N-[2-(4-Acetyl-1-Piperazinyl)Phenyl]-2-(3-Methylphenoxy)Acetamide (NAPMA) Inhibits Osteoclast Differentiation and Protects against Ovariectomy-Induced Osteoporosis

 , ,
, ,  ,
,
Abstract
:
1. Introduction
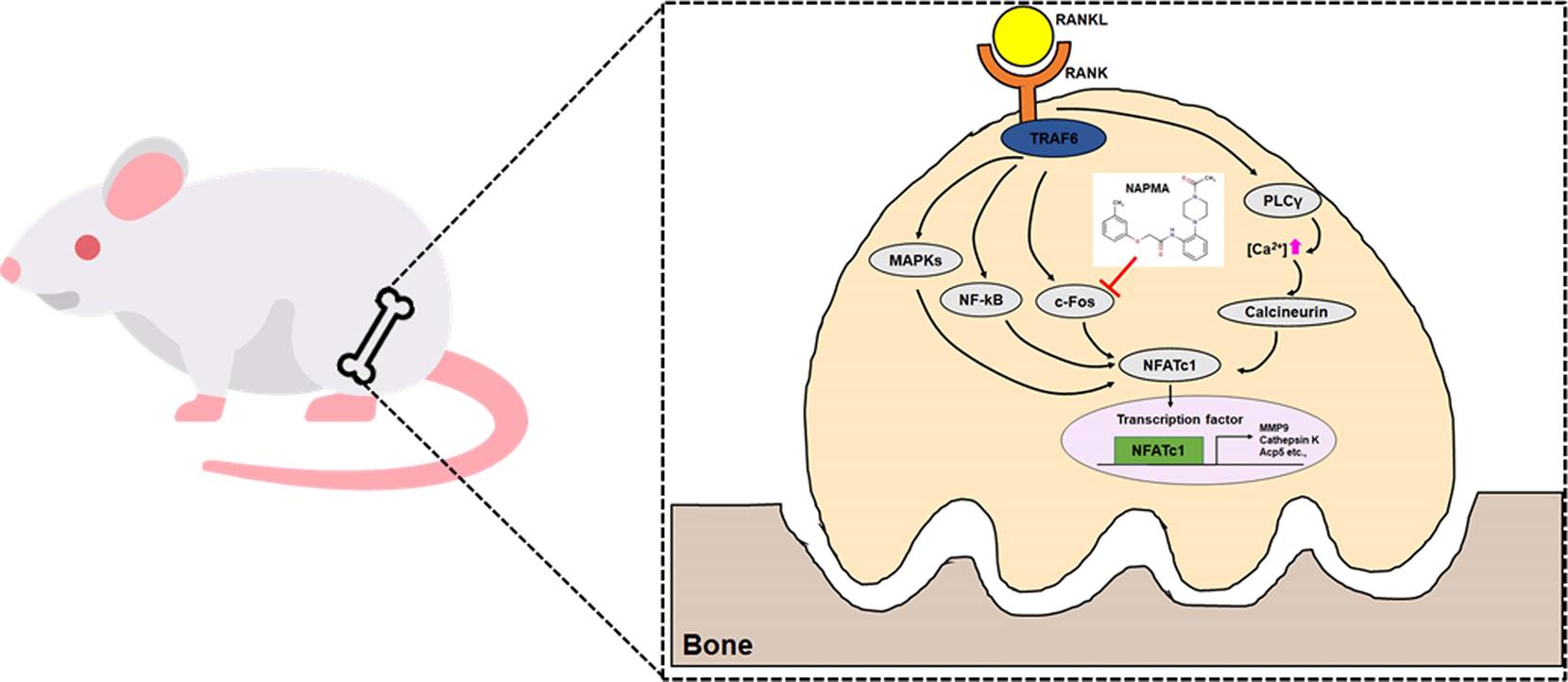
2. Results
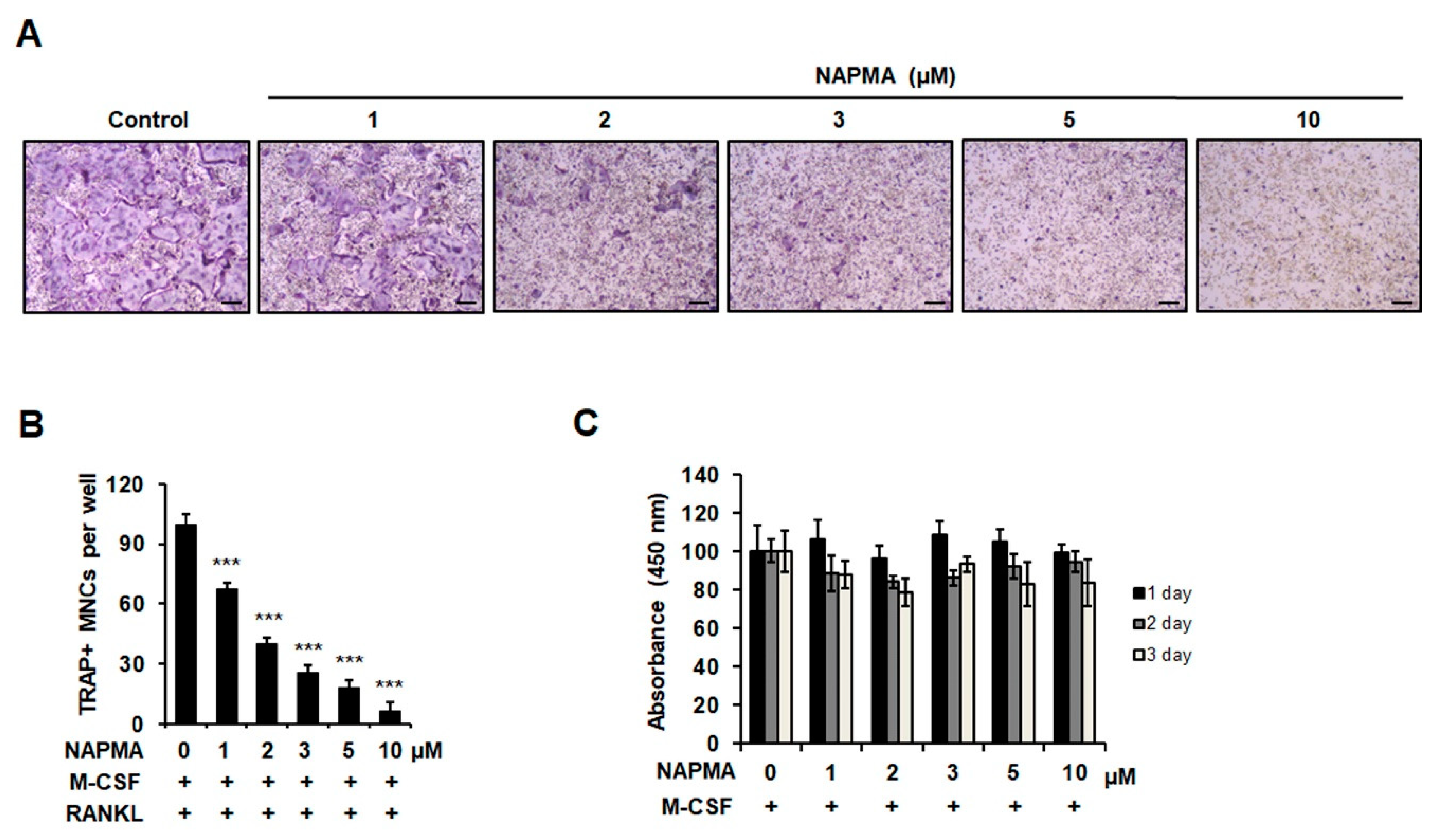
2.1. NAPMA Inhibits RANKL-Induced Osteoclastogenesis In Vitro but Does Not Affect the Differentiation of Primary Calvarial Cells Into Osteoblasts
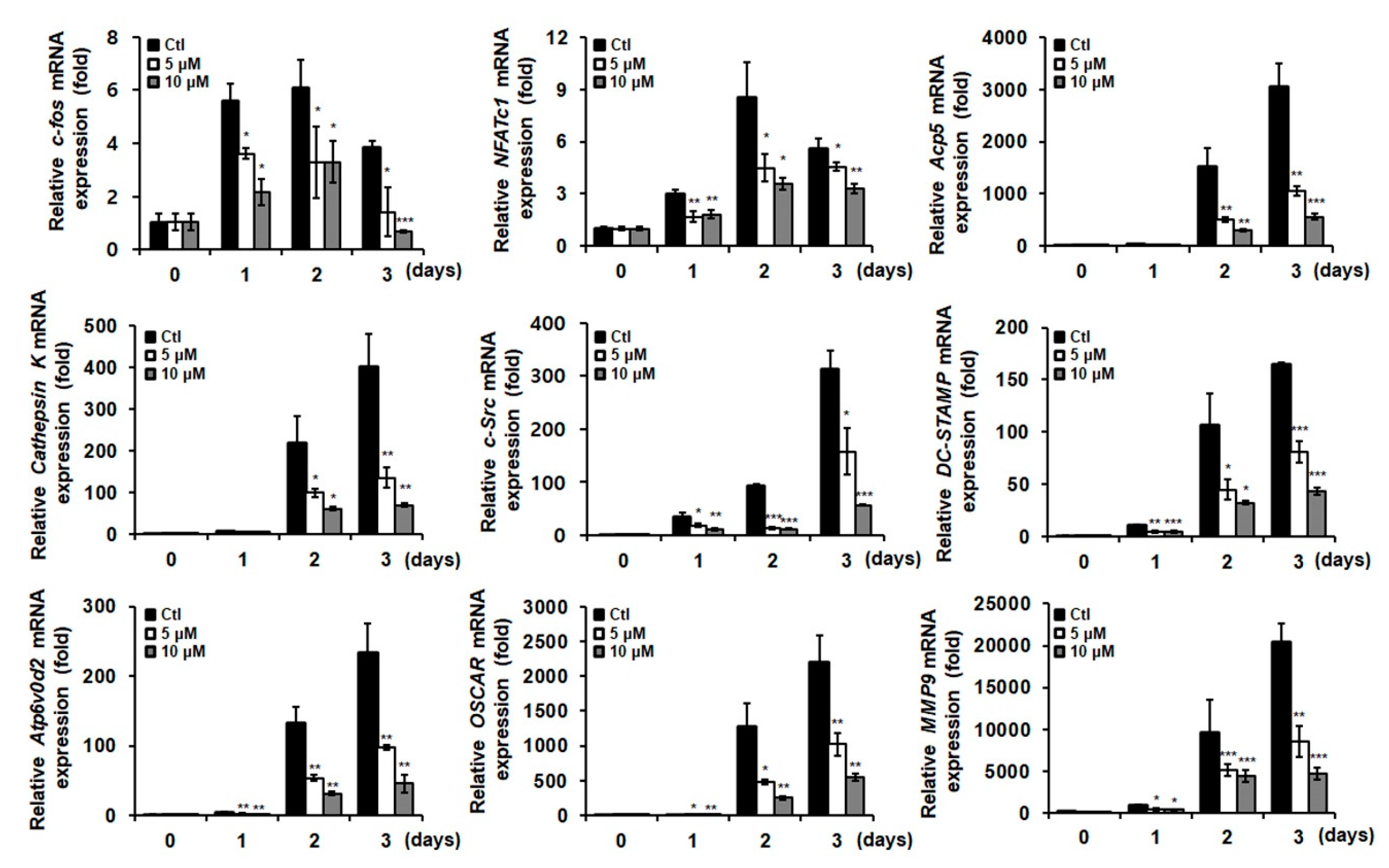
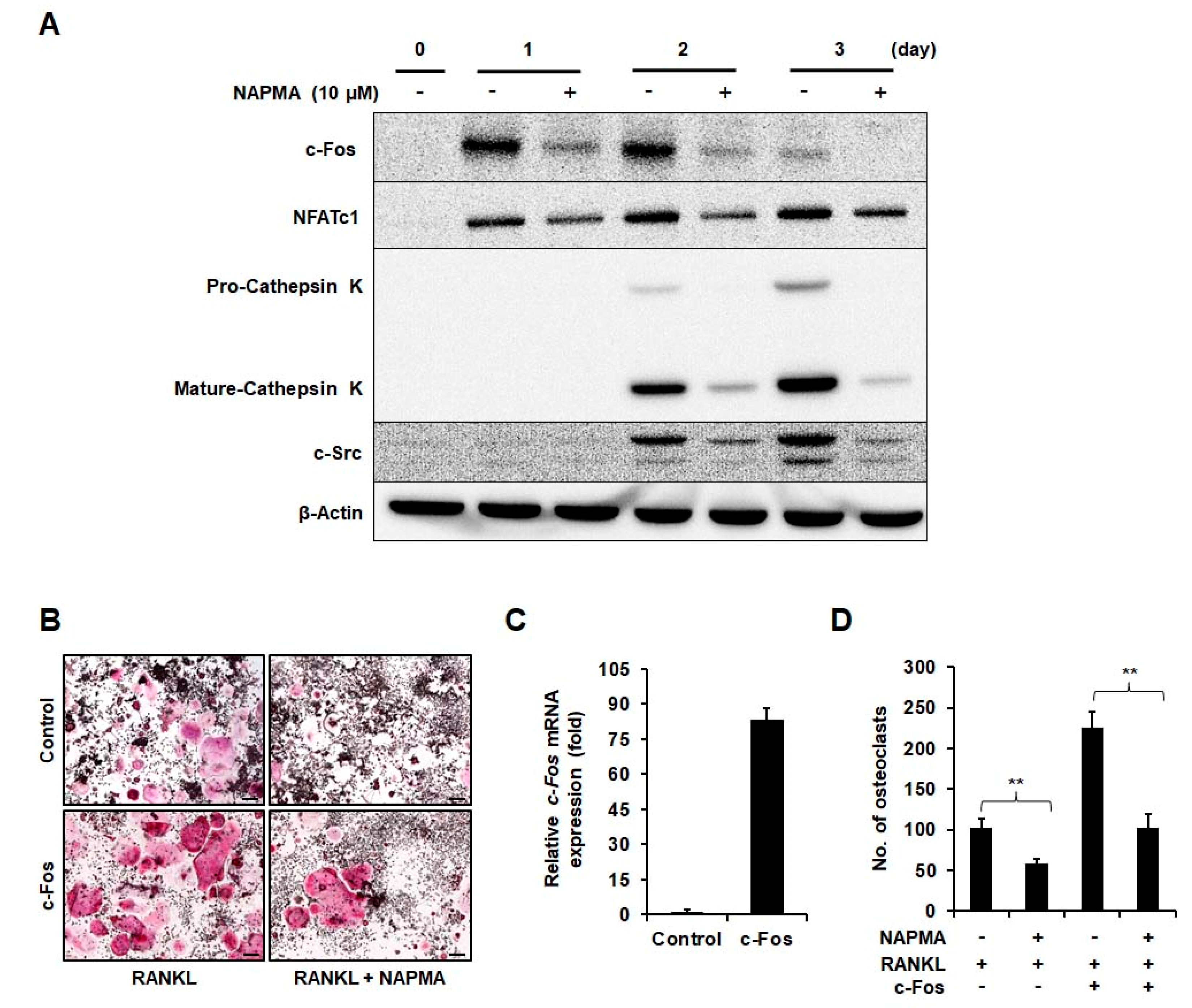
2.2. NAPMA Suppresses the Expression of Osteoclast Marker Genes
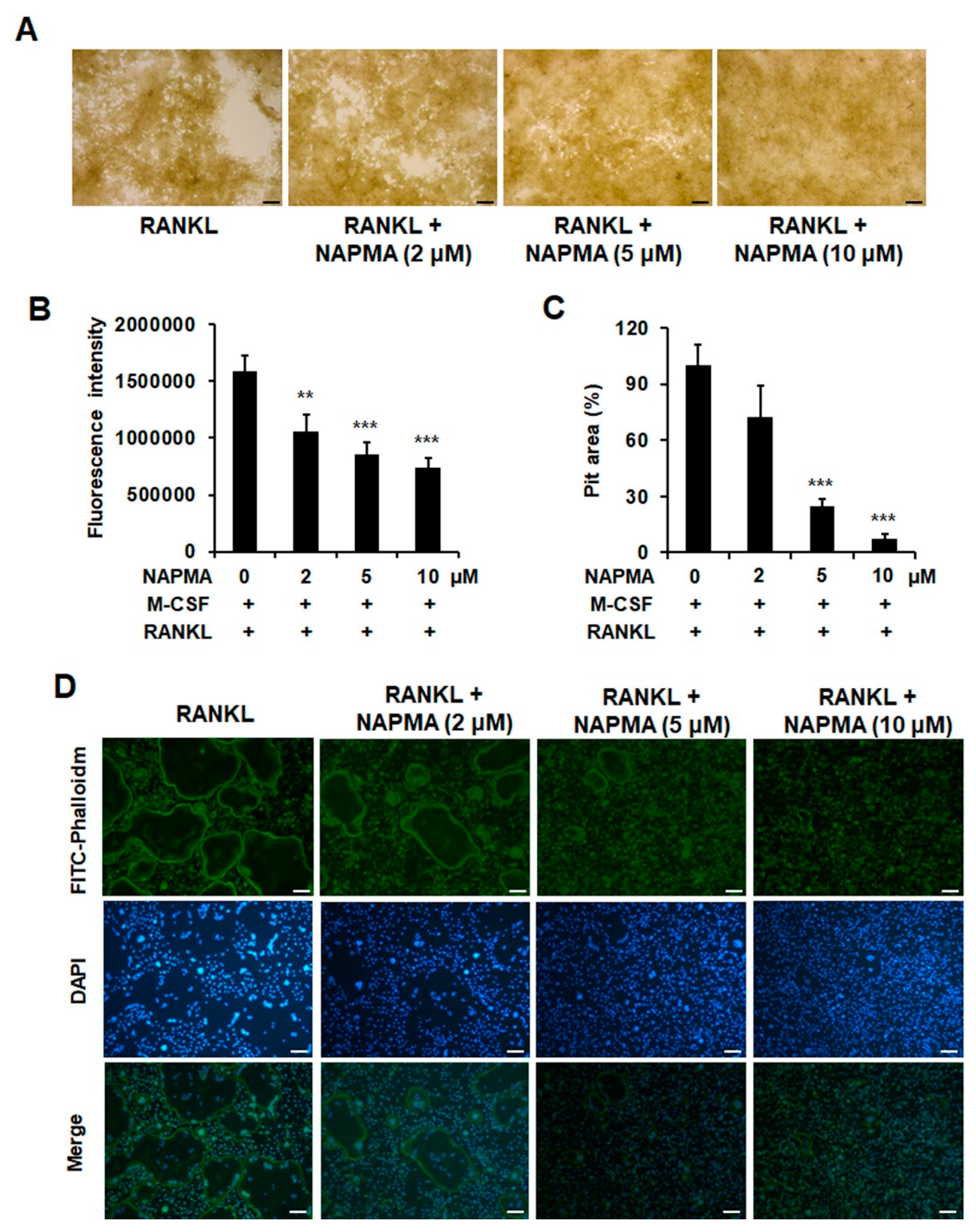
2.3. NAPMA Inhibits Bone Resorption and Actin Ring Formation in Osteoclasts
2.4. NAPMA Inhibits Osteoclast Differentiation to a Greater Extent than PPOA Derivatives
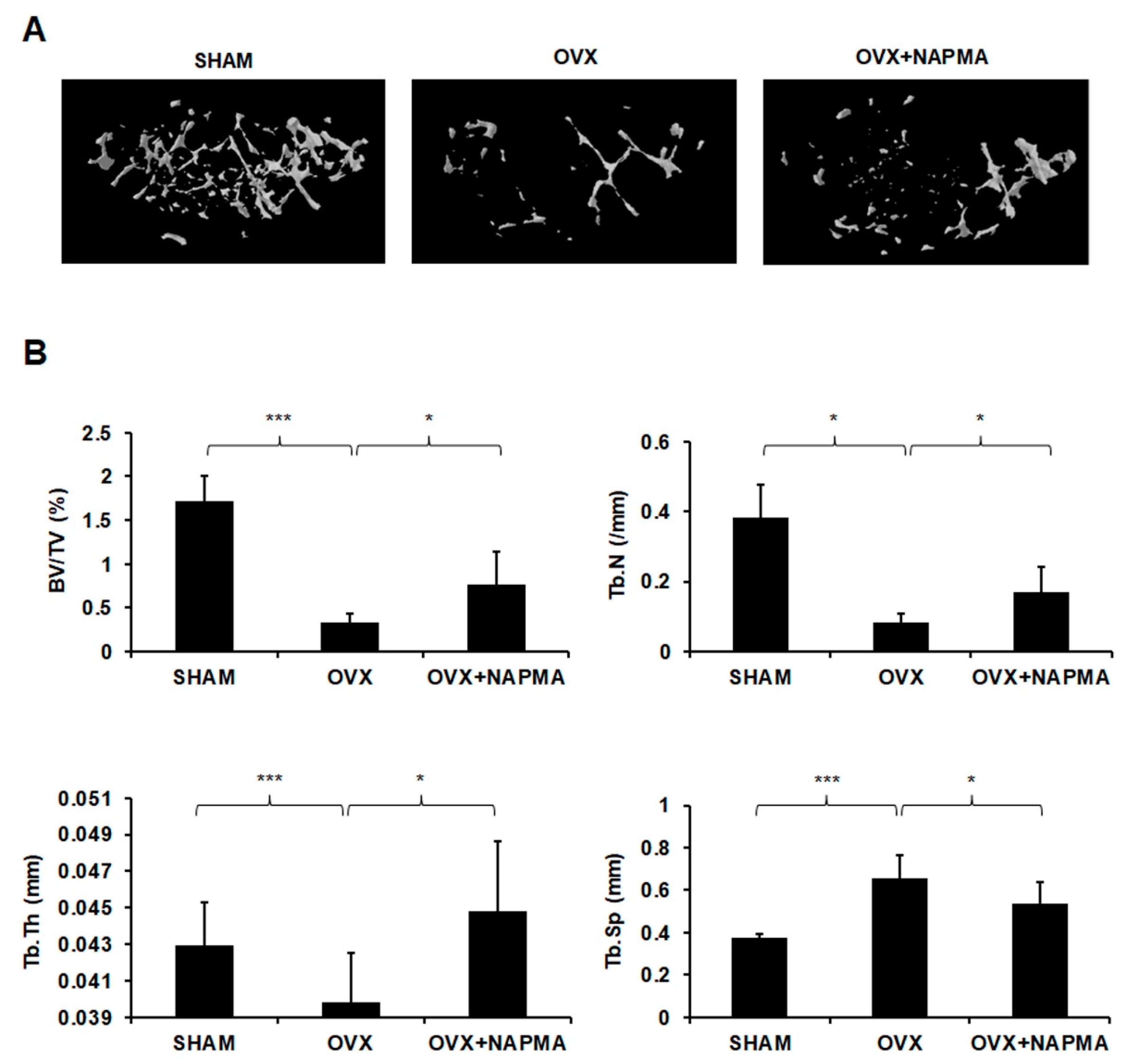
2.5. NAPMA Attenuates OVX-Induced Bone Loss in Mice
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Reagents
4.3. In Vitro Osteoclastogenesis Assay
4.4. Cell Viability Assay
4.5. Osteoclast Precursor Proliferation Assay
4.6. In Vitro Osteoblastogenesis Assay
4.7. Plasmid Transfection
4.8. Real-Time PCR
4.9. Western Blotting
4.10. Resorption Pit Assay
4.11. Actin Ring Formation Assay
4.12. Calcineurin Activity Assay
4.13. In Vivo Animal Model
4.14. Micro-CT Imaging and Data Analysis
4.15. Micro-CT
4.16. Tissue Preparation and Histomorphometric Analysis
4.17. Measurement of Serum CTX-1, Osteocalcin, RANKL, OPG, IL-6 Levels
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RANKL | receptor activator of nuclear factor-kappa B ligand |
| NAPMA | N-[2-(4-acetyl-1-piperazinyl)phenyl]-2-(3-methylphenoxy)acetamide |
| TRAP | tartrate-resistant acid phosphatase |
| BMMs | bone marrow-derived macrophages |
| MNCs | multinucleated cells |
| M-CSF | macrophage colony-stimulating factor |
| RANK | receptor activator of nuclear factor-kappa Β |
| TRAF6 | tumor necrosis factor receptor-associated factor 6 |
| MAPKs | mitogen-activated protein kinases |
| ERK | extracellular signal-regulated |
| JNK | c-Jun N-terminus kinase |
| AP-1 | activator protein 1 |
