
Solvent Deuterium Oxide Isotope Effects on the Reactions of Organophosphorylated Acetylcholinesterase †


Abstract
1. Introduction
2. Results
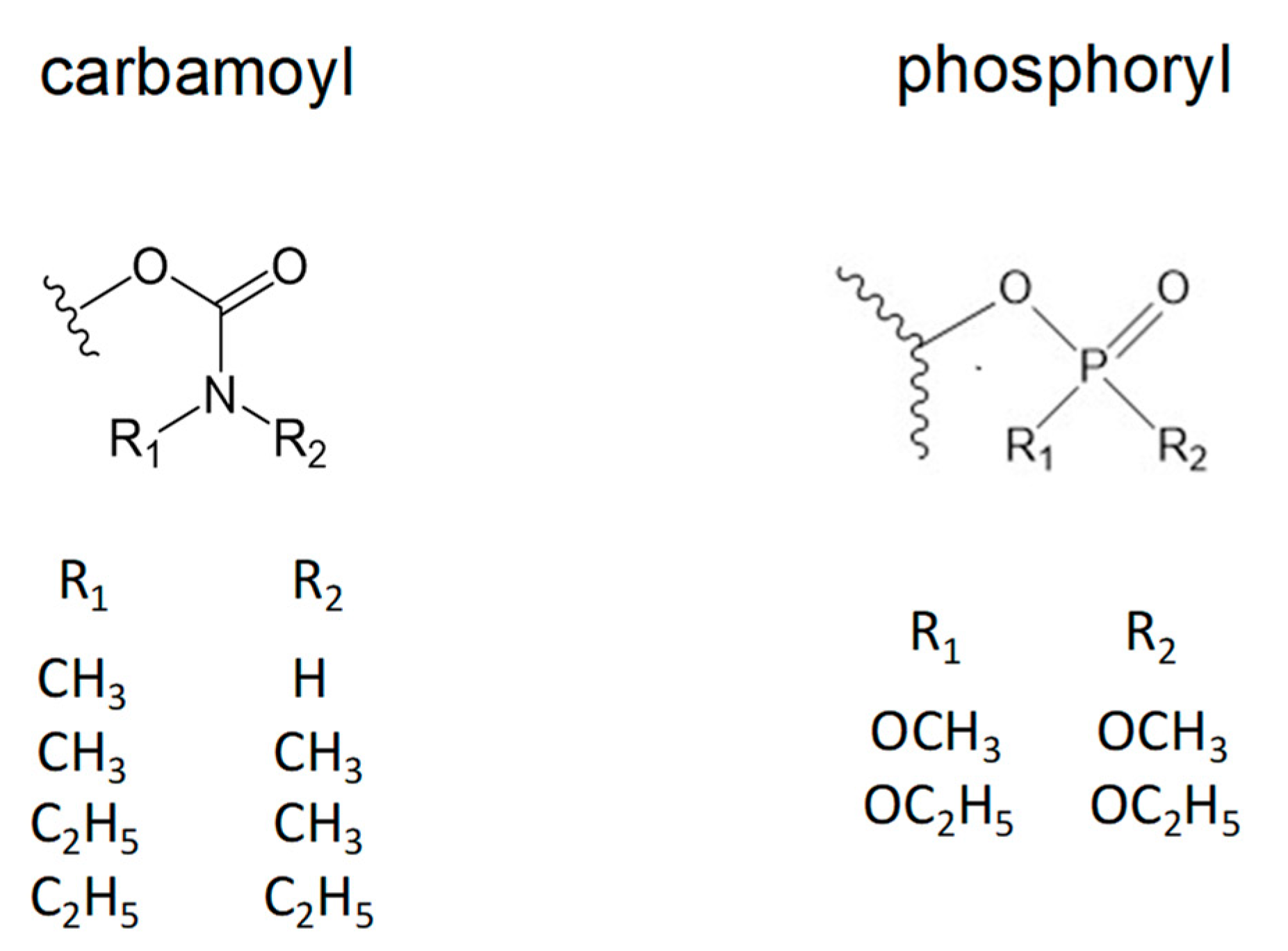
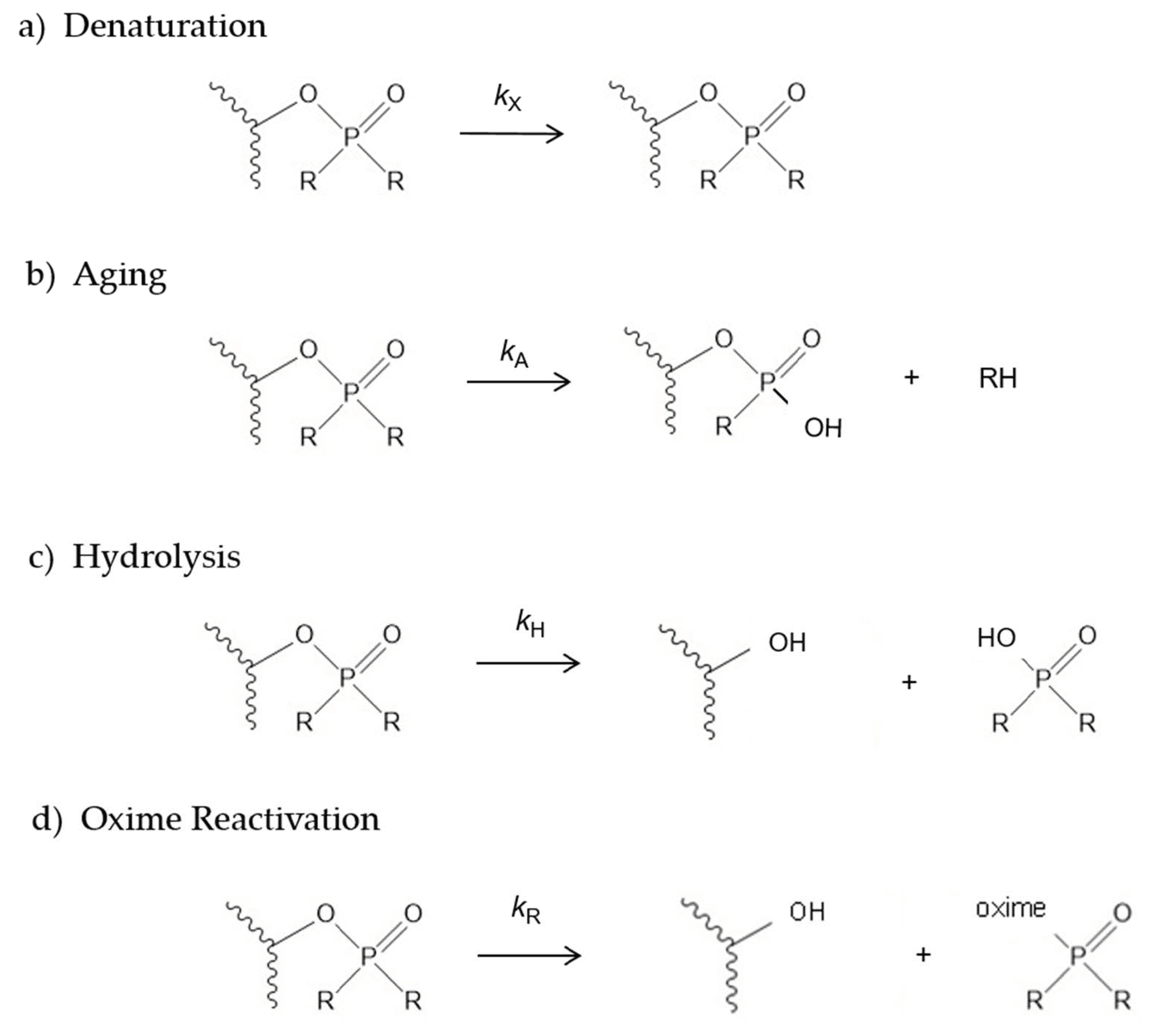
2.1. Denaturation Rate Constants kX for Organophosphorylated AChE
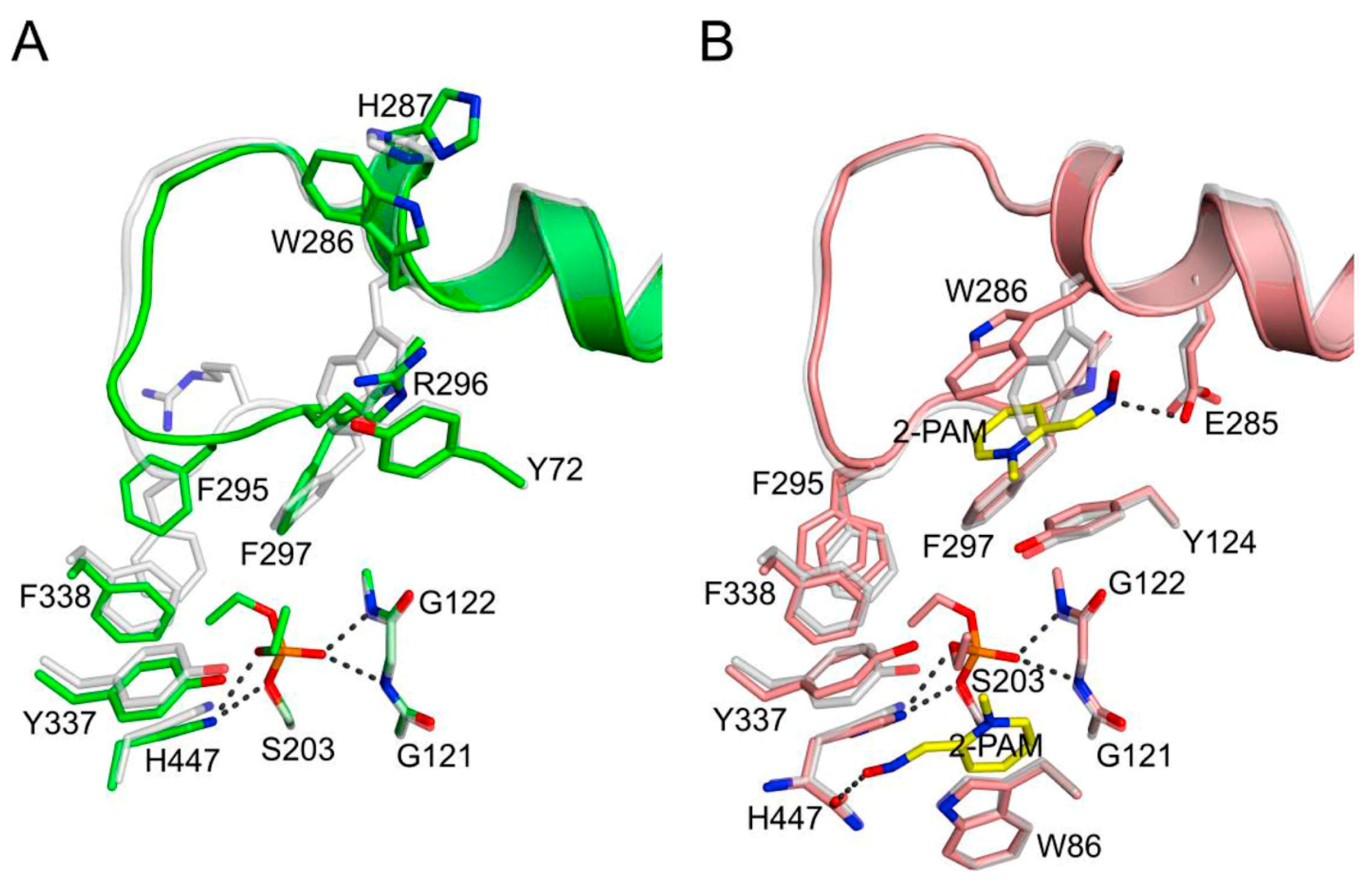
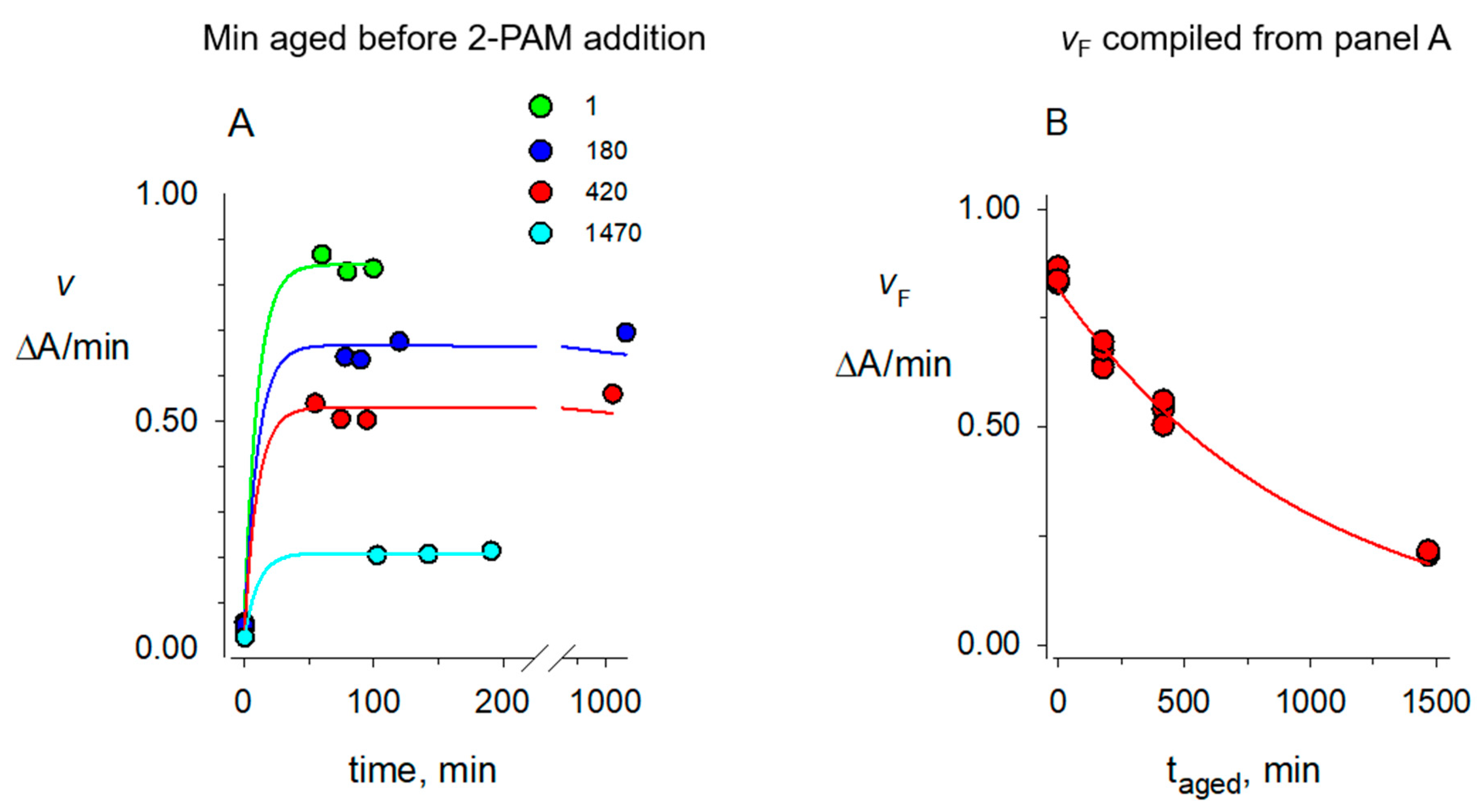
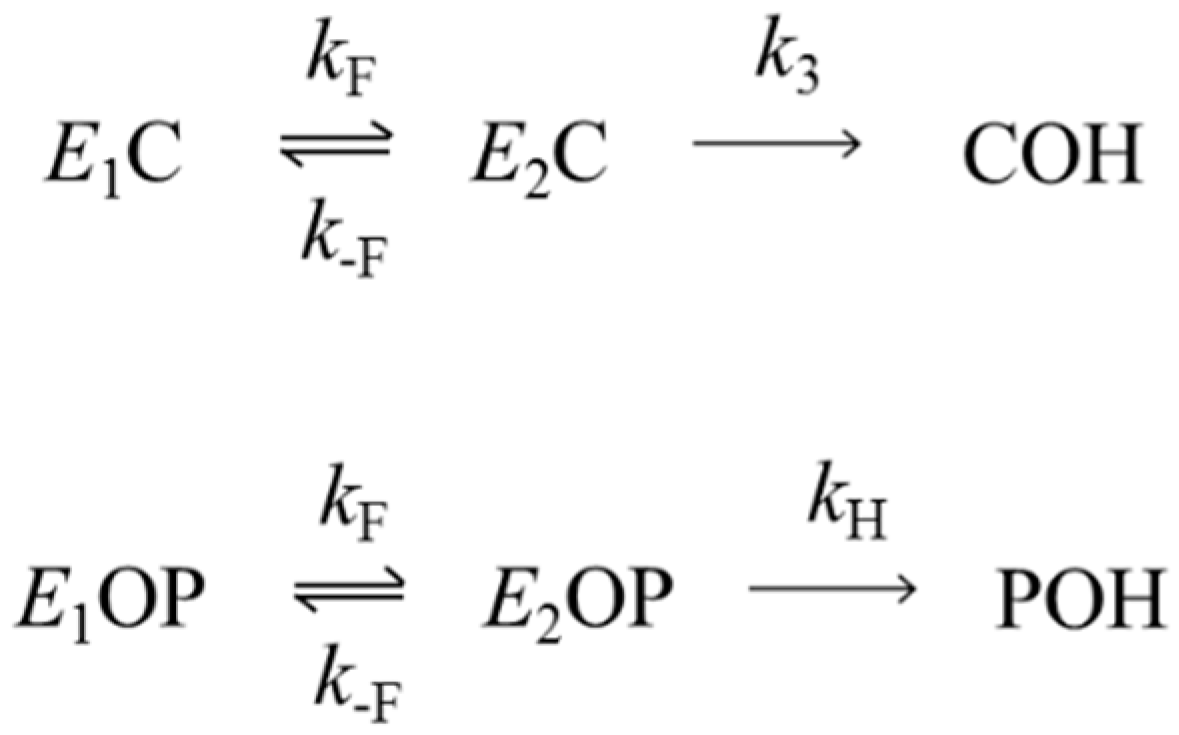
2.2. Aging Rate Constants kA for Organophosphorylated AChE
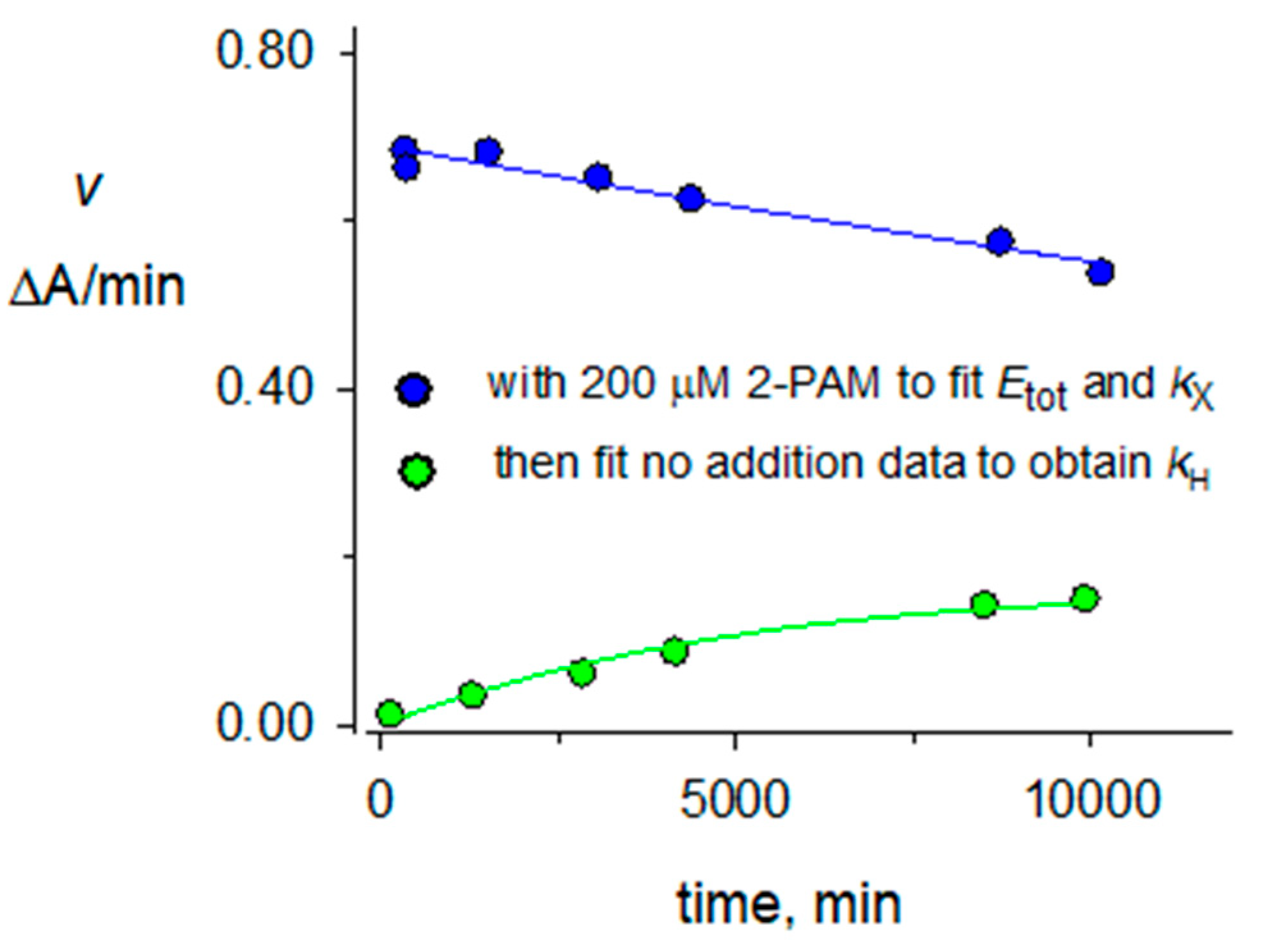
2.3. Hydrolysis Rate Constants kH for Organophosphorylated AChE
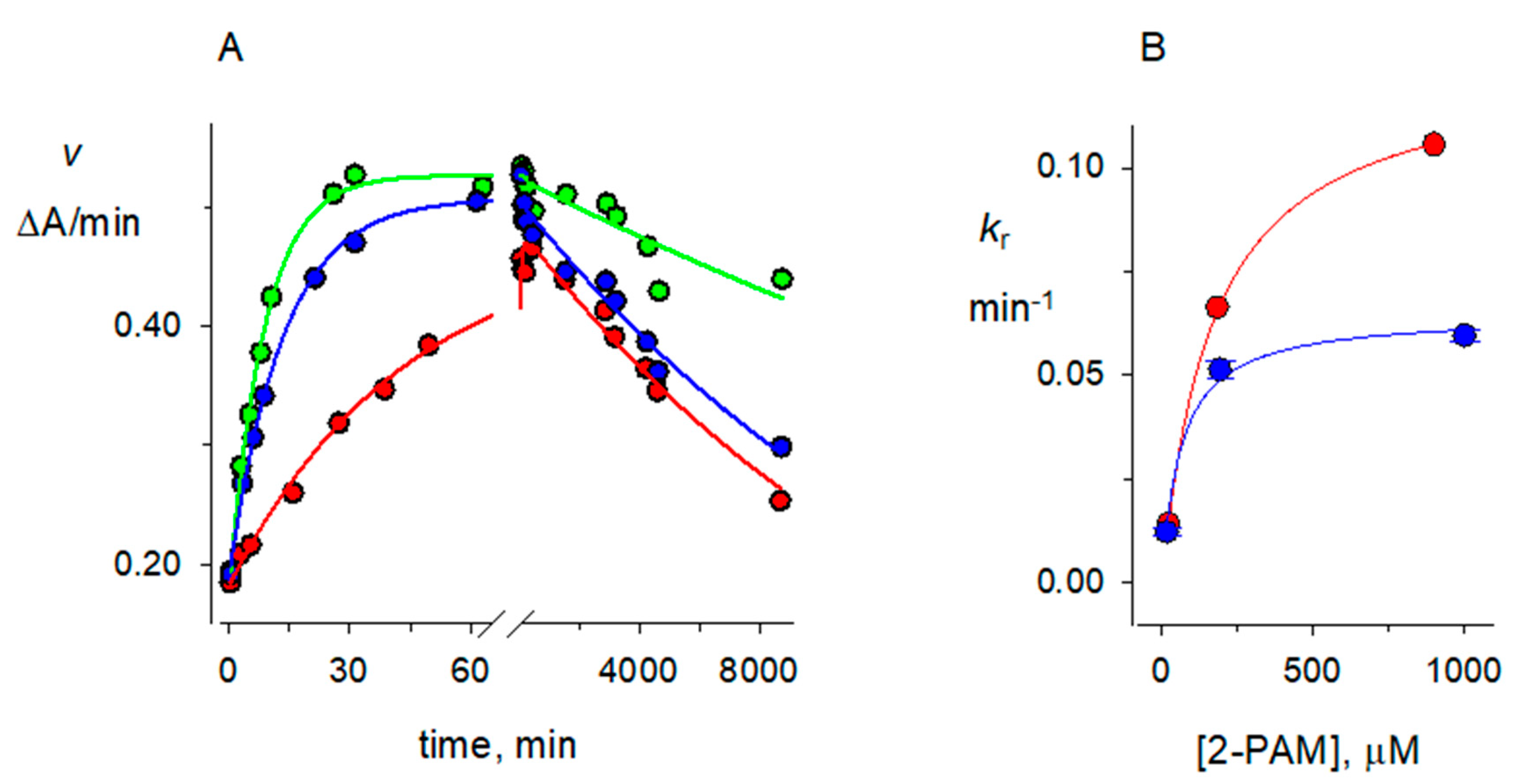
2.4. 2-PAM Reactivation Rate Constants kR for Organophosphorylated AChE
2.5. Accuracy of Rate Constants for Reactions of Organophosphorylated hAChE
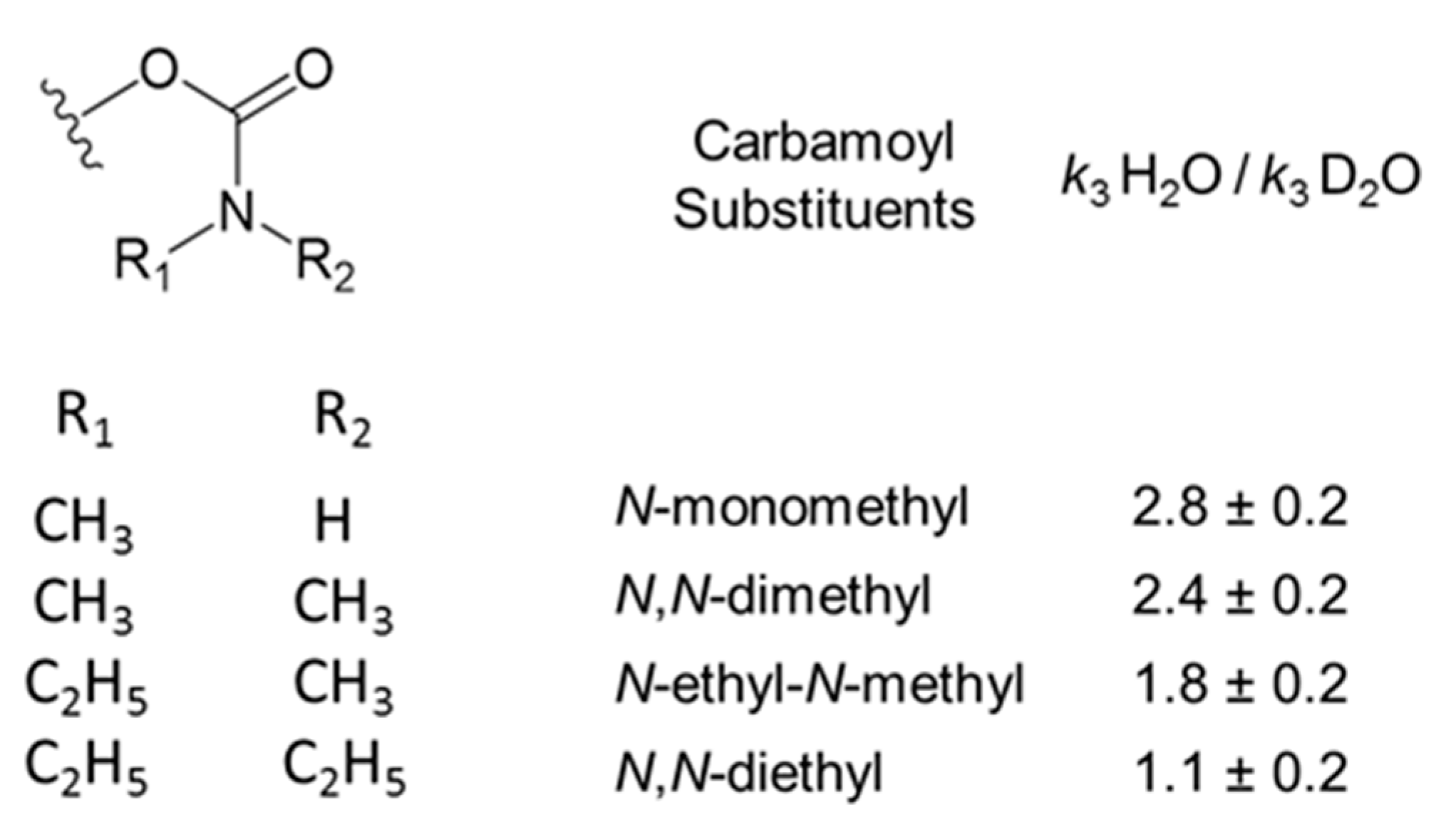
2.6. Solvent Deuterium Oxide Isotope Effects on Reaction Rate Constants
3. Discussion
3.1. Kinetics of Decarbamoylation and of the Hydrolysis of Organophosphorylated AChE
3.2. Solvent Deuterium Oxide Isotope Effects on Rate Constants for Reactions of Organophosphorylated AChE
4. Materials and Methods
4.1. Reagents
4.2. Assay of Substrate Hydrolysis
4.3. Reactions of Organophosphorylated hAChE
