. Appl. Pyrolysis 2013, 100, 192–198. [Google Scholar] [CrossRef]
- Nasr, M.F.; Mosleh, S.E.-S.; Ali, S.S.M.; Abo El-Ola, S.M. Tailored removal of zinc and chrome ions by the adsorption onto nonwoven ACF (activated carbon fiber) prepared from PAN (polyacrylonitrile) waste. Desalin. Water Treat. 2019, 162, 252–259. [Google Scholar] [CrossRef]
- Lorenc-Grabowska, E.; Gryglewicz, G.; Diez, M.A. Kinetics and equilibrium study of phenol adsorption on nitrogen-enriched activated carbons. Fuel 2013, 114, 235–243. [Google Scholar] [CrossRef]
- Gonsalvesh, L.; Marinov, S.P.; Gryglewicz, G.; Carleer, R.; Yperman, J. Preparation, characterization and application of polystyrene based activated carbons for Ni(II) removal from aqueous solution. Fuel Process. Technol. 2016, 149, 75–85. [Google Scholar] [CrossRef]
- Hu, W.; Cheng, S.; Xia, H.; Zhang, L.; Jiang, X.; Zhang, Q.; Chen, Q. Waste phenolic resin derived activated carbon by microwave-assisted KOH activation and application to dye wastewater treatment. Green Process. Synth. 2019, 8, 408–415. [Google Scholar] [CrossRef]
- Choma, J.; Osuchowski, Ł.; Marszewski, M.; Jaroniec, M. Highly microporous polymer-based carbons for CO2 and H2 adsorption. RSC Adv. 2014, 4, 14795. [Google Scholar] [CrossRef]
- Yue, Z.; Dunya, H.; Ashuri, M.; Kucuk, K.; Aryal, S.; Antonov, S.; Alabbad, B.; Segre, C.U.; Mandal, B.K. Synthesis of a very high specific surface area active carbon and its electrical double-layer capacitor properties in organic electrolytes. ChemEngineering 2020, 4, 43. [Google Scholar] [CrossRef]
- Lorenc-Grabowska, E.; Diez, M.A.; Gryglewicz, G. Influence of pore size distribution on the adsorption of phenol on PET-based activated carbons. J. Colloid Interface Sci. 2016, 469, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Grzyb, B.; Machnikowski, J.; Weber, J.V. Mechanism of co-pyrolysis of coal-tar pitch with polyvinylpyridine. J. Anal. Appl. Pyrolysis 2004, 72, 121–130. [Google Scholar] [CrossRef]
- Carrott, P.J.M.; Ribeiro Carrott, M.M.L.; Correia, P.F.M.M. Evolution of porosity of activated carbon fibres prepared from pre-oxidized acrylic fibres. Microporous Mesoporous Mater. 2018, 264, 176–180. [Google Scholar] [CrossRef]
- Franklin, R.E. Crystallite growth in graphitizing and non-graphitizing carbons. Proc. R. Soc. Lond. A 1951, 209, 196–218. [Google Scholar] [CrossRef]
- Marsh, H.; Walker, P.L., Jr. The Formation of Graphitizable Carbons via Mesophase: Chemical and Kinetic Considerations. In Chemistry and Physics of Carbon; Walker, P.L., Jr., Thrower, P.A., Eds.; Marcel Dekker Inc.: New York, NY, USA, 1994; Volume 15, pp. 230–299. [Google Scholar]
- Stoeckli, H.F. Microporous carbons and their characterization: The present state of the art. Carbon 1990, 28, 1–6. [Google Scholar] [CrossRef]
- Marsh, H.; Wynne-Jones, W.F.K. The surface properties of carbon-I the effect of activated diffusion in the determination of surface area. Carbon 1964, 1, 269–279. [Google Scholar] [CrossRef]
- Shcherban, N.D.; Yaremov, P.S.; Ilyin, V.G.; Ovcharova, M.V. Influence of the method of activation on the structural and sorption properties of the products of carbonization of sucrose. J. Anal. Appl. Pyrolysis 2014, 107, 155–164. [Google Scholar] [CrossRef]
- Lin, S.-H.; Hsu, L.-Y.; Chou, C.-S.; Jhang, J.-W.; Wu, P. Carbonization process of Moso bamboo (Phyllostachys pubescens) charcoal and its governing thermodynamics. J. Anal. Appl. Pyrolysis 2014, 107, 9–16. [Google Scholar] [CrossRef]
- Rouzaud, J.-N.; Clinard, C. Quantitative high-resolution transmission electron microscopy: A promising tool for carbon materials characterization. Fuel Process. Technol. 2002, 77–78, 229–235. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F.; Molina-Sabio, M. Textural and chemical characterization of microporous carbons. Adv. Colloid Interface Sci. 1998, 76–77, 271–294. [Google Scholar] [CrossRef]
- Shi, H. Activated carbons and double layer capacitance. Electrochim. Acta 1996, 41, 1633–1639. [Google Scholar] [CrossRef]
- Neimark, A.V.; Lin, Y.; Ravikovitch, P.I.; Thommes, M. Quenched solid density functional theory and pore size analysis of micro-mesoporous carbons. Carbon 2009, 47, 1617–1628. [Google Scholar] [CrossRef]
- Bardestani, R.; Patience, G.S.; Kaliaguine, S. Experimental methods in chemical engineering: Specific surface area and pore size distribution measurements—BET, BJH, and DFT. Can. J. Chem. Eng. 2019, 97, 2781–2791. [Google Scholar] [CrossRef]
- Jagiello, J.; Thommes, M. Comparison of DFT characterization methods based on N2, Ar, CO2, and H2 adsorption applied to carbons with various pore size distributions. Carbon 2004, 42, 1227–1232. [Google Scholar] [CrossRef]
- Stoeckli, F.; López-Ramón, M.V.; Hugi-Cleary, D.; Guillot, A. Micropore sizes in activated carbons determined from the Dubinin–Radushkevich equation. Carbon 2001, 39, 1115–1116. [Google Scholar] [CrossRef]
- Denoyel, R.; Fernandez-Colinas, J.; Grillet, Y.; Rouquerol, J. Assessment of the surface area and microporosity of activated charcoals from immersion calorimetry and nitrogen adsorption data. Langmuir 1993, 9, 515–518. [Google Scholar] [CrossRef]
- Stoeckli, F.; Hugi-Cleary, D. On the mechanisms of phenol adsorption by carbons. Russ. Chem. Bull. 2001, 50, 2060–2063. [Google Scholar] [CrossRef]
- Stoeckli, H.F.; Kraehenbuehl, F. The enthalpies of immersion of active carbons, in relation to the Dubinin theory for the volume filling of micropores. Carbon 1981, 19, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Ruike, M.; Kasu, T.; Setoyama, N.; Suzuki, T.; Kaneko, K. Inaccessible pore characterization of less-crystalline microporous solids. J. Phys. Chem. 1994, 98, 9594–9600. [Google Scholar] [CrossRef]
- Kierzek, K.; Machnikowski, J. Cellulose-derived carbons as a high performance anodic material for Na-ion battery. Ionics 2018, 24, 1313–1320. [Google Scholar] [CrossRef]