| NFAT | nuclear factor of activated T-cells |
| Cts K | cathepsin K |
| MMP-9 | matrix metalloprotease 9 |
| DC-STAMP | dendritic cell-specific transmembrane protein |
| FACS | fluoresceinamine-labeled chondroitin sulfate |
| CaP | calcium phosphate |
| DMSO | dimethyl sulfoxide |
| OVX | ovariectomized |
| BV/TV | trabecular bone volume |
| Tb.N | trabecular number |
| Tb.Th | trabecular thickness |
| Tb.Sp | trabecular separation |
| CTX-1 | type 1 collagen cross-linked C-terminal telopeptide |
| OPG | osteoprotegerin |
References
- Eriksen, E.F. Cellular mechanisms of bone remodeling. Rev. Endocr. Metab. Disord. 2010, 11, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Xiu, Y.; Boyce, B.F. Osteoclast fusion and regulation by RANKL-dependent and independent factors. World J. Orthop. 2012, 3, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef]
- Bi, H.; Chen, X.; Gao, S.; Yu, X.; Xiao, J.; Zhang, B.; Liu, X.; Dai, M. Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review. Front. Med. 2017, 4, 234. [Google Scholar] [CrossRef]
- Sato, K.; Takayanagi, H. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 2006, 18, 419–426. [Google Scholar] [CrossRef]
- Cooper, C. Epidemiology of osteoporotic fracture: Looking to the future. Rheumatology 2005, 44 (Suppl. 4), iv36–iv40. [Google Scholar] [CrossRef] [Green Version]
- Kling, J.M.; Clarke, B.L.; Sandhu, N.P. Osteoporosis prevention, screening, and treatment: A review. J. Womens Health 2014, 23, 563–572. [Google Scholar] [CrossRef]
- Darnay, B.G.; Haridas, V.; Ni, J.; Moore, P.A.; Aggarwal, B.B. Characterization of the intracellular domain of receptor activator of NF-kappaB (RANK). Interaction with tumor necrosis factor receptor-associated factors and activation of NF-kappab and c-Jun N-terminal kinase. J. Biol. Chem. 1998, 273, 20551–20555. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef]
