Abstract
In order to prepare, at low cost, new compounds active against Plasmodium falciparum, and with a less side-effects, we have designed and synthesized a library of 1,4-disubstituted piperidine derivatives from 4-aminopiperidine derivatives 6. The resulting compound library has been evaluated against chloroquine-sensitive (3D7) and chloroquine-resistant (W2) strains of P. falciparum. The most active molecules—compounds 12d (13.64 nM (3D7)), 13b (4.19 nM (3D7) and 13.30 nM (W2)), and 12a (11.6 nM (W2))—were comparable to chloroquine (22.38 nM (3D7) and 134.12 nM (W2)).
1. Introduction
This year’s World Health Organization (WHO) report shows that after an unprecedented period of success in global malaria control, progress has stalled [1]. In 2016, there were an estimated 216 million cases of malaria, an increase of about 5 million cases over 2015. Deaths reached 445,000, a similar number to the previous year.
Malaria-related mortality followed the same trend, i.e., a decline from 2010 to 2014, and then an increase in 2015 and 2016. According to this report, it is in the WHO African region that the increase in cases of malaria and associated deaths was the most significant. The African region still accounts for some 90% of worldwide malaria cases and related deaths. Fifteen countries, all but one in sub-Saharan Africa, account for 80% of the global burden of malaria.
One of the biggest challenges facing malaria chemotherapy is the rapid emergence of resistance to existing antimalarial drugs [2]. Chloroquine was replaced as first line therapy by the sulfonamide antimalarials and, later on, artemisinin combination therapy (ACT), following the development of widespread resistance against the drug by Plasmodium falciparum [3]. This challenge underscores the need for the continued search for new antimalarials.
The 4-arylaminopiperidine is a structural moiety found in many alkaloids [4,5,6,7,8,9,10,11] and pharmaceutical products such as fentanyl and structurally-related analgesic opioids or H1-antihistamines agents such as bamipine [12,13,14,15,16,17] and neurokinin 1 (NK1) receptor antagonists [18,19,20]. Studies have shown that compounds with piperidine rings [4,8,21,22,23,24,25,26] have good selectivity and activity for the P. falciparum strain.
Research is being pursued for the discovery of new antimalarials with less side effects, a faster onset of action and a better rate of response [4,8,21,22,23,24,25,26]. In the process of searching for new small molecules interacting with the P. falciparum strain, we have identified the target compounds A with various R1, R2 and R3 substituents (Figure 1). In this paper, we describe the synthesis of some new derivatives with potential antimalarial properties.
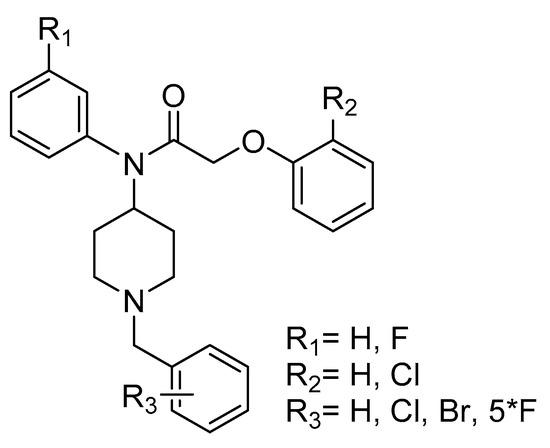
Figure 1.
target compounds A.
2. Results and Discussion
2.1. Chemistry
The key compound 6 has been synthesized through a three-step process according to the Scheme 1 [27]. Thus, reductive amination [28,29,30,31] of N-boc-piperidin-4-one (1) with anilines 2a, b in CH2Cl2, gave compounds 3a, b in 75–85% yield. Acylation of the sodium salts of 3 with phenoxyacetyl chlorides 4 in CH2Cl2 at 0 °C furnished compounds 5 (85–90%). Final deprotection [32,33] of 5 using trifluoroacetic acid at room temperature provided compounds 6a–b (50–95%) (Scheme 1).
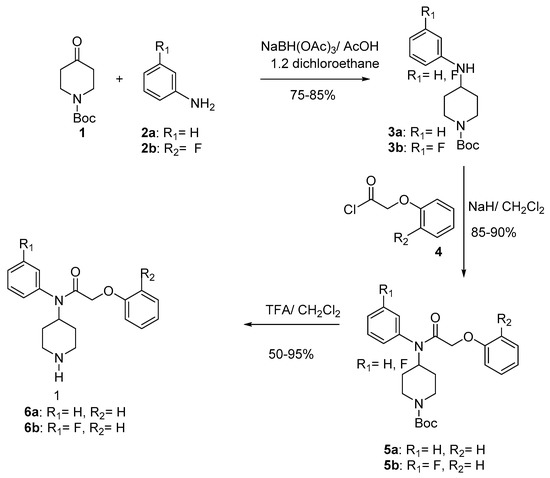
Scheme 1.
Synthesis of compounds 6a–b.
The alkylation of 3 with the acetyl chloride supplied compounds 7 in 85% yield. The condensation of 7 with the phenol 8 gave compounds 9 (78–80%). Final deprotection [32,33] of 9 using trifluoroacetic acid at room temperature provided compounds 6c–d (50–95%) (Scheme 2). The Table 1 gives the overall yields of compounds 6a–d.
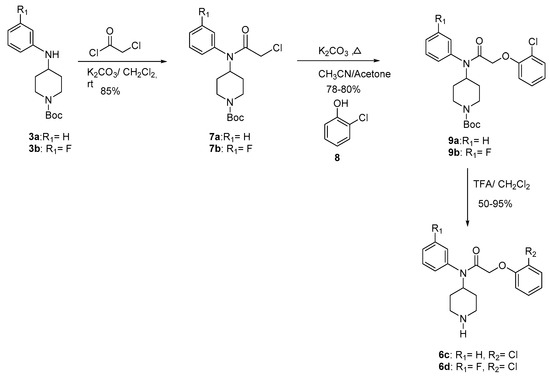
Scheme 2.
Synthesis of compound 6c and 6d.

Table 1.
The products of the synthesis of 6 recorded.
Condensation of phenoxyacetyl chloride with compound 3a in the presence of triethylamine at room temperature in acetone gave compounds 5a (55%) and 10 (30%) (Scheme 3). In this reaction we used excess phenoxyacetyl chloride, and we think that this excess probably made the reaction medium acidic which cause the cleavage of the N-Boc protective group. To avoid this side reaction, we used NaH in CH2Cl2 which furnished compound 5a (Scheme 1).
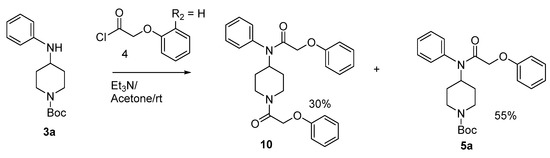
Scheme 3.
Synthesis of compounds 5a and 10.
A pharmaco-modulation has been achieved on the parent molecule 6 taking advantage of the nucleophilicity of the piperidine nitrogen leading to compounds 17–34 in good yield. Thus, reductive amination [28,29,30,31] of 6 with benzaldehyde derivatives in 1,2-dichloroethane (ClCH2CH2Cl), gave compounds A (Scheme 4) (Table 2).
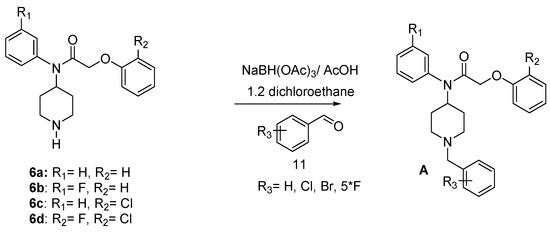
Scheme 4.
Synthesis of target compounds A.
Table 2.
The target compounds A.
2.2. The Antimalarial Activity of Derivatives 6 and Target Compounds A
Studies have shown that compounds with piperidine rings [4,8,21,22,23,24,25,26] have good selectivity and activity for the P. falciparum strain. This prompted us to assess their antiplasmodial activity against the chloroquine-sensitive 3D7 and chloroquine-resistant W2 strains of P. falciparum as well as their cytotoxic activity against HUVEC cells (Table 3 and Table 4). Solutions of the 22 synthetic products and the negative control (chloroquine (CQ)) were prepared by two-fold dilution, in a dose-titration range of 0.098–100 µg/mL, to obtain 11 concentrations each, and all of them were inactive against W2 (IC50 > 100). The compounds exhibited activities in the nanomolar range against both parasitic strains. Their cytotoxicity against HUVEC ranged from CC50 0.052 ± 0.004 to >100 mM, thus resulting in varied selectivity indexes (SI), 26 for 13b in the 3D7 strain and >11.3 for 14c in the W2 strain. Compared with chloroquine (IC50 = 22.38 (3D7) and 134.12 (W2)), the compounds 13b (IC50 = 13.30 nM) and 12a (IC50 = 11.06 nM) showed a strong activity against W2. Molecules 13b (IC50 = 4.19 nM), 12d (IC50 = 13.64 nM), 14d (IC50 = 14.85 nM) and 6b (IC50 = 17, 42 nM) had the highest activity against 3D7.
Table 3.
The antimalarial activity of compounds derivatives 6.
Table 4.
The antimalarial activity of the target compounds.
Interestingly, compounds 6c and 6d are inactive against both strains. However, after pharmacomodulation on the nitrogen atom, their derivatives 12d, 14d, 17c, 13c and 14c showed good activity against both strains.