- Hoinkis, E. Small Angle Scattering of Neutrons and X-rays from Carbons and Graphites. In Chemistry and Physics of Carbon; Thrower, P.A., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1997; Volume 25, pp. 71–241. [Google Scholar]
- Nishikawa, K. Pore Structure Analyses of Carbons by Small-Angle X-ray Scattering. In Carbon Alloys: Novel Concepts to Develop Carbon Science and Technology; Yusada, E., Inagaki, M., Kaneko, K., Endo, M., Oya, A., Tanabe, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2003; pp. 175–188. [Google Scholar]
- Li, Y.; Lu, Y.; Meng, Q.; Jensen, A.C.S.; Zhang, Q.; Zhang, Q.; Tong, Y.; Qi, Y.; Gu, L.; Titirici, M.-M.; et al. Regulating pore structure of hierarchical porous waste cork-derived hard carbon anode for enhanced na storage performance. Adv. Energy Mater. 2019, 9, 1902852. [Google Scholar] [CrossRef]
- Calo, J.M.; Hall, P.J.; Antxustegi, M. Carbon porosity characterization via small angle neutron scattering. Colloids Surf. A Physicochem. Eng. 2001, 187–188, 219–232. [Google Scholar] [CrossRef]
- Sakurovs, R.; Koval, L.; Grigore, M.; Sokolova, A.; de Campo, L.; Rehm, C. Nanostructure of cokes. Int. J. Coal Geol. 2018, 188, 112–120. [Google Scholar] [CrossRef]
- Biniak, S.; Świątkowski, A.; Pakuła, M. Electrochemical Studies of Phenomena at Active Carbon-Electrolyte Solutions Interfaces. In Chemistry and Physics of Carbon; Radovic, L.R., Ed.; Marcel Dekker Inc.: New York, NY, USA, 2001; Volume 27, pp. 125–225. [Google Scholar]
- Guan, T.; Zhao, J.; Zhang, G.; Zhang, D.; Han, B.; Tang, N.; Wang, J.; Li, K. Insight into controllability and predictability of pore structures in pitch-based activated carbons. Microporous Mesoporous Mater. 2018, 271, 118–127. [Google Scholar] [CrossRef]
- Rodríguez-Reinoso, F.; Molina-Sabio, M.; González, M.T. The use of steam and CO2 as activating agents in the preparation of activated carbons. Carbon 1995, 33, 15–23. [Google Scholar] [CrossRef]
- Molina-Sabio, M.; Gonzalez, M.T.; Rodriguez-Reinoso, F.; Sepúlveda-Escribano, A. Effect of steam and carbon dioxide activation in the micropore size distribution of activated carbon. Carbon 1996, 34, 505–509. [Google Scholar] [CrossRef]
- Lillo-Ródenas, M.A.; Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A. Preparation of activated carbons from Spanish anthracite. Carbon 2001, 39, 751–759. [Google Scholar] [CrossRef]
- Lillo-Ródenas, M.A.; Cazorla-Amorós, D.; Linares-Solano, A. Understanding chemical reactions between carbons and NaOH and KOH. Carbon 2003, 41, 267–275. [Google Scholar] [CrossRef]
- Maciá-Agulló, J.A.; Moore, B.C.; Cazorla-Amorós, D.; Linares-Solano, A. Activation of coal tar pitch carbon fibres: Physical activation vs. chemical activation. Carbon 2004, 42, 1367–1370. [Google Scholar] [CrossRef]
- Linares-Solano, A.; Lozano-Castello, D.; Lillo-Ródenas, M.A.; Cazorla-Amorós, D. Carbon Activation by Alkaline Hydroxides Preparation and Reactions, Porosity and Performance. In Chemistry and Physics of Carbon; Radovic, L.R., Ed.; Marcel Dekker: New York, NY, USA, 2008; Volume 30, pp. 1–62. [Google Scholar]
- Kierzek, K.; Frackowiak, E.; Lota, G.; Gryglewicz, G.; Machnikowski, J. Electrochemical capacitors based on highly porous carbons prepared by KOH activation. Electrochim. Acta 2004, 49, 515–523. [Google Scholar] [CrossRef]
- Lillo-Ródenas, M.A.; Juan-Juan, J.; Cazorla-Amorós, D.; Linares-Solano, A. About reactions occurring during chemical activation with hydroxides. Carbon 2004, 42, 1371–1375. [Google Scholar] [CrossRef]
- Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A.; Quinn, D.F. Influence of pore size distribution on methane storage at relatively low pressure: Preparation of activated carbon with optimum pore size. Carbon 2002, 40, 989–1002. [Google Scholar] [CrossRef]
- Xing, B.-L.; Guo, H.; Chen, L.-J.; Chen, Z.-F.; Zhang, C.-X.; Huang, G.-X.; Xie, W.; Yu, J.-L. Lignite-derived high surface area mesoporous activated carbons for electrochemical capacitors. Fuel Process. Technol. 2015, 138, 734–742. [Google Scholar] [CrossRef]
- Tellez-Juárez, M.C.; Fierro, V.; Zhao, W.; Fernández-Huerta, N.; Izquierdo, M.T.; Reguera, E.; Celzard, A. Hydrogen storage in activated carbons produced from coals of different ranks: Effect of oxygen content. Int. J. Hydrog. Energy 2014, 39, 4996–5002. [Google Scholar] [CrossRef]
- Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A. Powdered activated carbons and activated carbon fibers for methane storage: A comparative study. Energy Fuels 2002, 16, 1321–1328. [Google Scholar] [CrossRef]
- Cheng, G.; Zhang, C.X.; Zhang, X.M.; Zhang, Z.L. Preparation of lignite-based activated carbon and its flue gas desulfurization performance. Mater. Res. Express 2019, 6, 25603. [Google Scholar] [CrossRef]
- Deng, J.; Peng, Z.; Xiao, Z.; Song, S.; Dai, H.; Li, L. Porous doped carbons from anthracite for high-performance supercapacitors. Appl. Sci. 2020, 10, 1081. [Google Scholar] [CrossRef] [Green Version]
- Alkathiri, D.S.S.; Sabri, M.A.; Ibrahim, T.H.; ElSayed, Y.A.; Jumean, F. Development of activated carbon fibers for removal of organic contaminants. Int. J. Environ. Sci. Technol. 2020, 39, 215. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Cazorla-Amorós, D.; Linares-Solano, A.; Delpeux, S.; Frackowiak, E.; Szostak, K.; Béguin, F. High surface area carbon nanotubes prepared by chemical activation. Carbon 2002, 40, 1614–1617. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Azaïs, P.; Cacciaguerra, T.; Cazorla-Amorós, D.; Linares-Solano, A.; Béguin, F. KOH and NaOH activation mechanisms of multiwalled carbon nanotubes with different structural organisation. Carbon 2005, 43, 786–795. [Google Scholar] [CrossRef]
- Lozano-Castelló, D.; Lillo-Ródenas, M.A.; Cazorla-Amorós, D.; Linares-Solano, A. Preparation of activated carbons from Spanish anthracite. Carbon 2001, 39, 741–749. [Google Scholar] [CrossRef]
- Di Blasi, C.; Galgano, A.; Branca, C. Effects of potassium hydroxide impregnation on wood pyrolysis. Energy Fuels 2009, 23, 1045–1054. [Google Scholar] [CrossRef]