- Walsh, M.C.; Kim, N.; Kadono, Y.; Rho, J.; Lee, S.Y.; Lorenzo, J.; Choi, Y. Osteoimmunology: Interplay between the immune system and bone metabolism. Annu. Rev. Immunol. 2006, 24, 33–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayanagi, H. Osteoimmunology: Shared mechanisms and crosstalk between the immune and bone systems. Nat. Rev. Immunol. 2007, 7, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H. The role of NFAT in osteoclast formation. Ann. N. Y. Acad. Sci. 2007, 1116, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Motyckova, G.; Weilbaecher, K.N.; Horstmann, M.; Rieman, D.J.; Fisher, D.Z.; Fisher, D.E. Linking osteopetrosis and pycnodysostosis: Regulation of cathepsin K expression by the microphthalmia transcription factor family. Proc. Natl. Acad. Sci. USA 2001, 98, 5798–5803. [Google Scholar] [CrossRef] [Green Version]
- Trzeciakiewicz, A.; Habauzit, V.; Mercier, S.; Lebecque, P.; Davicco, M.J.; Coxam, V.; Demigne, C.; Horcajada, M.N. Hesperetin stimulates differentiation of primary rat osteoblasts involving the BMP signalling pathway. J. Nutr. Biochem. 2010, 21, 424–431. [Google Scholar] [CrossRef]
- Yamashita, M.; Otsuka, F.; Mukai, T.; Yamanaka, R.; Otani, H.; Matsumoto, Y.; Nakamura, E.; Takano, M.; Sada, K.E.; Makino, H. Simvastatin inhibits osteoclast differentiation induced by bone morphogenetic protein-2 and RANKL through regulating MAPK, AKT and Src signaling. Regul. Pept. 2010, 162, 99–108. [Google Scholar] [CrossRef]
- Kwak, H.B.; Lee, B.K.; Oh, J.; Yeon, J.T.; Choi, S.W.; Cho, H.J.; Lee, M.S.; Kim, J.J.; Bae, J.M.; Kim, S.H.; et al. Inhibition of osteoclast differentiation and bone resorption by rotenone, through down-regulation of RANKL-induced c-Fos and NFATc1 expression. Bone 2010, 46, 724–731. [Google Scholar] [CrossRef]
- Kim, H.J.; Chang, E.J.; Kim, H.M.; Lee, S.B.; Kim, H.D.; Su Kim, G.; Kim, H.H. Antioxidant alpha-lipoic acid inhibits osteoclast differentiation by reducing nuclear factor-kappaB DNA binding and prevents in vivo bone resorption induced by receptor activator of nuclear factor-kappaB ligand and tumor necrosis factor-alpha. Free Radic. Biol. Med. 2006, 40, 1483–1493. [Google Scholar] [CrossRef]
