
Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells

 ,
, 

Abstract
1. Introduction
2. Results and Discussion
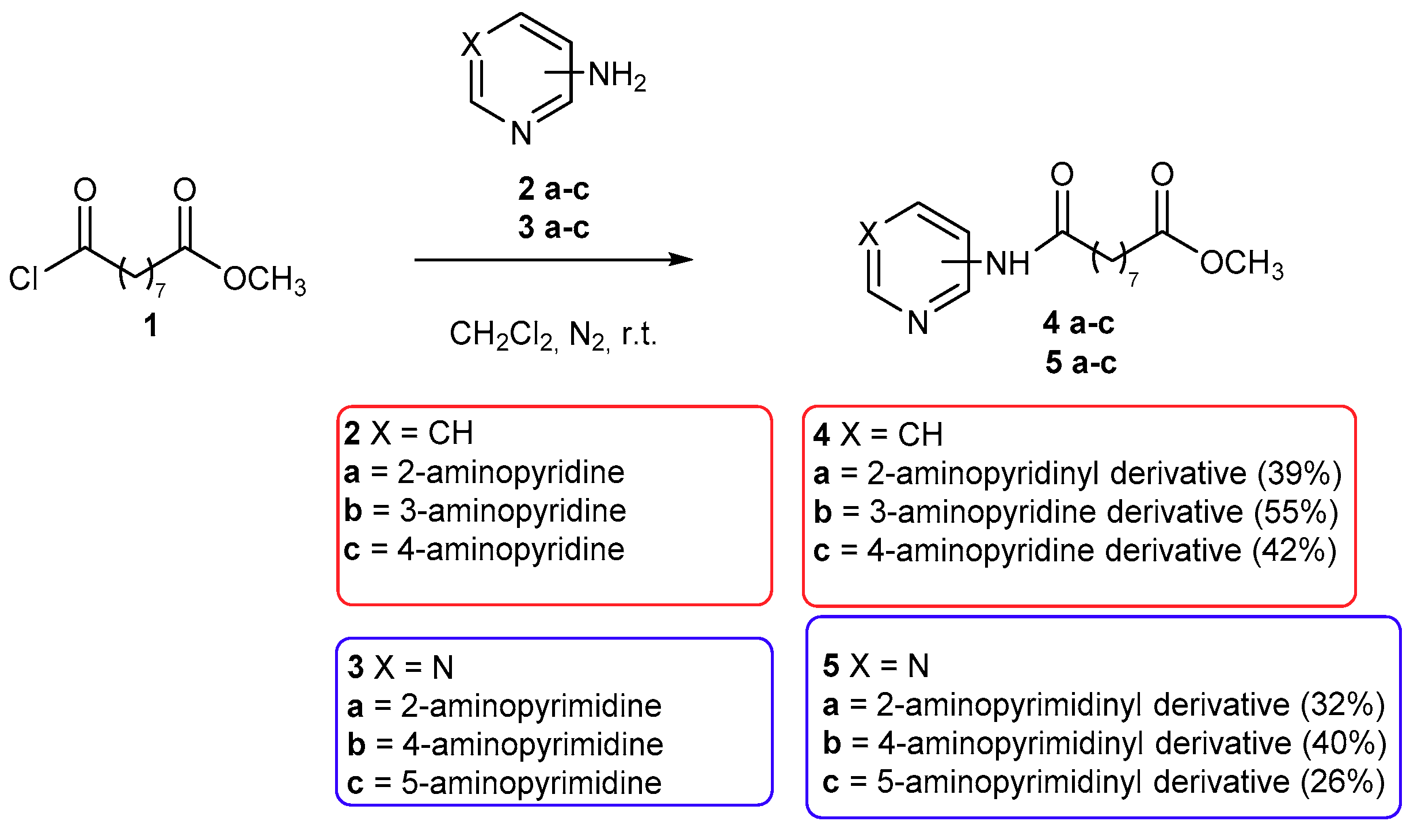
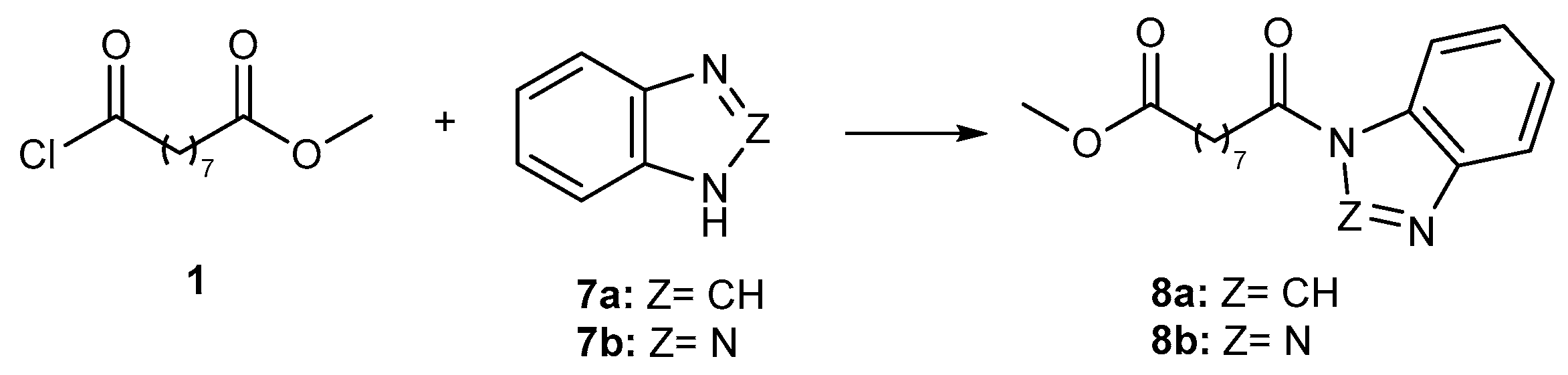
2.1. Chemistry
2.2. Biological Activity
2.2.1. In Vitro Effects on Cell Viability
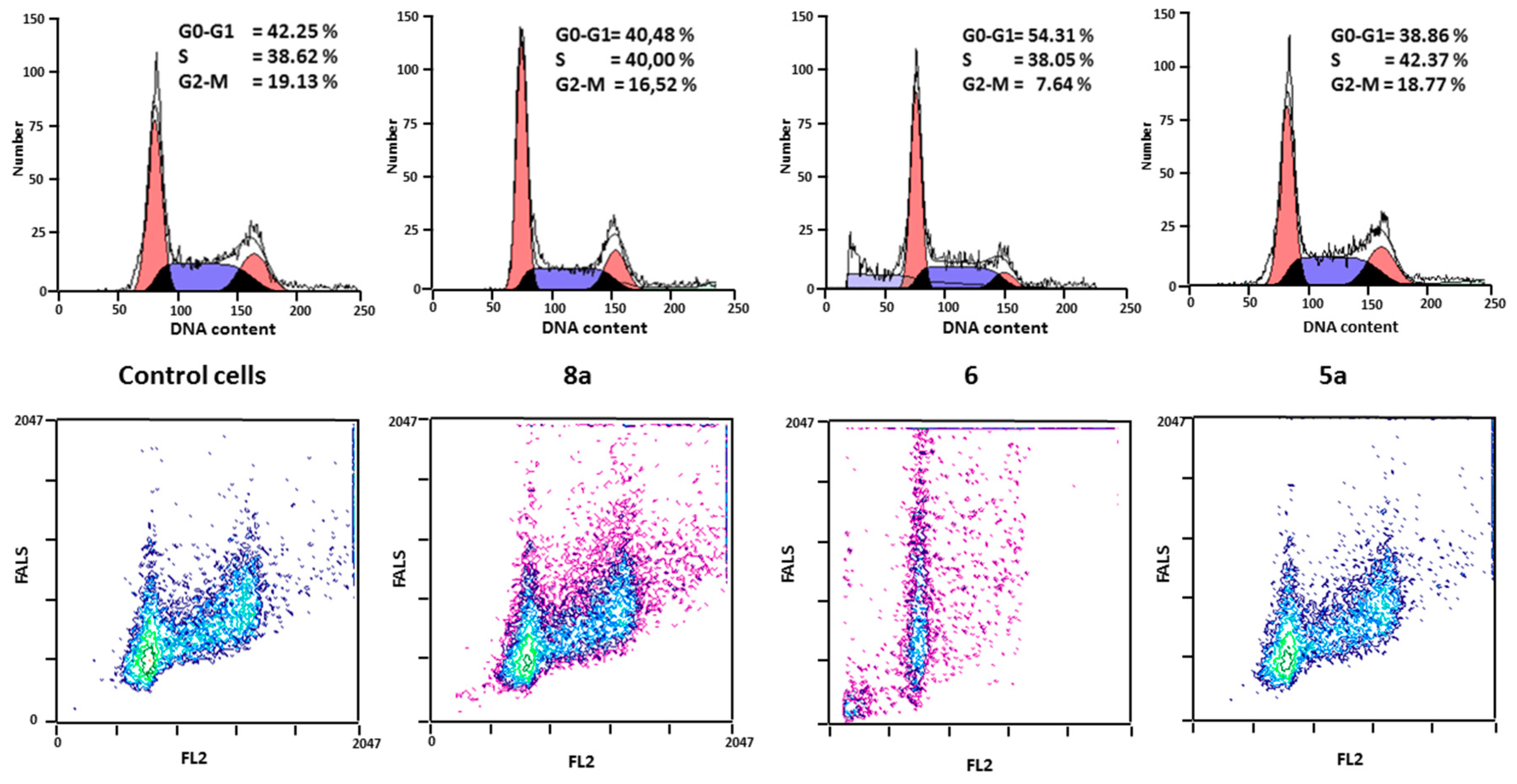
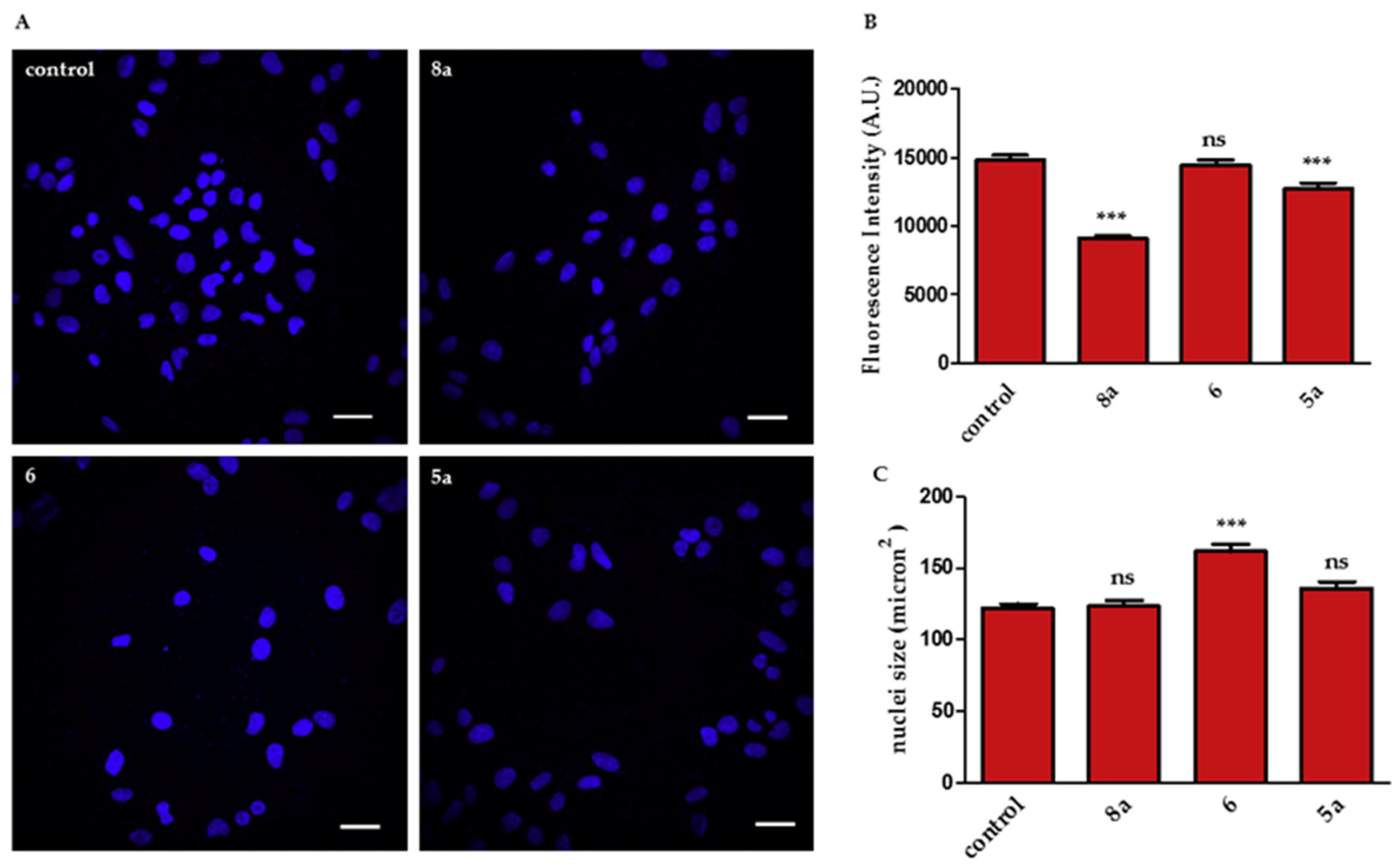
2.2.2. Effects on Cell Proliferation
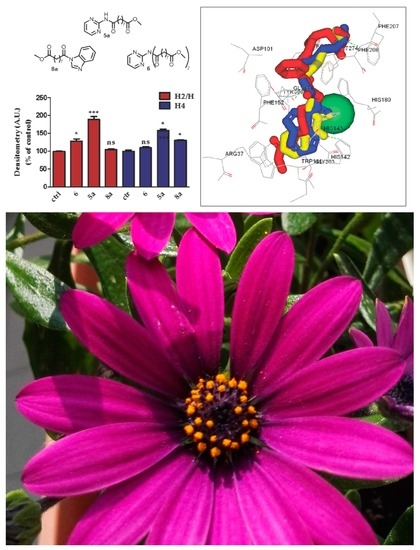
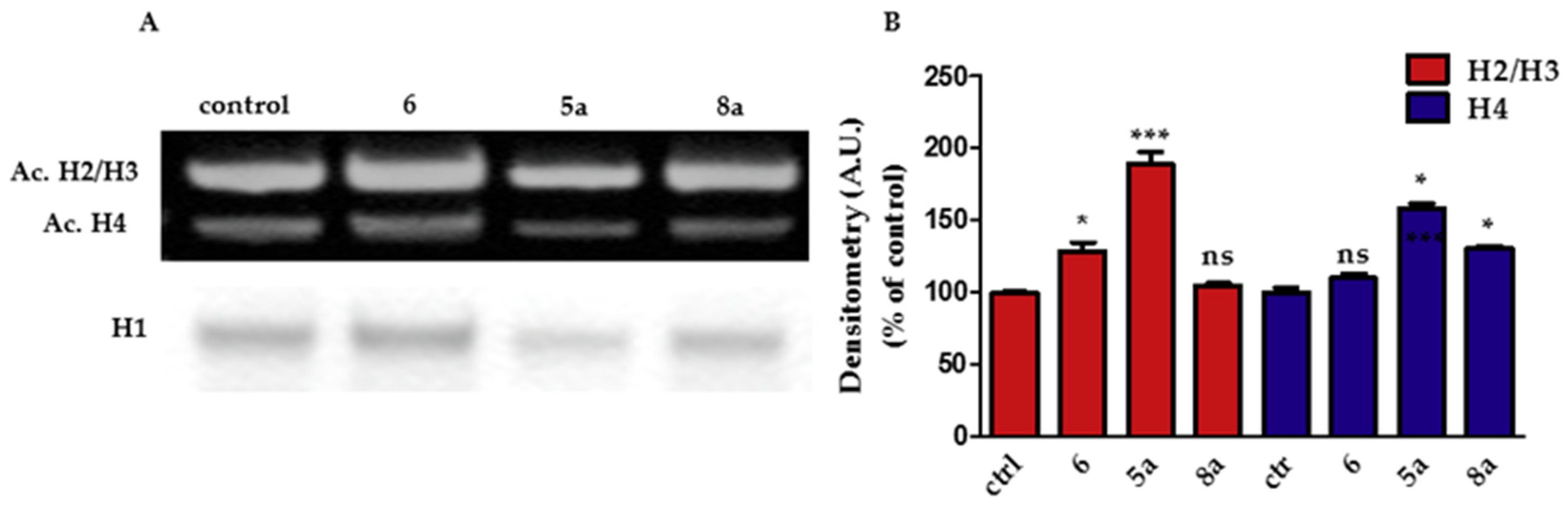
2.2.3. Effect on Histone Acetylation
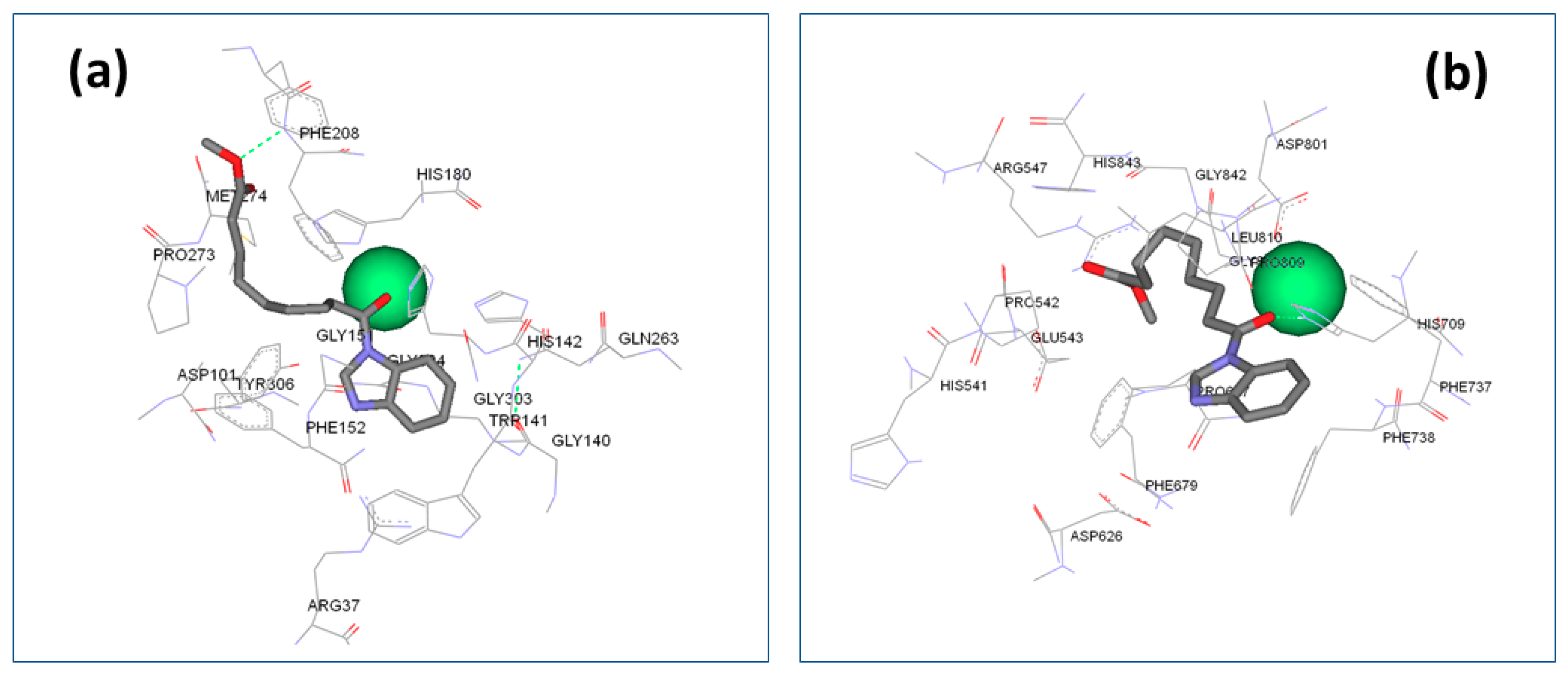
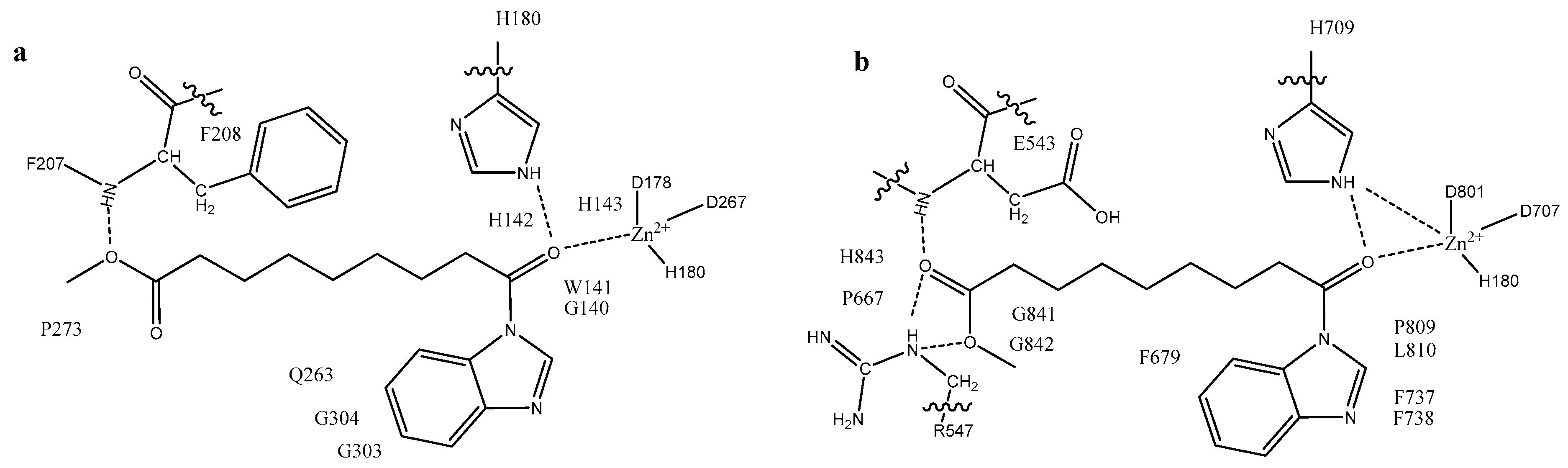
2.3. Docking Evaluation
Analysis of the Binding Mode
3. Materials and Methods
3.1. Chemical Syntheses
3.1.1. Synthesis of Methyl 9-Chloro-9-oxononanoate (1)
3.1.2. General Procedure for the Synthesis of Compounds 4a–c, 5a–c, 6, and 8a,b
3.2. Cell Culture and Treatments
3.2.1. Cell Culture
3.2.2. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) Assay
3.2.3. Cell Cycles
3.2.4. Histone Extraction, SDS-PAGE, and Western Blot
3.2.5. Hoechst 33,342 Staining
3.3. Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anderson, T. Ueber die Producte der trocknen Destillation thierischer Materien. Justus Liebigs Ann. Chem. 1851, 80, 44–55. [Google Scholar] [CrossRef]
- Moen, M.D.; McKeage, K.; Plosker, G.L.; Siddiqui, M.A. Imatinib: A review of its use in chronic myeloid leukaemia. Drugs 2007, 67, 299–320. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Scott, L.J. Imatinib: A review of its use in the management of gastrointestinal stromal tumours. Drugs 2007, 67, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Zwergel, C.; Stazi, G.; Valente, S.; Mai, A. Histone Deacetylase Inhibitors: Updated Studies in Various Epigenetic-Related Diseases. J. Clin. Epigen. 2016, 2, 1–15. [Google Scholar] [CrossRef]