Compound 13b exhibits 5-fold more activity against strain 3D7 and 10-fold more against strain W2 with very low cytotoxicity (CC50 = 112 nM), resulting in a high selectivity index (SI = 26.7 (3D7) and 8.4 (W2), respectively) relative to chloroquine (CC50 = 37.56 nM, SI = 1.7 (3D7) and 0.3 (W2)).
Substitution of the piperidine nitrogen atom with a pentafluorobenzyl moiety did not significantly alter the activity of its derivative molecules against both strains. Indeed, the compounds 17b and 17d, derived from 6b and 6d, respectively, remained inactive while the activity of 17a (37.63 nM (3D7) and 47.84 nM (W2)) and 17c (14.65 nM (3D7) and 36.88 nM (W2)) respectively, and 6a (34.46 nM (3D7) and 61.37 nM (W2)) and 6c (17.42 nM (3D7) and 30.35 nM (W2)) varied slightly.
3. Materials and Methods
3.1. Apparatus, Materials, and Analytical Reagents
All chemical reagents and anhydrous solvents were obtained from commercial sources and used without further purification. The 1H- and 13C-NMR spectra were recorded in CDCl3 at ambient temperature on an AMX 500 spectrometer (Bruker, Palaiseau, France). Some product structures were confirmed by DEPT 135, HMQC and HMBC experiments. Chemical shifts are given in δ (ppm) and coupling constants J (Hz) relative to TMS used as internal standard; multiplicities were recorded as s (singlet), d (doublet), dd (double doublet), t (triplet), dt (double triple), q (quartet) or m (multiplet). Reactions involving anhydrous conditions were conducted in dry glassware under a nitrogen atmosphere. The infrared spectra have been recorded on a model 842 spectrometer (Perkin-Elmer, 842) using polystyrene as reference. The melting points have been measured on a Tottoli S Bucchi device (Buchi, Rungis, France). Microanalysis have been done on a Perkin-Elmer 2400-CMN apparatus (Perkin ElmerVillebon-sur-Yvette, France). GC/MS conditions: Analyses were performed using a 5890 gas chromatogram connected to a G 1019 A mass spectrometer (both from Hewlett Packard, Alpharetta, GA, USA) operating in the electrospray ionization mode (ESI).
3.2. Chemistry
3.2.1. General procedure for the Synthesis of tert-Butyl 4-(phenylamino) Piperidine-1-carboxylates 3a–b
A solution of aniline (1 equiv) in 1,2-dichloroethane (100 mL) containing t-butyl-4-oxo-1-piperidine carboxylate (1 equiv), sodium triacetoxyborohydride (1.5 equiv) and acetic acid (1.5 equiv) was stirred for 24 h at 20 °C. 1N NaOH (50 mL, 50 mmol) and 50 mL of ethyl acetate were added. The phases were separated and the aqueous layer was extracted with ethyl acetate (3x25 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by crystallization (ether petroleum/ethyl acetate (8:2)).
tert-Butyl 4-(phenylamino) piperidine-1-carboxylate (3a). Aniline (2.8 g, 30.11 mmol) in 1.2-dichloroethane (100 mL) containing t-butyl-4-oxo-1-piperidine carboxylate (6 g, 30.11 mmol), sodium triacetoxyborohydride (9.57 g, 45.1 mmol) and acetic acid (2.71 g, 45.16 mmol) gave compound 3a (7.07 g, 85%), m.p.: 105 °C. 1H-NMR (CDCl3): 1.3 (m, 2H, CH2); 1.49 (s, 9H, 3CH3); 2.0 (m, 2H, CH2); 2.9 (m, 2H, CH2); 3.3 (m, 1H, CH); 3.7 (broadband, 1H, NH); 4.1 (m, 2H, CH2); 6.5–7.5 (m, 5H aromatic). 13C-NMR (CDCl3) δ: 28.57 (3× CH3); 32.52 (2× CH2); 42.30 (2× CH2); 50.22 (CH); 79.72 (C); 113.42 (2× CHAr); 117.61 (CHAr); 129.49 (2× CHAr); 149.89 (C); 154.92 (C). MS (m/z): calcd. for C16H24N2O2 276.2 found 277.1 [M + 1]; IR cm−1: 1763.07 (CO carbamate); 1671.6 (CO, amide).
tert-Butyl 4-(3-fluorophenylamino) piperidine-1-carboxylate (3b). 3-Fluoroaniline (3.34 g, 30.11 mmol) in 1,2-dichloroethane (100 mL) containing t-butyl-4-oxo-1-piperidine carboxylate (6 g, 30.11 mmol), sodium triacetoxyborohydride (9.57 g, 45.1 mmol) and acetic acid (2.71 g, 45.16 mmol) gave compound 3b (6.58 g, 74%); m.p.: 114 °C. 1H-NMR (CDCl3): 1.3 (m, 2H, CH2); 1.49 (s, 9H, 3CH3); 2.0 (m, 2H, CH2); 2.9 (m, 2H, CH2); 3,4 (m, 1H, CH); 3.8 broad band, 1H, NH); 4.1 (m, 2H, CH2); 6.5–7.5 (m, 4H aromatic). 13C-NMR (CDCl3) δ: 28.56 (3× CH3); 32.36 (2× CH2); 42.73 (2× CH2); 50.26 (CH); 79.81 (C); 99.95 (d, J = 25.26 Hz, CHAr); 103.84 (d, J = 21.25 Hz, CHAr); 109.19 (d, J = 2.2 Hz, CHAr); 130.58 (d, J = 10.30 Hz, CHAr); 148.73 (d, J = 10.55 Hz, C); 154.896 (C), 163.34 (d, J = 242.72 Hz, C). ESI (m/z) calcd for C16H23FN2O2 294.2; found 294.1 [M + 1]; IR cm−1: 1760.07 (CO carbamate); 1670.6 (CO, amide).
3.2.2. General Procedure for the Coupling with Phenoxyacetyl chloride: Synthesis of Compounds 5a–b
To an ice-cooled suspension of sodium hydride (60% in mineral oil, 2 equiv) in CH2Cl2 (10 mL) was added dropwise a solution of compound 3 (1 equiv), in CH2Cl2 (15 mL). After stirring 15 min phenoxyacetyl chloride 4 (2 equiv) was added. The reaction mixture was stirred for 1 h at 0 °C, and the temperature was raised to room temperature during 3 h. 20 mL of saturated solution of NaHCO3 was carefully added. The aqueous layer was extracted with CH2Cl2 (3 × 20 mL). The combined organic phases were dried over MgSO4, and concentrated in vacuum. The crude product was purified by crystallisation (ether petroleum/ethyl acetate (8:2)).
tert-Butyl 4-(2-phenoxy-N-phenylacetamido) piperidine-1-carboxylate (5a). Following the general procedure, sodium hydride (60% in mineral oil, 0.723 g, 18.1 mmol) in CH2Cl2 (10 mL) was added dropwise a solution of compound 3a (2.5 g, 9.05 mmol), in CH2Cl2 (15 mL). After stirring 15 min phenoxyacetyl chloride (2.5 mL, 18.1 mmol) was added to give compound 5a (3.06 g, 82%). 1H-NMR (CDCl3): 1.25 (m, 2H, CH2); 1.4 (s, 9H); 1.8 (m, 2H, CH2); 2.9 (m, 2H, CH2); 4.1 (m, 2H, CH2); 4.25 (m, 2H, CH2); 4.8 (m, 1H, CH); 6.7–7.5 (m, 10H aromatic). 13C-NMR (CDCl3) δ: 28.456 (3× CH3, C(CH3)3); 30.32 (2× CH2); 43.315 (2× CH2); 52.96 (CH); 66.7 (CH2); 79.73 (C); 114.8 (2× CHAr); 121.47 (CHAr); 129.34 (CH); 129.46 (2× CHAr); 129.88 (2× CHAr); 130.09 (2× CHAr); 136.89 (C); 154.63 (C); 158.14 (C); 167.6 (C). ESI (m/z) calcd for C24H30N2O4 410, 2 found 411.1 [M + 1], IR cm−1: 1744.95 (CO carbamate); 1652.06 (CO, amide).
tert-Butyl 4-(N-(3-fluorophenyl)-2-phenoxyacetamido) piperidine-1-carboxylate (5b). Following the general procedure, sodium hydride (60% in mineral oil, 0.677 g, 16.93 mmol) in CH2Cl2 (10 mL) was added dropwise a solution of compound 3b (2.49 g, 8.46 mmol), in CH2Cl2 (15 mL). After stirring 15 min phenoxyacetyl chloride (2.88 g, 16.93 mmol) was added to give compound 5b (2.34 g, 65%). 1H-NMR (CDCl3): 1.25 (m, 2H, CH2); 1.4 (s, 9H, 3× CH3); 1.8 (d, 2H, CH2); 2.8 (m, 2H, CH2); 4.15 (m, 2H, CH2); 4.30 (s, 2H, CH2); 4.75 (m, H, CH); 6.7–7.6 (m, 9 H aromatic). 13C-NMR (CDCl3) δ: 28.44 (3× CH3, C(CH3)3); 30.284 (2× CH2); 43.16 (2× CH2); 53.188 (CH); 66.82 (CH2); 79.81 (C); 114.76 (2× CHAr); 116.53 (d, J = 20.74 Hz, CHAr); 117.63 (d, J = 21.49 Hz, CHAr); 121.60 (CHAr); 125.97 (d, J = 3.14 Hz, CHAr); 129.50 (2× CHAr);130.92 (d, J = 7.3 Hz, CHAr); 138.52 (d, J = 9.17 Hz, C); 154.58 (C); 157.97 (C); 161.91 (d, J = 250.14 Hz, C); 167.19 (C). ESI (m/z): calcd for C24H29FN2O4 428.2, found 429.0 [M + 1]; IR cm−1: 1745.07 (CO carbamate); 1660.6 (CO, amide).