- Gallagher, J.C.; Tella, S.H. Controversies in osteoporosis management: Antiresorptive therapy for preventing bone loss: When to use one or two antiresorptive agents? Clin. Obstet. Gynecol. 2013, 56, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Koehne, T.; Kahl-Nieke, B.; Amling, M.; Korbmacher-Steiner, H. Inhibition of bone resorption by bisphosphonates interferes with orthodontically induced midpalatal suture expansion in mice. Clin. Oral Investig. 2018, 22, 2345–2351. [Google Scholar] [CrossRef]
- Kennel, K.A.; Drake, M.T. Adverse effects of bisphosphonates: Implications for osteoporosis management. Mayo Clin. Proc. 2009, 84, 632–637. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.H.; Chen, Z.; Lee, J.; Lee, S.W.; Min, S.H.; Kim, N.D.; Lee, T.H. Inhibitory Effects of 2N1HIA (2-(3-(2-Fluoro-4-Methoxyphenyl)-6-Oxo-1(6H)-Pyridazinyl)-N-1H-Indol-5-Ylacetamide) on Osteoclast Differentiation via Suppressing Cathepsin K Expression. Molecules 2018, 23, 3139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, E.; Lee, J.K.; Lee, J.Y.; Chen, Z.; Ahn, S.H.; Kim, N.D.; Kook, M.S.; Min, S.H.; Park, B.J.; Lee, T.H. BCPA {N,N’-1,4-Butanediylbis[3-(2-chlorophenyl)acrylamide]} Inhibits Osteoclast Differentiation through Increased Retention of Peptidyl-Prolyl cis-trans Isomerase Never in Mitosis A-Interacting 1. Int. J. Mol. Sci. 2018, 19, 3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Cho, E.; Lee, J.; Lee, S.; Lee, T.H. Inhibitory Effects of N-[2-(4-acetyl-1-piperazinyl) phenyl]-2-(2-chlorophenoxy) acetamide on Osteoclast Differentiation in Vitro via the Downregulation of TRAF6. Int. J. Mol. Sci. 2019, 20, 5196. [Google Scholar] [CrossRef] [Green Version]
- Han, S.Y.; Lee, K.H.; Kim, Y.K. Poligoni Multiflori Radix enhances osteoblast formation and reduces osteoclast differentiation. Int. J. Mol. Med. 2018, 42, 331–345. [Google Scholar]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Asagiri, M.; Sato, K.; Usami, T.; Ochi, S.; Nishina, H.; Yoshida, H.; Morita, I.; Wagner, E.F.; Mak, T.W.; Serfling, E.; et al. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J. Exp. Med. 2005, 202, 1261–1269. [Google Scholar] [CrossRef] [Green Version]