- Dolezal, M.; Zitko, J. Pyrazine derivatives: A patent review (June 2012 – present). Expert Opin. Ther. Patents 2015, 25, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Miniyar, P.B.; Murumkar, P.R.; Patil, P.S.; Barmade, M.A.; Bothara, K.G. Unequivocal Role of Pyrazine Ring in Medicinally Important Compounds: A Review. Mini-Rev. Med. Chem. 2013, 13. [Google Scholar] [CrossRef]
- Xi, N.; Huang, Q.; Liu, L. Imidazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Volume 4 (Five-membered Rings with Two Heteroatoms, each with their Fused Carbocyclic Derivatives); Elsevier: Amsterdam, The Netherlands, 2008; pp. 143–364. [Google Scholar]
- Chen, B.; Heal, H. Thiazoles. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Volume 4 (Five-membered Rings with Two Heteroatoms, each with their Fused Carbocyclic Derivatives); Elsevier: Amsterdam, The Netherlands, 2008; pp. 635–754. [Google Scholar]
- Forlani, L.; Lugli, A.; Boga, C.; Bonamartini Corradi, A.; Sgarabotto, P. Mechanism of the formation of 1,2,4-Thiadiazoles by Condensation of Aromatic Thioamides and of N-Substituted Thioureas. J. Heterocycl. Chem. 2000, 37, 63–69. [Google Scholar] [CrossRef]
- Boga, C.; Stengel, R.; Abdayem, R.; Del Vecchio, E.; Forlani, L.; Todesco, P.E. Regioselectivity in the Addition of Vinylmagnesium Bromide to Heteroarylic Ketones: C- versus O-Alkylation. J. Org. Chem. 2004, 69, 8903–8909. [Google Scholar] [CrossRef]
- Boga, C.; Micheletti, G. Regioselectivity in the Addition of Grignard Reagents to Bis(2-benzothiazolyl) ketone. C- versus O-alkylation Using Aryl Grignard Reagents. Eur. J. Org. Chem 2010, 5659–5665. [Google Scholar]
