Cobalt Catalyzed C-P Bond Formation by Cross-Coupling of Boronic Acids with P(O)H Compounds in Presence of Zinc


Abstract
:
1. Introduction
2. Result and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Aryl Phosphonates
3.2.1. Compound 8a: Diethyl Phenylphosphonate
3.2.2. Compound 8b: Diethyl 2-Methylphenylphosphonate
3.2.3. Compound 8c: Diethyl 4-Methoxyphenylphosphonate
3.2.4. Compound 8d: Diethyl 4-Ethylphenylphosphonate
3.2.5. Compound 8e: Diethyl 3-Nitrophenylphosphonate
3.2.6. Compound 8f: Diethyl 4-Fluorophenylphosphonate
3.2.7. Compound 8h: Diethyl 2,6-Difluorophenylphosphonate
3.2.8. Compound 9a: Dimethyl Phenylphosphonate
3.2.9. Compound 9b: Dimethyl 4-Methoxyphosphonate
3.2.10. Compound 9c: Dimethyl 3-Nitrophenylphosphonate
3.2.11. Compound 10a: Diisopropyl Phenylphosphonate
3.2.12. Compound 10b: Diisopropyl 4-Methoxyphosphonate
3.2.13. Compound 10c: Diisopropyl 3-Nitrophenylphosphonate
3.2.14. Compound 11a: Diphenyl Phenylphosphonate
3.2.15. Compound 11b: Diphenyl 4-Methoxyphosphonate
3.2.16. Compound 11c: Diphenyl 3-Nitrophenylphosphonate
3.2.17. Compound 12a: Dibenzyl Phenylphosphonate
3.2.18. Compound 12b: Dibenzyl 4-Methoxyphosphonate
3.2.19. Compound 12c: Dibenzyl 3-Nitrophenylphosphonate
3.2.20. Compound 12d: Dibenzyl 4-(Trifluoromethyl)phenylphosphonate
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giannousis, P.P.; Bartlett, P.A. Phosphorus amino acid analogs as inhibitors of leucine aminopeptidase. J. Am. Chem. Soc. 1987, 30, 1603. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Spörri, H. Organic Phosphorous Compounds 96. Resolution of 1-amino-2-(4-fluorophenyl) ethylphosphonic acid as well as some di- and tri-peptides. Phosphorus Sulfur Silicon Relat. Elem. Tetrahedron Lett. 1991, 61, 69. [Google Scholar] [CrossRef]
- Sikorski, J.A.; Miller, M.J.; Braccolino, D.S.; Cleary, D.G.; Corey, S.; Font, J.L.; Gruys, K.J.; Han, C.Y.; Lin, K.; Pansegrau, P.D.; et al. EPSP Synthase: The Design and Synthesis of Bisubstrate Inhibitors Incorporating Novel 3-Phosphate Mimics. Phosphorus Sulfur Silicon Relat. Elem. 1993, 76, 115. [Google Scholar] [CrossRef]
