Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry



Abstract
:
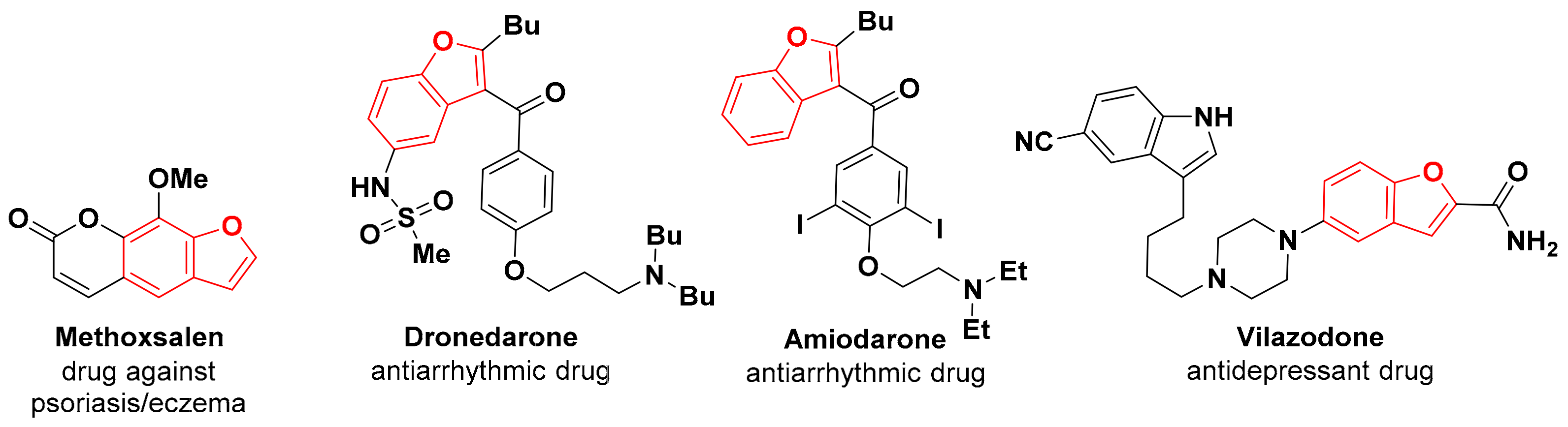
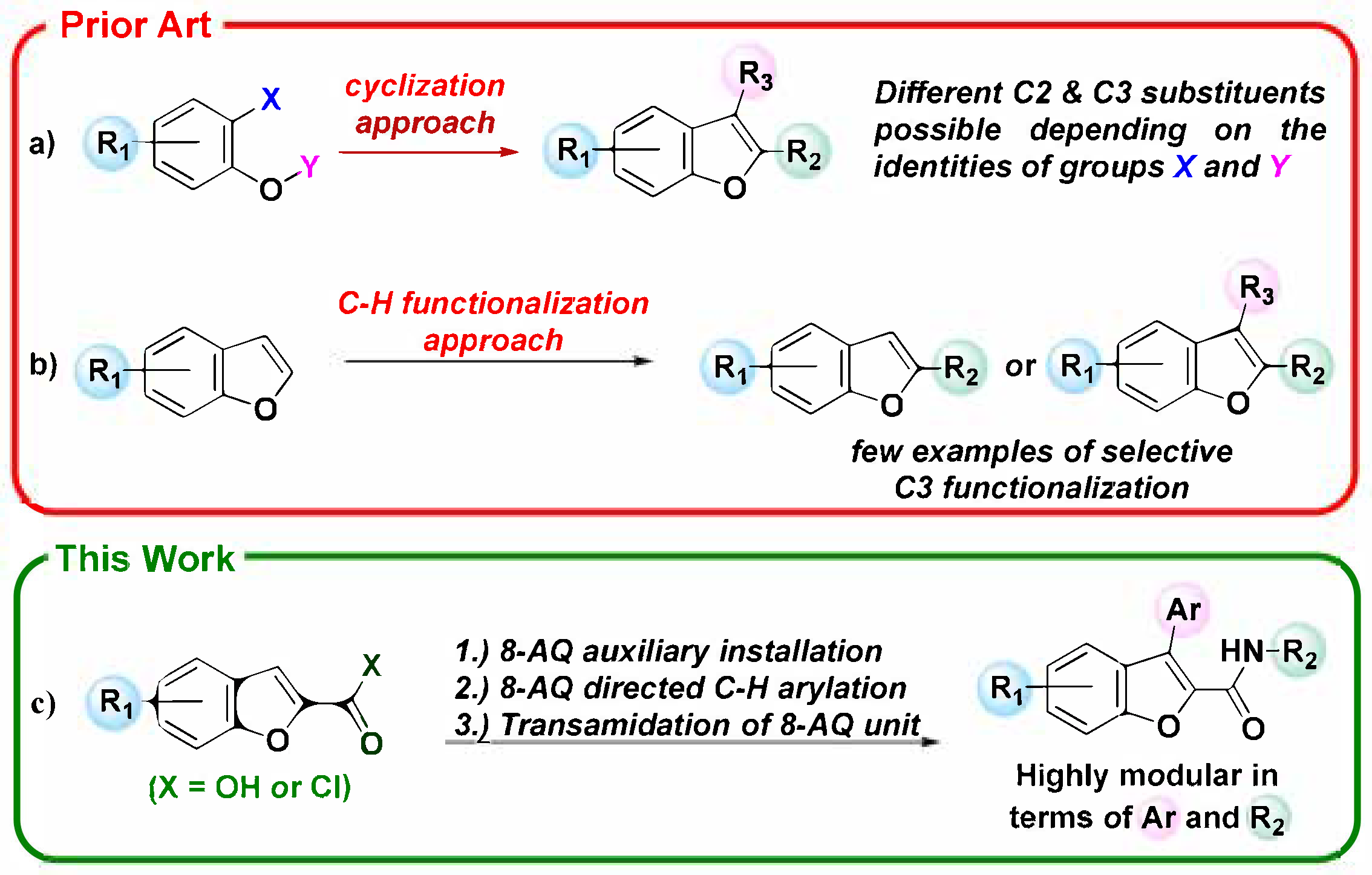
1. Introduction
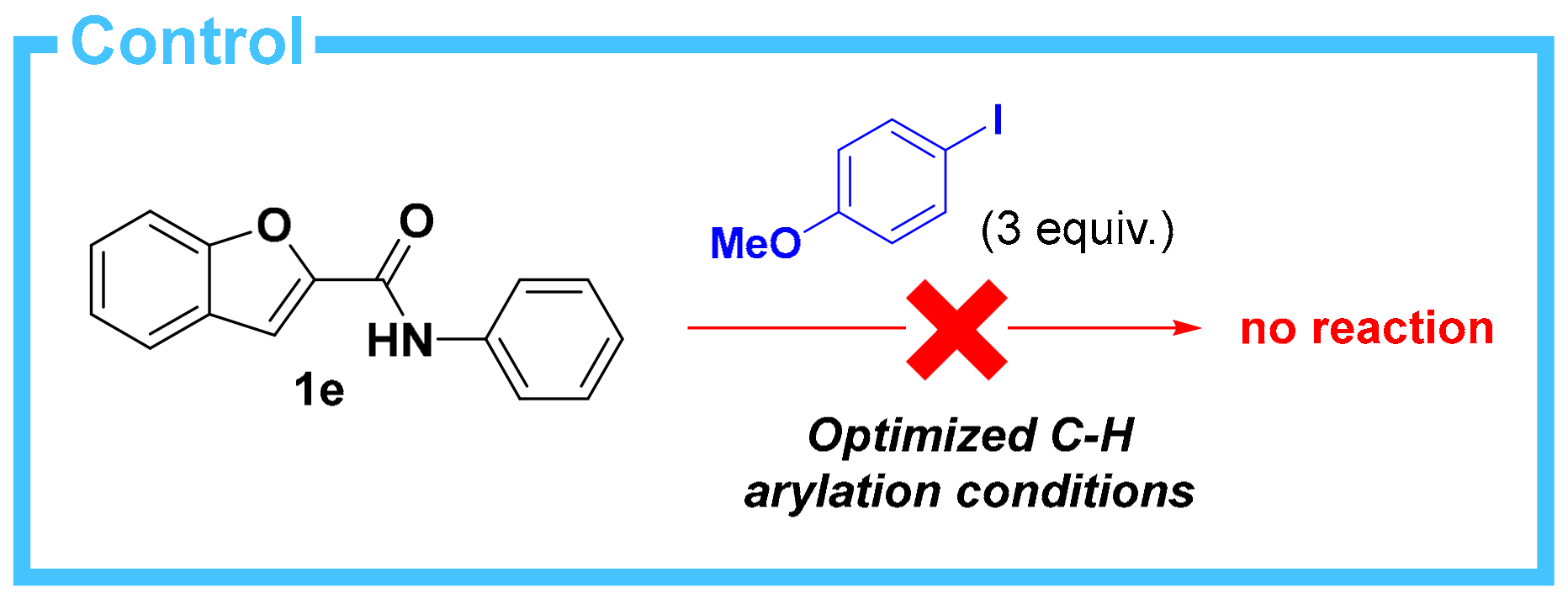
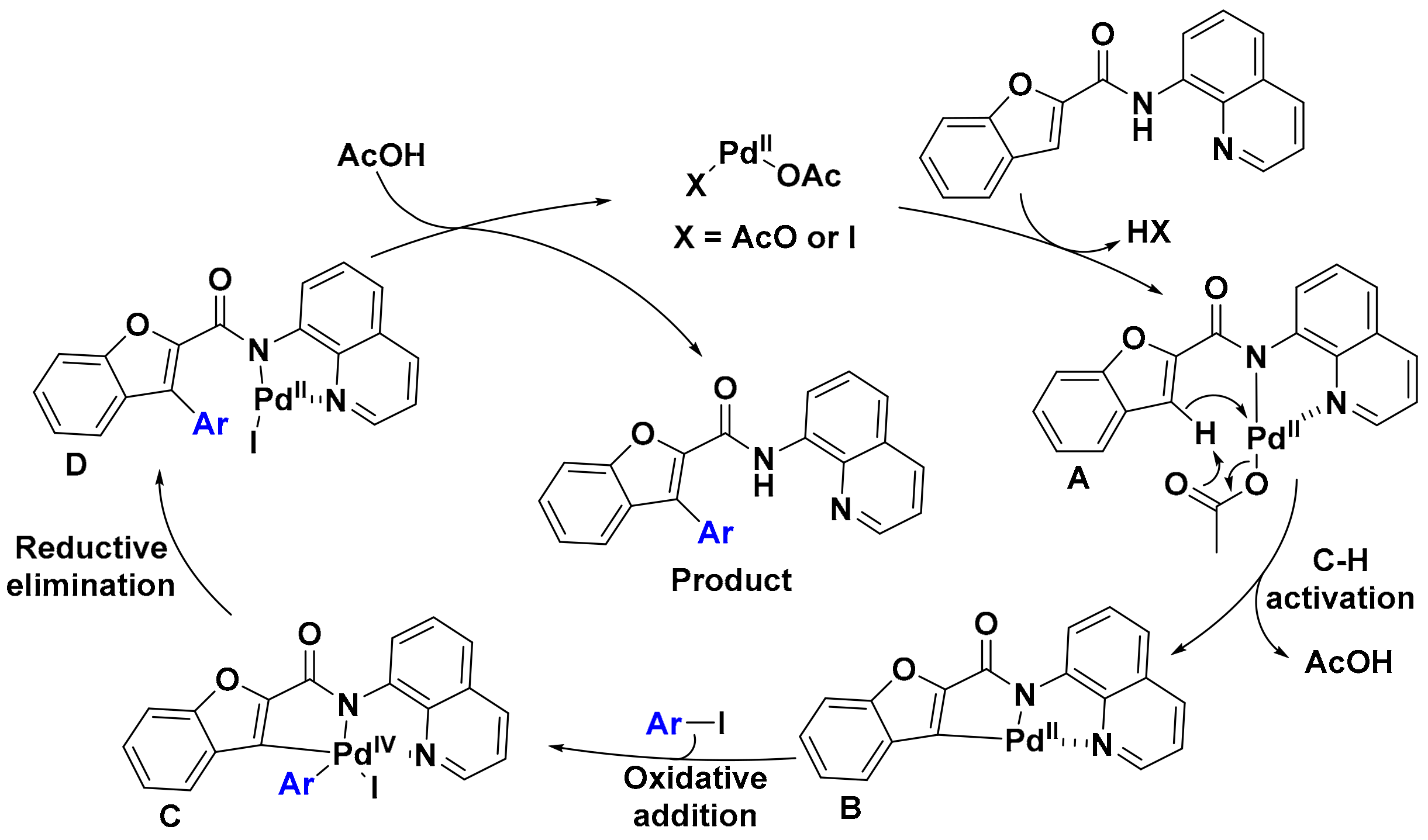
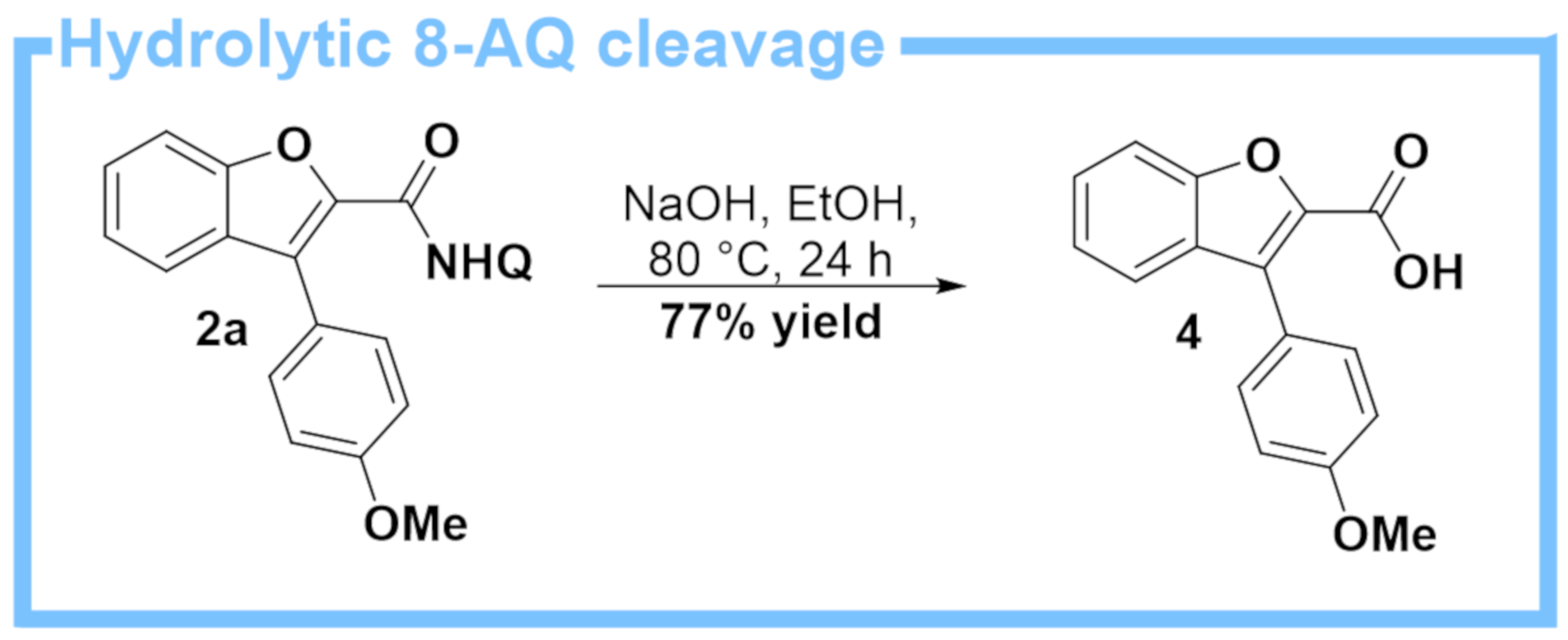
2. Results
3. Discussion
4. Materials and Methods
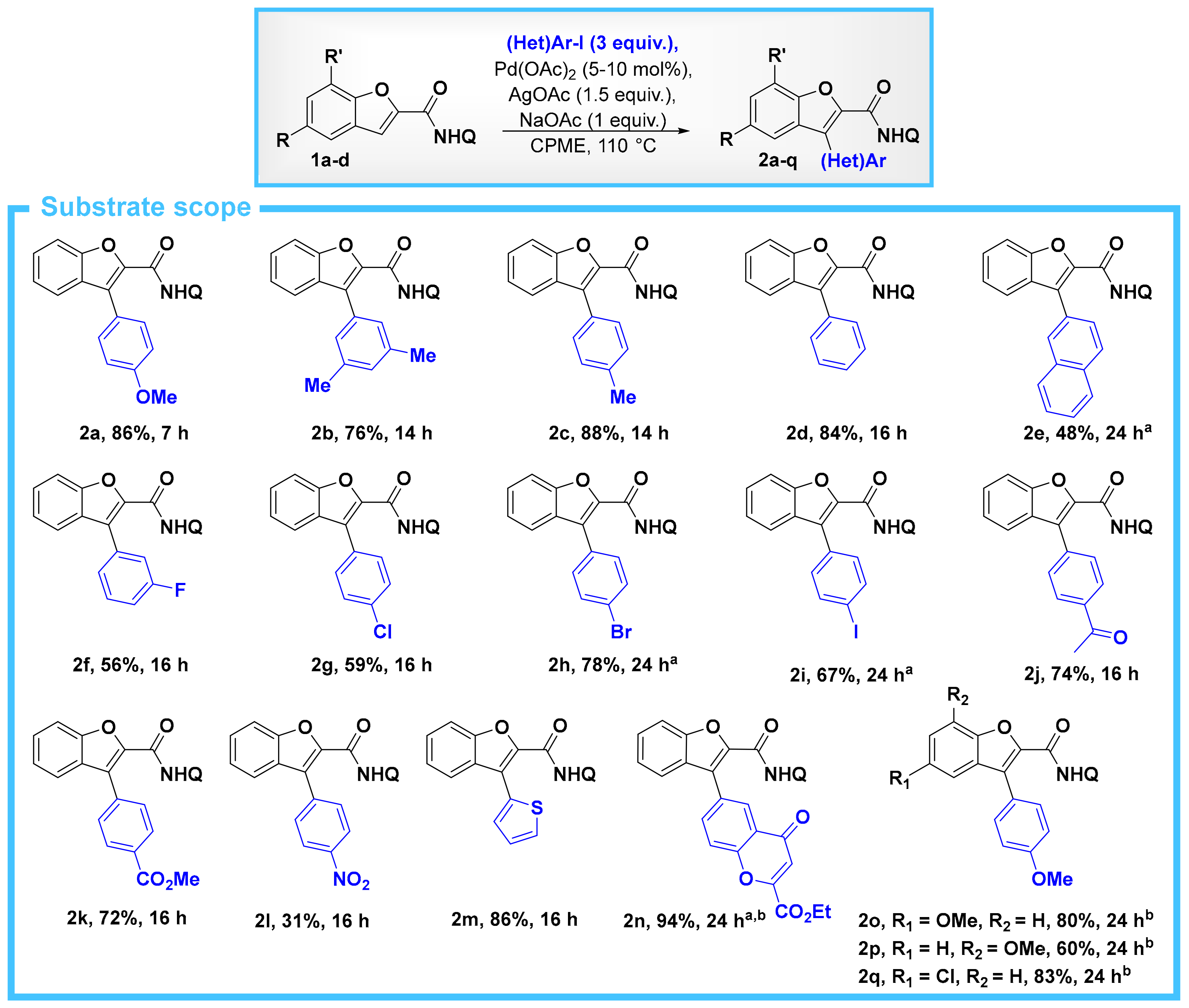
4.1. General Procedure for the C–H Arylation of Substrates 1a–e
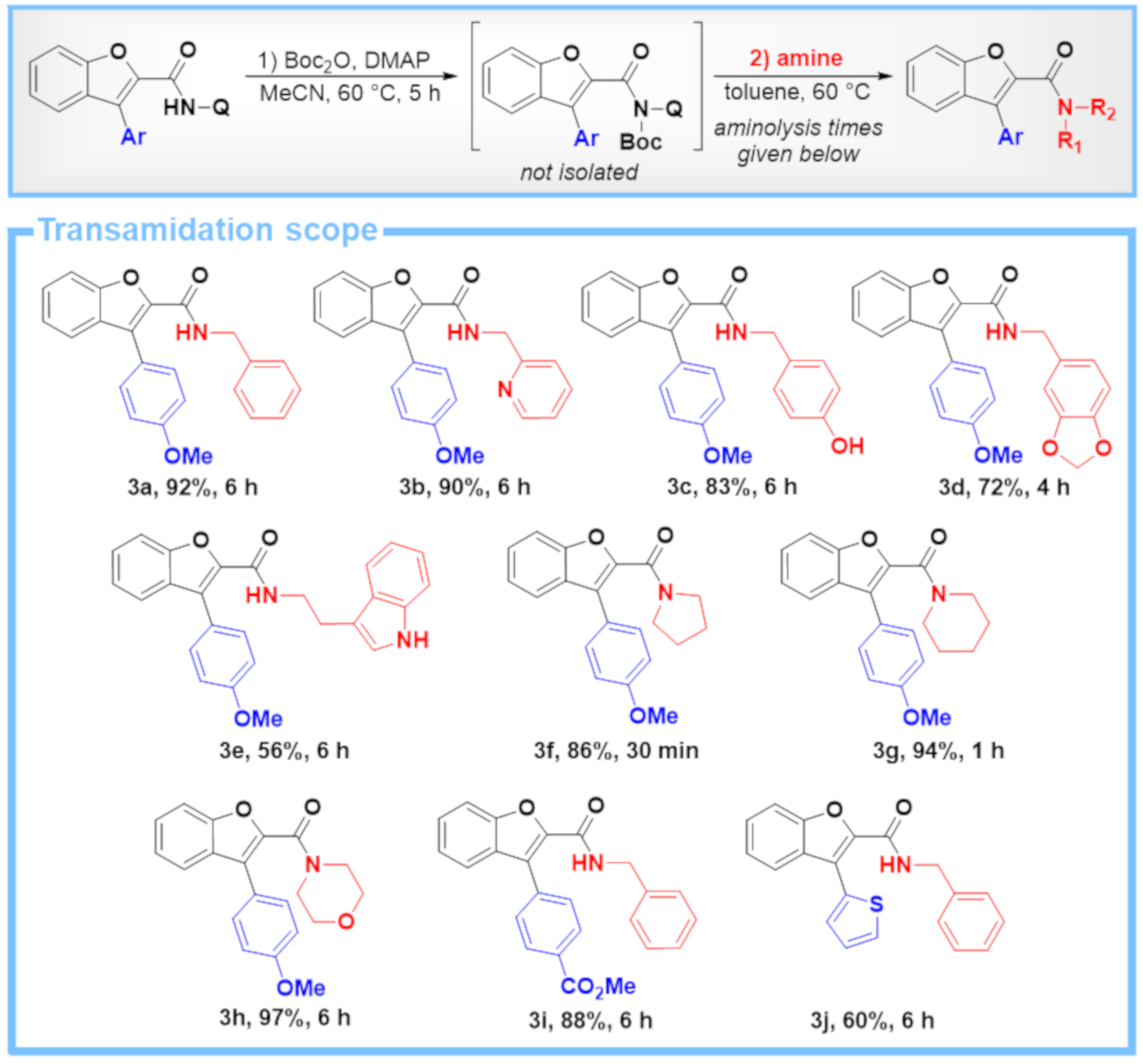
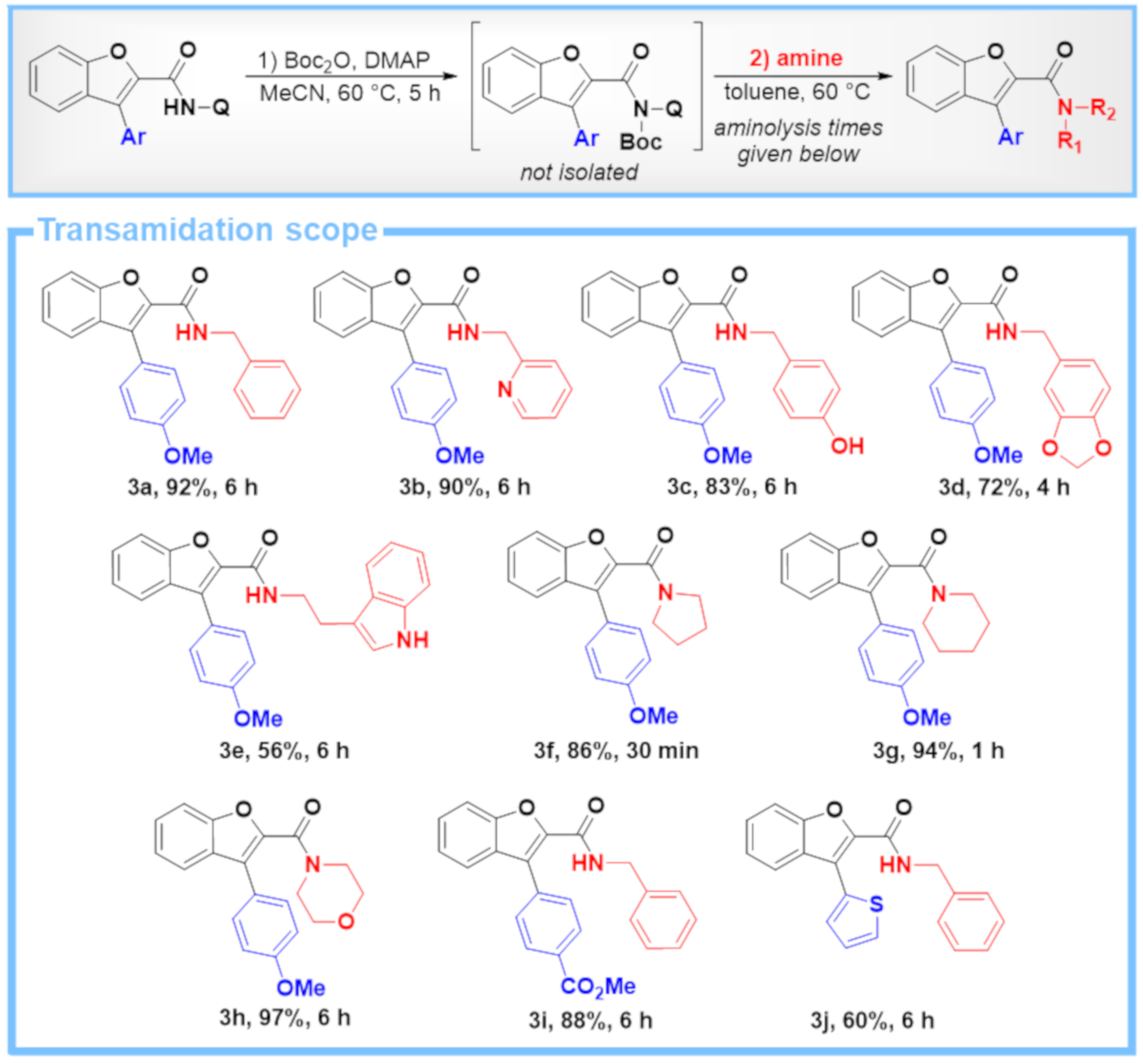
4.2. General Procedure C: Two-Step One-Pot Transamidation of C–H Arylation Products with Different Amines
4.2.1. Boc Activation
4.2.2. Aminolysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Radadiya, A.; Shah, A. Bioactive benzofuran derivatives: An insight on lead developments, radioligands and advances of the last decade. Eur. J. Med. Chem. 2015, 97, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Patil, M.R.; Chethana, K.R.; Chand, K.; Amelia Santos, M.; Keri, R.S. Benzofuran: An emerging scaffold for antimicrobial agents. RSC Adv. 2015, 5, 96809–96828. [Google Scholar] [CrossRef]
- Khanam, H. Shamsuzzaman Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.D.; MacCoss, M.; Lawson, A.D. Rings in drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef] [PubMed]
- Khodarahmi, G.; Asadi, P.; Hazzanzadeh, F.; Khodarahmi, E. Benzofuran as a promising scaffold for the synthesis of antimicrobial and antibreast cancer agents: A review. J. Res. Med. Sci. 2015, 20, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Salomé, C.; Ribeiro, N.; Chavagnan, T.; Thuaud, F.; Serova, M.; de Gramont, A.; Faivre, S.; Raymond, E.; Désaubry, L. Benzofuran derivatives as anticancer inhibitors of mTOR signaling. Eur. J. Med. Chem. 2014, 81, 181–191. [Google Scholar] [CrossRef]
- Abdelhafez, O.M.; Amin, K.M.; Ali, H.I.; Abdalla, M.M.; Ahmed, E.Y. Design, synthesis and anticancer activity of benzofuran derivatives targeting VEGFR-2 tyrosine kinase. RSC Adv. 2014, 4, 11569–11579. [Google Scholar] [CrossRef]
- More, K.R. Review on Synthetic Routes for Synthesis of Benzofuran-Based Compounds. J. Chem. Pharm. Res. 2017, 9, 210–220. [Google Scholar]
- Heravi, M.M.; Zadsirjan, V. Chapter Five-Recent Advances in the Synthesis of Benzo[b]furans. Adv. Heterocycl. Chem. 2015, 117, 261–376. [Google Scholar]
- Abu-Hashem, A.A.; Hussein, H.A.R.; Aly, A.S.; Gouda, M.A. Synthesis of Benzofuran Derivatives via Different Methods. Synth. Commun. 2014, 44, 2285–2312. [Google Scholar] [CrossRef]
- Huang, W.; Xu, J.; Liu, C.; Chen, Z.; Gu, Y. Lewis Acid-Catalyzed Synthesis of Benzofurans and 4,5,6,7-Tetrahydrobenzofurans from Acrolein Dimer and 1,3-Dicarbonyl Compounds. J. Org. Chem. 2019, 84, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Merkushev, A.A.; Strelnikov, V.N.; Uchuskin, M.G.; Trushnov, I.V. A simple synthesis of benzofurans by acid-catalyzed domino reaction of salicyl alcohols with N-tosylfurfurylamine. Tetrahedron 2017, 73, 6523–6529. [Google Scholar] [CrossRef]
