
The Pathology of Parkinson’s Disease and Potential Benefit of Dietary Polyphenols





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Parkinson’s
1.1. Prevalence and Incidence of Parkinson’s Disease
1.2. Symptoms and Diagnostic Criteria
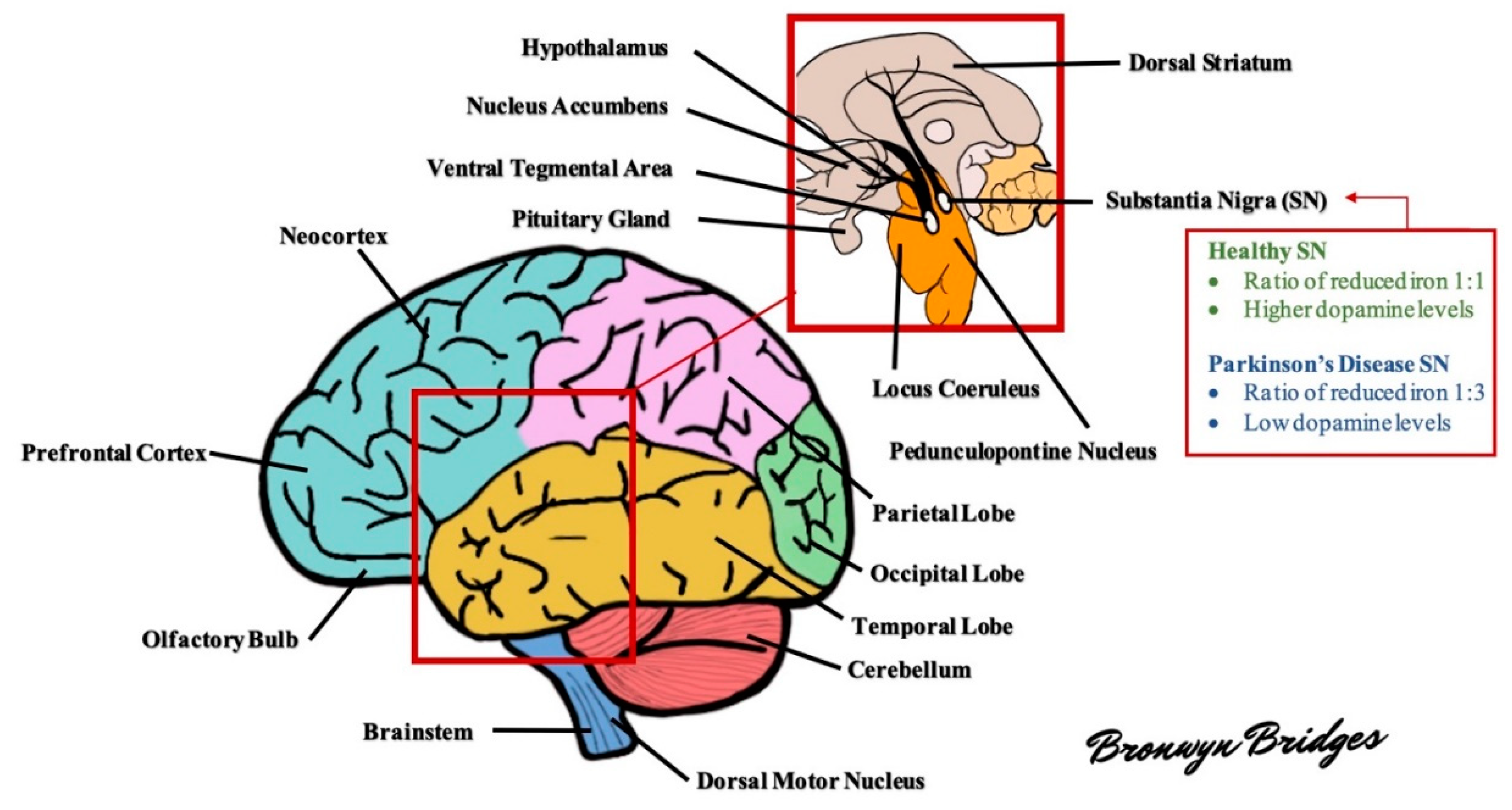
1.3. Pathology
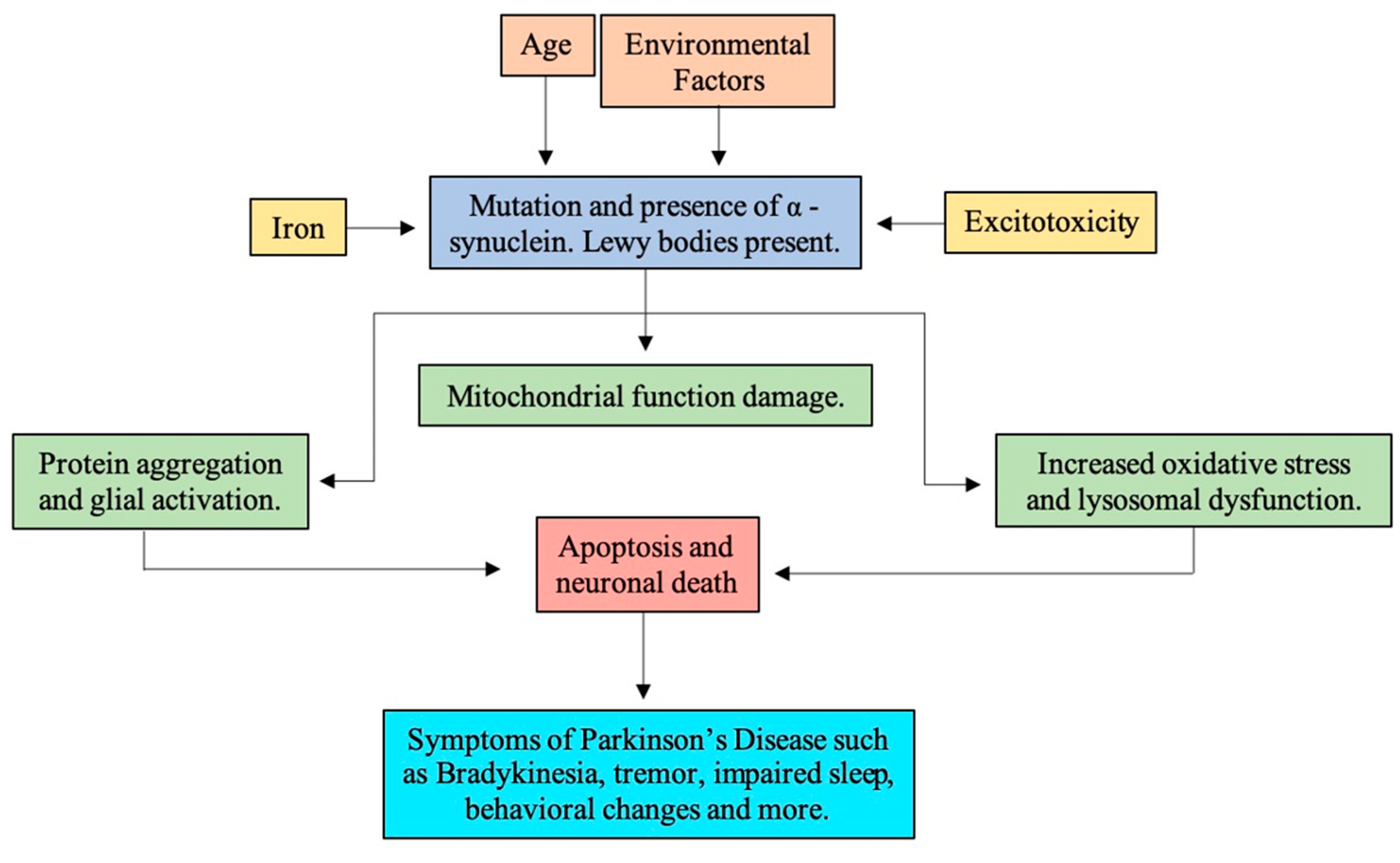
1.4. Pathogenesis
1.4.1. Reactive Oxidative Stress
- Neuronal NOS, localized in neurons, neutrophils, astrocytes.
- Endothelial NOS, present in vascular endothelium and astrocytes.
- Inducible NOS, expressed in macrophages, hepatocytes, glial cells, and neuronal cultures.
Dopamine
Iron
Neuromelanin
Lipids
Glutathione
1.4.2. Mitochondria Dysfunction
1.4.3. α-Synuclein
1.4.4. Neuroinflammation
1.5. Risk Factors
1.5.1. Genetic Risk Factors
1.5.2. Environmental Risk Factors
2. Polyphenols
2.1. Phenolic Acids
2.2. Flavonoids
2.3. Stilbenes
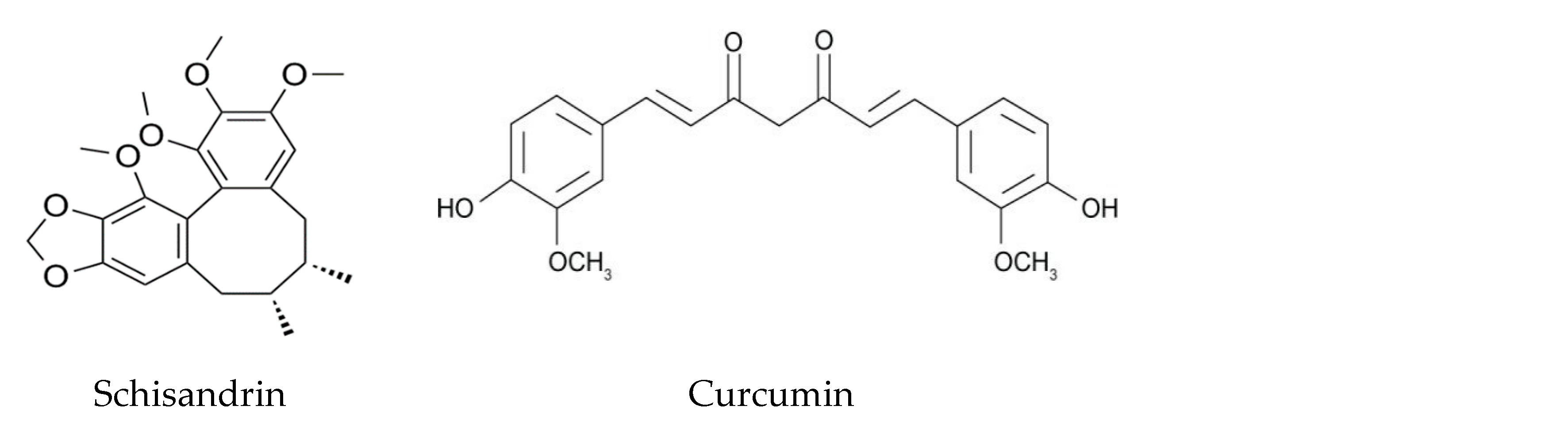
2.4. Lignans
3. Mechanisms of Neuroprotection
3.1. Phenolic Acids
3.2. Flavonoids
3.3. Stilbenoids
3.4. Lignans
3.5. Notable Polyphenols
4. Bioavailability
4.1. The Gut Microbiome and Brain Connection: Current Perspectives
4.2. Polyphenols and the Gut Microbiome
4.3. Polyphenols, Plasma, and the Brain
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, Present, and Future of Parkinson’s Disease: A Special Essay on the 200th Anniversary of the Shaking Palsy: The Shaking Palsy: Past, Present and Future. Mov. Disord. 2017, 32, 1264–1310. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Berg, D.; Postuma, R.B.; Adler, C.H.; Bloem, B.R.; Chan, P.; Dubois, B.; Gasser, T.; Goetz, C.G.; Halliday, G.; Joseph, L.; et al. MDS research criteria for prodromal Parkinson’s disease. Mov. Disord. 2015, 30, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Błaszczyk, J.W. Motor deficiency in Parkinson’s disease. Acta Neurobiol. Exp. 1998, 58, 79–93. [Google Scholar]
- Pfeiffer, R.F. Non-Motor Symptoms in Parkinson’s Disease. Parkinsonism Relat. Disord. 2016, 22, S119–S122. [Google Scholar] [CrossRef]
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, Regional, and National Age-Sex-Specific Mortality for 282 Causes of Death in 195 Countries and Territories, 1980–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, Regional, and National Burden of Parkinson’s Disease, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s Disease across North America. NPJ Parkinson’s Dis. 2018, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Kim, D.H.; Kwon, D.-Y.; Choi, M.; Kim, S.; Jung, J.-H.; Han, K.; Park, Y.-G. Trends in the incidence and prevalence of Parkinson’s disease in Korea: A nationwide, population-based study. BMC Geriatr. 2019, 19, 320. [Google Scholar] [CrossRef] [Green Version]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D.L. The Prevalence of Parkinson’s Disease: A Systematic Review and Meta-Analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Eusebi, P.; Franchini, D.; De Giorgi, M.; Abraha, I.; Montedori, A.; Casucci, P.; Calabresi, P.; Tambasco, N. Incidence and prevalence of Parkinson’s disease in the Italian region of Umbria: A population-based study using healthcare administrative databases. Neurol. Sci. 2019, 40, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Nerius, M.; Fink, A.; Doblhammer, G. Parkinson’s disease in Germany: Prevalence and incidence based on health claims data. Acta Neurol. Scand. 2016, 136, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.L.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Parkinson’s Disease and Parkinsonism: Neuropathology. Cold Spring Harb. Perspect. Med. 2012, 2, a009258. [Google Scholar] [CrossRef] [Green Version]
- Bridi, J.C.; Hirth, F. Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Mol. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Rüb, U.; De Vos, R.A.; Steur, E.N.J.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L.; Stoker, T.B.; Greenland, J.C. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Codon Publications: Brisbane, Australia, 2018; pp. 3–26. [Google Scholar]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G. Parkinson’s Disease Is Not Simply a Prion Disorder. J. Neurosci. 2017, 37, 9799–9807. [Google Scholar] [CrossRef] [Green Version]