- Meyer, J.H.; Bartlett, P.A. Macrocyclic Inhibitors of Penicillopepsin. 1. Design, Synthesis, and Evaluation of an Inhibitor Bridged between P1 and P3. J. Am. Chem. Soc. 1998, 120, 4600. [Google Scholar] [CrossRef]
- Stowasser, B.; Budt, K.; Jian-Qi, L.; Peyman, A.; Ruppert, D. New hybrid transition state analog inhibitors of HIV protease with peripheric C2-symmetry. Tetrahedron Lett. 1992, 33, 6625. [Google Scholar] [CrossRef]
- Pratt, R. Inhibition of a class C beta-lactamase by a specific phosphonate monoester. Science 1989, 246, 917. [Google Scholar] [CrossRef]
- Dhawan, B.; Redmor, D. Optically active 1-aminoalkylphosphonic acids. Phosphorous Sulfur Relat. Elem. 1987, 32, 119. [Google Scholar] [CrossRef]
- Duncan, K.; Walsh, C.T. ATP-dependent inactivation and slow binding inhibition of Salmonella typhimurium D-alanine:D-alanine ligase (ADP) by (aminoalkyl)phosphinate and aminophosphonate analogues of D-alanine. Biochemistry 1988, 27, 3709. [Google Scholar] [CrossRef]
- Alonso, E.; Alonso, E.; Solis, A.; del Pozo, C. Synthesis of N-Alkyl-(α-Aminoalkyl)Phosphine Oxides and Phosphonic Esters as Potential HIV-Protease Inhibitors, Starting from α-Aminoacids. Synlett 2000, 5, 698. [Google Scholar]
- Hirschmann, R.; Smith, A.; Taylor, C.; Benkovic, P.; Taylor, S.; Yager, K.; Sprengler, P.; Benkovic, S. Peptide synthesis catalyzed by an antibody containing a binding site for variable amino acids. Science 1994, 265, 234. [Google Scholar] [CrossRef]
- Allen, J.; Atherton, F.; Hall, M.; Hassall, C.; Holmes, S.; Lambert, R.; Nisbet, L.; Ringrose, P. Phosphonopeptides, a new class of synthetic antibacterial agents. Nature 1978, 272, 56–58. [Google Scholar] [CrossRef] [PubMed]
- Atherton, F.R.; Hassall, C.H.; Lambert, R.W. Synthesis and structure-activity relationships of antibacterial phosphonopeptides incorporating (1-aminoethyl)phosphonic acid and (aminomethyl)phosphonic acid. J. Med. Chem. 1986, 29, 29. [Google Scholar] [CrossRef] [PubMed]
- Sienczyc, M.; Oleksyszyn, J. Irreversible inhibition of serine proteases - design and in vivo activity of diaryl alpha-aminophosphonate derivatives. Curr. Med. Chem. 2009, 16, 1673. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wan, Q.; Yang, S.; Song, B.; Bhadury, P.; Jin, L.; Yan, K.; Liu, F.; Chen, Z.; Xue, W. Synthesis and Antiviral Activities of Amide Derivatives Containing the α-Aminophosphonate Moiety. J. Agric. Food Chem. 2008, 56, 998. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Szostak, M. Decarbonylative Phosphorylation of Amides by Palladium and Nickel Catalysis: The Hirao Cross-Coupling of Amide Derivatives. Angew. Chem. Int. Ed. Engl. 2017, 56, 12718. [Google Scholar] [CrossRef]
- Reddy, M.V.N.; Annar, S.; Krishna, A.B.; Reddy, G.C.S.; Reddy, C.S. Tetramethyl guanidine (TMG) catalyzed synthesis of novel α-amino phosphonates by one-pot reaction. Org. Commun. 2010, 3, 39. [Google Scholar]
- Rao, A.J.; Rao, P.V.; Rao, V.K.; Mohan, C.; Raju, C.N.; Reddy, C.S. Microwave Assisted One-pot Synthesis of Novel α-Aminophosphonates and Their Biological Activity. Bull. Korean Chem. Soc. 2010, 31, 1863. [Google Scholar] [CrossRef] [Green Version]
- Rezaei, Z.; Firouzabadi, H.; Iranpoor, N.; Ghaderi, A.; Jafari, M.; Jafari, A.; Zare, H. Design and one-pot synthesis of α -aminophosphonates and bis(alpha-aminophosphonates) by iron(III) chloride and cytotoxic activity. Eur. J. Med. Chem. 2009, 44, 4266. [Google Scholar] [CrossRef]
- Rao, X.; Song, Z.; He, L. Synthesis and antitumor activity of novel α-aminophosphonates from diterpenic dehydroabietylamine. Heteroatom Chem. 2008, 19, 512. [Google Scholar] [CrossRef]
- Kraicheva, I.; Bogomilova, A.; Tsacheva, I.; Momekov, G.; Troev, K. Synthesis, NMR characterization and in vitro antitumor evaluation of new aminophosphonic acid diesters. Eur. J. Med. Chem. 2009, 44, 3363. [Google Scholar] [CrossRef]
- Xu, Y.; Yan, K.; Song, B.; Xu, G.; Yang, S.; Xue, W.; Hu, D.; Lu, P.; Ouyang, G.; Jin, L.; et al. Synthesis and antiviral bioactivities of α -aminophosphonates containing alkoxyethyl moieties. Molecules 2006, 11, 666. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Faiger, H.; Belakhov, V.; Baasov, T. Towards the development of novel antibiotics: Synthesis and evaluation of a mechanism-based inhibitor of Kdo8P synthase. Bioorg. Med. Chem. 1999, 7, 2671. [Google Scholar] [CrossRef]
- Wu, H.C.; Yu, J.Q.; Spencer, J.B. Stereospecific Deoxygenation of Phosphine Oxides with Retention of Configuration Using Triphenylphosphine or Triethyl Phosphite as an Oxygen Acceptor. Org. Lett. 2004, 25, 4675. [Google Scholar] [CrossRef]