- Boga, C.; Del Vecchio, E.; Forlani, L.; Goumont, R.; Terrier, F.; Tozzi, S. Evidence for the Intermediacy of Wheland–Meisenheimer Complexes in SEAr Reactions of Aminothiazoles with 4,6-Dinitrobenzofuroxan. Chem. Eur. J. 2007, 13, 9600–9607. [Google Scholar] [CrossRef]
- Forlani, L.; Boga, C.; Mazzanti, A.; Zanna, N. Trapping and Analysing Wheland–Meisenheimer σ Complexes, Usually Labile and Escaping Intermediates. Eur. J. Org. Chem. 2012, 1123–1129. [Google Scholar]
- Boga, C.; Cino, S.; Micheletti, G.; Padovan, D.; Prati, L.; Mazzanti, A.; Zanna, N. New azo-decorated N-pyrrolidinylthiazoles: Synthesis, properties and an unexpected remote substituent effect transmission. Org. Biomol. Chem. 2016, 14, 7061–7068. [Google Scholar] [CrossRef] [PubMed]
- Boga, C.; Bordoni, S.; Casarin, L.; Micheletti, G.; Monari, M. Regioselectivity in Reactions between Bis(2-benzothiazolyl)ketone and Vinyl Grignard Reagents: C- versus O-alkylation—Part III. Molecules 2018, 23, 171. [Google Scholar] [CrossRef] [PubMed]
- Chugunova, E.; Boga, C.; Sazykin, I.; Cino, S.; Micheletti, G.; Mazzanti, A.; Sazykina, M.; Burilov, A.; Khmelevtsova, L.; Kostina, N. Synthesis and antimicrobial activity of novel structural hybrids of benzofuroxan and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 93, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Boga, C.; Micheletti, G.; Orlando, I.; Strocchi, E.; Vitali, B.; Verardi, L.; Sartor, G.; Calonghi, N. New Hybrids with 2-aminobenzothiazole and Azelayl Scaffolds: Synthesis, Molecular Docking and Biological Evaluation. Curr. Org. Chem. 2018, 22, 1649–1660. [Google Scholar] [CrossRef]
- Bertucci, C.; Hudaib, M.; Boga, C.; Calonghi, N.; Cappadone, C.; Masotti, L. Gas chromatography/mass spectrometry assay of endogenous cellular lipid peroxidation products: Quantitative analysis of 9- and 10-hydroxystearic acids. Rapid Commun. Mass Spectrom. 2002, 16, 859–864. [Google Scholar] [CrossRef]