- Giguère, N.; Nanni, S.B.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Kalia, L.V.; Brotchie, J.M.; Fox, S.H. Novel Nondopaminergic Targets for Motor Features of Parkinson’s Disease: Review of Recent Trials: Nondopaminergic Targets for Motor Features of PD. Mov. Disord. 2013, 28, 131–144. [Google Scholar] [CrossRef]
- Miller, D.B.; O’Callaghan, J.P. Biomarkers of Parkinson’s disease: Present and future. Metabosilm 2014, 64, S40–S46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Jenner, P. Oxidative Stress in Parkinson’s Disease. Ann. Neurol.: Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2003, 53, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Umeno, A.; Biju, V.; Yoshida, Y. In vivo ROS production and use of oxidative stress-derived biomarkers to detect the onset of diseases such as Alzheimer’s disease, Parkinson’s disease, and diabetes. Free Radic. Res. 2017, 51, 413–427. [Google Scholar] [CrossRef] [PubMed]
- Olguín, H.J.; Guzmán, D.C.; García, E.H.; Mejía, G.B. The Role of Dopamine and Its Dysfunction as a Consequence of Oxidative Stress. Oxidative Med. Cell. Longev. 2016, 2016, 9730467. [Google Scholar] [CrossRef] [Green Version]
- Perfeito, R.; Cunha-Oliveira, T.; Rego, A.C. Revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease—Resemblance to the effect of amphetamine drugs of abuse. Free Radic. Biol. Med. 2012, 53, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, S.; Haylett, W.L.; Johnson, G.; Carr, J.A.; Bardien, S. Antioxidant effects of curcumin in models of neurodegeneration, aging, oxidative and nitrosative stress: A review. Neuroscience 2019, 406, 1–21. [Google Scholar] [CrossRef]
- Kavya, R.; Saluja, R.; Singh, S.; Dikshit, M. Nitric oxide synthase regulation and diversity: Implications in Parkinson’s disease. Nitric Oxide 2006, 15, 280–294. [Google Scholar] [CrossRef]
- Weiss, B. Evidence for Mutagenesis by Nitric Oxide during Nitrate Metabolism in Escherichia coli. J. Bacteriol. 2006, 188, 829–833. [Google Scholar] [CrossRef] [Green Version]
- Maguire-Zeiss, K.A.; Short, D.W.; Federoff, H.J. Synuclein, dopamine and oxidative stress: Co-conspirators in Parkinson’s disease? Mol. Brain Res. 2005, 134, 18–23. [Google Scholar] [CrossRef]
- Hauser, D.N.; Hastings, T.G. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol. Dis. 2012, 51, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochman, A.; Kośka, C.; Metodiewa, D. Submolecular adventures of brain tyrosine: What are we searching for now? Amino Acids 2002, 23, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Dichtl, S.; Haschka, D.; Nairz, M.; Seifert, M.; Volani, C.; Lutz, O.; Weiss, G. Dopamine promotes cellular iron accumulation and oxidative stress responses in macrophages. Biochem. Pharmacol. 2018, 148, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Hofer, T.; Nurchi, V.; Aaseth, J. Iron and other metals in the pathogenesis of Parkinson’s disease: Toxic effects and possible detoxification. J. Inorg. Biochem. 2019, 199, 110717. [Google Scholar] [CrossRef] [PubMed]
- Mounsey, R.B.; Teismann, P. Chelators in the Treatment of Iron Accumulation in Parkinson’s Disease. Int. J. Cell Biol. 2012, 2012, 983245. [Google Scholar] [CrossRef] [Green Version]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Muñoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2015, 155, 96–119. [Google Scholar] [CrossRef]
- Smeyne, M.; Smeyne, R. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Zucca, F.A.; Basso, E.; Cupaioli, F.A.; Ferrari, E.; Sulzer, D.; Casella, L.; Zecca, L. Neuromelanin of the Human Substantia Nigra: An Update. Neurotox. Res. 2013, 25, 13–23. [Google Scholar] [CrossRef]
- Wakamatsu, K.; Nakao, K.; Tanaka, H.; Kitahori, Y.; Tanaka, Y.; Ojika, M.; Ito, S. The Oxidative Pathway to Dopamine-Protein Conjugates and Their Pro-Oxidant Activities: Implications for the Neurodegeneration of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 2575. [Google Scholar] [CrossRef] [Green Version]