- Al-Masum, M.; Livinghouse, T. Pd(0) Cu(I) cocatalyzed coupling of methylphenylphosphine-borane with aryl halides and aryl nonaflates. Tetrahedron Lett. 1999, 40, 7731. [Google Scholar] [CrossRef]
- Oshiki, T.; Imamoto, T. Unprecedented stereochemistry of the electrophilic arylation at chiral phosphorus. J. Am. Chem. Soc. 1992, 114, 3975. [Google Scholar] [CrossRef]
- Ley, S.; Thomas, A. Modern synthetic methods for copper-mediated C(aryl)[bond]O, C(aryl)[bond]N, and C(aryl)[bond]S bond formation. Angew. Chem. Int. Ed. 2003, 42, 5400. [Google Scholar] [CrossRef]
- Andaloussi, M.; Lindh, J.; Savmarker, J.; Sjoberg, P.; Larhed, M. Microwave-Promoted Palladium(II)-Catalyzed C-P Bond Formation by Using Arylboronic Acids or Aryltrifluoroborates. Chem. Eur. J. 2009, 15, 13069. [Google Scholar] [CrossRef]
- Piontek, A.; Bisz, E.; Szostak, M. Iron-Catalyzed Cross-Couplings in the Synthesis of Pharmaceuticals: In Pursuit of Sustainability. Angew. Chem. Int. Ed. 2018, 57, 11116. [Google Scholar] [CrossRef]
- Kalek, M.; Ziadi, A.; Stawinski, J. Microwave-Assisted Palladium-Catalyzed Cross-Coupling of Aryl and Vinyl Halides with H-Phosphonate Diesters. Org. Lett. 2008, 10, 4637. [Google Scholar] [CrossRef]
- Zhuang, R.; Xu, J.; Cai, Z.; Tang, G.; Fang, M.; Zhao, Y. Copper-Catalyzed C-P Bond Construction via Direct Coupling of Phenylboronic Acids with H-Phosphonate Diesters. Org. Lett. 2011, 13, 2110–2113. [Google Scholar] [CrossRef]
- Hu, G.; Chen, W.; Fu, T.; Peng, Z.; Qiao, H.; Gao, Y.; Zhao, Y. Nickel-Catalyzed C-P Cross-Coupling of Arylboronic Acids with P(O)H Compounds. Org. Lett. 2013, 15, 5362–5365. [Google Scholar] [CrossRef]
- Ohmiya, H.; Wakabayashi, K.; Yorimitsu, H.; Oshima, K. Cobalt-catalyzed cross-coupling reactions of alkyl halides with aryl Grignard reagents and their application to sequential radical cyclization/cross-coupling reactions. Tetrahedron 2006, 62, 2207. [Google Scholar] [CrossRef]
- Shinokubo, H.; Oshima, K. Transition Metal-Catalyzed Carbon−Carbon Bond Formation with Grignard Reagents—Novel Reactions with a Classic Reagent. Eur. J. Org. Chem. 2004, 10, 2081. [Google Scholar] [CrossRef]