4.4. Solvent Deuterium Oxide Isotope Effects
Funding
Conflicts of Interest
Abbreviations
References
- Rosenberry, T.L. Quantitative simulation of endplate currents at neuro-muscular junctions based on the reactions of acetylcholine with acetylcholine receptor and acetylcholinesterase. Biophys. J. 1979, 26, 263–290. [Google Scholar] [CrossRef]
- Venkatasubban, K.S.; Johnson, J.L.; Thomas, J.L.; Fauq, A.; Cusack, B.; Rosenberry, T.L. Decarbamoylation of acetylcholinesterases is markedly slowed as carbamoyl groups increase in size. Arch. Biochem. Biophys. 2018, 655, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Reiner, E.; Aldridge, W.N. Effect of pH on inhibition and spontaneous reactivation of acetylcholinesterase treated with esters of phosphorus acids and of carbamic acids. Biochem. J. 1967, 105, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Fukuto, T.R. Mechanism of action of organophosphorus and carbamate insecticides. Environ. Health Perspect. 1990, 87, 245–254. [Google Scholar] [CrossRef]
- Myers, D.K. Studies on cholinesterase. 10. Return of cholinesterase activity in the rat after inhibition by carbamoyl fluorides. Biochem. J. 1956, 62, 556–563. [Google Scholar]