- Voshavar, C.; Shah, M.; Xu, L.; Dutta, A.K. Assessment of Protective Role of Multifunctional Dopamine Agonist D-512 Against Oxidative Stress Produced by Depletion of Glutathione in PC12 Cells: Implication in Neuroprotective Therapy for Parkinson’s Disease. Neurotox. Res. 2015, 28, 302–318. [Google Scholar] [CrossRef]
- Genestra, M. Oxyl radicals, redox-sensitive signalling cascades and antioxidants. Cell. Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Kaidery, N.A.; Thomas, B. Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem. Int. 2018, 117, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Puspita, L.; Chung, S.Y.; Shim, J.-W. Oxidative stress and cellular pathologies in Parkinson’s disease. Mol. Brain 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Bose, A.; Beal, M.F. Mitochondrial Dysfunction in Parkinson’s Disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic Human iPSC Parkinson’s Model Shows Nitrosative Stress-Induced Dysfunction in MEF2-PGC1α Transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta (BBA)—Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K. α-Synuclein and Lewy pathology in Parkinson’s disease. Curr. Opin. Neurol. 2015, 28, 375–381. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Edwards, R.H. The physiological role of α-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.L.; Etherton, M.R.; Südhof, T.C. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; Van Der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.J.; Flood, P.M. Oxidative Stress and Microglial Cells in Parkinson’s Disease. Mediat. Inflamm. 2012, 2012, 401264. [Google Scholar] [CrossRef] [Green Version]
- Schetters, S.T.T.; Gomez-Nicola, D.; Garcia-Vallejo, J.J.; Van Kooyk, Y. Neuroinflammation: Microglia and T Cells Get Ready to Tango. Front. Immunol. 2018, 8, 1905. [Google Scholar] [CrossRef] [Green Version]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2008, 119, 182–192. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.P.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. The relation between α-synuclein and microglia in Parkinson’s disease: Recent developments. Neuroscience 2015, 302, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Grozdanov, V.; Bousset, L.; Hoffmeister, M.; Bliederhaeuser, C.; Meier, C.; Madiona, K.; Pieri, L.; Kiechle, M.; McLean, P.J.; Kassubek, J.; et al. Increased Immune Activation by Pathologic α-Synuclein in Parkinson’s Disease. Ann. Neurol. 2019, 86, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilms, H.; Zecca, L.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Lucius, R. Inflammation in Parkinson’s diseases and other neurodegenerative diseases: Cause and therapeutic implications. Curr. Pharm. Des. 2007, 13, 1925–1928. [Google Scholar] [CrossRef]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Schapira, A.H.V. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delamarre, A.; Meissner, W.G. Epidemiology, Environmental Risk Factors and Genetics of Parkinson’s Disease. La Presse Med. 2017, 46, 175–181. [Google Scholar] [CrossRef]
- Cruces-Sande, A.; Rodríguez-Pérez, A.I.; Herbello-Hermelo, P.; Bermejo-Barrera, P.; Mendez-Alvarez, E.; Labandeira-García, J.L.; Soto-Otero, R. Copper Increases Brain Oxidative Stress and Enhances the Ability of 6-Hydroxydopamine to Cause Dopaminergic Degeneration in a Rat Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 56, 2845–2854. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.P.; Chen, J.; Chai, Z.F.; Hu, Y. The neurotoxicity of iron, copper and cobalt in Parkinson’s disease through ROS-mediated mechanisms. BioMetals 2016, 29, 665–678. [Google Scholar] [CrossRef]
- Martins-Junior, A.D.C.; Morcillo, P.; Ijomone, O.M.; Venkataramani, V.; Harrison, F.E.; Lee, E.-S.; Bowman, A.B.; Aschner, M. New Insights on the Role of Manganese in Alzheimer’s Disease and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 3546. [Google Scholar] [CrossRef]