3.2.3. General Procedure for the Coupling with Chloroacetyl chloride: Synthesis of Compounds 7a–b
One equiv of compound 3 was dissolved in 25 mL of CH2Cl2, 2 equiv. of potassium carbonate were added and the mixture was cooled to 0 °C. Two equiv. of chloroacetyl chloride were added to 0 °C, and the mixture was stirred overnight. The reaction was quenched by addition of a saturated solution of NaHCO3 (25 mL), the aqueous phase was decanted and extracted twice with 15 mL of CH2Cl2. Combined organic phases were washed with water and brine, dried over MgSO4 and concentrated in vacuum. The crude product was purified by crystallisation (ether petroleum/ethyl acetate (8:2)).
tert-Butyl-4-(2-chloro-N-phenylacetamido) piperidine-1-carboxylate (7a). Following the general procedure, 2.5 g (9.03 mmol) of compound 3a were dissolved in 25 mL of CH2Cl2, 2.5 g (18.06 mmol) of potassium carbonate were added and the mixture was cooled to 0 °C. 2.04 g (18.06 mmol) of chloroacetyl chloride were added, and the mixture was stirred overnight to afford 7a as a white solid (2.55 g, 80%), m.p.: 110 °C. 1H-NMR (CDCl3, 500 MHz): 1.25 (m, 2H, CH2); 1.4 (s, 9 H, 3× CH3); 1.8 (m, 2H, CH2); 3.7 (s, 2H, CH2); 2.8 (m, 2H, CH2); 4.1 (m, 2H, CH2); 4.75 (m, 1H, CH); 7–7.5 (m, 5H aromatic). 13C-NMR (125 MHz, CDCl3) δ: 28.485 (C(CH3)3); 30.32 (2× CH2); 42.55 (CH2); 43.41 (2× CH2); 53.5 (CH); 79.8 (C); 129.47 (2× CHAr); 129.9 (CHAr); 130.2 (2× CH); 137.3 (C); 154.64 (C); 166.04 (C). ESI (m/z): calcd for C18H25ClN2O3 352.2, found 353.0 [M + 1]; IR cm−1: 1730.5 (CO carbamate); 1699.03 (CO, amide).
tert-Butyl 4-(2-chloro-N-(3-fluorophenyl) acetamido)piperidine-1-carboxylate (7b). Following the general procedure, 2.5 g (8.46 mmol) of compound 3b were dissolved in 25 mL of CH2Cl2, then 2.34 g (16.93 mmol) of potassium carbonate were added and the mixture was cooled to 0 °C. Chloroacetyl chloride (1.91 g, 18.06 mmol) was added, and the mixture was stirred overnight to afford 7b (2.66 g, 85%), m.p.: 90 °C. 1H-NMR (CDCl3, 500 MHz): 1.25 (m, 2H, CH2); 1.4 (s, 9 H, 3CH3); 1.8 (m, 2H, CH2); 3.7 (s, 2H, CH2); 2.8 (m, 2H, CH2); 4.1 (m, 2H, CH2); 4.75 (m, 1H, CH); 7–7.5 (m, 5H aromatic). 13C-NMR (125 MHz, CDCl3) δ: 28.44 (3× CH3); 30.27 (2× CH2); 42.26 (CH2); 42.97 (2× CH2); 53.69 (CH); 79.878 (C); 116.72 (d, J = 20.74 Hz, CHAr); 117.79 (d, J = 21.49 Hz, CHAr); 126.14 (CHAr); 131.08 (d, J = 9.17 Hz, CHAr); 138.8 (d, J = 9.17 Hz, C); 154.58 (C); 161.926 (C); 163.92 (d, J = 234,80 Hz, C). ESI (m/z): calcd for C18H24ClFN2O3 370.1 found 371.0 [M + 1]; IR cm−1: 1730.5 (CO carbamate); 1699.08 (CO, amide).
3.2.4. General Procedure for Synthesis of Compounds 9a–b
To a mixed solution of acetonitrile/acetone (50/50) were added 1 equiv of compound 7; 1 equiv of 2-chlorophenol and 2 equiv of potassium carbonate. After 12 h of stirring under reflux the mixture was concentrated in vacuum. The residual was dissolved in 15 mL of ethyl acetate and (1N) of NaOH (15 mL), the aqueous phase was decanted and extracted twice with 15 mL of ethyl acetate. Combined organic phases were washed with water and brine, dried over MgSO4 and concentrated in vacuum. The crude product was purified by crystallisation (ether petroleum/ethyl acetate (8:2)).
tert-Butyl 4-(2-(2-chlorophenoxy)-N-phenylacetamido) piperidine-1-carboxylate (9a). Following the general procedure, 2.83 g (8.04 mmol) of compound 7; 1.032 g (8.04 mmol) of 2-chlorophenol and 2.22 g (16.09 mmol) of potassium carbonate. (3.57 g, 99%); m.p.: 123 °C; 1H-NMR (CDCl3, 600 MHz): 1.3 (m, 2H, CH2); 1.5 (s, 9H, 3× CH3); 1.80 (m, 2H, CH2); 2.85 (m, 2H, CH2); 4.12 (m, 1H, CH); 4.33 (s, 2H, CH2); 4.76 (m, H, CH); 6.4–7.5 (m, 9H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 28.46(3× CH3); 29.83 (2× CH2); 42.75 (2× CH2) 53.04 (CH); 67.62 (CH2); 79.78 (C); 113.95 (2× CHAr); 122.27 (2× CHAr); 123.27 (C); 127.61 (CHAr); 129.45(CHAr); 129.97 (CHAr); 130.14 (CHAr); 130.45 (CHAr); 136.64 (C); 153.9 (C); 154.64 (C); 166.84 (C). ESI (m/z): calcd for C24H29ClN2O4 444.1, found 445.0 [M + 1]; IR cm−1: 1727.5 (CO carbamate); 1693.33 (CO, amide).
tert-Butyl 4-(2-(2-chlorophenoxy)-N-(3-fluorophenyl) acetamido) piperidine-1-carboxylate (9b). Following the general procedure, 1.80 g (4.87 mmol) of compound 7; 0.626 g (4.87 mmol) of 2-chlorophenol and 1.34 g (9.75 mmol) of potassium carbonate were reacted to give 9b (2.01 g, 89%); m.p.: 125°C; 1H-NMR (CDCl3, 500 MHz): 1.18 (m, 2H, CH2); 1.21 (s, 9H, 3CH3); 1.74 (m, 2H, CH2); 2.71 (t, J = 12 Hz, 2H, CH2); 4.06 (m, 2H, CH2); 4.31 (s, 2H, CH2); 4.67 (m, 1H, CH) 6.4–7.5 (m, 8H aromatic). 13C-NMR (125 MHz, CDCl3) δ: 27.12 (2× CH2); 28.36 (3CH3); 30.21 (2× CH2); 53.32 (CH); 67.75 (CH2); 79.75 (C); 114.0 (CHAr); 116.43 (d, J = 31.25 Hz, CHAr); 117.40 (d, J = 26.25 Hz, CHAr); 122.36 (CHAr); 123.27 (C); 125.94 (CHAr); 127.55 (CHAr); 130.52 (CHAr); 130.84 (d, J = 11.25 Hz, CHAr); 138.20 (d, J = 20 Hz, C); 153.69 (C); 154.51 (C); 161.41 (d, J = 281.25 Hz, C); 166.60 (C). ESI (m/z): calcd for C24H28ClFN2O4 462.2, found 463.0 [M + 1]; IR cm−1: 1727.5 (CO carbamate); 1693.33 (CO, amide).
3.2.5. General Procedure for Deprotection: Synthesis of 6a–b
One equiv of compound 5 was dissolved in 15 mL of CH2Cl2, and 13 equiv of trifluoroacetic acid were added. After 2 h of stirring at room temperature, the reaction mixture was concentrated under vacuum. The residue was dissolved in 5 mL of ethyl acetate then neutralized with NaHCO3 (5%). The aqueous layer was extracted with ethyl acetate (4x5mL). The combined organic phases were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by crystallisation (ether petroleum/ethyl acetate (8:2)).
2-Phenoxy-N-Phenyl-N-(piperidin-4-yl) acetamide (6a). Following the general procedure, 3.06 g (7.48 mmol) of compound 5a were dissolved in 15 mL of CH2Cl2, then 7.49 mL (97.24 mmol) of trifluoroacetic acid were added to produce compound 6a (1.15 g, 50%); m.p.: 59 °C. 1H-NMR (CDCl3, 600 MHz): 1.5 (m, 2H, CH2); 1.9 (m, 2H, CH2); 2.7 (m, 2H, CH2); 3.4 (m, 2H, CH2); 3.4 (broad, 1H, NH); 4.6 (s, 2H, O-CH2); 4.75 (m, 1H, CH), 6.5–7.7 (m, 10H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30 (2× CH2); 45.06 (2× CH2); 52.15 (CH); 66.74 (CH2); 114.76 (2× CHAr); 121.53 (CHAr); 129.17 (CHAr); 129.46 (2× CHAr); 129.50 (CHAr); 130.00 (2× CHAr); 130.17 (CHAr); 136.63 (C); 158.07 (C); 167.48 (C). ESI (m/z): calcd for C19H22N2O2 310.2, found 311.0 [M + 1]; IR cm−1: 3443 (NH), 1691.33 (CO).