- Jagtoyen, M.; Derbyshire, F. Some considerations of the origins of porosity in carbons from chemically activated wood. Carbon 1993, 31, 1185–1192. [Google Scholar] [CrossRef]
- Suárez-García, F.; Martínez-Alonso, A.; Tascón, J.M.D. Activated carbon fibers from Nomex by chemical activation with phosphoric acid. Carbon 2004, 42, 1419–1426. [Google Scholar] [CrossRef]
- Castro-Muñiz, A.; Suárez-García, F.; Martínez-Alonso, A.; Tascón, J.M.D. Activated carbon fibers with a high content of surface functional groups by phosphoric acid activation of PPTA. J. Colloid Interface Sci. 2011, 361, 307–315. [Google Scholar] [CrossRef]
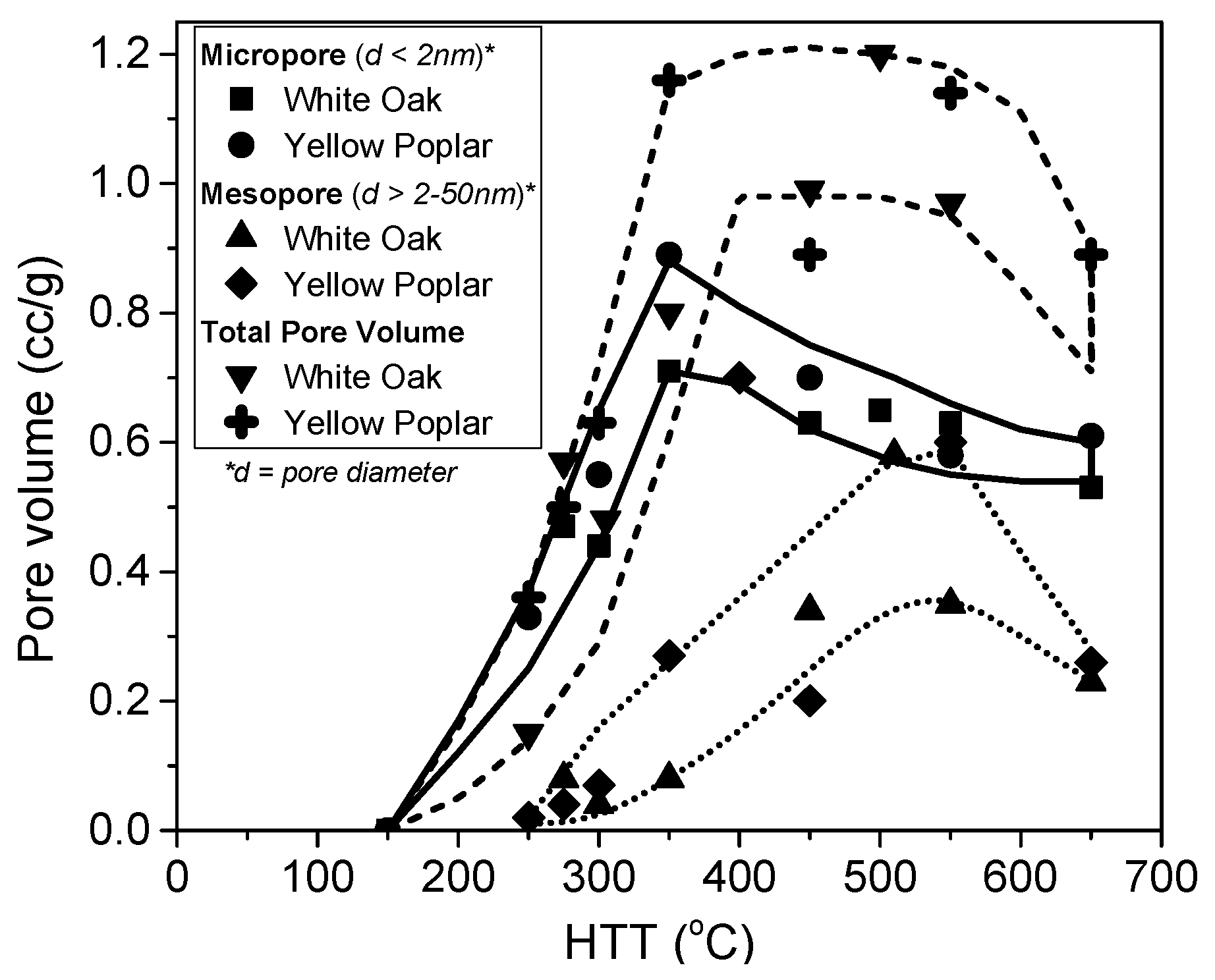
- Jagtoyen, M.; Derbyshire, F. Activated carbons from yellow poplar and white oak by H3PO4 activation. Carbon 1998, 36, 1085–1097. [Google Scholar] [CrossRef]
- Solum, M.S.; Pugmire, R.J.; Jagtoyen, M.; Derbyshire, F. Evolution of carbon structure in chemically activated wood. Carbon 1995, 33, 1247–1254. [Google Scholar] [CrossRef]
- Huang, C.; Puziy, A.M.; Sun, T.; Poddubnaya, O.I.; Suárez-García, F.; Tascón, J.M.D.; Hulicova-Jurcakova, D. Capacitive behaviours of phosphorus-rich carbons derived from lignocelluloses. Electrochim. Acta 2014, 137, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Puziy, A.M.; Poddubnaya, O.I.; Socha, R.P.; Gurgul, J.; Wisniewski, M. XPS and NMR studies of phosphoric acid activated carbons. Carbon 2008, 46, 2113–2123. [Google Scholar] [CrossRef]
- Benaddi, H.; Bandosz, T.J.; Jagiello, J.; Schwarz, J.A.; Rouzaud, J.N.; Legras, D.; Béguin, F. Surface functionality and porosity of activated carbons obtained from chemical activation of wood. Carbon 2000, 38, 669–674. [Google Scholar] [CrossRef]
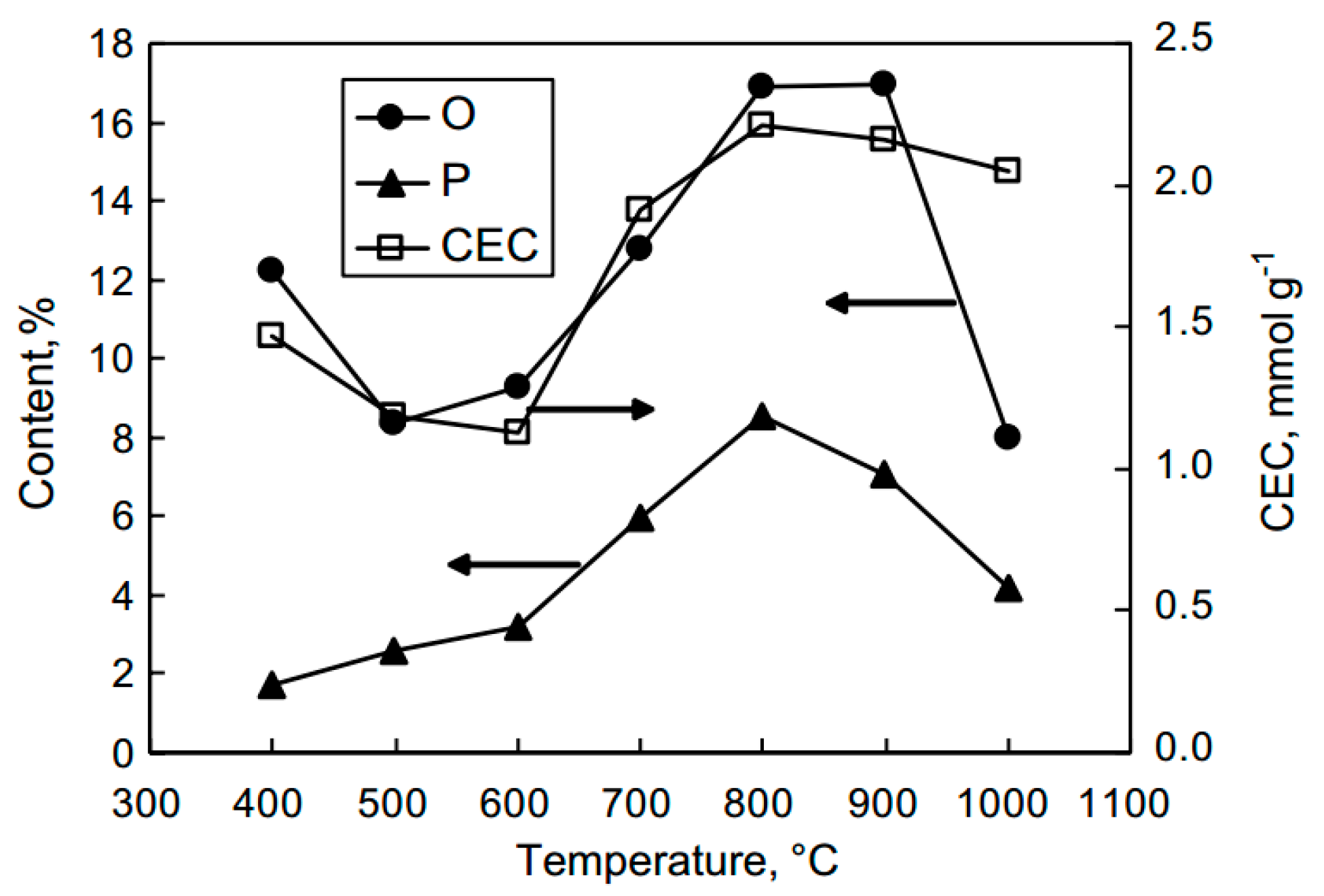
- Puziy, A.M.; Poddubnaya, O.I.; Martínez-Alonso, A.; Castro-Muñiz, A.; Suárez-García, F.; Tascón, J.M.D. Oxygen and phosphorus enriched carbons from lignocellulosic material. Carbon 2007, 45, 1941–1950. [Google Scholar] [CrossRef]
- Puziy, A.M.; Poddubnaya, O.I.; Martínez-Alonso, A.; Suárez-García, F.; Tascón, J.M.D. Surface chemistry of phosphorus-containing carbons of lignocellulosic origin. Carbon 2005, 43, 2857–2868. [Google Scholar] [CrossRef]
- Suárez-García, F.; Villar-Rodil, S.; Blanco, C.G.; Martínez-Alonso, A.; Tascón, J.M.D. Effect of phosphoric acid on chemical transformations during Nomex pyrolysis. Chem. Mater. 2004, 16, 2639–2647. [Google Scholar] [CrossRef]
- Imamura, R.; Matsui, K.; Takeda, S.; Ozaki, J.; Oya, A. A new role for phosphorus in graphitization of phenolic resin. Carbon 1999, 37, 261–267. [Google Scholar] [CrossRef]
- Sych, N.V.; Trofymenko, S.I.; Poddubnaya, O.I.; Tsyba, M.M.; Sapsay, V.I.; Klymchuk, D.O.; Puziy, A.M. Porous structure and surface chemistry of phosphoric acid activated carbon from corncob. Appl. Surf. Sci. 2012, 261, 75–82. [Google Scholar] [CrossRef]
- Molina-Sabio, M.; Rodríguez-Reinoso, F.; Caturla, F.; Sellés, M.J. Porosity in granular carbons activated with phosphoric acid. Carbon 1995, 33, 1105–1113. [Google Scholar] [CrossRef]
- Molina-Sabio, M.; Rodríguez-Reinoso, F. Role of chemical activation in the development of carbon porosity. Colloids Surf. A Physicochem. Eng. 2004, 241, 15–25. [Google Scholar] [CrossRef]
- Girgis, B.S.; Ishak, M.F. Activated carbon from cotton stalks by impregnation with phosphoric acid. Mater. Lett. 1999, 39, 107–114. [Google Scholar] [CrossRef]
- Guo, Y.; Rockstraw, D.A. Physicochemical properties of carbons prepared from pecan shell by phosphoric acid activation. Bioresour. Technol. 2007, 98, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Macías-García, A.; Carrasco-Amador, J.P.; Encinas-Sánchez, V.; Díaz-Díez, M.A.; Torrejón-Martín, D. Preparation of activated carbon from kenaf by activation with H3PO4. Kinetic study of the adsorption/electroadsorption using a system of supports designed in 3D, for environmental applications. J. Environ. Chem. Eng. 2019, 7, 103196. [Google Scholar] [CrossRef]
- Han, Q.; Wang, J.; Goodman, B.A.; Xie, J.; Liu, Z. High adsorption of methylene blue by activated carbon prepared from phosphoric acid treated eucalyptus residue. Powder Technol. 2020, 366, 239–248. [Google Scholar] [CrossRef]
- Guo, Y.; Rockstraw, D.A. Physical and chemical properties of carbons synthesized from xylan, cellulose, and Kraft lignin by H3PO4 activation. Carbon 2006, 44, 1464–1475. [Google Scholar] [CrossRef]
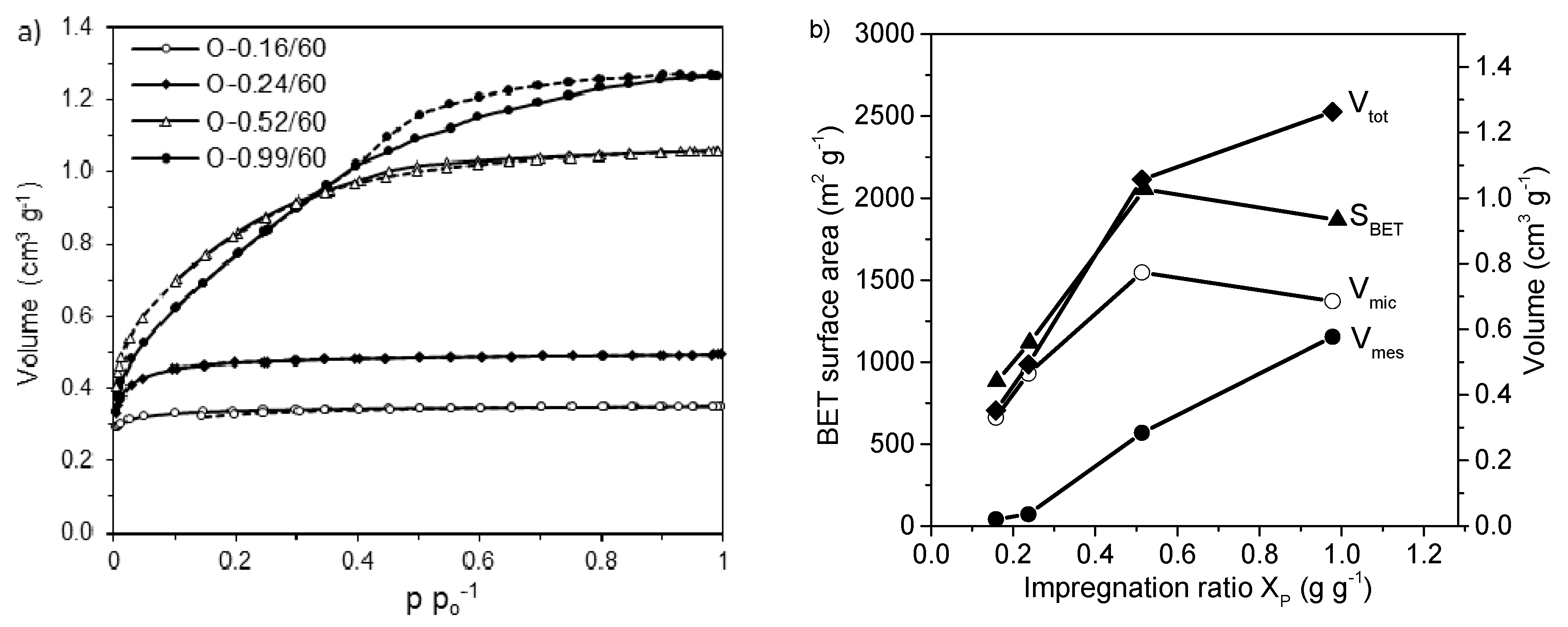
- Fierro, V.; Torné-Fernández, V.; Celzard, A. Kraft lignin as a precursor for microporous activated carbons prepared by impregnation with ortho-phosphoric acid: Synthesis and textural characterization. Microporous Mesoporous Mater. 2006, 92, 243–250. [Google Scholar] [CrossRef]
- Benaddi, H.; Legras, D.; Rouzaud, J.N.; Beguin, F. Influence of the atmosphere in the chemical activation of wood by phosphoric acid. Carbon 1998, 36, 306–309. [Google Scholar] [CrossRef]
- Danish, M.; Ahmad, T.; Hashim, R.; Said, N.; Akhtar, M.N.; Mohamad-Saleh, J.; Sulaiman, O. Comparison of surface properties of wood biomass activated carbons and their application against rhodamine B and methylene blue dye. Surf. Interfaces 2018, 11, 1–13. [Google Scholar] [CrossRef]
- Zhang, Z.; Lei, Y.; Li, D.; Zhao, J.; Wang, Y.; Zhou, G.; Yan, C.; He, Q. Sudden heating of H3PO4-loaded coconut shell in CO2 flow to produce super activated carbon and its application for benzene adsorption. Renew. Energy 2020, 153, 1091–1099. [Google Scholar] [CrossRef]
- Nahil, M.A.; Williams, P.T. Pore characteristics of activated carbons from the phosphoric acid chemical activation of cotton stalks. Biomass Bioenergy 2012, 37, 142–149. [Google Scholar] [CrossRef]
- Yavuz, R.; Akyildiz, H.; Karatepe, N.; Çetinkaya, E. Influence of preparation conditions on porous structures of olive stone activated by H3PO4. Fuel Process. Technol. 2010, 91, 80–87. [Google Scholar] [CrossRef]