- Sanjay, A.; Houghton, A.; Neff, L.; DiDomenico, E.; Bardelay, C.; Antoine, E.; Levy, J.; Gailit, J.; Bowtell, D.; Horne, W.C.; et al. Cbl associates with Pyk2 and Src to regulate Src kinase activity, alpha(v)beta(3) integrin-mediated signaling, cell adhesion, and osteoclast motility. J. Cell Biol. 2001, 152, 181–195. [Google Scholar] [CrossRef]
- Lee, K.; Chung, Y.H.; Ahn, H.; Kim, H.; Rho, J.; Jeong, D. Selective Regulation of MAPK Signaling Mediates RANKL-dependent Osteoclast Differentiation. Int. J. Biol. Sci. 2016, 12, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Miyauchi, S.; Anada, T.; Imaizumi, H.; Suzuki, O. Evaluation of osteoclastic resorption activity using calcium phosphate coating combined with labeled polyanion. Anal. Biochem. 2011, 410, 7–12. [Google Scholar] [CrossRef]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florencio-Silva, R.; Sasso, G.R.; Sasso-Cerri, E.; Simoes, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed. Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.H.; Kim, J.H.; Kwak, H.B.; Huang, H.; Han, S.H.; Ha, H.; Lee, S.W.; Woo, E.R.; Lee, Z.H. Inhibition of osteoclast differentiation and bone resorption by tanshinone IIA isolated from Salvia miltiorrhiza Bunge. Biochem. Pharmacol. 2004, 67, 1647–1656. [Google Scholar] [CrossRef]
- Dobbs, M.B.; Buckwalter, J.; Saltzman, C. Osteoporosis: The increasing role of the orthopaedist. Iowa Orthop. J. 1999, 19, 43–52. [Google Scholar] [PubMed]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Khosla, S. Increasing options for the treatment of osteoporosis. N. Engl. J. Med. 2009, 361, 818–820. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Ryu, S.Y.; Choi, J.S.; Min, Y.K.; Kim, S.H. Saurolactam inhibits osteoclast differentiation and stimulates apoptosis of mature osteoclasts. J. Cell. Physiol. 2009, 221, 618–628. [Google Scholar] [CrossRef]
- Qin, S.; Ang, E.; Dai, L.; Yang, X.; Ye, D.; Chen, H.; Zhou, L.; Yang, M.; Teguh, D.; Tan, R.; et al. Natural Germacrane Sesquiterpenes Inhibit Osteoclast Formation, Bone Resorption, RANKL-Induced NF-kappaB Activation, and IkappaBalpha Degradation. Int. J. Mol. Sci. 2015, 16, 26599–26607. [Google Scholar] [CrossRef] [Green Version]