- Korn, T.J.; Schade, M.A.; Wirth, S.; Knochel, P. Cobalt(II)-Catalyzed Cross-Coupling between Polyfunctional Arylcopper Reagents and Aryl Fluorides or Tosylates. Org. Lett. 2006, 8, 725. [Google Scholar] [CrossRef]
- Amatore, M.; Gosmini, C.; Perichon, J. CoBr2(Bpy): An Efficient Catalyst for the Direct Conjugate Addition of Aryl Halides or Triflates onto Activated Olefins. J. Org. Chem. 2006, 71, 6130. [Google Scholar] [CrossRef]
- Yorimitsu, H.; Oshima, K. New synthetic reactions catalyzed by cobalt complexes. Pure Appl. Chem. 2006, 78, 441. [Google Scholar] [CrossRef]
- Holzer, B.; Hoffman, R. Kumada–Corriu coupling of Grignard reagents, probed with a chiral Grignard reagent. Chem. Commun. 2003, 6, 732–733. [Google Scholar] [CrossRef]
- Datilus, V.; Abdulkarim, M.; Patrizio, A.; Kaur, P. Ter-pyridine catalyzed allylation of aldehydes and ketones under metal-free condition. Tetrahedron Lett. 2016, 57, 2778. [Google Scholar] [CrossRef]
- Cahiez, G.; Moyexic, A. Cobalt catalyzed cross-coupling reactions. Chem. Rev. 2010, 110, 1435. [Google Scholar] [CrossRef]
- Xu, K.; Hu, H.; Yang, F.; Wu, Y. Synthesis of Aryl and Arylmethyl Phosphonates by Cross-Coupling of Aryl or Arylmethyl Halides (X = I, Br and Cl) with Diisopropyl H-Phosphonate. Eur. J. Org. Chem. 2013, 2, 319–325. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, D.; Mo, F.; Zhang, Y.; Wang, J. Metal-Free Aromatic Carbon–Phosphorus Bond Formation via a Sandmeyer-Type Reaction. J. Org. Chem. 2016, 81, 11603–11611. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 8a–8m, 9a–9c, 10a–10c, 11a–11c and 12a–12d are available from the authors. |


{kind=link}
{kind=link}
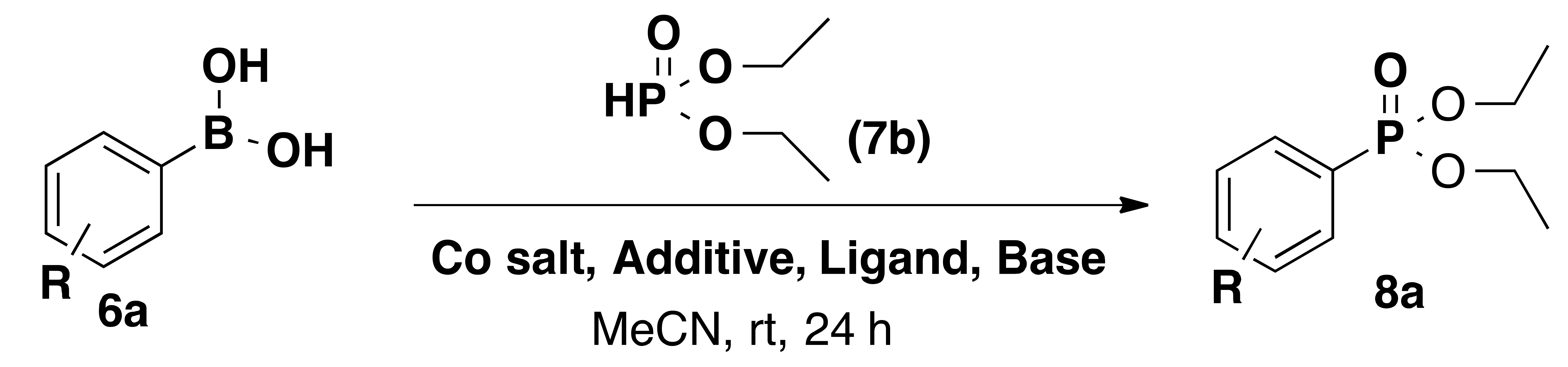
| Entry | Ligand | Cobalt Source | Additive | Base | Yield% |
|---|---|---|---|---|---|
| 1 | 1 | CoCl2 | - | Cs2CO3 | 19 |
| 2 | 1 | CoCl2 | - | K2CO3 | 32 |
| 3 | 1 | CoCl2 | Zn | DIPEA | 34 |
| 4 | 1 | CoCl2 | Zn | K2CO3 | 38 |
| 5 | 1 | CoCl2 | Zn | Et3N | 44 |
| 6 | 1 | CoCl2 | Zn | - | 42 |
| 7 | 2 | CoCl2 | Zn | - | 56 |
| 8 | 3 | CoCl2 | Zn | - | 65 |
| 9 | 4 | CoCl2 | Zn | - | 79 |
| 10 | 5 | CoCl2 | Zn | - | 60 |
| 11 | 4 | Co(NO3)2 | Zn | - | 61 |
| 12 | 4 | CoI2 | Zn | - | 84 |
| 13 | 4 | Co(OAc)2 | Zn | - | 56 |