- Froede, H.C.; Wilson, I.B. Acetylcholinesterase. In The Enzymes, 3rd ed.; Boyer, P.D., Ed.; Academic Press: New York, NY, USA, 1971; pp. 87–114. [Google Scholar]
- Wilson, I.B. Acetylcholinesterase. XI. Reversibility of tetraethyl pyrophosphate inhibiton. J. Biol. Chem. 1951, 190, 111–117. [Google Scholar]
- Rosenberry, T.L.; Cheung, J. Rate-limiting step in the decarbamoylation of acetylcholinesterases with large carbamoyl groups. Chem. Biol. Interact. 2019, 308, 392–395. [Google Scholar] [CrossRef]
- Franklin, M.C.; Rudolph, M.J.; Ginter, C.; Cassidy, M.S.; Cheung, J. Structures of paraoxon-inhibited human acetylcholinesterase reveal perturbations of the acyl loop and the dimer interface. Proteins 2016, 84, 1246–1256. [Google Scholar] [CrossRef]
- Hörnberg, A.; Tunemalm, A.K.; Ekström, F. Crystal structures of acetylcholinesterase in complex with organophosphorus compounds suggest that the acyl pocket modulates the aging reaction by precluding the formation of the trigonal bipyramidal transition state. Biochemistry 2007, 46, 4815–4825. [Google Scholar] [CrossRef]