- Guo, Y.; Rockstraw, D.A. Activated carbons prepared from rice hull by one-step phosphoric acid activation. Microporous Mesoporous Mater. 2007, 100, 12–19. [Google Scholar] [CrossRef]
- Castro, J.B.; Bonelli, P.R.; Cerrella, E.G.; Cukierman, A.L. Phosphoric acid activation of agricultural residues and bagasse from sugar cane: Influence of the experimental conditions on adsorption characteristics of activated carbons. Ind. Eng. Chem. Res. 2000, 39, 4166–4172. [Google Scholar] [CrossRef]
- Caturla, F.; Molina-Sabio, M.; Rodríguez-Reinoso, F. Preparation of activated carbon by chemical activation with ZnCl2. Carbon 1991, 29, 999–1007. [Google Scholar] [CrossRef]
- Marsh, H.; Rodriguez-Reinoso, F. Activated Carbon, 1st ed.; Elsevier Science: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Kante, K.; Nieto-Delgado, C.; Rangel-Mendez, J.R.; Bandosz, T.J. Spent coffee-based activated carbon: Specific surface features and their importance for H2S separation process. J. Hazard. Mater. 2012, 201–202, 141–147. [Google Scholar] [CrossRef]
- Angin, D. Production and characterization of activated carbon from sour cherry stones by zinc chloride. Fuel 2014, 115, 804–811. [Google Scholar] [CrossRef]
- Kumar, A.; Mohan Jena, H. High surface area microporous activated carbons prepared from Fox nut (Euryale ferox) shell by zinc chloride activation. Appl. Surf. Sci. 2015, 356, 753–761. [Google Scholar] [CrossRef]
- Ozdemir, I.; Şahin, M.; Orhan, R.; Erdem, M. Preparation and characterization of activated carbon from grape stalk by zinc chloride activation. Fuel Process. Technol. 2014, 125, 200–206. [Google Scholar] [CrossRef]
- Sun, Y.; Webley, P.A. Preparation of activated carbons from corncob with large specific surface area by a variety of chemical activators and their application in gas storage. Chem. Eng. J. 2010, 162, 883–892. [Google Scholar] [CrossRef]
- Olivares-Marín, M.; Fernández-González, C.; Macías-García, A.; Gómez-Serrano, V. Preparation of activated carbon from cherry stones by chemical activation with ZnCl2. Appl. Surf. Sci. 2006, 252, 5967–5971. [Google Scholar] [CrossRef]
- Abatan, O.G.; Oni, B.A.; Agboola, O.; Efevbokhan, V.; Abiodun, O.O. Production of activated carbon from African star apple seed husks, oil seed and whole seed for wastewater treatment. J. Clean. Prod. 2019, 232, 441–450. [Google Scholar] [CrossRef]
- Shrestha, L.K.; Shrestha, R.G.; Maji, S.; Pokharel, B.P.; Rajbhandari, R.; Shrestha, R.L.; Pradhananga, R.R.; Hill, J.P.; Ariga, K. High surface area nanoporous graphitic carbon materials derived from lapsi seed with enhanced supercapacitance. Nanomaterials 2020, 10, 728. [Google Scholar] [CrossRef] [Green Version]
- Ahmadpour, A.; Do, D.D. The preparation of activated carbon from macadamia nutshell by chemical activation. Carbon 1997, 35, 1723–1732. [Google Scholar] [CrossRef]
- Torregrosa, R.; Martín-Martínez, J. Activation of lignocellulosic materials: A comparison between chemical, physical and combined activation in terms of porous texture. Fuel 1991, 70, 1173–1180. [Google Scholar] [CrossRef]
- Üner, O.; Geçgel, Ü.; Bayrak, Y. Preparation and characterization of mesoporous activated carbons from waste watermelon rind by using the chemical activation method with zinc chloride. Arab. J. Chem. 2019, 12, 3621–3627. [Google Scholar] [CrossRef] [Green Version]
- Nasrullah, A.; Saad, B.; Bhat, A.H.; Khan, A.S.; Danish, M.; Isa, M.H.; Naeem, A. Mangosteen peel waste as a sustainable precursor for high surface area mesoporous activated carbon: Characterization and application for methylene blue removal. J. Clean. Prod. 2019, 211, 1190–1200. [Google Scholar] [CrossRef]
- Spagnoli, A.A.; Giannakoudakis, D.A.; Bashkova, S. Adsorption of methylene blue on cashew nut shell based carbons activated with zinc chloride: The role of surface and structural parameters. J. Mol. Liq. 2017, 229, 465–471. [Google Scholar] [CrossRef]
- Hoseinzadeh Hesas, R.; Arami-Niya, A.; Wan Daud, W.M.A.; Sahu, J.N. Comparison of oil palm shell-based activated carbons produced by microwave and conventional heating methods using zinc chloride activation. J. Anal. Appl. Pyrolysis 2013, 104, 176–184. [Google Scholar] [CrossRef]
- He, X.; Ling, P.; Qiu, J.; Yu, M.; Zhang, X.; Yu, C.; Zheng, M. Efficient preparation of biomass-based mesoporous carbons for supercapacitors with both high energy density and high power density. J. Power Sources 2013, 240, 109–113. [Google Scholar] [CrossRef]
- Rufford, T.E.; Hulicova-Jurcakova, D.; Khosla, K.; Zhu, Z.; Lu, G.Q. Microstructure and electrochemical double-layer capacitance of carbon electrodes prepared by zinc chloride activation of sugar cane bagasse. J. Power Sources 2010, 195, 912–918. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Zhang, G.; Liu, W.; Wang, D.; Dong, Y. Preparation of activated carbon from willow leaves and evaluation in electric double-layer capacitors. Mater. Lett. 2016, 176, 60–63. [Google Scholar] [CrossRef]
- Cheng, L.; Guo, P.; Wang, R.; Ming, L.; Leng, F.; Li, H.; Zhao, X.S. Electrocapacitive properties of supercapacitors based on hierarchical porous carbons from chestnut shell. Colloids Surf. A Physicochem. Eng. 2014, 446, 127–133. [Google Scholar] [CrossRef]
- Timur, S.; Kantarli, I.C.; Onenc, S.; Yanik, J. Characterization and application of activated carbon produced from oak cups pulp. J. Anal. Appl. Pyrolysis 2010, 89, 129–136. [Google Scholar] [CrossRef]
- Lin, H.; Liu, Y.; Chang, Z.; Yan, S.; Liu, S.; Han, S. A new method of synthesizing hemicellulose-derived porous activated carbon for high-performance supercapacitors. Microp. Mesop. Mat. 2020, 292, 109707. [Google Scholar] [CrossRef]
- Ramirez-Castro, C.; Schütter, C.; Passerini, S.; Balducci, A. Microporous carbonaceous materials prepared from biowaste for supercapacitor application. Electrochim. Acta 2016, 206, 452–457. [Google Scholar] [CrossRef]
- Rawal, S.; Joshi, B.; Kumar, Y. Synthesis and characterization of activated carbon from the biomass of Saccharum bengalense for electrochemical supercapacitors. J. Energy Storage 2018, 20, 418–426. [Google Scholar] [CrossRef]