| 14 15 | 4 4 | CoBr2 Co(acac)2 | Zn Zn | - - | 91 63 |
| Entry | Ligand | % Yield |
|---|---|---|
| 1 | Pyridine (ligand 1) | 54 |
| 2 | 2-picolylamine (ligand 2) | 56 |
| 3 | di-(2-picolyl)amine (ligand 3) | 65 |
| 4 | ter-pyridine (ligand 4) | 79 |
| 5 | 2,2′-dipyridylamine (ligand 5) | 60 |
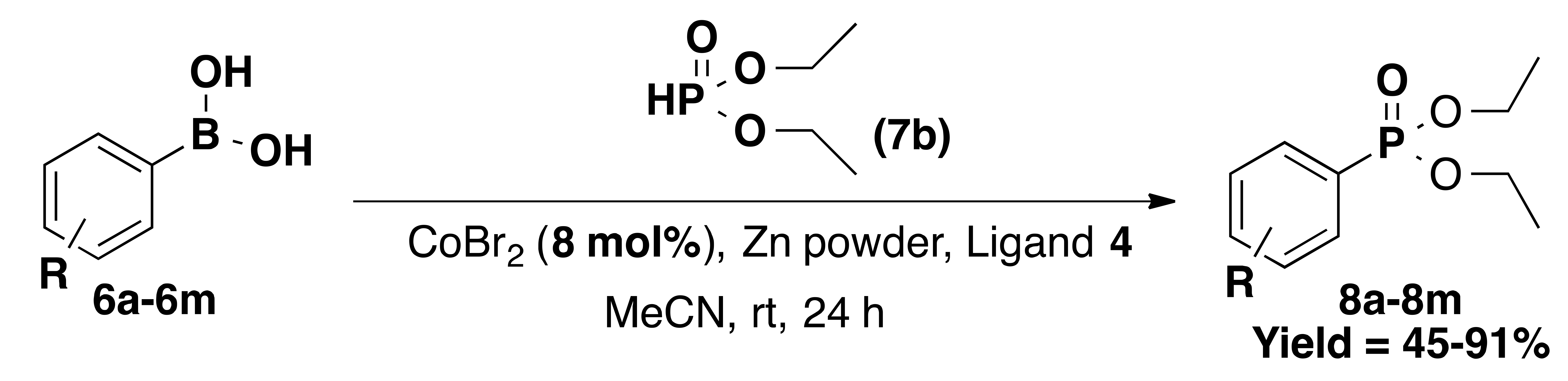
| Entry | R | Product | Yield% |
|---|---|---|---|
| 1 | H (6a) | 8a | 91 |
| 2 | 2-methyl (6b) | 8b | 89 |
| 3 | 4-methoxy (6c) | 8c | 87 |
| 4 | 4-ethynyl (6d) | 8d | 78 |
| 5 | 3-nitro (6e) | 8e | 66 |
| 6 | 4-fluoro (6f) | 8f | 65 |
| 7 | 2-fluoro (6g) | 8g | 82 |
| 8 | 2,6-difluoro (6h) | 8h | 63 |
| 9 | 4-trifluoromethyl (6i) | 8i | 45 |
| 10 11 12 13 | 1-Naphthyl (6j) 4-nitro (6k) 4-cyano (6l) 4-pyridyl (6m) | 8j 8k 8l 8m | 76 74 68 57 |
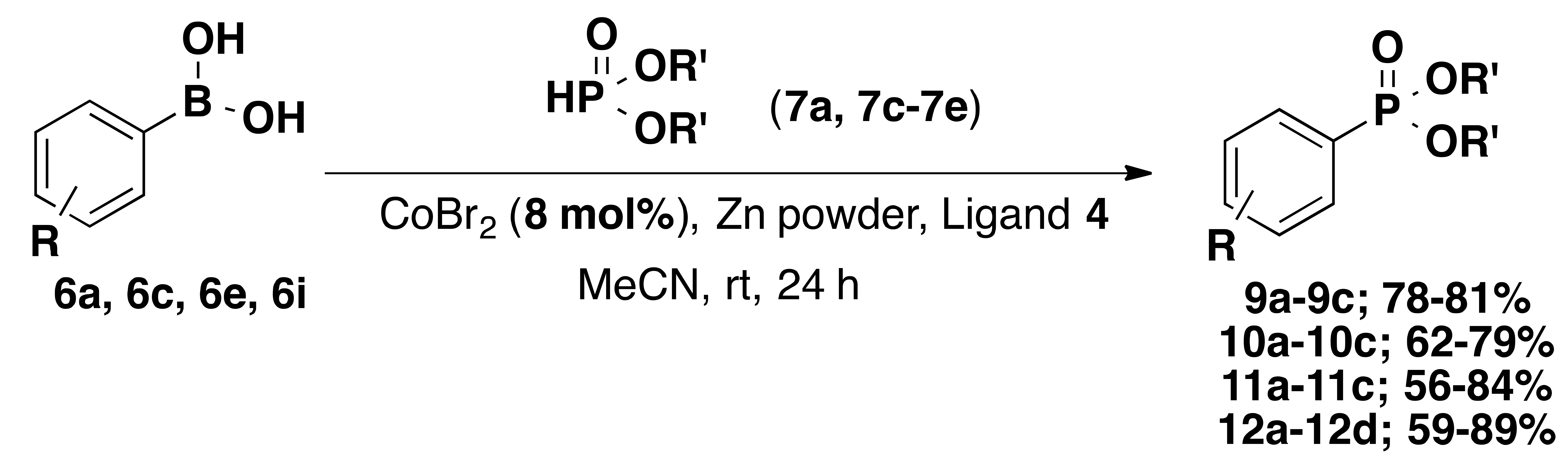
| Entry | R | R′ | Product | Yield% |
|---|---|---|---|---|
| 1 | H (6a) | Me (7a) | 9a | 79 |
| 2 | 4-methoxy (6c) | Me (7a) | 9b | 78 |
| 3 | 3-nitro (6e) | Me (7a) | 9c | 81 |
| 4 | H (6a) | iPr (7c) | 10a | 79 |
| 5 | 4-methoxy (6c) | iPr (7c) | 10b | 62 |
| 6 | 3-nitro (6e) | iPr (7c) | 10c | 71 |
| 7 | H (6a) | Ph (7d) | 11a | 84 |
| 8 | 4-methoxy (6c) | Ph (7d) | 11b | 74 |
| 9 | 3-nitro (6e) | Ph (7d) | 11c | 56 |
| 10 | H (6a) | Bn (7e) | 12a | 89 |
| 11 | 4-methoxy (6c) | Bn (7e) | 12b | 78 |
| 12 | 3-nitro (6e) | Bn (7e) | 12c | 67 |
| 13 | 4-trifluoro(methyl) (6i) | Bn (7e) | 12d | 59 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hicks, I.; McTague, J.; Hapatsha, T.; Teriak, R.; Kaur, P. Cobalt Catalyzed C-P Bond Formation by Cross-Coupling of Boronic Acids with P(O)H Compounds in Presence of Zinc. Molecules 2020, 25, 290. https://doi.org/10.3390/molecules25020290
Hicks I, McTague J, Hapatsha T, Teriak R, Kaur P. Cobalt Catalyzed C-P Bond Formation by Cross-Coupling of Boronic Acids with P(O)H Compounds in Presence of Zinc. Molecules. 2020; 25(2):290. https://doi.org/10.3390/molecules25020290
Chicago/Turabian StyleHicks, Ian, Jonathan McTague, Tatiana Hapatsha, Rania Teriak, and Parminder Kaur. 2020. "Cobalt Catalyzed C-P Bond Formation by Cross-Coupling of Boronic Acids with P(O)H Compounds in Presence of Zinc" Molecules 25, no. 2: 290. https://doi.org/10.3390/molecules25020290