- Warner, A.J.; Churn, A.; McGough, J.S.; Ingleson, M.J. BCl3-Induced Annulative Oxo- and Thioboration for the Formation of C3-Borylated Benzofurans and Benzothiophenes. Angew. Chem. Int. Ed. 2017, 56, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Bankar, S.K.; Mathew, J.; Ramasastry, S.S.V. Synthesis of benzofurans via an acid catalysed transacetalisation/Fries-type O → C rearrangement/Michael addition/ring-opening aromatisation cascade of β-pyrones. Chem. Commun. 2016, 52, 5569–5572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contiero, F.; Jones, K.M.; Matts, E.A.; Porzelle, A.; Tomkinson, N.C.O. Direct Preparation of Benzofurans from O-Arylhydroxylamines. Synlett 2009, 3003–3006. [Google Scholar]
- Witczak, M.; Kwiecien, H. Efficient Cyclization of 2-Phenoxyalkanals to 2-Alkylbenzo[b]furans. Synth. Commun. 2005, 35, 2223–2230. [Google Scholar] [CrossRef]
- Ishibashi, K.; Nakajima, K.; Sugioka, Y.; Sugiyama, M.; Hamata, T.; Horikoshi, H.; Nishi, T. Synthesis of 2-Phenylbenzofuran Derivatives as Testosterone 5α-Reductase Inhibitor. Chem. Pharm. Bull. 1999, 47, 226–240. [Google Scholar] [CrossRef] [Green Version]
- Bruneau, A.; Gustafson, K.P.J.; Yuan, N.; Tai, C.-W.; Persson, I.; Zou, X.; Backvall, J.E. Synthesis of Benzofurans and Indoles from Terminal Alkynes and Iodoaromatics Catalyzed by Recyclable Palladium Nanoparticles Immobilized on Siliceous Mesocellular Foam. Chem. Eur. J. 2017, 23, 12886–12891. [Google Scholar] [CrossRef]
- Bosiak, M.J. A Convenient Synthesis of 2-Arylbenzo[b]furans from Aryl Halides and 2-Halophenols by Catalytic One-Pot Cascade Method. ACS Catal. 2016, 6, 2429–2434. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Akiyama, T.; Sasou, H.; Katsumata, H.; Manabe, K. One-Pot Synthesis of Substituted Benzo[b]furans and Indoles from Dichlorophenols/Dichloroanilines Using a Palladium-Dihydroxyterphenylphosphine Catalyst. J. Org. Chem. 2016, 81, 5450–5463. [Google Scholar] [CrossRef]
- Bochicchio, A.; Chiummiento, L.; Funicello, M.; Lopardo, M.T.; Lupattelli, P. Efficient synthesis of 5-nitro-benzo[b]furans via 2-bromo-4-nitro-phenyl acetates. Tetrahedron Lett. 2010, 51, 2824–2827. [Google Scholar] [CrossRef]
- Lingam, V.S.P.R.; Vinodkumar, R.; Mukkanti, K.; Thomas, A.; Gopalan, B. A simple approach to highly functionalized benzo[b]furans from phenols and aryl iodides via aryl propargyl ethers. Tetrahedron Lett. 2008, 49, 4260–4264. [Google Scholar] [CrossRef]