- He, X.; Ling, P.; Yu, M.; Wang, X.; Zhang, X.; Zheng, M. Rice husk-derived porous carbons with high capacitance by ZnCl2 activation for supercapacitors. Electrochim. Acta 2013, 105, 635–641. [Google Scholar] [CrossRef]
- Demiral, H.; Gündüzoğlu, G. Removal of nitrate from aqueous solutions by activated carbon prepared from sugar beet bagasse. Bioresour. Technol. 2010, 101, 1675–1680. [Google Scholar] [CrossRef]
- Rufford, T.E.; Hulicova-Jurcakova, D.; Fiset, E.; Zhu, Z.; Lu, G.Q. Double-layer capacitance of waste coffee ground activated carbons in an organic electrolyte. Electrochem. Commun. 2009, 11, 974–977. [Google Scholar] [CrossRef]
- Wu, Y.; Xia, C.; Cai, L.; Shi, S.Q. Controlling pore size of activated carbon through self-activation process for removing contaminants of different molecular sizes. J. Colloid Interface Sci. 2018, 518, 41–47. [Google Scholar] [CrossRef]
- Sun, K.; Leng, C.-Y.; Jiang, J.-C.; Bu, Q.; Lin, G.-F.; Lu, X.-C.; Zhu, G.-Z. Microporous activated carbons from coconut shells produced by self-activation using the pyrolysis gases produced from them, that have an excellent electric double layer performance. New Carbon Mater. 2017, 32, 451–459. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Cadek, M.; Béguin, F. Tuning carbon materials for supercapacitors by direct pyrolysis of seaweeds. Adv. Funct. Mater. 2009, 19, 1032–1039. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Leroux, F.; Béguin, F. A high-performance carbon for supercapacitors obtained by carbonization of a seaweed biopolymer. Adv. Mater. 2006, 18, 1877–1882. [Google Scholar] [CrossRef]
- Kleszyk, P.; Ratajczak, P.; Skowron, P.; Jagiello, J.; Abbas, Q.; Frąckowiak, E.; Béguin, F. Carbons with narrow pore size distribution prepared by simultaneous carbonization and self-activation of tobacco stems and their application to supercapacitors. Carbon 2015, 81, 148–157. [Google Scholar] [CrossRef]
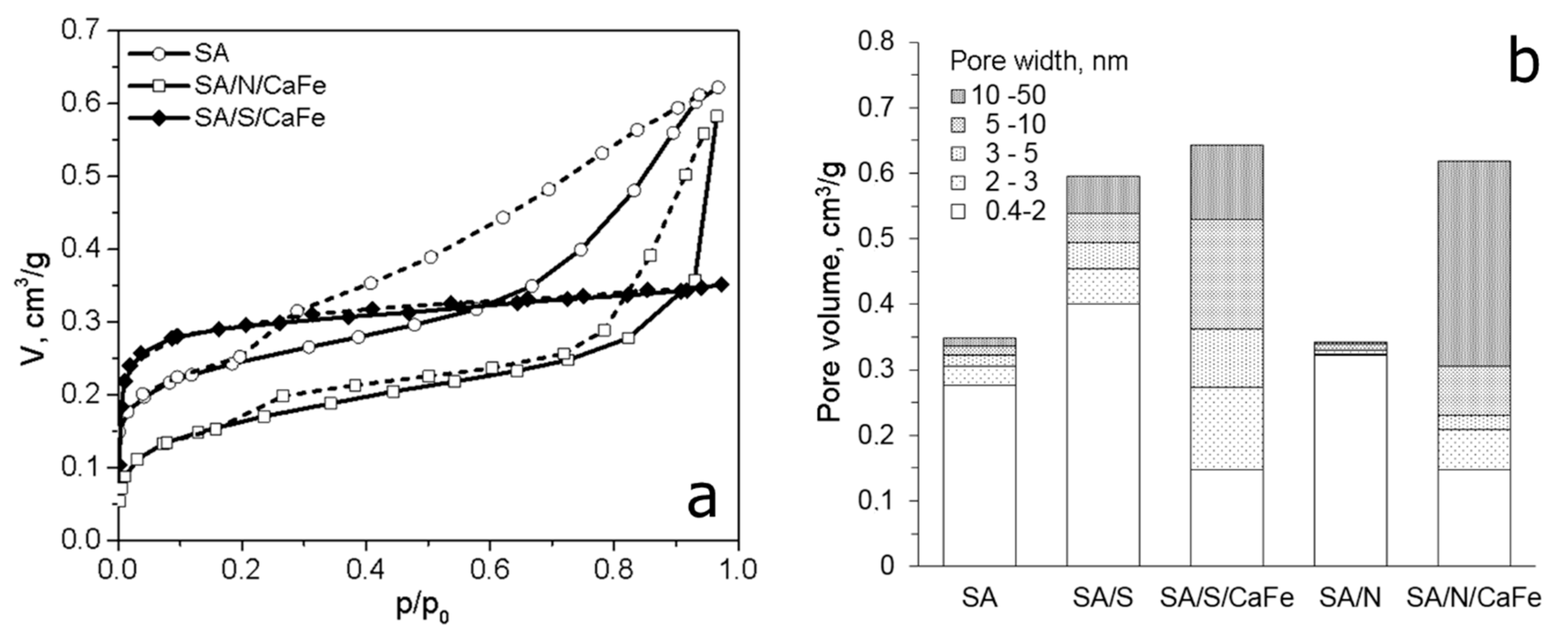
- Lorenc-Grabowska, E.; Gryglewicz, G.; Gryglewicz, S. Development of mesoporosity in activated carbons via coal modification using Ca- and Fe-exchange. Microp. Mesop. Mat. 2004, 76, 193–201. [Google Scholar] [CrossRef]
- Gryglewicz, G.; Lorenc-Grabowska, E. Mesoporous activated carbons from Ca and Fe exchanged sub-bituminous and bituminous coals. Carbon 2004, 42, 688–691. [Google Scholar] [CrossRef]
- Yoshizawa, N.; Yamada, Y.; Furuta, T.; Shiraishi, M.; Kojima, S.; Tamai, H.; Yasuda, H. Coal-based activated carbons prepared with organometallics and their mesoporous structure. Energy Fuels 1997, 11, 327–330. [Google Scholar] [CrossRef]
- Wang, L.; Sun, F.; Hao, F.; Qu, Z.; Gao, J.; Liu, M.; Wang, K.; Zhao, G.; Qin, Y. A green trace K2CO3 induced catalytic activation strategy for developing coal-converted activated carbon as advanced candidate for CO2 adsorption and supercapacitors. Chem. Eng. J. 2020, 383, 123205. [Google Scholar] [CrossRef]
- Liu, Z.; Ling, L.; Qiao, W.; Lu, C.; Wu, D.; Liu, L. Effects of various metals and their loading methods on the mesopore formation in pitch-based spherical activated carbon. Carbon 1999, 37, 1333–1335. [Google Scholar] [CrossRef]
- Liu, Z.; Ling, L.; Qiao, W.; Liu, L. Preparation of pitch-based spherical activated carbon with developed mesopore by the aid of ferrocene. Carbon 1999, 37, 663–667. [Google Scholar] [CrossRef]
- Tamai, H.; Kakii, T.; Hirota, Y.; Kumamoto, T.; Yasuda, H. Synthesis of extremely large mesoporous activated carbon and its unique adsorption for giant molecules. Chem. Mater. 1996, 8, 454–462. [Google Scholar] [CrossRef]
- El-Merraoui, M.; Tamai, H.; Yasuda, H.; Kanata, T.; Mondori, J.; Nadai, K.; Kaneko, K. Pore structures of activated carbon fibers from organometallics/pitch composites by nitrogen adsorption. Carbon 1998, 36, 1769–1776. [Google Scholar] [CrossRef]
- Diez, N.; Alvarez, P.; Granda, M.; Blanco, C.; Gryglewicz, G.; Wróbel-Iwaniec, I.; Śliwak, A.; Machnikowski, J.; Menendez, R. Tailoring micro-mesoporosity in activated carbon fibers to enhance SO2 catalytic oxidation. J. Colloid Interface Sci. 2014, 428, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basova, Y.V.; Edie, D.D.; Badheka, P.Y.; Bellam, H.-C. The effect of precursor chemistry and preparation conditions on the formation of pore structure in metal-containing carbon fibers. Carbon 2005, 43, 1533–1545. [Google Scholar] [CrossRef]
- Oya, A.; Yoshida, S.; Alcaniz-Monge, J.; Linares-Solano, A. Formation of mesopores in phenolic resin-derived carbon fiber by catalytic activation using cobalt. Carbon 1995, 33, 1085–1090. [Google Scholar] [CrossRef]
- Marsh, H.; Diez, M.A.; Kuo, K. Specific Reactivities of Pure Carbon of Diverse Origins. In Fundamental Issues in Control of Carbon Gasification Reactivity; Lahaye, J., Ehrburgor, P., Eds.; Springer: Dordrecht, The Netherlands, 1991; pp. 205–220. [Google Scholar]
- Kaneko, K.; Ishii, C. Superhigh surface area determination of microporous solids. Colloids Surfaces 1992, 67, 203–212. [Google Scholar] [CrossRef]
- Barbieri, O.; Hahn, M.; Herzog, A.; Kötz, R. Capacitance limits of high surface area activated carbons for double layer capacitors. Carbon 2005, 43, 1303–1310. [Google Scholar] [CrossRef]
- Salitra, G.; Soffer, A.; Eliad, L.; Cohen, Y.; Aurbach, D. Carbon electrodes for double-layer capacitors i. relations between ion and pore dimensions. J. Electrochem. Soc. 2000, 147, 2486. [Google Scholar] [CrossRef]
- Eliad, L.; Salitra, G.; Soffer, A.; Aurbach, D. Ion sieving effects in the electrical double layer of porous carbon electrodes: Estimating effective ion size in electrolytic solutions. J. Phys. Chem. B 2001, 105, 6880–6887. [Google Scholar] [CrossRef]
- Eliad, L.; Pollak, E.; Levy, N.; Salitra, G.; Soffer, A.; Aurbach, D. Assessing optimal pore-to-ion size relations in the design of porous poly(vinylidene chloride) carbons for EDL capacitors. Appl. Phys. A 2006, 82, 607–613. [Google Scholar] [CrossRef]