- Thummuri, D.; Naidu, V.G.M.; Chaudhari, P. Carnosic acid attenuates RANKL-induced oxidative stress and osteoclastogenesis via induction of Nrf2 and suppression of NF-kappaB and MAPK signalling. J. Mol. Med. 2017, 95, 1065–1076. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Wang, X.; Liu, Y.; He, A.; Jia, R. NFATc1: Functions in osteoclasts. Int. J. Biochem. Cell Biol. 2010, 42, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Arai, A.; Mizoguchi, T.; Harada, S.; Kobayashi, Y.; Nakamichi, Y.; Yasuda, H.; Penninger, J.M.; Yamada, K.; Udagawa, N.; Takahashi, N. Fos plays an essential role in the upregulation of RANK expression in osteoclast precursors within the bone microenvironment. J. Cell Sci. 2012, 125 Pt 12, 2910–2917. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.Y.; Park, C.H.; Na, Y.H.; Bai, H.W.; Cho, J.Y.; Chung, B.Y. Inhibition of RANKL-induced osteoclast differentiation through the downregulation of c-Fos and NFATc1 by Eremochloa ophiuroides (centipedegrass) extract. Mol. Med. Rep. 2016, 13, 4014–4022. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P.; Montgomery, C.; Geske, R.; Bradley, A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell 1991, 64, 693–702. [Google Scholar] [CrossRef]
- Chen, W.; Xie, Z.; Tang, P.; Wang, Y.; Jie, Z.; Qin, A.; Jiang, X.; Hu, Z.; Fan, S. The emerging role of IMD 0354 on bone homeostasis by suppressing osteoclastogenesis and bone resorption, but without affecting bone formation. Cell Death Dis. 2019, 10, 654. [Google Scholar] [CrossRef]
- Wang, J.; Chen, G.; Zhang, Q.; Zhao, F.; Yu, X.; Ma, X.; Liu, M. Phillyrin Attenuates Osteoclast Formation and Function and Prevents LPS-Induced Osteolysis in Mice. Front. Pharmacol. 2019, 10, 1188. [Google Scholar] [CrossRef] [Green Version]
- Kohn, S.R.; Morgan, W.W.; Carrillo, A.J. Effect with time of a norepinephrine synthesis inhibitor (U-14,624) on hypothalamic catecholamine and plasma gonadotrophin concentrations in the ovariectomized rat. J. Reprod. Fertil. 1982, 66, 185–188. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Qian, Z.; Zhang, X.; Chen, F.; Ni, S.; Kang, Z.; Zhang, F.; Li, D.; Yu, B. Tetrandrine Prevents Bone Loss in Ovariectomized Mice by Inhibiting RANKL-Induced Osteoclastogenesis. Front. Pharmacol. 2019, 10, 1530. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, D.; van Rietbergen, B.; Laib, A.; Ruegsegger, P. The ability of three-dimensional structural indices to reflect mechanical aspects of trabecular bone. Bone 1999, 25, 55–60. [Google Scholar] [CrossRef]
- Yun, I.G.; Ahn, S.H.; Yoon, W.J.; Kim, C.S.; Lim, Y.K.; Kook, J.K.; Jung, S.; Choi, C.H.; Lee, T.H. Litsea japonica Leaf Extract Suppresses Proinflammatory Cytokine Production in Periodontal Ligament Fibroblasts Stimulated with Oral Pathogenic Bacteria or Interleukin-1beta. Int. J. Mol. Sci. 2018, 19, E2494. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.H.; Lee, J.K.; Kim, N.D.; Kim, S.H.; Lee, S.; Jung, S.; Chay, K.O.; Lee, T.H. DPIE [2-(1,2-diphenyl-1H-indol-3-yl)ethanamine] Augments Pro-Inflammatory Cytokine Production in IL-1beta-Stimulated Primary Human Oral Cells. Int. J. Mol. Sci. 2018, 19, E1835. [Google Scholar] [CrossRef] [Green Version]
- Bai, S.; Kopan, R.; Zou, W.; Hilton, M.J.; Ong, C.T.; Long, F.; Ross, F.P.; Teitelbaum, S.L. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J. Biol. Chem. 2008, 283, 6509–6518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellows, C.G.; Aubin, J.E.; Heersche, J.N.; Antosz, M.E. Mineralized bone nodules formed in vitro from enzymatically released rat calvaria cell populations. Calcif. Tissue Int. 1986, 38, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ye, L.; Hui, T.Q.; Yang, D.M.; Huang, D.M.; Zhou, X.D.; Mao, J.J.; Wang, C.L. Bone morphogenetic protein 2-induced human dental pulp cell differentiation involves p38 mitogen-activated protein kinase-activated canonical WNT pathway. Int. J. Oral Sci. 2015, 7, 95–102. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′→3′) | |
|---|---|---|
| GAPDH | Forward | TGTGTCCGTCGTGGATCTGA |
| Reverse | GATGCCTGCTTCACCACCTT | |
| c-Fos | Forward | CGAAGGGAACGGAATAAGATG |
| Reverse | GCTGCCAAAATAAACTCCAG | |
| NFATc1 | Forward | ACCACCTTTCCGCAACCA |