- Caudle, W.M.; Guillot, T.S.; Lazo, C.R.; Miller, G.W. Industrial toxicants and Parkinson’s disease. NeuroToxicology 2012, 33, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Weuve, J.; Press, D.Z.; Grodstein, F.; Wright, R.O.; Hu, H.; Weisskopf, M.G. Cumulative Exposure to Lead and Cognition in Persons with Parkinson’s Disease: Exposure to Lead and Cognition in PD. Mov. Disord. 2013, 28, 176–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochmanski, J.; E VanOeveren, S.; Patterson, J.R.; Bernstein, A.I. Developmental Dieldrin Exposure Alters DNA Methylation at Genes Related to Dopaminergic Neuron Development and Parkinson’s Disease in Mouse Midbrain. Toxicol. Sci. 2019, 169, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Radad, K.; Al-Shraim, M.; Al-Emam, A.; Wang, F.; Kranner, B.; Rausch, W.-D.; Moldzio, R. Rotenone: From modelling to implication in Parkinson’s disease. Folia Neuropathol. 2019, 57, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Singla, R.K.; Dubey, A.K.; Garg, A.; Sharma, R.K.; Fiorino, M.; Ameen, S.M.; Haddad, M.A.; Al-Hiary, M. Natural Polyphenols: Chemical Classification, Definition of Classes, Subcategories, and Structures. J. AOAC Int. 2019, 102, 1397–1400. [Google Scholar] [CrossRef]
- Scalbert, A.; Williamson, G. Dietary intake and bioavailability of polyphenols. J. Nutr. 2000, 130, 2073S–2085S. [Google Scholar] [CrossRef]
- Han, X.; Shen, T.; Lou, H.-X. Dietary Polyphenols and Their Biological Significance. Int. J. Mol. Sci. 2007, 8, 950–988. [Google Scholar] [CrossRef] [Green Version]
- González-Vallinas, M.; González-Castejón, M.; Rodríguez-Casado, A.; Molina, A.R.-D. Dietary phytochemicals in cancer prevention and therapy: A complementary approach with promising perspectives. Nutr. Rev. 2013, 71, 585–599. [Google Scholar] [CrossRef]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jiménez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Santos-Buelga, C.; Feliciano, A.S. Flavonoids: From Structure to Health Issues. Molecules 2017, 22, 477. [Google Scholar] [CrossRef]
- Zakaryan, H.; Arabyan, E.; Oo, A.; Zandi, K. Flavonoids: Promising natural compounds against viral infections. Arch. Virol. 2017, 162, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Kasiotis, K.M.; Pratsinis, H.; Kletsas, D.; Haroutounian, S.A. Resveratrol and related stilbenes: Their anti-aging and anti-angiogenic properties. Food Chem. Toxicol. 2013, 61, 112–120. [Google Scholar] [CrossRef]
- Durazzo, A.; Lucarini, M.; Camilli, E.; Marconi, S.; Gabrielli, P.; Lisciani, S.; Gambelli, L.; Aguzzi, A.; Novellino, E.; Santini, A.; et al. Dietary Lignans: Definition, Description and Research Trends in Databases Development. Molecules 2018, 23, 3251. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.-Y.; Chen, S.-L.; Yang, M.; Wu, J.; Sinkkonen, J.; Zou, K. An update on lignans: Natural products and synthesis. Nat. Prod. Rep. 2009, 26, 1251–1292. [Google Scholar] [CrossRef]
- Langston, J.W. The MPTP Story. J. Park. Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchet, J.; Longpre, F.; Bureau, G.; Morissette, M.; DiPaolo, T.; Bronchti, G.; Martinoli, M.-G. Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-S.; Heng, Y.; Mou, Z.; Huang, J.-Y.; Yuan, Y.-H.; Chen, N.-H. Reassessment of subacute MPTP-treated mice as animal model of Parkinson’s disease. Acta Pharmacol. Sin. 2017, 38, 1317–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tieu, K. A Guide to Neurotoxic Animal Models of Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef] [PubMed]
- Ciulla, M.; Marinelli, L.; Cacciatore, I.; Di Stefano, A.; Stefano, A. Role of Dietary Supplements in the Management of Parkinson’s Disease. Biomolecules 2019, 9, 271. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Tripathi, P.; Yadawa, A.K.; Singh, S. Promising Polyphenols in Parkinson’s Disease Therapeutics. Neurochem. Res. 2020, 45, 1731–1745. [Google Scholar] [CrossRef]