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Farruggia, G.; Buontempo, F.; Boga, C.; Brusa, G.L.; Santucci, M.A.; Masotti, L. 9-Hydroxystearic acid upregulates p21WAF1 in HT29 cancer cells. Biochem. Biophys. Res. Commun. 2004, 314, 138–142. [Google Scholar] [CrossRef]
- Calonghi, N.; Pagnotta, E.; Parolin, C.; Tognoli, C.; Boga, C.; Masotti, L. 9-Hydroxystearic acid interferes with EGF signalling in a human colon adenocarcinoma. Biochem. Biophys. Res. Commun. 2006, 342, 585–588. [Google Scholar] [CrossRef]
- Busi, A.; Aluigi, A.; Guerrini, A.; Boga, C.; Sartor, G.; Calonghi, N.; Sotgiu, G.; Posati, T.; Corticelli, F.; Fiori, J.; et al. Unprecedented behavior of (9R)-9-hydroxystearic acid loaded keratin nanoparticles on cancer cell cycle. Mol. Pharm. 2019, 16, 931–942. [Google Scholar] [CrossRef]
- Calonghi, N.; Pagnotta, E.; Parolin, C.; Molinari, C.; Boga, C.; Dal Piaz, F.; Brusa, G.L.; Santucci, M.A.; Masotti, L. Modulation of apoptotic signalling by 9-hydroxystearic acid in osteosarcoma cells. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids 2007, 1771, 139–146. [Google Scholar] [CrossRef]
- Boanini, E.; Torricelli, P.; Boga, C.; Micheletti, G.; Cassani, M.C.; Fini, M.; Bigi, A. (9R)-9-Hydroxystearate-Functionalized Hydroxyapatite as Anti-Proliferative and Cytotoxic Agent towards Osteosarcoma Cells. Langmuir 2016, 32, 188–194. [Google Scholar] [CrossRef]
- Calonghi, N.; Boga, C.; Telese, D.; Bordoni, S.; Sartor, G.; Torsello, C.; Micheletti, G. Synthesis of 9-Hydroxystearic Acid Derivatives and Their Antiproliferative Activity on HT 29 Cancer Cells. Molecules 2019, 24, 3714. [Google Scholar] [CrossRef] [PubMed]