N-(3-Fluorophenyl)-2-phenoxy-N-(piperidin-4-yl) acetamide (6b). Following the general procedure, 4.65 g (10.87 mmol) of compound 5b were dissolved in 15 mL of CH2Cl2, 10.81 mL (141.31 mmol) of trifluoroacetique acid were added. (2.5 g, 70%); m.p.: 200 °C; 1H-NMR (CDCl3, 500 MHz): 1.3 (m, 2H, CH2); 1.80 (m, 2H, CH2); 2.85 (m, 2H, CH2); 3.7 (broad, 1H, NH); 4.06 (m, 2H, CH2); 4.31 (m, 2H, CH2); 4.67 (m, 1H, CH) 6.4–7.5 (m, 9H aromatic). 13C-NMR (125 MHz, CDCl3) δ: 27.14 (2× CH2); 43.99 (2× CH2); 51.11 (CH); 66.69 (CH2); 114.74 (2× CHAr); 117.34 (d, J = 50.53 Hz, CHAr); 121.97 (CHAr); 129.68 (2× CHAr); 129.86 (CHAr); 131.71 (CHAr); 138.23 (C); 153.69 (C); 164.11–161.27 (C); 168.211 (C). ESI (m/z): calcd for C19H21FN2O2 328.2; found 329.166 [M + 1]; IR cm−1: 3445 (NH), 1693.01 (CO).
2-(2-Chlorophenoxy)-N-Phenyl-N-(piperidin-4-yl) acetamide (6c). Compound 9 (3.90 g, 8.78 mmol) was dissolved in 15 mL of CH2Cl2, then 8.73 mL (114.14 mmol) of trifluoroacetic acid were added, to give 6c (3.02 g, 95%); m.p.: 161 °C; 1H-NMR (CDCl3, 600 MHz): 1.70 (m, 2H, CH2); 1.93 (broad, 1H, NH); 2.06 (m, 2H, CH2); 2.97 (m, 2H, CH2); 3.38 (m, 2H, CH2); 4.37 (s, 2H, CH2); 4.77 (m, 1H, CH); 6.5–7.5 (m, 9H Ar). 13C-NMR (150 MHz, CDCl3) δ: 27.12 (2× CH2); 43.55 (2× CH2); 50.68 (CH); 67.53 (CH2); 114.01 (2× CHAr); 122.27 (CHAr); 123.27 (C); 127.55 (CHAr); 129.55(2× CHAr); 129.84 (CHAr); 130.24 (CHAr); 130.55 (CHAr); 135.76 (C); 153.67 (C); 167.27 (C). ESI (m/z): calcd for C19H21ClN2O2 344.1; found 345.13 [M + 1]; IR cm−1: 3445 (NH), 1691.33 (CO).
2-(2-Chlorophenoxy)-N-(3-fluorophenyl)-N-(piperidin-4-yl) acetamide (6d). Following the general procedure, 1.99 g (4.327 mmol) of compound 9 were dissolved in 15 mL of CH2Cl2, and 4.30 mL (56.25 mmol) of trifluoroacetic acid were added to afford 6d (2.4 g 60%); m.p.: 140°C; 1H-NMR (CDCl3, 600 MHz): 1.40 (m, 2H, CH2); 2.06 (m, 2H, CH2); 2.7 (broad band, 1H, NH) 2.97 (m, 2H, CH2); 3.2 (m, 2H, CH2); 4.37 (s, 2H, CH2); 4.77 (m, 1H, CH); 6.5–7.5 (m, 8H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.9 (2× CH2); 45.7 (2× CH2); 53.11 (CH); 67.79 (CH2); 114.05 (CHAr); 116.55 (d, J = 24.9 Hz, CHAr); 117.74 (d, J = 106.5 Hz, CHAr); 122.447 (CHAr); 123.27 (CHAr); 126.068 (d, J = 3.75 Hz, CHAr); 127.673 (CHAr); 130.619 (CHAr); 131.958 (d, J = 10.95 Hz, CHAr); 138.292 (C); 153.780 (C); 161.963 (d, J = 298.5 Hz, C); 166.60 (C). ESI (m/z): calcd for C19H20ClFN2O2 362.1; found 363.12 [M + 1]; IR cm−1: 3445 (NH), 1691.33 (CO).
3.2.6. General Procedure for Synthesis of Target Compounds A
A solution of benzaldehyde derivatives (1 equiv) in 1,2-dichloroethane containing compound 6 (1 equiv), sodium triacetoxyborohydride (1.5 equiv) and acetic acid (1.5 equiv) was stirred for 24 h at 20 °C. 1N NaOH (15 mL) and 15 mL of ethyl acetate were added. The phases were separated and the aqueous layer was extracted with ethyl acetate (3 × 15 mL). The combined organic layers were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by chromatography on silica gel (petroleum ether/EtOAc (8:2)).
N-(1-Benzylpiperidin-4-yl)-2-phenoxy-N-phenylacetamide (12a). Following the general procedure for reductive amination, using benzaldehyde derivative 11 (R3 = H) (34 mg, 0.322 mmol); compound 6a (100 mg, 0.322 mmol); sodium triacetoxyborohydride (102 mg, 0.4838 mmol) and acetic acid (29 mg, 0.4838 mmol) in 1,2-dichloroethane (5 mL) give compound 12a (0.1291 g, 52%); m.p. 89 °C. 1H-NMR (CDCl3, 600 MHz): 1.32 (m, 2H, CH2); 1.74 (m, 2H, CH2); 2.06 (m, 2H, CH2); 2.83 (m, 2H, CH2); 3.42 (s, 2H, CH2); 4.15 (s, 2H, CH2); 4.62 (m, H, CH); 6.68–7.38 (m, 15H aromatic); 13C-NMR (150 MHz, CDCl3) δ: 32.17 (2× CH2); 43.72 (2× CH2); 52.84 (CH); 62.89 (CH2); 66.73 (CH2); 114.74 (4× CHAr); 121.32 (CHAr); 128.25 (CHAr); 129.08 (CHAr); 129.34 (4× CHAr); 129.68 (2× CHAr); 130.14 (2× CHAr); 135.30 (C) 136.69 (C); 158.14 (C); 167.25 (C). ESI (m/z): calcd for C26H28N2O2 400.02; found 401.22 [M + 1]; IR cm−1: 1680 (CO).
N-(1-Benzylpiperidin-4-yl)-N-(3-fluorophenyl)-2-phenoxyacetamide (12b). Following the general procedure, benzaldehyde derivative 11 (R3 = H(o)) (48.4 mg, 0.457 mmol); compound 6b (150 mg, 0.457 mmol); sodium triacetoxyborohydride (145.3 mg, 0.6855 mmol) and acetic acid (41.6 mg, 0.6855 mmol) in 1.2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to furnish 12b (0.103 g, 54%); m.p.: 90 °C. 1H-NMR (CDCl3, 500 MHz): 1.36 (m, 2H, CH2); 1.83 (m, 2H, CH2); 2.15 (m, 2H, CH2); 2.94 (m, 2H, CH2); 3.50 (s, 2H, CH2,); 4.30 (s, 2H, CH2); 4.67 (m, H, CH); 6.78–7.47 (m, 14H aromatic). 13C-NMR (125 MHz, CDCl3) δ: 30.26 (2× CH2); 43.72 (2× CH2); 52.84 (CH); 62.80 (CH2); 66.81 (CH2); 114.73 (4× CHAr); 117.50 (d, J = 22 Hz, CHAr); 121.47 (2× CHAr); 126.03 (CHAr); 127.16 (C); 128.24 (2× CHAr); 129.16 (CHAr); 129.40 (2× CHAr); 130.66 (CHAr); 138.67 (C); 157.99 (C); 161.59 (d, J = 251 Hz, C); 167.06 (C). ESI (m/z): calcd for C26H27FN2O2 418.2; found 419.21 [M + 1]; IR cm−1: 1687.
2-(2-Chlorophenoxy)-N-(1-benzylpiperidin-4-yl)-N-phenylacetamide (12c). Following the general procedure, benzaldehyde derivative 11 (R3 = H) (46.2 mg, 0.435 mmol); compound 6c (150 mg, 0.435 mmol); sodium triacetoxyborohydride (138.5 mg, 0.6538 mmol) and acetic acid (39.5 mg, 0.6538 mmol) in 1.2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to afford 12c (0.0946 g, 50%); m.p.: 83 °C; 1H-NMR (CDCl3, 400 MHz): 1.47 (m, 2H, CH2); 1.84 (m, 2H, CH2); 2.14 (m, 2H, CH2); 2.94 (m, 2H, CH2); 3.48 (s, 2H, CH2); 4.35 (s, 2H, CH2); 4.64 (m, H, CH); 6.73–7.47 (m, 14H aromatic). 13C-NMR (100 MHz, CDCl3) δ: 30.23 (2× CH2); 43.37 (2× CH2); 52.88 (CH); 62.90 (CH2); 67.66 (CH2); 114.01 (2× CHAr); 122.09 (2× CHAr).; 123.26 (C); 127.11 (C); 127.46 (2× CHAr); 128,21 (2× CHAr); 129.12 (CHAr); 129.71 (2× CHAr); 130.18 (2× CHAr); 130.41 (CHAr); 136.99 (C); 153.94 (C); 166.67 (C). ESI (m/z): calcd for C26H27ClN2O2 434.2; found 435.18 [M + 1]; IR cm−1: 1683 (CO).