- Bernini, R.; Cacchi, S.; De Salve, I.; Fabrizi, G. Palladium-Catalyzed Synthesis of Lipophilic Benzo[b]furans from Cardanol. Synthesis 2007, 6, 873–882. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Wang, L.; Pagni, R.M. Sonogashira coupling and cyclization reactions on alumina: A route to aryl alkynes, 2-substituted-benzo[b]furans and 2-substituted-indoles. Tetrahedron 2001, 57, 8017–8028. [Google Scholar] [CrossRef]
- More, K.R.; Mali, R.S. An alternate method for the synthesis of 2-aryl/alkyl-5-bromo-7methoxy benzofurans; application to the synthesis of Egonol, Homoegonol, and analogs via Heck reaction. Tetrahedron 2016, 72, 7496–7504. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar Gangwar, M.; Prakasham, A.P.; Mhatre, D.; Kalita, A.C.; Ghosh, P. Accessing a Biologically Relevant Benzofuran Skeleton by a One-Pot Tandem Heck Alkynylation/Cyclization Reaction Using Well-Defined Palladium N-Heterocyclic Carbene Complexes. Inorg. Chem. 2016, 55, 2882–2893. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, W.; Chen, H.; Wu, W.; Peng, J.; Gao, Y.; Jiang, H. Pd-Catalyzed Highly Regio- and Stereoselective Formation of C-C Double Bonds: An Efficient Method for the Synthesis of Benzofuran-, Dihydrobenzofuran-, and Indoline-Containing Alkenes. J. Org. Chem. 2015, 80, 7456–7467. [Google Scholar] [CrossRef]
- Yuan, H.; Bi, K.J.; Li, B.; Yue, R.C.; Ye, J.; Shen, Y.H.; Shan, L.; Jin, H.Z.; Sun, Q.Y.; Zhang, W.D. Construction of 2-Substituted-3-Functionalized Benzofurans via Intramolecular Heck Coupling: Application to Enantioselective Total Synthesis of Daphnodorin. B. Org. Lett. 2013, 15, 4742–4745. [Google Scholar] [CrossRef]
- Heravi, M.M.; Fazeli, A. Recent Advances in the Application of the Heck Reaction in the Synthesis of Heterocyclic Compounds. Heterocycles 2010, 81, 1979–2026. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Wang, T.; Yao, X.; Wang, P.; Wang, P.; Jing, S.; Liang, Y.; Zhang, Z. Oxidant and Transition-Metal-Free Photoinduced Direct Oxidative Annulation of 1-Aryl-2-(furan/thiophen-2-yl)butane-1,3-diones. J. Org. Chem. 2017, 82, 12097–12105. [Google Scholar] [CrossRef]
- Xia, Z.; Khaled, O.; Mouries-Mansuy, V.; Ollivier, C.; Fensterbank, L. Dual Photoredox/Gold Catalysis Arylative Cyclization of o-Alkynylphenols with Aryldiazonium Salts: A Flexible Synthesis of Benzofurans. J. Org. Chem. 2016, 81, 7182–7190. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Das, J. A novel photochemical Wittig reaction for the synthesis of 2-aryl/alkylbenzofurans. Tetrahedron Lett. 2011, 52, 1112–1116. [Google Scholar] [CrossRef]
- Sumanthi, T.; Balasubramanian, K.K. A photochemical route to 2-alkenyl- and 2-ethynylbenzofurans. Tetrahedron Lett. 1990, 31, 3775–3778. [Google Scholar] [CrossRef]
- Pandey, G.; Krishna, A.; Bhalerao, U.T. A one step synthesis of 2-substituted benzofurans from 2-aryl-1-substituted ethane-1-ones by photoinduced set reactions. Tetrahedron Lett. 1989, 30, 1867–1870. [Google Scholar] [CrossRef]