- Park, H.-A.; Ellis, A. Dietary Antioxidants and Parkinson’s Disease. Antioxidants 2020, 9, 570. [Google Scholar] [CrossRef]
- Leri, M.; Scuto, M.; Ontario, M.L.; Calabrese, V.; Calabrese, V.; Bucciantini, M.; Stefani, M. Calabrese Healthy Effects of Plant Polyphenols: Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, M.; Rigacci, S. Beneficial properties of natural phenols: Highlight on protection against pathological conditions associated with amyloid aggregation. BioFactors 2014, 40, 482–493. [Google Scholar] [CrossRef]
- Amro, M.S.; Teoh, S.L.; Norzana, A.G.; Srijit, D. The potential role of herbal products in the treatment of Parkinson’s disease. La Clin. Ter. 2018, 169, e23–e33. [Google Scholar]
- Liu, H.; Zhang, W.; Luo, X.; Ye, Y.; Zhu, X. Paeoniflorin Attenuates Neuroinflammation and Dopaminergic Neurodegeneration in the MPTP Model of Parkinson’s Disease by Activation of Adenosine A1 Receptor. Br. J. Pharmacol. 2009, 148, 314–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.-Z.; Zhu, J.; Jin, D.-Z.; Zhang, L.; Ji, X.-Q.; Ye, Y.; Tang, C.-P.; Zhu, X.-Z. Behavioral recovery following sub-chronic paeoniflorin administration in the striatal 6-OHDA lesion rodent model of Parkinson’s disease. J. Ethnopharmacol. 2007, 112, 327–332. [Google Scholar] [CrossRef]
- Cao, B.-Y.; Yang, Y.-P.; Luo, W.-F.; Mao, C.-J.; Han, R.; Sun, X.; Cheng, J.; Liu, C.-F. Paeoniflorin, a potent natural compound, protects PC12 cells from MPP+ and acidic damage via autophagic pathway. J. Ethnopharmacol. 2010, 131, 122–129. [Google Scholar] [CrossRef]
- Lu, Z.; Nie, G.; Belton, P.S.; Tang, H.; Zhao, B. Structure–activity relationship analysis of antioxidant ability and neuroprotective effect of gallic acid derivatives. Neurochem. Int. 2006, 48, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Cassidy, A.; Schwarzschild, M.A.; Rimm, E.B.; Ascherio, A. Habitual intake of dietary flavonoids and risk of Parkinson disease. Neurology 2012, 78, 1138–1145. [Google Scholar] [CrossRef] [Green Version]
- Debnath-Canning, M.; Unruh, S.; Vyas, P.; Daneshtalab, N.; Igamberdiev, A.U.; Weber, J.T. Fruits and leaves from wild blueberry plants contain diverse polyphenols and decrease neuroinflammatory responses in microglia. J. Funct. Foods 2020, 68, 103906. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Madathil, S.K.; Pandey, M.; Haobam, R.; Rajamma, U.; Mohanakumar, K.P. Quercetin up-regulates mitochondrial complex-I activity to protect against programmed cell death in rotenone model of Parkinson’s disease in rats. Neuroscience 2013, 236, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Shi, C.; Cao, J.; Sun, Y.; Zhao, X.; Guo, Y.; Wang, C.; Lei, H.; Jiang, H.; Ablat, N.; et al. Neuroprotective Effects of a Standardized Flavonoid Extract of Safflower against Neurotoxin-Induced Cellular and Animal Models of Parkinson’s Disease. Sci. Rep. 2016, 6, 22135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, W.; Fan, L.; Kim, Y.-C.; Ishikawa, S.; Iguchi-Ariga, S.M.; Pu, X.-P.; Ariga, H. Kaempferol Derivatives Prevent Oxidative Stress—Induced Cell Death in a DJ-1—Dependent Manner. J. Pharmacol. Sci. 2009, 110, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.T.; Ock, J.; Kwon, B.-M.; Suk, K. Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int. Immunopharmacol. 2008, 8, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.S.; Ahn, T.-B. The role of quercetin on the survival of neuron-like PC12 cells and the expression of α-synuclein. Neural Regen. Res. 2015, 10, 1113. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Quercetin, not caffeine, is a major neuroprotective component in coffee. Neurobiol. Aging 2016, 46, 113–123. [Google Scholar] [CrossRef]
- Maher, P. Protective effects of fisetin and other berry flavonoids in Parkinson’s disease. Food Funct. 2017, 8, 3033–3042. [Google Scholar] [CrossRef]
- Zenkov, N.; Chechushkov, A.V.; Kozhin, P.M.; Kandalintseva, N.V.; Martinovich, G.G.; Menshchikova, E. Plant phenols and autophagy. Biochemistry (Mosc.) 2016, 81, 297–314. [Google Scholar] [CrossRef]