2-(2-Chlorophenoxy)-N-(1-benzylpiperidin-4-yl)-N-(3-fluorophenyl) acetamide (12d). Following the general procedure, benzaldehyde derivatives 11 (R3 = H(o)) (43.91 mg, 0.4142 mmol); compound 6d (150, 0.4142 mmol); sodium triacetoxyborohydride (131.6 mg, 0.6213 mmol) and acetic acid (37.3 mg, 0.6213 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C, giving 12d (0.0936 g, 54%); m.p.: 73 °C. 1H-NMR (CDCl3, 600 MHz): 1.25 (m, 2H, CH2); 1.43 (m, 2H, CH2); 1.78 (m, 2H, CH2); 2.14 (m, 2H, CH2); 2.92 (s, 2H, CH2); 3.49 (s, 2H, CH2).; 4.36 (s, 2H, CH2); 4.63 (m, H, CH); 6.73–7.47 (m, 13H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.12 (2× CH2); 43.37 (2× CH2); 52.82 (CH); 62.90 (CH2); 67.77 (CH2); 114.03 (2× CHAr); 116.50 (CHAr); 116.67 (C); 117.63 (d, J = 21.36 Hz, CHAr); 122.39 (2× CHAr); 123.31 (C); 126.11 (d, J = 2.51 Hz, CHAr); 127.65 (CHAr); 128.43 (CHAr); 129.41 (CHAr); 130.60 (2× CHAr); 130.93 (d, J = 8.79 Hz, CHAr); 138.36 (C); 153.81 (C); 161.95 (d, J = 250.14 Hz, C); 166.67 (C). ESI (m/z): calcd for C26H26ClFN2O2 452.2; found 453.18 [M + 1]; IR cm−1: 1689 (CO).
N-(1-(2-Bromobenzyl)piperidin-4-yl)-2-phenoxy-N-phenylacetamide (13a). Following the general procedure for reductive amination using benzaldehyde derivative 11 (R3 = Br (o)) (75.3 mg, 0.410 mmol); compound 6a (127 mg, 0.410 mmol); sodium triacetoxyborohydride (130.15 mg, 0.615 mmol) and acetic acid (36.9 mg, 0.615 mmol)) in 1,2-dichloroethane (5 mL) to give compound 13a (0.119 g, 60%); m.p. 66°C. 1H-NMR (CDCl3, 600 MHz): 1.44 (m, 2H, CH2); 1.64 (m, 2H, CH2); 2.25 (m, 2H, CH2); 2.90 (m, 2H, CH2); 3.53 (s, 2H, CH2,); 4.23 (s, 2H, CH2); 4.70 (m, H, CH); 6.73–7.47 (m, 14H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.50 (2× CH2); 53.07 (CH); 53.13 (2× CH2); 61.77 (CH2); 66.77 (CH2); 114.80 (2× CHAr); 121.42 (CHAr); 124.69 (C); 127.27 (CHAr); 128.44 (CHAr); 129.20 (CHAr); 129.46 (2× CHAr); 129.82 (2× CHAr); 130.24 (2× CHAr); 130.64 (CHAr); 132.82 (CHAr); 137.12 (C); 137.21 (C); 158.19 (C); 167.33 (C). ESI (m/z): calcd for C26H27BrN2O2 478.1; found 479.13 [M + 1]; IR cm−1: 1678 (CO).
N-(1-(2-Bromobenzyl)piperidin-4-yl)-N-(3-fluorophenyl)-2-phenoxyacetamide (13b). Following the general procedure, benzaldehyde derivatives 11 (R3 = Br (o)) (84.5 mg; 0.457 mmol); compound 6b (150 mg, 0.457 mmol); sodium triacetoxyborohydride (142.29 mg, 0.685 mmol) and acetic acid (41.16 mg, 0.685 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C. Yield of 13b: 0.176 g (58%); IR cm−1: 1687; MS: 469.14 [M + 1]; m.p.: 55 °C; H1-NMR (CDCl3, 500 MHz): 1.36 (m, 2H, CH2); 1.83 (m, 2H, CH2); 2.15 (m, 2H, CH2); 2.94 (m, 2H, CH2); 3.50 (s, 2H, CH2,); 4.30 (s, 2H, CH2); 4.67 (m, H, CH); 6.78–7.47 (m, 13H aromatic). 13C-NMR (125 MHz, CDCl3) δ: 30.26 (2× CH2); 42.89 (2× CH2); 52.98 (CH); 61.65 (CH2); 66.88 (CH2); 114.81 (4× CHAr); 116.42 (d, J = 20 Hz, CHAr); 117.52 (d, J = 20 Hz, CHAr); 121.47 (2× CHAr); 126.06 (CHAr); 128.56 (C); 129.53 (4× CHAr); 130.22 (d, J = 11,25 Hz, C); 132.90 (CHAr); 138.73 (C); 158.05 (C); 161.59 (d, J = 247,5 Hz, C); 167.23 (C). ESI (m/z): calcd for C26H26BrFN2O2 496.1; found 497.14 [M + 1]. IR cm−1: 1687 (CO).
N-(1-(2-Bromobenzyl)piperidin-4-yl)-2-(2-chlorophenoxy)-N-phenylacetamide (13c). Following the general procedure, benzaldehyde derivatives 11 (R3 = Br (o)) (80.5 mg; 0.435 mmol); compound 6c (150 mg, 0.435 mmol); sodium triacetoxyborohydride (138 mg, 0.653 mmol) and acetic acid (39.2 mg, 0.653 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to give 13c (0.122 g, 55%); m.p.: 64 °C. 1H-NMR (CDCl3, 400 MHz): 1.36 (m, 2H, CH2); 1.86 (m, 2H, CH2); 2.27 (m, 2H, CH2); 2.94 (m, 2H, CH2); 3.57 (s, 2H, CH2); 4.35 (s, 2H, CH2); 4.742 (m, H, CH); 6.73–747 (m, 13H aromatic). 13C-NMR (100 MHz, CDCl3) δ: 30.34 (2× CH2); 42.98 (2× CH2); 52.97 (CH); 61.63 (CH2); 67.66 (2H, CH2); 113.99 (2× CHAr); 122.10 (CHAr); 123.35 (C); 124.62 (C); 127.21 (C); 127.47 (2× CHAr); 128.82 (2× CHAr); 129.74 (2× CHAr); 130.14 (CHAr); 130.42 (CHAr); 132.72 (2× CHAr); 136.91 (C); 153.93 (C); 166.70 (C). ESI (m/z): calcd for C26H6BrClN2O2 512.1; found 513.09 [M + 1]; IR cm−1: 1651 (CO).
N-(1-(2-Bromobenzyl)piperidin-yl)-2-(2-chlorophenoxy)-N-(3-fluorophenyl) acetamide (13d). Following the general procedure, benzaldehyde derivative 11 (R3 = Br (o)) (76.63 mg, 0.4142 mmol); compound 6d (150 mg, 0.4142 mmol); sodium triacetoxyborohydride (131.6 mg, 0.6213 mmol) and acetic acid (37.3 mg, 0.6213 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to furnish 13d (0.1295 g, 59%); m.p.: 100 °C; 1H-NMR (CDCl3, 600 MHz): 1.33 (m, 2H, CH2); 1.80 (m, 2H, CH2); 2.23 (m, 2H, CH2); 2.92 (m, 2H, CH2); 3.54 (s, 2H, CH2,); 4.37 (s, 2H, CH2); 4.74 (m, H, CH); 6.73–7.47 (m, 12H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.45 (2× CH2); 43.41 (2× CH2); 53.04 (CH); 61.72 (CH2); 67.74 (CH2); 113.95 (2× CHAr); 116.50 (d, J = 21 Hz, CHAr); 117.78 (d, J = 21 Hz, CHAr); 122.38 (CHAr); 123.25 (C); 124.70 (C); 126.17 (CHAr); 127.32 (CHAr); 127.66 (CHAr); 128.51 (CHAr); 130.60 (CHAr); 130.92 (d, J = 9 Hz, CHAr); 132.86 (CHAr);138.60 (C); 138.51 (C); 153.78 (C); 162.10 (d, J = 249 Hz, C); 166.68 (C). ESI (m/z): calcd for C26H25BrClFN2O2 530.1; found 531.08 [M + 1]; IR cm−1: 1689 (CO).
N-(1-(2-Chlorobenzyl)piperidin-4-yl)-2-phenoxy-N-phenylacetamide (14a). Following the general procedure for reductive amination using benzaldehyde derivative 11 (R3 = Cl (o)) (43.5 mg, 0.4838 mmol); compound 6a (150 mg, 0.4838 mmol); sodium triacetoxyborohydride (153.8 mg, 0.7253 mmol) and acetic acid (43.5 mg, 0.7253 mmol) in 1,2-dichloroethane (5 mL) gave title compound 14a (0.109 g, 52%); m.p.: 69 °C. 1H-NMR (CDCl3, 500 MHz): 1.47 (m, 2H, CH2); 1.81 (m, 2H, CH2); 2.28 (m, 2H, CH2); 2.94 (m, 2H, CH2); 3.59 (s, 2H, CH2); 4.23 (s, 2H, CH2); 4.67 (m, H, CH); 6.73–7.47 (m, 14H aromatic). 13C-NMR (125 MHz, CDCl3) δ: 30.32 (2× CH2); 42.30 (2× CH2); 53.03 (CH2); 66.82 (CH2); 114.84 (4× CHAr); 121.45 (2× CHAr); 126.73 (C); 129.25 (CHAr); 129.47 (4× CHAr); 129.58 (CHAr): 129.85 (CHAr); 130.20 (CHAr); 134.50 (C); 137.02 (C); 158.22 (C); 167.40 (C). ESI (m/z): calcd for C26H27ClN2O2 434.2; found 435.18 [M + 1]; IR cm−1: 1673 (CO).