- Deng, G.; Li, M.; Yu, K.; Liu, C.; Liu, Z.; Duan, S.; Chen, W.; Yang, X.; Zhang, H.; Walsh, P.J. Synthesis of Benzofuran Derivatives through Cascade Radical Cyclization/Intermolecular Coupling of 2-Azaallyls. Angew. Chem. Int. Ed. 2019, 58, 2826–2830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, S.; Cai, Z.; Liu, P.; Sun, P. Radical Addition Cascade Cyclization of 1,6-Enynes with DMSO To Access Methylsulfonylated and Carbonylated Benzofurans under Transition-Metal-Free Conditions. J. Org. Chem. 2018, 83, 9344–9352. [Google Scholar] [CrossRef]
- Zheng, H.X.; Shan, X.H.; Qu, J.P.; Kang, Y.B. Strategy for Overcoming Full Reversibility of Intermolecular Radical Addition to Aldehydes: Tandem C-H and C-O Bonds Cleaving Cyclization of (Phenoxymethyl)arenes with Carbonyls to Benzofurans. Org. Lett. 2018, 20, 3310–3313. [Google Scholar] [CrossRef]
- Rueping, M.; Leiendecker, M.; Das, A.; Poisson, T.; Bui, L. Potassium tert-butoxide mediated Heck-type cyclization/isomerization–benzofurans from organocatalytic radical cross-coupling reactions. Chem. Commun. 2011, 47, 10629–10631. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Meijs, G.F. Formation of dihydrobenzofurans by radical cyclization. J. Chem. Soc. Chem. Comm. 1981, 136–137. [Google Scholar] [CrossRef]
- Yi, W.; Chen, W.; Liu, F.X.; Zhong, Y.; Wu, D.; Zhou, Z.; Gao, H. Rh(III)-Catalyzed and Solvent-Controlled Chemoselective Synthesis of Chalcone and Benzofuran Frameworks via Synergistic Dual Directing Groups Enabled Regioselective C–H Functionalization: A Combined Experimental and Computational Study. ACS Catal. 2018, 8, 9508–9519. [Google Scholar] [CrossRef]
- Ichake, S.S.; Konala, A.; Kavala, V.; Kuo, C.W.; Yao, C.F. Palladium-Catalyzed Tandem C-H Functionalization/Cyclization Strategy for the Synthesis of 5-Hydroxybenzofuran Derivatives. Org. Lett. 2017, 19, 54–57. [Google Scholar] [CrossRef]
- Agasti, S.; Sharma, U.; Naveen, T.; Maiti, D. Orthogonal selectivity with cinnamic acids in 3-substituted benzofuran synthesis through C–H olefination of phenols. Chem. Commun. 2015, 51, 5375–5378. [Google Scholar] [CrossRef] [PubMed]
- Agasti, S.; Maity, S.; Szabo, K.J.; Maiti, D. Palladium-Catalyzed Synthesis of 2,3-Disubstituted Benzofurans: An Approach Towards the Synthesis of Deuterium Labeled Compounds. Adv. Synth. Catal. 2015, 357, 2331–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, M.; Thirupathi, N.; Reddy, T.J.; Kanojiya, S.; Sridhar, M. Pd-Catalyzed Isocyanide Assisted Reductive Cyclization of 1-(2-Hydroxyphenyl)-propargyl Alcohols for 2-Alkyl/Benzyl Benzofurans and Their Useful Oxidative Derivatization. J. Org. Chem. 2015, 80, 12311–12320. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Naveen, T.; Maji, A.; Manna, S.; Maiti, D. Palladium-Catalyzed Synthesis of Benzofurans and Coumarins from Phenols and Olefins. Angew. Chem. Int. Ed. 2013, 52, 12669–12673. [Google Scholar] [CrossRef]