- Tu, W.; Wang, H.; Li, S.; Liu, Q.; Sha, H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of The Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019, 10, 637. [Google Scholar] [CrossRef] [Green Version]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-κB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2019, 21, 101059. [Google Scholar] [CrossRef]
- Ren, H.; Hao, J.; Liu, T.; Zhang, N.; Lv, H.; Song, E.; Zhu, C. Hesperetin Suppresses Inflammatory Responses in Lipopolysaccharide-Induced RAW 264.7 Cells via the Inhibition of NF-κB and Activation of Nrf2/HO-1 Pathways. Inflammation 2016, 39. [Google Scholar] [CrossRef]
- Cui, B.; Zhang, S.; Wang, Y.; Guo, Y. Farrerol Attenuates Β-Amyloid-Induced Oxidative Stress and Inflammation Through Nrf2/Keap1 Pathway in A Microglia Cell Line. Biomed. Pharmacother. 2019, 109, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, H.; Ding, K.; Zhang, L.; Wang, C.; Li, T.; Wei, W.; Lu, X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2–ARE pathway. Free Radic. Biol. Med. 2014, 71, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.P.; Jain, P.D.; Sancheti, J.S.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective and neurotrophic effects of Apigenin and Luteolin in MPTP induced parkinsonism in mice. Neuropharmacology 2014, 86, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Sagara, Y.; Vanhnasy, J.; Maher, P. Induction of PC12 cell differentiation by flavonoids is dependent upon extracellular signal-regulated kinase activation. J. Neurochem. 2004, 90, 1144–1155. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, A.; Schluesener, H. Natural polyphenols against neurodegenerative disorders: Potentials and pitfalls. Ageing Res. Rev. 2012, 11, 329–345. [Google Scholar] [CrossRef]
- Caruana, M.; Cauchi, R.; Vassallo, N. Putative Role of Red Wine Polyphenols against Brain Pathology in Alzheimer’s and Parkinson’s Disease. Front. Nutr. 2016, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Wu, Q.; Lu, Y.; Gong, Q.; Shi, J. Neuroprotective Effect of Resveratrol on 6-OHDA-Induced Parkinson’s Disease in Rats. Eur. J. Pharmacol. 2008, 600, 78–82. [Google Scholar] [CrossRef]
- Ramassamy, C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: A review of their intracellular targets. Eur. J. Pharmacol. 2006, 545, 51–64. [Google Scholar] [CrossRef]
- Jang, J.; Surh, Y. Protective Effect of Resveratrol on Β-Amyloid-Induced Oxidative PC12 Cell Death. Free Radic. Biol. Med. 2003, 34, 1100–1110. [Google Scholar] [CrossRef]
- Abolaji, A.O.; Adedara, A.O.; Adie, M.A.; Vicente-Crespo, M.; Farombi, E.O. Resveratrol prolongs lifespan and improves 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced oxidative damage and behavioural deficits in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2018, 503, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-Y.; Hsieh, M.-T.; Wu, C.-R.; Tsai, F.-H.; Lu, T.-C.; Hsieh, C.-C.; Li, W.-C.; Lin, Y.-T.; Peng, W.-H. Schizandrin protects primary cultures of rat cortical cells from glutamate-induced excitotoxicity. J. Pharmacol. Sci. 2008, 107, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Zhi, Y.; Jin, Y.; Pan, L.; Zhang, A.; Liu, F. Schisandrin A ameliorates MPTP-induced Parkinson’s disease in a mouse model via regulation of brain autophagy. Arch. Pharmacal Res. 2019, 42, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Watzlawik, J.O.; Fiesel, F.C.; Springer, W. Autophagy in Parkinson’s Disease. J. Mol. Biol. 2020, 432, 2651–2672. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, C.; Siani, F.; Mus, L.; Ghezzi, C.; Cerri, S.; Pacchetti, B.; Bigogno, C.; Blandini, F. Neuroprotective Effects of Lignan 7-Hydroxymatairesinol (HMR/Lignan) in A Rodent Model of Parkinson’s Disease. Nutrition 2020, 69, 110494. [Google Scholar] [CrossRef]
- Lahaie-Collins, V.; Bournival, J.; Plouffe, M.; Carange, J.; Martinoli, M.-G. Sesamin modulates tyrosine hydroxylase, superoxide dismutase, catalase, inducible NO synthase and interleukin-6 expression in dopaminergic cells under MPP+-induced oxidative stress. Oxidative Med. Cell. Longev. 2009, 1, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef]