- Calonghi, N.; Cappadone, C.; Pagnotta, E.; Boga, C.; Bertucci, C.; Fiori, J.; Tasco, G.; Casadio, R.; Masotti, L. Histone deacetylase 1: A target of 9-hydroxystearic acid in the inhibition of cell growth in human colon cancer. J. Lipid Res. 2005, 46, 1596–1603. [Google Scholar] [CrossRef] [PubMed]
- Parolin, C.; Calonghi, N.; Presta, E.; Boga, C.; Caruana, P.; Naldi, M.; Andrisano, V.; Masotti, L.; Sartor, G. Mechanism and stereoselectivity of HDAC I inhibition by (R)-9-hydroxystearic acid in colon cancer. Biochim. Biophys. Acta 2012, 1821, 1334–1340. [Google Scholar]
- Albadri, S.; Naso, F.; Gauron, C.; Parolin, C.; Duroure, K.; Fiori, J.; Boga, C.; Vriz, S.; Calonghi, N.; Del Bene, F. Redox signaling via lipid peroxidation regulates retinal progenitor cell differentiation. Dev. Cell 2019, 50, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M. Cancer biology: Mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Br. J. Cancer. 2006, 95 (Suppl. 1), S2–S6. [Google Scholar] [CrossRef]
- Harris, M.G.; Stewart, R. Amino group acidity in aminopyridines and aminopyrimidines. Can. J. Chem. 1977, 55, 3800–3806. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, J.; Ji, M.; Gai, P.; Lv, Q. Inhibitory effect of 5-Fluorouracil on the proliferation of human osteosarcoma cells in vitro. J. BUON 2019, 24, 1706–1711. [Google Scholar]
- Shin, S.H.; Choi, Y.J.; Lee, H.; Han-Soo Kim, H.-S.; Seo, S.W. Oxidative stress induced by low-dose doxorubicin promotes the invasiveness of osteosarcoma cell line U2OS in vitro. Tumor Biol. 2016, 37, 1591–1598. [Google Scholar] [CrossRef]
- Wu, Z.; Ma, C.; Shan, Z.; Ju, Y.; Li, S.; Zhao, Q. Histone deacetylase inhibitors suppress the growth of human osteosarcomas in vitro and in vivo. J. BUON. 2013, 18, 1032–1037. [Google Scholar]
- Bai, Y.; Chen, Y.; Chen, X.; Jiang, J.; Wang, X.; Wang, L.; Wang, J.; Zhang, J.; Gao, L. Trichostatin A activates FOXO1 and induces autophagy in osteosarcoma. Arch. Med. Sci. 2019, 15, 204–213. [Google Scholar] [CrossRef]