- Thürmer, S.; Kobayashi, Y.; Ohba, T.; Kanoh, H. Pore-size dependent effects on structure and vibrations of 1-ethyl-3-methylimidazolium tetrafluoroborate in nanoporous carbon. Chem. Phys. Lett. 2015, 636, 129–133. [Google Scholar] [CrossRef]
- Raymundo-Piňero, E.; Gao, Q.; Béguin, F. Carbons for supercapacitors obtained by one-step pressure induced oxidation at low temperature. Carbon 2013, 61, 278–283. [Google Scholar] [CrossRef]
- Ania, C.O.; Pernak, J.; Stefaniak, F.; Raymundo-Piñero, E.; Béguin, F. Solvent-free ionic liquids as in situ probes for assessing the effect of ion size on the performance of electrical double layer capacitors. Carbon 2006, 44, 3126–3130. [Google Scholar] [CrossRef] [Green Version]
- Mysyk, R.; Gao, Q.; Raymundo-Piñero, E.; Béguin, F. Microporous carbons finely-tuned by cyclic high-pressure low-temperature oxidation and their use in electrochemical capacitors. Carbon 2012, 50, 3367–3374. [Google Scholar] [CrossRef]
- Ruiz, V.; Pandolfo, A.G. Polyfurfuryl alcohol derived activated carbons for high power electrical double layer capacitors. Electrochim. Acta 2010, 55, 7495–7500. [Google Scholar] [CrossRef]
- Pohlmann, S.; Lobato, B.; Centeno, T.A.; Balducci, A. The influence of pore size and surface area of activated carbons on the performance of ionic liquid based supercapacitors. Phys. Chem. Chem. Phys. 2013, 15, 17287–17294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, M.P.S.; Wilson, B.E.; Kashefolgheta, S.; Anderson, E.L.; He, S.; Bühlmann, P.; Stein, A. Ionic liquids as electrolytes for electrochemical double-layer capacitors: Structures that optimize specific energy. ACS Appl. Mater. Interfaces 2016, 8, 3396–3406. [Google Scholar] [CrossRef]
- Decaux, C.; Matei Ghimbeu, C.; Dahbi, M.; Anouti, M.; Lemordant, D.; Béguin, F.; Vix-Guterl, C.; Raymundo-Piñero, E. Influence of electrolyte ion–solvent interactions on the performances of supercapacitors porous carbon electrodes. J. Power Sources 2014, 263, 130–140. [Google Scholar] [CrossRef]
- Raymundo-Piñero, E.; Kierzek, K.; Machnikowski, J.; Béguin, F. Relationship between the nanoporous texture of activated carbons and their capacitance properties in different electrolytes. Carbon 2006, 44, 2498–2507. [Google Scholar] [CrossRef]
- Qu, D.; Shi, H. Studies of activated carbons used in double-layer capacitors. J. Power Source 1998, 74, 99–107. [Google Scholar] [CrossRef]
- Endo, M.; Maeda, T.; Takeda, T.; Kim, Y.J.; Koshiba, K.; Hara, H.; Dresselhaus, M.S. Capacitance and pore-size distribution in aqueous and nonaqueous electrolytes using various activated carbon electrodes. J. Power Source 2001, 148, A910. [Google Scholar] [CrossRef]
- Boujibar, O.; Ghamouss, F.; Ghosh, A.; Achak, O.; Chafik, T. Activated carbon with exceptionally high surface area and tailored nanoporosity obtained from natural anthracite and its use in supercapacitors. J. Power Source 2019, 436, 226882. [Google Scholar] [CrossRef]
- Kandasamy, A.; Ramasamy, T.; Samrin, A.; Narayanasamy, P.; Mohan, R.; Bazaka, O.; Levchenko, I.; Bazaka, K.; Mohandas, M. Hierarchical doped gelatin-derived carbon aerogels: Three levels of porosity for advanced supercapacitors. Nanomaterials 2020, 10, 1178. [Google Scholar] [CrossRef]
- Hirose, E.; Nomoto, S.; Yoshioka, K. Activated Carbon for Use in Electric Double Layer Capacitor and Method of Producing the Same. Patent EP-1094478-A2, 25 April 2001. [Google Scholar]
- Endo, M.; Koyama, S.; Noguchi, M.; Oki, N. Manufacturing Method of Electrolytic Capacitor, Rectifier, Detector, Switchgear, Light-Sensitive Device or Thermosensitive Device. Patent jp2001189244, 10 July 2001. [Google Scholar]
- Sonobe, N.; Nagai, A.; Aida Tomoyuki, T.; Noguchi, M.; Iwaida, M.; Komazawa, E. Method of Producing A Carbon Material for An Electric double Layer Capacitor Electrode. Patent jp4117056, 9 July 2008. [Google Scholar]
- Qu, D. Studies of the activated carbons used in double-layer supercapacitors. J. Power Source 2002, 109, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Torchała, K.; Kierzek, K.; Machnikowski, J. Capacitance behavior of KOH activated mesocarbon microbeads in different aqueous electrolytes. Electrochim. Acta 2012, 86, 260–267. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Horie, Y.; Matsuzawa, Y.; Ozaki, S.; Endo, M.; Dresselhaus, M.S. Structural features necessary to obtain a high specific capacitance in electric double layer capacitors. Carbon 2004, 42, 2423–2432. [Google Scholar] [CrossRef]
- Menéndez, J.A.; Phillips, J.; Xia, B.; Radovic, L.R. On the modification and characterization of chemical surface properties of activated carbon: In the search of carbons with stable basic properties. Langmuir 1996, 12, 4404–4410. [Google Scholar] [CrossRef]
- Menéndez, J.A.; Xia, B.; Phillips, J.; Radovic, L.R. On the modification and characterization of chemical surface properties of activated carbon: Microcalorimetric, electrochemical, and thermal desorption probes. Langmuir 1997, 13, 3414–3421. [Google Scholar] [CrossRef]
- Tian, Y.; Song, Y.; Tang, Z.; Guo, Q.; Liu, L. Influence of high temperature treatment of porous carbon on the electrochemical performance in supercapacitor. J. Power Source 2008, 184, 675–681. [Google Scholar] [CrossRef]
- Ruch, P.W.; Hahn, M.; Rosciano, F.; Holzapfel, M.; Kaiser, H.; Scheifele, W.; Schmitt, B.; Novák, P.; Kötz, R.; Wokaun, A. In situ X-ray diffraction of the intercalation of (C2H5)4N+ and BF4− into graphite from acetonitrile and propylene carbonate based supercapacitor electrolytes. Electrochim. Acta 2007, 53, 1074–1082. [Google Scholar] [CrossRef]
- Hahn, M.; Barbieri, O.; Campana, F.P.; Kötz, R.; Gallay, R. Carbon based double layer capacitors with aprotic electrolyte solutions: The possible role of intercalation/insertion processes. Appl. Phys. A 2006, 82, 633–638. [Google Scholar] [CrossRef] [Green Version]
- Iwaida, M.; Koyama, S.; Murakami, K. Polarizable Electrode for Electric Double Layer Capacitor and Electric Double Layer Capacitor Using it. Patent JP2004193569, 8 July 2004. [Google Scholar]