- Millard, C.B.; Kryger, G.; Ordentlich, A.; Greenblatt, H.M.; Harel, M.; Raves, M.L.; Segall, Y.; Barak, D.; Shafferman, A.; Silman, I.; et al. Crystal structures of aged phosphonylated acetylcholinesterase: Nerve agent reaction products at the atomic level. Biochemistry 1999, 38, 7032–7039. [Google Scholar] [CrossRef]
- Millard, C.B.; Koellner, G.; Ordentlich, A.; Shafferman, A.; Silman, I.; Sussman, J.L. Reaction products of acetylcholinesterase and VX reveal a mobile histidine in the catalytic triad. J. Am. Chem. Soc. 1999, 121, 9883–9884. [Google Scholar] [CrossRef]
- Kovalevsky, A.; Blumenthal, D.K.; Cheng, X.; Taylor, P.; Radic, Z. Limitations in current acetylcholinesterase structure–based design of oxime antidotes for organophosphate poisoning. Ann. N. Y. Acad. Sci. 2016, 1378, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Michel, H.O.; Hackley, B.E.; Berkowitz, L.; List, G.; Hackley, E.B.; Gillilan, W.; Pankau, M. Ageing and dealkylation of soman (pinacolylmethylphosphonofluoridate)-Inactivated eel cholinesterase. Arch. Biochem. Biophys. 1967, 121, 29–34. [Google Scholar] [CrossRef]
- Rosenberry, T.L. Catalysis by acetylcholinesterase. Evidence that the rate-limiting step for acylation with certain substrates precedes general acid-base catalysis. Proc. Natl. Acad. Sci. USA 1975, 72, 3834–3838. [Google Scholar]