- Murugaiyah, V.; Mattson, M.P. Neurohormetic phytochemicals: An evolutionary-bioenergetic perspective. Neurochem. Int. 2015, 89, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Banerjee, S.; Sil, P.C. The beneficial role of curcumin on inflammation, diabetes and neurodegenerative disease: A recent update. Food Chem. Toxicol. 2015, 83, 111–124. [Google Scholar] [CrossRef]
- Ghasemi, F.; Bagheri, H.; Barreto, G.E.; Read, M.I.; Sahebkar, A. Effects of Curcumin on Microglial Cells. Neurotox. Res. 2019, 36, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.; Yoshioka, Y.; Terao, J. Role of Intestinal Microbiota in the Bioavailability and Physiological Functions of Dietary Polyphenols. Molecules 2019, 24, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raval, U.; Harary, J.; Zeng, E.; Pasinetti, G.M. The dichotomous role of the gut microbiome in exacerbating and ameliorating neurodegenerative disorders. Expert Rev. Neurother. 2020, 20, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; De Angelis, A.L.H.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakõiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Perez-Pardo, P.; Kliest, T.; Dodiya, H.; Broersen, L.; Garssen, J.; Keshavarzian, A.; Kraneveld, A. The Gut-Brain Axis in Parkinson’s Disease: Possibilities for Food-Based Therapies. Eur. J. Pharmacol. 2017, 817, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- Collins, S.M.; Surette, M.; Bercik, P. The interplay between the intestinal microbiota and the brain. Nat. Rev. Genet. 2012, 10, 735–742. [Google Scholar] [CrossRef]
- Heijtz, R.D.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [Green Version]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Toth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Dinan, T.G.; Cryan, J.F. Mood by microbe: Towards clinical translation. Genome Med. 2016, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Tillisch, K.; Labus, J.; Kilpatrick, L.; Jiang, Z.; Stains, J.; Ebrat, B.; Guyonnet, D.; Legrain–Raspaud, S.; Trotin, B.; Naliboff, B.; et al. Consumption of Fermented Milk Product with Probiotic Modulates Brain Activity. Gastroenterology 2013, 144, 1394–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, N.; Arboleya, S.; Valdés, L.; Stanton, C.; Ross, R.P.; Ruiz, L.; Gueimonde, M.; Reyes-Gavilán, C.G.D.L. The human intestinal microbiome at extreme ages of life. Dietary intervention as a way to counteract alterations. Front. Genet. 2014, 5, 406. [Google Scholar] [CrossRef] [PubMed]
- Salazar, N.; Lopez, P.; Valdés, L.; Margolles, A.; Suárez, A.; Patterson, Á.M.; Cuervo, A.; Reyes-Gavilán, C.G.D.L.; Ruas-Madiedo, P.; González, S.; et al. Microbial Targets for the Development of Functional Foods Accordingly with Nutritional and Immune Parameters Altered in the Elderly. J. Am. Coll. Nutr. 2013, 32, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.; Thuret, S. Gut Microbiota: A Modulator of Brain Plasticity and Cognitive Function in Ageing. Healthcare 2015, 3, 898–916. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.P.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Varannes, S.B.D.; Naveilhan, P.; Nguyên, J.-M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Serra, D.; Almeida, L.M.; Dinis, T.C. Polyphenols in the management of brain disorders: Modulation of the microbiota-gut-brain axis. In Advances in Food and Nutrition Research; Academic Press: Cambridge, UK, 2019; pp. 1–27. [Google Scholar]
- Scheperjans, F.; Aho, V.; Pereira, P.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2014, 30, 350–358. [Google Scholar] [CrossRef]
- Marín, L.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Bioavailability of Dietary Polyphenols and Gut Microbiota Metabolism: Antimicrobial Properties. BioMed Res. Int. 2015, 2015, 905215. [Google Scholar] [CrossRef] [Green Version]
- Cardona, F.; Andres-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [Green Version]