- Roberts, W.E.; Mozsary, P.G.; Klinger, E. Nuclear size as a cell-kinetic marker for osteoblast differentiation. Am. J. Anat. 1982, 165, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Jevtic’, P.; Edens, L.J.; Vukovic’, L.D.; Levy, D.L. Sizing and shaping the nucleus: Mechanisms and significance. Curr. Opin. Cell Bio.l 2014, 28, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Chow, K.-H.; Factor, R.E.; Ullman, K.S. The nuclear envelope environment and its cancer connections. Nat. Rev. Cancer 2012, 12, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.B.; Park, K.R.; Kim, S.Y.; Han, S.Y. Induction of nuclear enlargement and senescence by sirtuin inhibitors in glioblastoma cells. Immune Netw. 2016, 16, 183–188. [Google Scholar] [CrossRef]
- Bang, M.; Kim, D.G.; Gonzales, E.L.; Kwon, K.J.; Shin, C.Y. Etoposide Induces Mitochondrial Dysfunction and Cellular Senescence in Primary Cultured Rat Astrocytes. Biomol. Ther. 2019, 27, 530–539. [Google Scholar] [CrossRef]
- Kong, J.Y.; Rabkin, S.W. Palmitate induces structural alterations in nuclei of cardiomyocytes. Tissue Cell 1999, 31, 473–479. [Google Scholar]
- Gordon, G.B. Saturated free fatty acid toxicity II. Lipid accumulation, ultrastructural alterations and toxicity in mammalian cells in culture. Exp. Molec. Path. 1977, 27, 262–276. [Google Scholar] [CrossRef]
- Pham, L.; Kaiser, B.; Romsa, A.; Schwarz, T.; Gopalakrishnan, R.; Jensen, E.D.; Mansky, K.C. HDAC3 and HDAC7 Have Opposite Effects on Osteoclast Differentiation. J. Biol. Chem. 2011, 286, 12056–12065. [Google Scholar] [CrossRef]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting Histone Deacetylases in Diseases: Where Are We? Antioxid. Redox Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef]