- Xu, G.; Liu, K.; Sun, J. Gold-Catalyzed Controllable C2-Functionalization of Benzofurans with Aryl Diazoesters. Org. Lett. 2018, 20, 72–75. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Jie, K.; Huang, L.; Guo, S.; Cai, H. Copper-Catalyzed C2 and C3 Phosphonation of Benzofuran and Benzothiophene with Trialkyl Phosphites. ChemCatChem 2018, 10, 716–719. [Google Scholar] [CrossRef]
- Fang, H.; Guo, L.; Zhang, Y.; Yao, W.; Huang, Z. A Pincer Ruthenium Complex for Regioselective C–H Silylation of Heteroarenes. Org. Lett. 2016, 18, 5624–5627. [Google Scholar] [CrossRef]
- Paymode, D.J.; Ramana, C.V. Ruthenium(II)-Catalyzed C3 Arylation of 2-Aroylbenzofurans with Arylboronic Acids/Aryltrifluoroborates via Carbonyl-Directed C–H Bond Activation. J. Org. Chem. 2015, 80, 11551–11558. [Google Scholar] [CrossRef]
- Schramm, Y.; Takeuchi, M.; Semba, K.; Nakao, Y.; Hartwig, J.F. Anti-Markovnikov Hydroheteroarylation of Unactivated Alkenes with Indoles, Pyrroles, Benzofurans, and Furans Catalyzed by a Nickel–N-Heterocyclic Carbene System. J. Am. Chem. Soc. 2015, 137, 12215–12218. [Google Scholar] [CrossRef]
- Yin, S.-C.; Zhou, Q.; Zhao, X.-Y.; Shao, L.-X. N-Heterocyclic Carbene-Palladium(II)-1-Methylimidazole Complex Catalyzed Direct C–H Bond Arylation of Benzo[b]furans with Aryl Chlorides. J. Org. Chem. 2015, 80, 8916–8921. [Google Scholar] [CrossRef] [PubMed]
- Loukotova, L.; Yuan, K.; Doucet, H. Regiocontroled Palladium-Catalysed Direct Arylation at Carbon C2 of Benzofurans using Benzenesulfonyl Chlorides as the Coupling Partners. ChemCatChem 2014, 6, 1303–1309. [Google Scholar] [CrossRef]
- Carrer, A.; Brinet, D.; Florent, J.-C.; Rousselle, P.; Bertounesque, E. Palladium-Catalyzed Direct Arylation of Polysubstituted Benzofurans. J. Org. Chem. 2012, 77, 1316–1327. [Google Scholar] [CrossRef]
- Dao-Huy, T.; Haider, M.; Glatz, F.; Schnürch, M.; Mihovilovic, M.D. Direct Arylation of Benzo[b]furan and Other Benzo-Fused Heterocycles. Eur. J. Org. Chem. 2014, 8119–8125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liégault, B.; Petrov, I.; Gorelsky, S.I.; Fagnou, K. Modulating Reactivity and Diverting Selectivity in Palladium-Catalyzed Heteroaromatic Direct Arylation Through the Use of a Chloride Activating/Blocking Group. J. Org. Chem. 2010, 75, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Ionita, M.; Roger, J.; Doucet, H. Palladium Catalyzed Direct 3-Arylation of Benzofurans using Low Catalyst Loadings. ChemSusChem 2010, 3, 367–376. [Google Scholar] [CrossRef]