N-(1-(2-Chlorobenzylpiperidin-4-yl)-2-(2-chlorophenoxy)-N-phenylacetamide (14c). Following the general procedure, benzaldehyde derivative 11 (R3 = Cl (o)) (61.02 mg; 0.4359 mmol); compound 6c (150 mg, 0.4359 mmol); sodium triacetoxyborohydride (138.58 mg, 0.6538 mmol) and acetic acid (39.26 mg, 0.6538 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to produce 14c (0.1326 g, 65%); m.p.: 55 °C; 1H-NMR (CDCl3, 400 MHz): 1.36 (m, 2H, CH2); 1.75 (m, 2H, CH2); 2.16 (m, 2H, CH2); 2.84 (m, 2H, CH2); 3.49 (s, 2H, CH2); 4.25 (s, 2H, CH2); 4.62 (m, H, CH); 6.64–7.37 (m, 13H aromatic). 13C-NMR (100 MHz, CDCl3) δ: 30.38 (2× CH2); 52.99 (CH); 53.03 (2× CH2); 59.09 (CH2); 67.64 (CH2); 113.97 (2× CHAr); 122.10 (2× CHAr); 123.24 (C); 127.47 (2× CHAr); 129.15 (C); 129.35 (CHAr); 129.44 (C); 129.75 (2× CHAr); 130.15 (2× CHAr); 130.42 (2× CHAr); 140.41 (C); 153.92 (C); 166.69 (C). ESI (m/z): calcd for C26H26Cl2N2O2 468.1; found 469.14 [M + 1]. IR cm−1: 1687 (CO).
N-(1-(2-Chlorobenzyl)piperidin-4-yl)-2-(2-chlorophenoxy)-N-(3-fluorophenyl) acetamide (14d). Following the general procedure, benzaldehyde derivative 11 (R3 = Cl(o)) (57.9 mg, 0.4142 mmol); compound 6d (150 mg, 0.4142 mmol); sodium triacetoxyborohydride (131.6 mg, 0.6213 mmol) and acetic acid (37.3 mg, 0.6213 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to give 14d (0.11 g, 55%); m.p.: 85 °C; 1H-NMR (CDCl3, 600 MHz): 1.44 (m, 2H, CH2); 1.78 (m, 2H, CH2); 2.22 (m, 2H, CH2); 2.92 (m, 2H, CH2); 3.56 (s, 2H, CH2); 4.36 (s, 2H, CH2); 4.64 (m, H, CH); 6.73–7.37 (m, 12H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.42 (2× CH2); 53.04 (CH); 53.37 (2× CH2); 59.17 (CH2); 67.74 (CH2); 113.95 (2× CHAr); 116.62 (d, J = 21 Hz, CHAr); 117.78 (d, J = 21 Hz, CHAr); 122.36 (CHAr); 123.25 (C); 126.16 (CHAr); 126.68 (CHAr); 127.66 (CHAr); 128.24 (CHAr); 129.55 (CHAr); 130.59 (CHAr); 130.91 (d, J = 10.5 Hz, CHAr); 134.34 (C); 136 (C); 138.44 (d, J = 7.5 Hz, C); 153.78 (C); 162.09 (d, J = 249 Hz, C); 166.62 (C). ESI (m/z): calcd for C26H25Cl2FN2O2 486.1; found 487.13 [M + 1]; IR cm−1: 1688 (CO).
N-(1-(2.4-Dihydroxybenzyl) piperidin-4-yl)-2-phenoxy-N-phenylacetamide (15a). Following the general procedure for reductive amination using benzaldehyde derivative 11 (R3 = OH (o), OH (p)) (66.86 mg, 0.4838 mmol); compound 6a (150 mg, 0.4838 mmol); sodium triacetoxyborohydride (153.8 mg, 0.725 mmol) and acetic acid (43 mg, 0.725 mmol) in 1,2-dichloroethane (5 mL) give compound 15a (0.1233 g, 59%); m.p. 68 °C. 1H-NMR (CDCl3, 600 MHz): 1.42 (m, 2H, CH2); 2.23 (m, 2H, CH2); 3.03 (m, 2H, CH2); 3.16 (m, 2H, CH2); 3.60 (s, 2H, CH2); 4.23 (s, 2H, CH2); 4.86 (m, H, CH); 5.30 (s, 2× OH); 6.73–7.42 (m, 13HAr). 13C-NMR (150 MHz, CDCl3) δ: 30.27 (2× CH2); 43.66 (2× CH2); 51.96 (CH2); 52.80 (CH); 67.24 (CH2); 114.57 (CHAr); 114.79 (4× CHAr); 121.57 (CHAr); 129.52 (4× CHAr); 129.73 (CHAr); 129.98 (CHAr); 130.09 (CHAr); 136.57 (C); 156.45 (C); 156.66 (C); 157.51 (C); 158.10 (C); 166.78 (C). ESI (m/z): calcd for C26H28O4 432.2; found 433.22 [M + 1]; IR cm−1: 1661 (CO).
N-(-1(2-Hydroxy-6-methoxybenzyl)piperidin-4-yl)-2-phenoxy-N-phenylacetamide (16a). Following the general procedure reductive amination using benzaldehyde derivative 11 (R3= OMe (o), OH (o)) (73.55 mg, 0.4838 mmol); compound 6a (150 mg, 0.4838 mmol); sodium triacetoxyborohydride (153.8 mg, 0.7253 mmol) and acetic acid (43.5 mg, 0.7253 mmol) in 1.2-dichloroethane (5 mL) give the title compound 16a (0.114 g, 53%); mp 68 °C. 1H-NMR (CDCl3, 600 MHz): 1.44 (m, 2H, CH2); 1.87 (m, 2H, CH2); 2.25 (m, 2H, CH2); 3.02 (m, 2H, CH2); 3.75 (s, 5H, CH2, CH3); 4.22 (s, 2H, CH2); 4.62 (m, H, CH); 6.68–7.38 (m, 13HAr). 13C-NMR (150 MHz, CDCl3) δ: 30.30 (2× CH2); 43.72 (2× CH2); 52.45 (CH2); 52.62 (CH); 55.61 (CH3); 66.68 (CH2); 101.61 (CHAr); 109.32 (C); 114.91 (2× CHAr); 121.49 (CHAr); 128.71 (CHAr); 129.49 (2× CHAr); 129.51 (2× CHAr); 130.03 (2× CHAr); 130.06 (2× CHAr); 136.81 (C); 157.71 (C); 158.14 (C); 159.11 (C); 167.54 (C). ESI (m/z): calcd for C27H30N2O4 446.2; found 447.29 [M + 1]; IR cm−1: 1678 (CO).
N-(1-(Perfluorobenzyl)piperidin-4-yl)-2-phenoxy-N-phenylacetamide (17a). Following the general procedure, benzaldehyde derivative 11 (R3 = 5xF (o,m,p)) (94.87 mg; 0.483 mmol); compound 6a (150 mg, 0.483 mmol); sodium triacetoxyborohydride (153 mg, 0.7258 mmol) and acetic acid (43 mg, 0.7258 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to afford 17a (0.118 g, 50%); m.p.: 104 °C. 1H-NMR (CDCl3, 600 MHz): 1.42 (m, 2H, CH2); 1.79 (m, 2H, CH2); 2.24 (m, 2H, CH2); 2.88 (m, 2H, CH2); 3.65 (s, 2H, CH2); 4.21 (s, 2H, CH2); 4.60 (m, H, CH); 6.73–7.42 (m, 10H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.27 (2× CH2); 48.64 (CH2); 52.18 (2× CH2); 52.40 (CH); 66.77 (CH2); 114.81 (2× CHAr); 121.46 (CHAr); 129.27 (CHAr); 129.47 (2× CHAr); 129.84 (2× CHAr); 110.16 (C); 130.22 (2× CHAr); 136.56 (C); 136.88 (C); 138.16 (C); 144.42 (2× C); 146.42 (C); 158.15 (C); 167.36 (C). ESI (m/z): calcd for C26H23F5 N2O2 490.2; found 491.17 [M +1]; IR cm−1: 1683 (CO).