- Bottomley, M.J.; Lo Surdo, P.; Di Giovine, P.; Cirillo, A.; Scarpelli, R.; Ferrigno, F.; Jones, P.; Neddermann, P.; De Francesco, R.; Steinkühler, C.; et al. Structural and Functional Analysis of the Human HDAC4 Catalytic Domain Reveals a Regulatory Structural Zinc-Binding Domain. J. Biol. Chem. 2008, 283, 26694–26704. [Google Scholar] [CrossRef]
- Miller, T.A.; Witter, D.J.; Belvedere, S. Histone Deacetylase Inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Amellem, O.; Stokke, T.; Sandvik, J.A.; Pettersen, E.O. The retinoblastoma gene product is reversibly dephosphorylated and bound in the nucleus in S and G2 phases during hypoxic stress. Exp. Cell Res. 1996, 227, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Strocchi, E.; Fornari, F.; Minguzzi, M.; Gramantieri, L.; Milazzo, M.; Rebuttini, V.; Breviglieri, S.; Camaggi, C.M.; Locatelli, E.; Bolondi, L.; et al. Design, synthesis and biological evaluation of pyrazole derivatives as potential multi-kinase inhibitors in hepatocellular carcinoma. Eur. J. Med. Chem. 2012, 48, 391–401. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the all synthesized compounds are available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solvent | U2OS IC50 (µM) | HDFa IC50 (µM) | HT29 IC50 (µM) | PC3 IC50 (µM) | IGROV1 IC50 (µM) |
|---|---|---|---|---|---|---|
 | DMSO 60 mM | >100 | n.a. | >100 | n.a. | n.a. |
 | DMSO 60 mM | >100 | n.a. | >100 | n.a. | n.a. |
 | DMSO 60 mM | >100 | n.a. | n.a. | n.a. | n.a. |
 | DMSO 60 mM | 50 | n.a. | n.a | n.a | n.a |
 | DMSO 60 mM | >100 | n.a. | n.a. | n.a. | >100 |
 | DMSO 60 mM | n.a. | n.a. | >100 | >100 | >100 |
 | DMSO 60 mM | 35 | n.a. | >100 | n.a. | n.a. |
 | DMSO 60 mM | 50 | n.a. | >100 | >100 | n.a. |