- Iwaida, M.; Koyama, S.; Murakami, K.; Otsuka, K.; Ozaki, K.; Tsutsui, M. Activated Carbon, Polarizable Electrode for Electric Double-Layer Capacitor, and Electric Double-Layer Capacitor Using the Same. Patent JP2004189586, 8 July 2004. [Google Scholar]
- Tanahashi, I. Electrochemical characterization of activated carbon-fiber cloth polarizable electrodes for electric double-layer capacitors. J. Electrochem. Soc. 1990, 137, 3052. [Google Scholar] [CrossRef]
- Ishikawa, M.; Sakamoto, A.; Monta, M.; Matsuda, Y.; Ishida, K. Effect of treatment of activated carbon fiber cloth electrodes with cold plasma upon performance of electric double-layer capacitors. J. Power Source 1996, 60, 233–238. [Google Scholar] [CrossRef]
- Hsieh, C.-T.; Teng, H. Influence of oxygen treatment on electric double-layer capacitance of activated carbon fabrics. Carbon 2002, 40, 667–674. [Google Scholar] [CrossRef]
- Babel, K.; Jurewicz, K. KOH activated carbon fabrics as supercapacitor material. J. Phys. Chem. Solids 2004, 65, 275–280. [Google Scholar] [CrossRef]
- Burke, A. R&D considerations for the performance and application of electrochemical capacitors. Electrochim. Acta 2007, 53, 1083–1091. [Google Scholar] [CrossRef]
- Pandolfo, A.G.; Hollenkamp, A.F. Carbon properties and their role in supercapacitors. J. Power Source 2006, 157, 11–27. [Google Scholar] [CrossRef]
- Wang, K.-P.; Teng, H. The performance of electric double layer capacitors using particulate porous carbons derived from PAN fiber and phenol-formaldehyde resin. Carbon 2006, 44, 3218–3225. [Google Scholar] [CrossRef]
- Torchała, K.; Kierzek, K.; Gryglewicz, G.; Machnikowski, J. Narrow-porous pitch-based carbon fibers of superior capacitance properties in aqueous electrolytes. Electrochim. Acta 2015, 167, 348–356. [Google Scholar] [CrossRef]
- Bleda-Martínez, M.J.; Lozano-Castelló, D.; Cazorla-Amorós, D.; Morallón, E. Kinetics of double-layer formation: Influence of porous structure and pore size distribution. Energy Fuels 2010, 24, 3378–3384. [Google Scholar] [CrossRef]
- Endo, M.; Kim, Y.J.; Maeda, T.; Koshiba, K.; Katayam, K.; Dresselhaus, M.S. Morphological effect on the electrochemical behavior of electric double-layer capacitors. J. Mater. Res. 2001, 16, 3402–3410. [Google Scholar] [CrossRef] [Green Version]
- Egashira, M.; Izumi, T.; Yoshimoto, N.; Morita, M. Determining water content in activated carbon for double-layer capacitor electrodes. J. Power Source 2016, 326, 635–640. [Google Scholar] [CrossRef]
- Kurzweil, P.; Chwistek, M. Electrochemical stability of organic electrolytes in supercapacitors: Spectroscopy and gas analysis of decomposition products. J. Power Source 2008, 176, 555–567. [Google Scholar] [CrossRef]
- Hahn, M.; Würsig, A.; Gallay, R.; Novák, P.; Kötz, R. Gas evolution in activated carbon/propylene carbonate based double-layer capacitors. Electrochem. Commun. 2005, 7, 925–930. [Google Scholar] [CrossRef]
- Nguyen, Q.D.; Wu, Y.-H.; Wu, T.-Y.; Deng, M.-J.; Yang, C.-H.; Chang, J.-K. Gravimetric/volumetric capacitances, leakage current, and gas evolution of activated carbon supercapacitors. Electrochim. Acta 2016, 222, 1153–1159. [Google Scholar] [CrossRef]
- Azaïs, P.; Duclaux, L.; Florian, P.; Massiot, D.; Lillo-Rodenas, M.-A.; Linares-Solano, A.; Peres, J.-P.; Jehoulet, C.; Béguin, F. Causes of supercapacitors ageing in organic electrolyte. J. Power Source 2007, 171, 1046–1053. [Google Scholar] [CrossRef]
- Morimoto, T.; Hiratsuka, K.; Sanada, Y.; Kurihara, K. Electric double-layer capacitor using organic electrolyte. J. Power Source 1996, 60, 239–247. [Google Scholar] [CrossRef]
- Jerabek, E.C.; Mansfield, S.F. Method of Making an Ultracapacitor Electrode. US Patent 6 084 766, 4 July 2000. [Google Scholar]
- Yoshida, A.; Tanahashi, I.; Nishino, A. Effect of concentration of surface acidic functional groups on electric double-layer properties of activated carbon fibers. Carbon 1990, 28, 611–615. [Google Scholar] [CrossRef]
- Xu, K.; Ding, M.S.; Richard Jow, T. A better quantification of electrochemical stability limits for electrolytes in double layer capacitors. Electrochim. Acta 2001, 46, 1823–1827. [Google Scholar] [CrossRef]
- Hahn, M.; Kötz, R.; Gallay, R.; Siggel, A. Pressure evolution in propylene carbonate based electrochemical double layer capacitors. Electrochim. Acta 2006, 52, 1709–1712. [Google Scholar] [CrossRef]
- Kötz, R.; Hahn, M.; Ruch, P.; Gallay, R. Comparison of pressure evolution in supercapacitor devices using different aprotic solvents. Electrochem. Commun. 2008, 10, 359–362. [Google Scholar] [CrossRef]
- Pillay, B. The influence of side reactions on the performance of electrochemical double-layer capacitors. J. Electrochem. Soc. 1996, 143, 1806. [Google Scholar] [CrossRef]
- Nakamura, M.; Nakanishi, M.; Yamamoto, K. Influence of physical properties of activated carbons on characteristics of electric double-layer capacitors. J. Power Source 1996, 60, 225–231. [Google Scholar] [CrossRef]
- Lu, B.; Hu, L.; Yin, H.; Mao, X.; Xiao, W.; Wang, D. Preparation and application of capacitive carbon from bamboo shells by one step molten carbonates carbonization. Int. J. Hydrog Energy 2016, 41, 18713–18720. [Google Scholar] [CrossRef]
- Sugo, N.; Iwasaki, H.; Uehara, G. Activated Carbon, A Method of Manufacturing the Same, A Polarizable Electrode and An Electric Double Layer Capacitor. Patent WO2004011371 A1, 5 February 2004. [Google Scholar]
- Sugo, N.; Iwasaki, H.; Uehara, G. Activated Carbon, Process for Producing the Same, Polarizable Electrode, and Electric Double Layer Capacitor. Patent EP1176617 A3, 16 August 2006. [Google Scholar]
- Qu, J.; Geng, C.; Lv, S.; Shao, G.; Ma, S.; Wu, M. Nitrogen, oxygen and phosphorus decorated porous carbons derived from shrimp shells for supercapacitors. Electrochim. Acta 2015, 176, 982–988. [Google Scholar] [CrossRef]