N-(3-Fluorophenyl)-N-(1-(perfluorobenzyl)piperidin-4-yl)-2-phenoxyacetamide (17b). Following the general procedure, benzaldehyde derivative 11 (R3 = 5xF (o,m,p)) (89.6 mg, 0.457 mmol); compound 6b (150 mg, 0.457 mmol); sodium triacetoxyborohydride (142.29 mg, 0.685 mmol) and acetic acid (41.16 mg, 0.685 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C, to furnish 17b (0.125 g, 54%); m.p.: 95 °C. 1H-NMR (CDCl3, 600 MHz): 1.42 (m, 2H, CH2); 1.9 (m, 2H, CH2); 2.24 (m, 2H, CH2); 2.93 (m, 2H, CH2); 3.69 (s, 2H, CH2); 4.28 (s, 2H, CH2); 4.58 (m, H, CH); 6.73–7.42 (m, 9H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.27 (2× CH2); 43.66 (2× CH2); 51.79 (CH2); 52.40 (CH); 67.24 (CH2); 113.98 (2× CHAr); 116.90 (d, J = 19.5 Hz, CHAr); 117.60 (d, J = 21 Hz, CHAr); 122.52 (CHAr); 122.66 (CHAr); 123.18 (C); 125.95 (CHAr); 127.69 (2× CHAr); 130.66 (CHAr); 131.15 (C); 131.29 (d, J = 10.5 Hz, CHAr); 137.81 (2× C); 138.26 (2× C); 146.67 (C); 153.70 (C); 162 (d, J = 249 Hz, C); 167.77 (C). ESI (m/z): calcd for C26H25Cl2FN2O2 508.2; found 509.11 [M + 1]; IR cm−1: 1676 (CO).
2-(2-Chlorophenoxy)-N-(1-(perfluorobenzyl)piperidin-4-yl)-N-phenylacetamide (17c). Following the general procedure, benzaldehyde derivatives 11 (R3 = 5xF (o,m,p)) (85.57 mg; 0.435mmol); compound 6c (150 mg, 0.435 mmol); sodium triacetoxyborohydride (138 mg, 0.653 mmol) and acetic acid (39.2 mg, 0.653 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C. Yield of 17c: 0.139 g (61%); m.p.: 78 °C; 1H-NMR (CDCl3, 600 MHz): 1.42 (m, 2H, CH2); 1.79 (m, 2H, CH2); 2.24 (m, 2H, CH2); 2.93 (m, 2H, CH2); 3.69 (s, 2H, CH2,); 4.28 (s, 2H, CH2); 4.58 (m, H, CH); 6.73–7.42 (m, 9H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.27 (2× CH2); 48.64 (CH2); 52.18 (2× CH2); 52.40 (CH); 67.24 (CH2); 113.85 (2× CHAr); 122.51 (CHAr); 123.18 (C); 127.61 (2× CHAr); 129.61 (CHAr); 130.11 (CHAr); 130.56 (2× CHAr); 136.27 (C); 136.50 (2× C); 138.47 (2× C); 146.49 (C); 146.31 (C); 153.88 (C); 167.77 (C). ESI (m/z): calcd for C26H22ClF5 N2O2 524.1; found 525.13 [M + 1]; IR cm−1: 1683 (CO).
2-(2-Chlorophenoxy)-N-(3-fluorophenyl)-N-(1-(perfluorobenzyl) piperidin-4-yl) acetamide (17d). Following the general procedure, benzaldehyde derivative 11 (R3 = 5xF (o, m,p)) (81.19 mg, 0.4142 mmol); compound 6d (150 mg, 0.4142 mmol); sodium triacetoxyborohydride (131.6 mg, 0.6213 mmol) and acetic acid (37.3 mg, 0.6213 mmol) in 1,2-dichloroethane (5 mL) was stirred for 24 h at 20 °C to provide 17d (0.1324 g, 59%); m.p.: 125 °C; 1H-NMR (CDCl3, 600 MHz): 1.42 (m, 2H, CH2); 1.9 (m, 2H, CH2); 2.24 (m, 2H, CH2); 2.93 (m, 2H, CH2); 3.69 (s, 2H, CH2); 4.28 (s, 2H, CH2); 4.58 (m, H, CH); 6.73–7.42 (m, 8H aromatic). 13C-NMR (150 MHz, CDCl3) δ: 30.27 (2× CH2); 43.66 (2× CH2); 51.79 (CH2); 52.40 (CH); 67.24 (CH2); 113.98 (CHAr); 116.90 (d, J = 19.5 Hz, CHAr); 117.60 (d, J = 21 Hz, CHAr); 122.52 (CHAr); 122.66 (C); 123.18 (C); 125.95 (CHAr); 127.69 (CHAr); 130.66 (CHAr); 131.15 (C); 131.29 (d, J = 10.5 Hz, CHAr); 137.81 (2× C); 138.26 (2× C); 146.67 (C); 153.70 (C); 162 (d, J = 249 Hz, C); 167.77 (C). ESI (m/z): calcd for C26H22ClF5 N2O2 542.1; found 543.13 [M + 1]; IR cm−1: 1695 (CO).
3.3. Biological Assays
3.3.1. Antiplasmodial Assay
The antimalarial activity of extracts/compounds was evaluated against P. falciparum 3D7 and P. falciparum W2 strains, using the fluorescence-based SYBR Green I assay approach in 96-well microplates as described by Smilkstein et al. [34] with some modifications. Positive control wells for each assay contained no inhibitor while negative controls contained Chloroquine (CQ). The CQ molecule was provided from World Wide Antimalarial Resistance Network (wwarn Network). Experiments were run in duplicate with both test and control drugs employed at varying concentrations. Stock solutions (extracts) were prepared in dimethyl-sulfoxide (DMSO) and diluted with culture medium to give a maximum DMSO concentration of 0.5% in a final well volume of 200 μL containing 1% parasitemia and 2.5% haematocrit. Extracts and negative control (chloroquine (CQ)) were prepared by two-fold dilution, in a dose-titration range of 0.098–100 µg/mL, to obtain 11 concentrations each, in duplicate. The concentrations used for CQ were between 0.5 and 1000 nM. After 48 h incubation, the plates were subjected to 3 freeze thaw cycles to achieve complete hemolysis. The parasite lysis suspension was diluted 1:5 in SYBR Green I lysis buffer (10 mM NaCl, 1 mM Tris HCl pH 8, 2.5 mM EDTA pH 8, 0.05% SDS, 0,01 mg/mL proteinase K and 10X SYBR Green I). Incorporation of SYBR Green I in parasite DNA amplification was measured using the Master epRealplex cycler® (Eppendorf, Montesson, France) according the following program to increase the SYBR green incorporation: 90 °C for 1 min, decrease in temperature from 90 °C to 10 °C for 5 min with reading the fluorescence 10 °C for 1 min and a new reading at 10 °C for 2 min. The IC50 was calculated by nonlinear regression using icestimator website 1.2 version: http://www.antimalarial-icestimator.net/MethodIntro.htm.
3.3.2. Cytotoxicity on HUVEC
HUVEC cells were cultured in Gibco™ RPMI 1640 medium (Life Technologies, Saint-Aubin, France) complemented with 10% Fetal Bovine Serum and 1 mM l-glutamine (Sigma-Aldrich, Lesquin, France) and incubated in 5% CO2 at 37 °C. The cytotoxicity of extracts was evaluated using the SYBR Green I assay as previously described. HUVEC were seeded in a 96-well plate at 100,000 cells/well and incubated for 24 h to adhere. After discarding the old medium, the cells were incubated in the medium containing eight concentrations (0.78–100 μg/mL) of each extract in duplicate. After 48 h incubation, cells were visualized using an inverted microscope to check their morphology or the cell viability. The medium was subsequently removed and replaced by lysis buffer without SYBR Green I and the plates were subjected to 3 freeze-thaw cycles. The cell lysis suspension was diluted 1:2 in SYBR Green I lysis buffer. The incorporation of SYBR Green I in cell DNA and the IC50 analysis were obtained as previously.
4. Conclusions
In this study, we have prepared a small library of new nitrogen heterocycles displaying piperidine scaffolds using a flexible synthetic approach. Eighteen new derivatives were prepared in good yield. The antimalarial activity of these compounds has been described. The compounds were tested against P. falciparum 3D7 strains and W2. The best result is observed with the compounds 13b against the 3D7 strain and 12a against the W2 one with a selectivity index greater than chloroquine. We observed that modification with different R groups, for example compound 12a (R1 = R2 = R3 = H) in 13b (R1 = F, R2 = H, R3 = Br) significantly modulated the activity of the tested molecules. These molecules could be further optimized to provide good malaria drug candidates.
Author Contributions
R.S. performed the synthetic, drew the molecules and searched the literatures, A.G. and C.C. designed the target compounds, provided guidance to optimization the synthesis process and wrote paper, S.C. conceived and performed the biological assay. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the French cooperation.