- Larbi, K.S.; Djebbar, S.; Soulé, J.-F.; Doucet, H. Reactivity of benzofuran and benzothiophene in palladium-catalysed direct C2,C3-diarylations. J. Organomet. Chem. 2017, 843, 32–39. [Google Scholar] [CrossRef]
- Shen, K.; Fu, Y.; Li, J.-N.; Liu, L.; Guo, Q.-X. What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO? Tetrahedron 2007, 63, 1568–1576. [Google Scholar] [CrossRef]
- Verho, O.; Pourghasemi Lati, M.; Oschmann, M. A Two-Step Procedure for the Overall Transamidation of 8-Aminoquinoline Amides Proceeding via the Intermediate N-Acyl-Boc-Carbamates. J. Org. Chem. 2018, 83, 4464–4476. [Google Scholar] [CrossRef]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of sp3 C-H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef]
- Shabashov, D.; Daugulis, O. Auxiliary-Assisted Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 Carbon-Hydrogen Bonds. J. Am. Chem. Soc. 2010, 132, 3965–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugulis, O.; Roane, J.; Tran, L.D. Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon-Hydrogen Bonds. Acc. Chem. Res. 2015, 48, 1053–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, A.J.; Ricke, A.; Oschmann, M.; Verho, O. Convenient Access to Chiral Cyclobutanes with Three Contiguous Stereocenters from Verbenone by Directed C(sp3)−H arylation. Chem. Eur. J. 2019, 25, 5154–5157. [Google Scholar] [CrossRef] [PubMed]
- Antermite, D.; Affron, D.P.; Bull, J.A. Regio- and Stereoselective Palladium-Catalyzed C(sp3)–H Arylation of Pyrrolidines and Piperidines with C(3) Directing Groups. Org. Lett. 2018, 20, 3948–3952. [Google Scholar] [CrossRef] [PubMed]
- Melillo, B.; Zoller, J.; Hua, B.K.; Verho, O.; Borghs, J.C.; Nelson, S.D., Jr.; Maetani, M.; Wawer, M.J.; Clemons, P.A.; Schreiber, S.L. Synergistic Effects of Stereochemistry and Appendages on the Performance Diversity of a Collection of Synthetic Compounds. J. Am. Chem. Soc. 2018, 140, 11784–11790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Sun, W.-W.; Yu, Q.-Y.; Cao, P.; Dong, X.-P.; Wu, B. Stereoselective synthesis of (−)-3-PPP through palladium-catalysed unactivated C(sp3)–H arylation at the C-3 position of l-pipecolinic acid. Tetrahedron Lett. 2017, 58, 606–609. [Google Scholar]
- Maetani, M.; Zoller, J.; Melillo, B.; Verho, O.; Kato, N.; Pu, J.; Comer, E.; Schreiber, S.L. Synthesis of a Bicyclic Azetidine with In Vivo Antimalarial Activity Enabled by Stereospecific, Directed C(sp3)–H Arylation. J. Am. Chem. Soc. 2017, 139, 11300–11306. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, Y.; Wang, Z.; Zeng, T.; Liu, P.; Engle, K.M. Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. J. Am. Chem. Soc. 2017, 139, 11261–11270. [Google Scholar] [CrossRef]