- Quinn, D.M. Acetylcholinesterase: Enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 1987, 87, 955–979. [Google Scholar] [CrossRef]
- Worek, F.; Diepold, C.; Eyer, P. Dimethylphosphoryl-inhibited human cholinesterases: Inhibition, reactivation, and aging kinetics. Arch. Toxicol. 1999, 73, 7–14. [Google Scholar] [CrossRef]
- Jennings, L.L.; Malecki, M.; Komives, E.A.; Taylor, P. Direct analysis of the kinetic profiles of organophosphate-acetylcholinesterase adducts by MALDI-TOF mass spectrometry. Biochemistry 2003, 42, 11083–11091. [Google Scholar] [CrossRef]
- Grosfeld, H.; Barak, D.; Ordentlich, A.; Velan, B.; Shafferman, A. Interactions of oxime reactivators with diethylphosphoryl adducts of human acetylcholinesterase and its mutant derivatives. Mol. Pharmacol. 1996, 50, 639–649. [Google Scholar]
- Bender, M.L.; Hamilton, G.A. Kinetic isotope effects of deuterium oxide on several a-chymotrypsin-catalyzed reactions. J. Am. Chem. Soc. 1962, 84, 2570–2576. [Google Scholar] [CrossRef]
- Bender, M.L.; Clement, G.E.; Kezdy, F.J.; Heck, H.D.A. The correlation of the pH (pD) dependence and the stepwise mechanism of α-chymotrypsin-catalyzed reactions. J. Am. Chem. Soc. 1964, 86, 3680–3690. [Google Scholar] [CrossRef]