| Reverse | GGTACTGGCTTCTCTTCCGTTTC | |
| Acp5 | Forward | CAGCTGTCCTGGCTCAAAA |
| Reverse | ACATAGCCCACACCGTTCTC | |
| Cathepsin K | Forward | GGACGCAGCGATGCTAACTAA |
| Reverse | CAGAGAGAAGGGAAGTAGAGTTGTCACT | |
| c-Src | Forward | CCAGGCTGAGGAGTGGTACT |
| Reverse | CAGCTTGCGGATCTTGTAGT | |
| DC-STAMP | Forward | CGCACGATGCTTCATTCTTC |
| Reverse | CAGTGCCAGCCGCAATC | |
| ATP6v0d2 | Forward | GTGAGACCTTGGAAGACCTGAAA |
| Reverse | TCCTCATCTCCGTGTCAATTTTG | |
| OSCAR | Forward | TGGCGGTTTGCACTCTTCA |
| Reverse | GGAAGAACTCAGCCAGCTCAA | |
| MMP9 | Forward | CTGGACAGCCAGACACTAAAG |
| Reverse | CTCGCGGCAAGTCTTCAGAG | |
Sample Availability: Samples of the compound NAPMA is available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Ahn, S.-H.; Chen, Z.; Kang, S.; Choi, D.K.; Moon, C.; Min, S.H.; Park, B.-J.; Lee, T.-H. N-[2-(4-Acetyl-1-Piperazinyl)Phenyl]-2-(3-Methylphenoxy)Acetamide (NAPMA) Inhibits Osteoclast Differentiation and Protects against Ovariectomy-Induced Osteoporosis. Molecules 2020, 25, 4855. https://doi.org/10.3390/molecules25204855
Lee J, Ahn S-H, Chen Z, Kang S, Choi DK, Moon C, Min SH, Park B-J, Lee T-H. N-[2-(4-Acetyl-1-Piperazinyl)Phenyl]-2-(3-Methylphenoxy)Acetamide (NAPMA) Inhibits Osteoclast Differentiation and Protects against Ovariectomy-Induced Osteoporosis. Molecules. 2020; 25(20):4855. https://doi.org/10.3390/molecules25204855
Chicago/Turabian StyleLee, Jinkyung, Sun-Hee Ahn, Zhihao Chen, Sohi Kang, Dong Kyu Choi, Changjong Moon, Sang Hyun Min, Byung-Ju Park, and Tae-Hoon Lee. 2020. "N-[2-(4-Acetyl-1-Piperazinyl)Phenyl]-2-(3-Methylphenoxy)Acetamide (NAPMA) Inhibits Osteoclast Differentiation and Protects against Ovariectomy-Induced Osteoporosis" Molecules 25, no. 20: 4855. https://doi.org/10.3390/molecules25204855
APA StyleLee, J., Ahn, S.-H., Chen, Z., Kang, S., Choi, D. K., Moon, C., Min, S. H., Park, B.-J., & Lee, T.-H. (2020). N-[2-(4-Acetyl-1-Piperazinyl)Phenyl]-2-(3-Methylphenoxy)Acetamide (NAPMA) Inhibits Osteoclast Differentiation and Protects against Ovariectomy-Induced Osteoporosis. Molecules, 25(20), 4855. https://doi.org/10.3390/molecules25204855