- Verho, O.; Maetani, M.; Melillo, B.; Zoller, J.; Schreiber, S.L. Stereospecific Palladium-Catalyzed C–H Arylation of Pyroglutamic Acid Derivatives at the C3 Position Enabled by 8-Aminoquinoline as a Directing Group. Org. Lett. 2017, 19, 4424–4427. [Google Scholar] [CrossRef]
- Affron, D.P.; Davis, O.A.; Bull, J.A. Regio- and Stereospecific Synthesis of C-3 Functionalized Proline Derivatives by Palladium Catalyzed Directed C(sp3)–H Arylation. Org. Lett. 2014, 16, 4956–4959. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.C.; Lacker, C.R.; Chapman, L.M.; Reisman, S.E. A modular approach to prepare enantioenriched cyclobutanes: Synthesis of (+)-rumphellaone A. Chem. Sci. 2019, 10, 2315–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, L.M.; Beck, J.C.; Lacker, C.R.; Wu, L.; Reisman, S.E. Evolution of a Strategy for the Enantioselective Total Synthesis of (+)-Psiguadial, B. J. Org. Chem. 2018, 83, 6066–6085. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.M.; Beck, J.C.; Wu, L.; Reisman, S.E. Enantioselective Total Synthesis of (+)-Psiguadial, B. J. Am. Chem. Soc. 2016, 138, 9803–9806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutekunst, W.R.; Baran, P.S. Applications of C–H Functionalization Logic to Cyclobutane Synthesis. J. Org. Chem. 2014, 79, 2430–2452. [Google Scholar] [CrossRef]
- Gutekunst, W.R.; Gianatassio, R.; Baran, P.S. Sequential Csp3–H Arylation and Olefination: Total Synthesis of the Proposed Structure of Pipercyclobutanamide A. Angew. Chem. Int. Ed. 2012, 51, 7507–7510. [Google Scholar] [CrossRef] [Green Version]
- Frébault, F.; Maulide, N. Total Synthesis and Structural Revision of the Piperarborenines: When Photochemistry Meets C–H Activation. Angew. Chem. Int. Ed. 2012, 51, 2815–2817. [Google Scholar] [CrossRef]
- Gutekunst, W.R.; Baran, P.S. Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation. J. Am. Chem. Soc. 2011, 133, 19076–19079. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Yi, J.; Xu, H.; Li, S.; Yuan, R. An Efficient, One-Pot Transamidation of 8-Aminoquinoline Amides Activated by Tertiary-Butyloxycarbonyl. Molecules 2019, 24, 1234. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Szostak, M. Highly selective transition-metal-free transamidation of amides and amidation of esters at room temperature. Nat. Commun. 2018, 9, 4165. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shi, S.; Achtenhagen, M.; Liu, R.; Szostak, M. Metal-Free Transamidation of Secondary Amides via Selective N–C Cleavage under Mild Conditions. Org. Lett. 2017, 19, 1614–1617. [Google Scholar] [CrossRef]
- The corresponding acid chloride, benzofuran-2-carbonyl chloride, is also commercially-available to a reasonable cost, and of course it is also possible to use this for the synthesis of 1a. Starting from this acid chloride, we were able to prepare 1a in 97% yield (see SM for experimental details).