- Qian, N.; Kovach, I.M. Key active site residues in the inhibition of acetylcholinesterases by soman. FEBS Lett. 1993, 336, 263–266. [Google Scholar] [CrossRef]
- Kovach, I.M.; Bennet, A.J. Comparative study of nucleophilic am) enzymic reactions of 2-propyl methylphosphonate derivatives. Phosphorus Sulfur Silicon Relat. Elem. 1990, 51, 51–56. [Google Scholar] [CrossRef]
- Wong, L.; Radic, Z.; Bruggemann, R.J.M.; Hosea, N.; Berman, H.A.; Taylor, P. Mechanism of oxime reactivation of acetylcholinesterase analyzed by chirality and mutagenesis. Biochemistry 2000, 39, 5750–5757. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Saxena, A.; Smith, M.; Garcia, G.; Radic, Z.; Taylor, P.; Doctor, B.P. Phosphoryl oxime inhibition of acetylcholinesterase during oxime reactivation is prevented by edrophonium. Biochemistry 1999, 38, 9937–9947. [Google Scholar] [CrossRef] [PubMed]
- Mallender, W.D.; Szegletes, T.; Rosenberry, T.L. Organophosphorylation of acetylcholinesterase in the presence of peripheral site ligands: Distinct effects of propidium and fasciculin. J. Biol. Chem. 1999, 274, 8491–8499. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Cusack, B.; Davies, M.P.; Fauq, A.; Rosenberry, T.L. Unmasking tandem site interaction in human acetylcholinesterase. Substrate activation with a cationic acetanilide substrate. Biochemistry 2003, 42, 5438–5452. [Google Scholar] [PubMed]
- De Ferrari, G.V.; Mallender, W.D.; Inestrosa, N.C.; Rosenberry, T.L. Thioflavin T is a fluorescent probe of the acetylcholinesterase peripheral site that reveals conformational interactions between the peripheral and acylation sites. J. Biol. Chem. 2001, 276, 23282–23287. [Google Scholar] [CrossRef]
- Rosenberry, T.L.; Scoggin, D.M. Structure of human erythrocyte acetylcholinesterase. Characterization of intersubunit disulfide bonding and detergent interaction. J. Biol. Chem. 1984, 259, 5643–5652. [Google Scholar]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
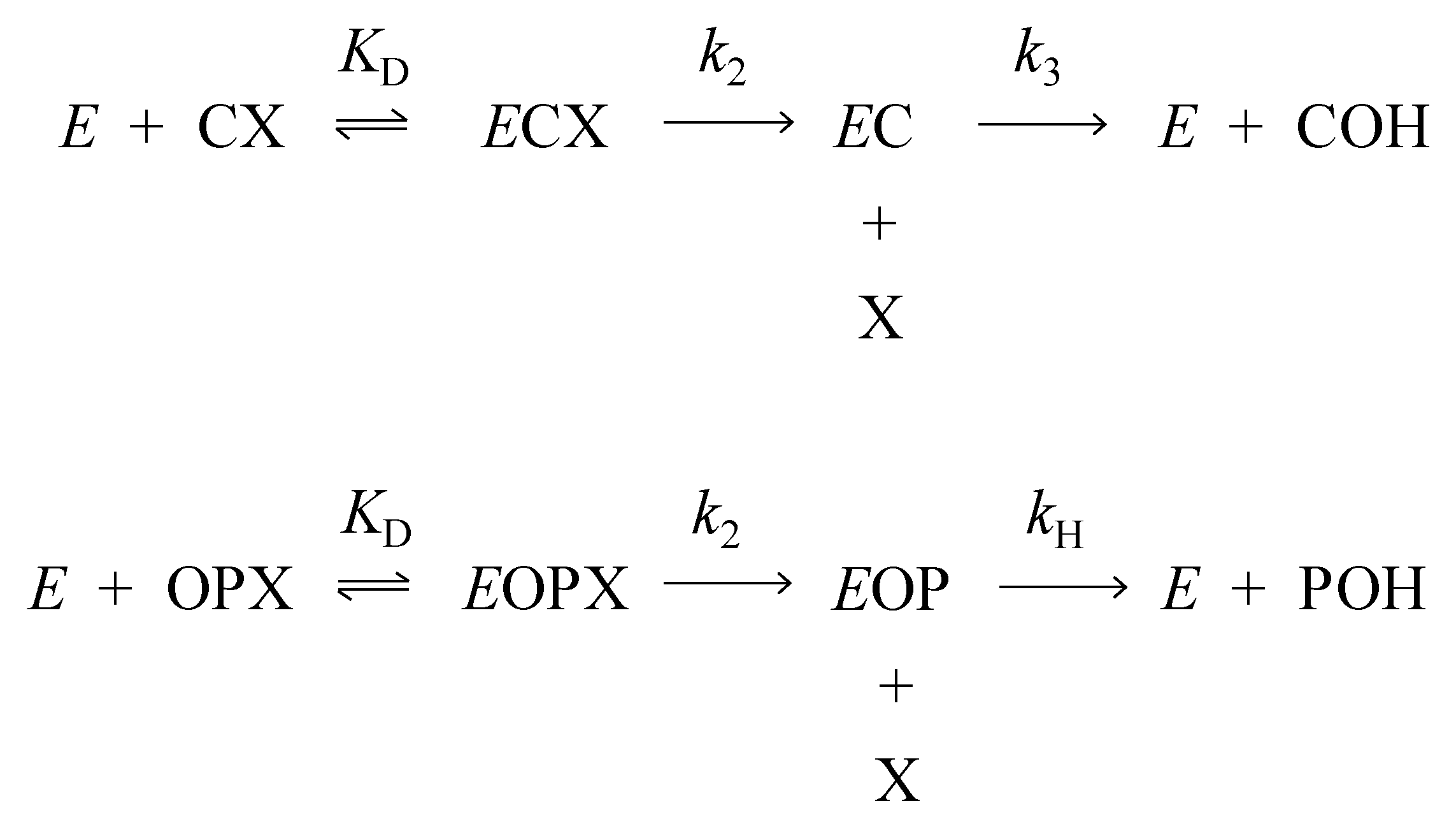{kind=link}
{kind=link}
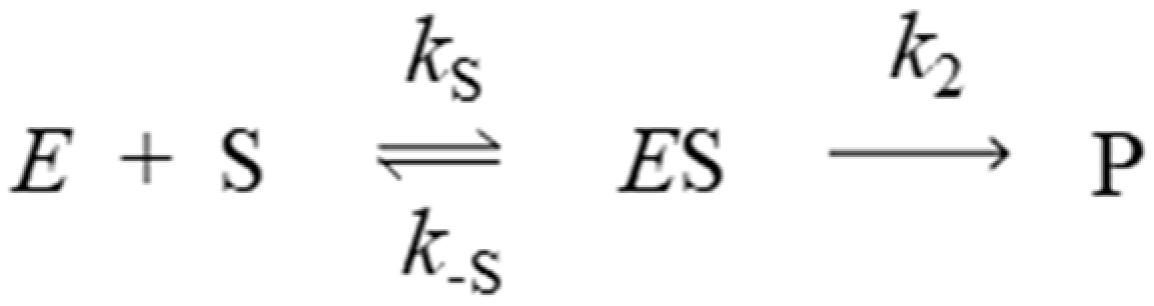{kind=link}
{kind=link}
| Phosphoryl Substituents | Reaction | k (10−6 min−1) H2O a | k (10−6 min−1) D2O | n | k H2O/k DO b |
|---|---|---|---|---|---|
| Dimethyl- | Aging (kA) | 1500 ± 500 | 1400 ± 500 | 2 | 1.1 ± 0.1 |