Acknowledgments
We thank Université de REIMS, Institut de chimie moléculaire de Reims (ICMR), France, for recording NMR, Université Paris XI, France, for bioactive tests and the French cooperation for the attribution of the bourse for Rokhyatou Seck.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Health Organization. World Malaria Report; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Noedl, H. The need for new antimalarial drugs less prone to resistance. Curr. Pharm. Des. 2013, 19, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Njuguna, N.; Ongarora, D.S.B.; Chibale, K. Artemisin derivatives: A patent review (2006-present). Expert. Opin. Ther. Pat. 2012, 22, 1179–1203. [Google Scholar] [CrossRef] [PubMed]
- Ongarora, D.S.B.; Strydom, N.; Wicht, K.; Njoroge, M.; Wiesner, L.; Egan, T.J.; Wittlin, S.; Jurva, U.; Masimirembwa, C.M.; Chibale, K. Antimalarial benzoheterocyclic 4-aminoquinolines: Structure-activity relationship, in vivo evaluation, mechanistic and bioactivation studies. Bioorg. Med. Chem. 2015, 23, 5419–5432. [Google Scholar] [CrossRef] [PubMed]
- Toshio, H.; Fumihiro, I. Shin-Ichi, Y. Direct Arylation of 2-Pyridones; Photostimulated SRN1 Reaction between Cesium Phenoxides and Chloro-2-pyridones. Heterocycles 2000, 52, 253–260. [Google Scholar]
- Kohnen-Johannsen, K.L.; Kayser, O. Tropane Alkaloids: Chemistry, Pharmacology, Biosynthesis and Production. Molecules 2019, 24, 796. [Google Scholar] [CrossRef]
- Huang, J.P.; Fang, C.; Ma, X.; Wang, L.; Yang, J.; Luo, J.; Yan, Y.; Zhang, Y.; Huang, S.X. Tropane alkaloids biosynthesis involves an unusual type III polyketide synthase and non-enzymatic condensation. Nat. Commun. 2019, 10, 4036. [Google Scholar] [CrossRef]
- Santos, A.S.; Lukens, K.A.; Coelho, L.; Noguero, F.; Wirth, D.F.; Mazitschek, R.; Moreira, R.; Paulo, A. Exploring the 3-piperidin-4-yl-1H-indole scaffold as a novel antimalarial chemotype. Eur. J. Med. Chem. 2015, 102, 320–333. [Google Scholar] [CrossRef]
- Ishiguro, Y.; Kubota, T.; Ishiuchi, K.; Fromont, J.; Kobayashi, J. A novel piperidine alkaloid from an Okinawan marine sponge Plakortis sp. Tetrahedron Lett. 2009, 50, 3202–3204. [Google Scholar] [CrossRef]
- Kubizna, P.; Spanik, I.; Kozisek, J.; Szolcsanyi, P. Synthesis of 2,6-disubstituted piperidine alkaloids from ladybird beetles Calvia 10-guttata and Calvia 14-guttata. Tetrahedron 2010, 66, 2351–2355. [Google Scholar] [CrossRef]
- Gassama, A.; Ernenwein, C.; Hoffmann, N. Photochemical Key Steps in the Synthesis of Surfactants; from Furfural-Derived Intermediates. ChemSusChem 2009, 2, 1130–1137. [Google Scholar] [CrossRef]
- Weis, R.; Schweiger, K.; Faist, J.; Rajkovic, E.; Kungl, A.J.; Fabian, W.M.F.; Schunack, W.; Seebacher, W. Antimycobacterial and H1-antihistaminic activity of 2-substituted piperidine derivatives. Bioorg. Med. Chem. 2008, 16, 10326–10331. [Google Scholar] [CrossRef] [PubMed]
- Dambuza, N.S.; Smith, P.; Evans, A.; Norman, J.; Taylor, D.; Andayi, A.; Egan, T.; Chibale, K.; Wiesner, L. Antiplasmodial activity, in vivo pharmacokinetics and anti-malarial efficacy evaluation of hydroxypyridinone hybrids in a mouse model. Malar. J. 2015, 14, 505. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Scott, O.D. Metabolism of 4-Aminopiperidine Drugs by Cytochrome P450s: Molecular and Quantum Mechanical Insights into Drug Design. Med. Chem. Lett. 2011, 2, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Heringa, M. Review on raloxifene: Profile of a selective estrogen receptor modulator. Int. J. Clin. Pharmacol. Ther. 2003, 41, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Vogel, V.; Constantino, J.P.; Wickerman, L. Effects of tamoxifen vs raloxifene on the risk of developing invasive breast cancer and other disease outcomes: The NSABP Study of Tamoxifen and Raloxifene. JAMA 2006, 295, 2727–2741. [Google Scholar] [CrossRef]
- Wei, X.; Nieves, K.; Rodríguez, A.D. Neopetrosiamine A, biologically active bis-piperidine alkaloid from the Caribbean Sea sponge Neopetrosia proxima. Bioorg. Med. Chem. Lett. 2010, 20, 5905–5908. [Google Scholar] [CrossRef] [PubMed]
- Varty, G.B.; Cohen-Williams, M.E.; Hunter, J.C. The antidepressant-like effects of neurokinin NK1 receptor antagonists in a gerbil tail suspension test. Behav. Pharmacol. 2003, 14, 87–95. [Google Scholar] [CrossRef]
- Varty, G.B.; Cohen-Williams, M.E.; Morgan, C.A. The gerbil elevated plus-maze II: Anxiolytic-like effects of selective neurokinin NK1 receptor antagonists. Neuropsychopharmacology 2002, 27, 371–379. [Google Scholar] [CrossRef]
- Watanabe, Y.; Asai, H.; Ishii, T.; Kiuchi, S.; Okamoto, M.; Taniguchi, H.; Nagasaki, M.; Saito, A. Pharmacological characterization of T-2328, 2-fluoro-4′-methoxy-3′-[[[(2S,3S)-2-phenylpiperidinyl]-amino]methyl]-[1,1′-biphenyl]-4-carbonitrile dihydrochloride, as a brain-penetrating antagonist of tachykinin NK1 receptor. J. Pharm. Sci. 2008, 106, 121–127. [Google Scholar] [CrossRef]
- Watson, P.S.; Jiang, B.; Scott, B. A Diastereoselective Synthesis of 2.4-Disubstituted Piperidines: Scaffolds for Drug Discovery. Org. Lett. 2000, 2, 3679–3681. [Google Scholar] [CrossRef]
- Padmanilayam, M.; Scorneaux, B.; Dong, Y.; Chollet, J.; Matile, H.; Charman, S.A.; Creek, D.J.; Charman, W.N.; Tomas, S.T.; Scheurer, C.; et al. Antimalarial activity of N-alkyl amine, carboxamide, sulfonamide and urea derivatives of a dispiro-1,2,4-trioxolane piperidine. Bioorg. Med. Chem. Lett. 2006, 16, 5542–5545. [Google Scholar] [CrossRef] [PubMed]
- Meyers, M.J.; Anderson, E.J.; McNitt, S.A.; Krenning, T.M.; Singh, M.; Xu, J.; Zeng, W.; Qin, L.; Xu, W.; Zhao, S.; et al. Evaluation of spiropipéridine hydantoins as a novel class of antimalarial agent. Bioorg. Med. Chem. 2015, 23, 5144–5150. [Google Scholar] [CrossRef] [PubMed]
- Misra, M.; Pandey, S.K.; Pandey, V.P.; Pandey, J.; Tripathi, R.; Tripathi, R.P. Organocatalyzed highly atom economic one pot synthesis of tetrehydropyridines as antimalarials. Bioorg. Med. Chem. 2009, 17, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Sabbani, S.; Stocks, P.A.; Ellis, G.L.; Davies, J.; Hedenstrom, E.; Ward, S.A. O’Neill, P.M. Piperidine dispiro-1,2,4-trioxane analogues. Bioorg. Med. Chem. Lett. 2008, 18, 5804–5808. [Google Scholar] [CrossRef]
- Kikuchi, H.; Tasaka, H.; Hirai, S.; Takaya, Y.; Iwabuchi, Y.; Ooi, H.; Hatakeyama, S.; Kim, H.S.; Wataya, Y.; Oshima, Y. Potent Antimalarial Febrifugine Analogues Against the Plasmodium Malaria Parasite. J. Med. Chem. 2002, 45, 2563–2570. [Google Scholar] [CrossRef]
- Gassama, A.; Diatta, A. Synthesis of N-Substituted piperidines from piperidone. J. Soc. Ouest-Afr. Chim. 2015, 39, 31–40. [Google Scholar]
- Amed, F.; Magid, A.; Cynthia, A.; Kenneth, M.; Carson, G. Reductive amination of aldehydes and ketones by using sodium triacetoxyborohydride. Tetrahedron Lett. 1990, 31, 5595–5598. [Google Scholar] [CrossRef]
- Borch, R.F.; Bernstein, M.D.; Durst, H.D. Cyanohydridoboration anion as a selective reducing agent. J. Am. Chem. Soc. 1971, 93, 2897–2904. [Google Scholar] [CrossRef]
- Morandi, G.; Kebir, N.; Campistron, I.; Gohier, F.; Laguerre, A.; Pilard, J.F. Direct selective reductive amination of carbonyl telechelic oligoisoprenes: Elaboration of promising tri and tetrafunctionalized oligoisooprene intermediates. Tetrahedron Lett. 2007, 48, 7726–7730. [Google Scholar] [CrossRef]
- Khan, S.N.; Bae, S.Y.; Kim, H.S. A highly stereoselective reductive amination of 3-ketosteroid with amines: Improved synthesis of 3α-aminosteroids. Tetrahedron Lett. 2005, 46, 7675–7678. [Google Scholar] [CrossRef]
- Adachi, K.; Tsuru, E.; Banjyo, S.E.K.; Yamashita, T. Selective BH 3-Reduction of Amide Carbonyl Groups of Lithium Salts of N-t-Butoxycarbonyl (S)-O-Benzyl Tyrosyl (S)-Proline and N,N’-Ethylene-Bridged Dipeptides. Synthesis 1998, 11, 1623–1626. [Google Scholar] [CrossRef]
- Beamson, G.; Papworth, A.J.; Philipps, C.; Smith, A.M.; Whyman, R. Selective hydrogenation of amides using Rh/Mo catalysts. J. Cat. 2010, 93, 269. [Google Scholar] [CrossRef]
- Komlaga, G.; Genta-Jouve, G.; Cojean, S.; Dickson, R.T.; Mensah, M.L.K.; Loiseau, P.M.; Champy, P.; Beniddir, M.A. Antiplasmodial Securinega alkaloids from Phyllanthus fraternus: Discovery of natural (+)− allonorsecurinine. Tetrahedron Lett. 2017, 58, 3754–3756. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).