- Padmavathi, R.; Sankar, R.; Gopalakrishnan, B.; Parella, R.; Babu, S.A. Pd(OAc)2/AgOAc Catalytic System Based Bidentate Ligand Directed Regiocontrolled C–H Arylation and Alkylation of the C-3 Position of Thiophene- and Furan-2-carboxamides. Eur. J. Org. Chem. 2015, 3727–3742. [Google Scholar] [CrossRef]
- Attempts to purify these products by column chromatography (with either pentane/EtOAc or pentane/CH2Cl2) resulted in poor material recovery and low yields.
- Rit, R.K.; Yadav, R.; Ghosh, K.; Sahoo, A.K. Reusable directing groups [8-aminoquinoline, picolianamide, sulfoximine] in C(sp3)-H bond activation: Present and future. Tetrahedron 2015, 71, 4450–4459. [Google Scholar] [CrossRef]
- Arroniz, C.; Denis, J.G.; Ironmonger, A.; Rassias, G.; Larrosa, I. An organic cation as a silver(I) analogue for the arylation of sp2 and sp3 C-H bonds with iodoarenes. Chem. Sci. 2014, 5, 3509–3514. [Google Scholar] [CrossRef] [Green Version]
- Daugulis, O.; Zaitsev, V.G. Anilide ortho-arylation by using C-H activation methodology. Angew. Chem. Int. Ed. 2005, 44, 4046–4048. [Google Scholar] [CrossRef]
- Pereira, K.C.; Porter, A.L.; Potavathri, S.; LeBris, A.P.; DeBoef, B. Insight into the palladium-catalyzed oxidative arylation of benzofuran: Heteropoly acid oxidants evoke a Pd(II)/Pd(IV) mechanism. Tetrahedron 2013, 69, 4429–4435. [Google Scholar] [CrossRef] [Green Version]
- Bhaskararao, B.; Singh, S.; Anand, M.; Verma, P.; Prakash, P.; Athira, C.; Malakar, S.; Schaefer, H.F.; Sunoj, R.B. Is silver a mere terminal oxidant in palladium catalyzed C-H bond activation reactions? Chem. Sci. 2020, 11, 208–216. [Google Scholar] [CrossRef] [Green Version]
- Guin, S.; Dolui, P.; Zhang, X.; Paul, S.; Singh, V.K.; Pradhan, S.; Chandrashekar, H.B.; Anjana, S.S.; Paton, R.S.; Maiti, D. Iterative Arylation of Amino Acids and Aliphatic Amines via δ-C(sp3)−H Activation: Experimental and Computational Exploration. Angew. Chem. Int. Ed. 2019, 58, 5633–5638. [Google Scholar] [CrossRef]
- Mudarra, Á.L.; de Salinas, S.M.; Pérez-Temprano, M. Beyond the traditional roles of Ag in catalysis: Transmetalating ability of organosilver(I) species in Pd-catalysed reactions. Org. Biomol. Chem. 2019, 17, 1655–1667. [Google Scholar] [CrossRef]
Sample Availability: Samples of all the reported compounds can be made available from the authors upon request. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Additive (equiv.) | Solvent | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | none | toluene | 110 | 7 | 46 |
| 2 | none | toluene | 110 | 16 | 65 |
| 3 | none | toluene | 120 | 7 | 30 |
| 4 | (BnO)2PO2H (0.2) | toluene | 110 | 7 | 28 |
| 5 | PivOH (0.2) | toluene | 110 | 7 | 61 |
| 6 | PivOH (0.5) | toluene | 110 | 7 | 48 |
| 7 | PivOH (1.0) | toluene | 110 | 7 | 17 |
| 8 | NaOAc (0.5) | toluene | 110 | 7 | 62 |
| 9 | NaOAc (1.0) | toluene | 110 | 7 | 78 |
| 10 | NaOAc (2.0) | toluene | 110 | 7 | 70 |
| 11 | PivOH+NaOAc (0.2 + 1.0) | toluene | 110 | 7 | 56 |
| 12 | NaOAc (1.0) | t-amyl OH | 110 | 7 | 91 |
| 13 | NaOAc (1.0) | MeTHF | 110 | 7 | 81 |
| 14 | NaOAc (1.0) | CPME | 110 | 7 | 93 |
| 15 | NaOAc (1.0) | DCE | 110 | 7 | 69 |
| 16 | NaOAc (1.0) | MeCN | 110 | 7 | 18 |
| 17 [b] | NaOAc (1.0) | CPME | 110 | 15 | 80 |
| 18 [b,c] | NaOAc (1.0) | CPME | 110 | 15 | 73 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oschmann, M.; Johansson Holm, L.; Pourghasemi-Lati, M.; Verho, O. Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry. Molecules 2020, 25, 361. https://doi.org/10.3390/molecules25020361
Oschmann M, Johansson Holm L, Pourghasemi-Lati M, Verho O. Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry. Molecules. 2020; 25(2):361. https://doi.org/10.3390/molecules25020361
Chicago/Turabian StyleOschmann, Michael, Linus Johansson Holm, Monireh Pourghasemi-Lati, and Oscar Verho. 2020. "Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry" Molecules 25, no. 2: 361. https://doi.org/10.3390/molecules25020361
APA StyleOschmann, M., Johansson Holm, L., Pourghasemi-Lati, M., & Verho, O. (2020). Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry. Molecules, 25(2), 361. https://doi.org/10.3390/molecules25020361