| Diethyl- | 39 ± 7 c | 41 ± 4 c | 1 | 1.0 ± 0.2 | |
| Dimethyl- | Hydrolysis (kH) | 6000 ± 530 | 3100 ± 360 | 2 | 1.9 ± 0.1 |
| Diethyl- | 83 ± 12 | 41 ± 5 | 3 | 2.0 ± 0.1 | |
| Dimethyl- | 2-PAM reactivation (kR) | 120,000 ± 6000 | 61,000 ± 4000 | 2 | 1.96 ± 0.04 |
| Diethyl- | 23,000 ± 1500 | 15,000 ± 300 | 2 | 1.68 ± 0.14 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosenberry, T.L. Solvent Deuterium Oxide Isotope Effects on the Reactions of Organophosphorylated Acetylcholinesterase. Molecules 2020, 25, 4412. https://doi.org/10.3390/molecules25194412
Rosenberry TL. Solvent Deuterium Oxide Isotope Effects on the Reactions of Organophosphorylated Acetylcholinesterase. Molecules. 2020; 25(19):4412. https://doi.org/10.3390/molecules25194412
Chicago/Turabian StyleRosenberry, Terrone L. 2020. "Solvent Deuterium Oxide Isotope Effects on the Reactions of Organophosphorylated Acetylcholinesterase" Molecules 25, no. 19: 4412. https://doi.org/10.3390/molecules25194412
APA StyleRosenberry, T. L. (2020). Solvent Deuterium Oxide Isotope Effects on the Reactions of Organophosphorylated Acetylcholinesterase. Molecules, 25(19), 4412. https://doi.org/10.3390/molecules25194412