- Huang, C.; Sun, T.; Hulicova-Jurcakova, D. Wide electrochemical window of supercapacitors from coffee bean-derived phosphorus-rich carbons. ChemSusChem 2013, 6, 2330–2339. [Google Scholar] [CrossRef]
- Hulicova-Jurcakova, D.; Puziy, A.M.; Poddubnaya, O.I.; Suárez-García, F.; Tascón, J.M.D.; Lu, G.Q. Highly stable performance of supercapacitors from phosphorus-enriched carbons. J. Am. Chem. Soc. 2009, 131, 5026–5027. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Material | Cellulose | Lignin | Hemicellulose | Ref. |
|---|---|---|---|---|
| Oak wood | 39 | 28 | 19 | [4] |
| Birch wood | 42 | 20 | 21 | [4] |
| Coconut shell * | 40 | 32 | 28 | [5] |
| Palm kernel shell | 33 | 46 | 24 | ** |
| Almond shell * | 51 | 20 | 29 | [5] |
| Hazelnut shell * | 27 | 43 | 30 | [5] |
| Groundnut shell | 36 | 30 | 19 | [6] |
| Rice husk | 31 | 14 | 24 | [6] |
| Bagasse | 41 | 18 | 23 | [6] |
| Coir pith | 29 | 31 | 15 | [6] |
| Olive stones | 32 | 27 | 22 | [7] |
| Peach stones | 46 | 33 | 14 | [8] |
| Hemp stem | 51 | 13 | 20 | [9] |
| Sugarcane bagasse | 32–44 | 19–24 | 27–32 | [10] |
| Wheat straw | 33–40 | 15–20 | 20–25 | [11] |
| Softwood | 35–40 | 27–30 | 25–30 | [11] |
| Hardwood | 45–50 | 20–25 | 20–25 | [11] |
| Raw Material | Temperature °C | Char Yield wt% |
|---|---|---|
| Cellulose | 800 | 19 |
| Lignin | 800 | 43 |
| Palm kernel shell | 900 | 33 |
| Walnut shells | 800 | 26 |
| Coconut shells | 800 | 29 |
| Hazelnut shells | 800 | 25 |
| Cherry stones | 800 | 25 |
| Chitozan | 800 | 30 |
| PFA | 800 | 47 |
| PET | 800 | 19 |
| PAN | 550 | 44 |
| PANox | 550 | 67 |
| Polyvinylpyridine (PVP) | 550 | 9 |
| PVPox | 550 | 60 |
| Phenol formaldehyde resin | 800 | 43 |
| Coal-tar pitch | 800 | 45 |
| Lignite | 800 | 45–55 |
| High-volatile bituminous coal | 800 | 60–70 |
| Conditions | Burn-Off (wt%) | SBET (m2 g−1) | VT (cm3 g−1) | VDR (cm3 g−1) | L0 (nm) |
|---|---|---|---|---|---|
| KOH activation | 25.5 | 1710 | 0.73 | 0.59 | 1.41 |
| Pyrolysis | 21.5 | 0 | 0 | n.d. | n.d. |
| Reaction Temperature (°C) | Burn-Off (wt%) | SBET (m2 g−1) | VT (cm3 g−1) | VDR (cm3 g−1) | L0 (nm) | VDR/VT |
|---|---|---|---|---|---|---|
| 600 | 21.6 | 1790 | 0.71 | 0.66 | 1.11 | 0.93 |
| 650 | 22.8 | 1960 | 0.76 | 0.71 | 1.18 | 0.93 |
| 700 | 22.7 | 2410 | 1.01 | 0.80 | 1.20 | 0.79 |
| 750 | 22.8 | 2500 | 1.04 | 0.82 | 1.21 | 0.79 |
| 800 | 27.5 | 2590 | 1.12 | 0.82 | 1.29 | 0.73 |
| 850 | 33.2 | 2710 | 1.18 | 0.84 | 1.36 | 0.71 |
| 900 | 45.3 | 2610 | 1.10 | 0.84 | 1.33 | 0.76 |
| Pre-Carbonization Temperature (°C) | Burn-Off (wt%) | SBET (m2 g−1) | L0 (nm) | VDR/VT |
|---|---|---|---|---|
| 520 | 34.5 | 3030 | 1.40 | 0.71 |
| 600 | 27.9 | 2550 | 1.34 | 0.82 |
| 700 | 24.9 | 2030 | 1.17 | 0.98 |
| 800 | 24.4 | 1550 | 0.94 | 0.91 |
| 900 | 26.2 | 1310 | 0.84 | 0.92 |
| 1000 | 18.6 | 990 | 0.79 | 0.92 |
| Pre-Carbonization Temperature (°C) | Burn-Off (wt%) | SBET (m2 g−1) | L0 (nm) | VDR/VT |
|---|---|---|---|---|
| 520 | 22.4 | 2640 | 1.36 | 0.76 |
| 600 | 18.8 | 1450 | 1.23 | 0.74 |
| 700 | 23.7 | 1410 | 1.06 | 0.81 |
| 800 | 17.0 | 190 | 2.53 | 0.55 |
| 900 | 16.2 | ~0 | n.d. | n.d. |
| 1000 | 9.3 | ~0 | n.d. | n.d. |
| Precursor | H3PO4 g/g Precursor | Temp. (°C) | Soaking Time (min) | SBET (m2 g−1) | Vmic (cm3 g−1) | Vmic/VT | Ref. |
|---|---|---|---|---|---|---|---|
| Birch | 3.1 | 480 | 20 | 1826 | 0.67 | 0.46 | [4] |
| Oak | 3.1 | 480 | 20 | 2553 | 0.94 | 0.59 | [4] |
| Hemp stem | 1.6 | 480 | 10 | 2215 | 0.859 | 0.52 | [9] |
| Corncob | 1 | 400 | 60 | 2071 | 0.78 | 0.69 | [81] |
| Eucalyptus residue | 2.5 | 400 | 180 | 1545 | 0.04 | 0.02 | [87] |
| Cotton stalks | 1.5 | 500 | 120 | 1720 | 0.71 | 0.80 | [93] |
| Olive stone | 1.3 | 400 | 120 | 1740 | 0.73 | 0.82 | [94] |
| Rice hull | 1.5 | 450 | - | 1295 | 0.46 | 0.63 | [95] |
| Sugar cane bagasse | 1.5 | 500 | 60 | 1132 | 0.61 | 0.54 | [96] |
| Precursor | g ZnCl2/g Precursor | Temp. (°C) | Soaking Time (min) | SBET (m2 g−1) | Vmic (cm3 g−1) | Vmic/VT | Ref. |
|---|---|---|---|---|---|---|---|
| Cherry stones | 3 | 700 | 120 | 1704 | 0.98 | 0.63 | [100] |
| Fox nut | 2 | 600 | 60 | 2869 | 1.68 | 0.86 | [101] |
| Cherry stones | 3 | 500 | 120 | 1566 | 0.65 | 0.81 | [104] |
| Macadamia nutshell | 1 | 500 | 60 | 1718 | 0.72 | 0.88 | [107] |
| Palm shell | 1.35 | 500 | 120 | 1672 | 0.87 | 0.88 | [112] |
| Chestnut shell | 2 | 800 | 90 | 1824 | 0.87 | 0.89 | [116] |
| Rice husk | 2 | 500 | 60 | 2434 | 0.59 | 0.44 | [121] |
| Sugar beet bagasse | 3 | 700 | 90 | 1826 | 0.71 | 0.74 | [122] |
| Waste coffee grounds | 1 | 850 | 60 | 1019 | 0.34 | 0.71 | [123] |
| Ion | Non-Solvated Ion Size (nm) | Solvated Ion Size (nm) | Solvation Free Energy (kJ mol−1) |
|---|---|---|---|
| Organic electrolyte | |||
| TEA+ | 0.67–0.75 | 1.30 (in acetonitrile) 1.74 (in propylene carbonate) | |
| BF4− | 0.48–0.50 | 1.16 (in acetonitrile) 1.54 (in propylene carbonate) | |
| TFSI− | 0.37 | 1.35 (in ethylene carbonate) | |
| Water-based electrolyte | |||
| Li+ | 0.15–0.18 | 0.68–0.76 | 515–544 |
| Na+ | 0.20–0.23 | 0.55–0.72 | 365–435 |
| K+ | 0.27–0.30 | 0.40–0.66 | 271–351 |
| NH4+ | 0.30–0.32 | 0.66 | |
| Mg2+ | 0.12–0.14 | 0.60–0.94 | 1828–1922 |
| Ca2+ | 0.20–0.25 | 0.82–0.84 | 1306–1650 |
| Cl− | 0.33–0.39 | 0.65–0.66 | 340–371 |
| I− | 0.41–0.43 | 0.60 | 254 |
| SO42− | 0.43–0.58 | 0.60–0.74 | 1145 |
| NO3− | 0.38 | 0.67–0.68 | 328 |
| ClO4− | 0.45–0.47 | 0.60 | 238 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kierzek, K.; Gryglewicz, G. Activated Carbons and Their Evaluation in Electric Double Layer Capacitors. Molecules 2020, 25, 4255. https://doi.org/10.3390/molecules25184255
Kierzek K, Gryglewicz G. Activated Carbons and Their Evaluation in Electric Double Layer Capacitors. Molecules. 2020; 25(18):4255. https://doi.org/10.3390/molecules25184255
Chicago/Turabian StyleKierzek, Krzysztof, and Grażyna Gryglewicz. 2020. "Activated Carbons and Their Evaluation in Electric Double Layer Capacitors" Molecules 25, no. 18: 4255. https://doi.org/10.3390/molecules25184255