- Crozier, A.; Del Rio, D.; Clifford, M.N. Bioavailability of dietary flavonoids and phenolic compounds. Mol. Asp. Med. 2010, 31, 446–467. [Google Scholar] [CrossRef]
- Dolara, P.; Luceri, C.; De Filippo, C.; Femia, A.P.; Giovannelli, L.; Caderni, G.; Cecchini, C.; Silvi, S.; Orpianesi, C.; Cresci, A. Red wine polyphenols influence carcinogenesis, intestinal microflora, oxidative damage and gene expression profiles of colonic mucosa in F344 rats. Mutat. Res. Mol. Mech. Mutagen. 2005, 591, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Penders, J.; Lucchesi, M.; Goossens, G.H.; E Blaak, E. Gut microbiota composition in relation to the metabolic response to 12-week combined polyphenol supplementation in overweight men and women. Eur. J. Clin. Nutr. 2017, 71, 1040–1045. [Google Scholar] [CrossRef]
- Ito, H.; Gonthier, M.-P.; Manach, C.; Morand, C.; Mennen, L.; Rémésy, C.; Scalbert, A. Polyphenol levels in human urine after intake of six different polyphenol-rich beverages. Br. J. Nutr. 2005, 94, 500–509. [Google Scholar] [CrossRef]
- Olthof, M.R.; Hollman, P.C.; Katan, M.B. Chlorogenic acid and caffeic acid are absorbed in humans. J. Nutr. 2001, 131, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourne, L.C.; Rice-Evans, C. Bioavailability of Ferulic Acid. Biochem. Biophys. Res. Commun. 1998, 253, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, M.; Morand, C.; Mathevon, T.; Besson, C.; Manach, C.; Scalbert, A.; Remesy, C. The bioavailability of polyphenols is highly governed by the capacity of the intestine and of the liver to secrete conjugated metabolites. Eur. J. Nutr. 2005, 45, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [Green Version]
- Turner, R.S.; Thomas, R.G.; Craft, S.; Van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S.; et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Bowtell, J.L.; Bakkar, Z.A.; Conway, M.E.; Adlam, A.; Fulford, J. Enhanced task-related brain activation and resting perfusion in healthy older adults after chronic blueberry supplementation. Appl. Physiol. Nutr. Metab. 2017, 42, 773–779. [Google Scholar] [CrossRef]
- Jones, M.B.; Stuart, R.C.; Okello, E.J.; Watson, A. Cognitive and mood improvements following acute supplementation with purple grape juice in healthy young adults. Eur. J. Nutr. 2017, 56, 2621–2631. [Google Scholar] [CrossRef] [Green Version]
- Ullmann, U.; Haller, J.; Decourt, J.; Girault, N.; Girault, J.; Richard-Caudron, A.; Pineau, B.; Weber, P. A Single Ascending Dose Study of Epigallocatechin Gallate in Healthy Volunteers. J. Int. Med. Res. 2003, 31, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Erlund, I.; Kosonen, T.; Alfthan, G.; Mäenpää, J.; Perttunen, K.; Kenraali, J.; Parantainen, J.; Aro, A. Pharmacokinetics of quercetin from quercetin aglycone and rutin in healthy volunteers. Eur. J. Clin. Pharmacol. 2000, 56, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-H.; Nakata, R.; Oshima, S.; Inakuma, T.; Terao, J. Accumulation of quercetin conjugates in blood plasma after the short-term ingestion of onion by women. Am. J. Physiol. Integr. Comp. Physiol. 2000, 279, R461–R467. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aryal, S.; Skinner, T.; Bridges, B.; Weber, J.T. The Pathology of Parkinson’s Disease and Potential Benefit of Dietary Polyphenols. Molecules 2020, 25, 4382. https://doi.org/10.3390/molecules25194382
Aryal S, Skinner T, Bridges B, Weber JT. The Pathology of Parkinson’s Disease and Potential Benefit of Dietary Polyphenols. Molecules. 2020; 25(19):4382. https://doi.org/10.3390/molecules25194382
Chicago/Turabian StyleAryal, Sunisha, Taylor Skinner, Bronwyn Bridges, and John T. Weber. 2020. "The Pathology of Parkinson’s Disease and Potential Benefit of Dietary Polyphenols" Molecules 25, no. 19: 4382. https://doi.org/10.3390/molecules25194382
APA StyleAryal, S., Skinner, T., Bridges, B., & Weber, J. T. (2020). The Pathology of Parkinson’s Disease and Potential Benefit of Dietary Polyphenols. Molecules, 25(19), 4382. https://doi.org/10.3390/molecules25194382