 | DMSO 60 mM | n.a. | n.a. | n.a. | n.a. | >100 |
| HDAC Class I | HDAC Class II | |||||
|---|---|---|---|---|---|---|
| HDAC1-5ICN | HDAC2-4LXZ | HDAC3-4A69 | HDAC8-4QA3 | HDAC4-2VQM | HDAC7-3C0Z | |
| BE_∆G (mol_5a) | −5.80 | −6.90 | −5.47 | −8.05 | −6.03 | −7.26 |
| ∆G (mol_6) | −6.36 | −7.24 | −5.92 | −8.14 | −7.61 | −7.00 |
| ∆G (mol_8a) | −6.83 | −7.73 | −6.38 | −8.63 | −6.70 | −7.61 |
| Ki (mol_5a) | 55.93 μM | 8.77 μM | 98.23 μM | 1.26 μM | 37.85 μM | 4.78 μM |
| Ki (mol_6) | 21.78 μM | 4.94 μM | 45.52 μM | 1.07 μM | 2.62 μM | 7.37 μM |
| Ki (mol_8a) | 9.92 μM | 2.17 μM | 21.08 μM | 472.15 nM | 12.29 μM | 2.62 μM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micheletti, G.; Calonghi, N.; Farruggia, G.; Strocchi, E.; Palmacci, V.; Telese, D.; Bordoni, S.; Frisco, G.; Boga, C. Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells. Molecules 2020, 25, 404. https://doi.org/10.3390/molecules25020404
Micheletti G, Calonghi N, Farruggia G, Strocchi E, Palmacci V, Telese D, Bordoni S, Frisco G, Boga C. Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells. Molecules. 2020; 25(2):404. https://doi.org/10.3390/molecules25020404
Chicago/Turabian StyleMicheletti, Gabriele, Natalia Calonghi, Giovanna Farruggia, Elena Strocchi, Vincenzo Palmacci, Dario Telese, Silvia Bordoni, Giulia Frisco, and Carla Boga. 2020. "Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells" Molecules 25, no. 2: 404. https://doi.org/10.3390/molecules25020404
APA StyleMicheletti, G., Calonghi, N., Farruggia, G., Strocchi, E., Palmacci, V., Telese, D., Bordoni, S., Frisco, G., & Boga, C. (2020). Synthesis of Novel Structural Hybrids between Aza-Heterocycles and Azelaic Acid Moiety with a Specific Activity on Osteosarcoma Cells. Molecules, 25(2), 404. https://doi.org/10.3390/molecules25020404