2.1. Structure-Based Virtual Screening
The crystal structure of bovine rhodopsin cocrystallised with 11-
cis-retinal (PDB ID: 1U19, 93% identity with the human rhodopsin) was used to perform a structure-based virtual screening of the SPECS library [
9,
10], a collection of 300,000 commercially available compounds with favourable drug-like properties. 11-
cis-retinal is bound to the chromophore active site by forming a Schiff base with Lys296 and an H-bond with Glu113. Interactions between the ionone ring and the hydrophobic portion of the binding site formed by Met207, Phe212, Phe261, Trp265 and Ala269 further stabilise the 11-
cis-retinal binding. The hydrophobic area is believed to be an essential recognition site for the ionone ring and for the binding to the chromophore pocket, as also confirmed by the ability of the shortened retinal derivative, β-ionone, to competitively inhibit the binding of 11-
cis-retinal to rhodopsin, but without causing any physiological effect on opsin trafficking (
Figure 2) [
11,
12,
13].
The glide high-throughput virtual screening tool (HTVS) [
14], which uses the Glide-HTVS scoring function, was employed to virtually screen the SPECS database against the selected binding area. The best 24 compounds according to this initial screening were then redocked in the 11-
cis-retinal binding site using the more accurate standard-precision glide docking mode (GlideScore SP). In order to avoid any potential bias associated with the use of a single docking program/scoring function, the docking results (docking poses) were then rescored using three different scoring functions: Glide XP, CHEMPLP (PLANTS) and FlexX Score (Seesar) [
14,
15,
16]. After applying a
consensus score procedure (see
Section 4.1.1) 1200 molecules were chosen and their potential interactions with the protein binding site were visually inspected. Twenty-four virtual hits were selected (
Figure 3), purchased and evaluated in a competitive-binding assay (see
Section 2.2). As an example, the potential binding for compound
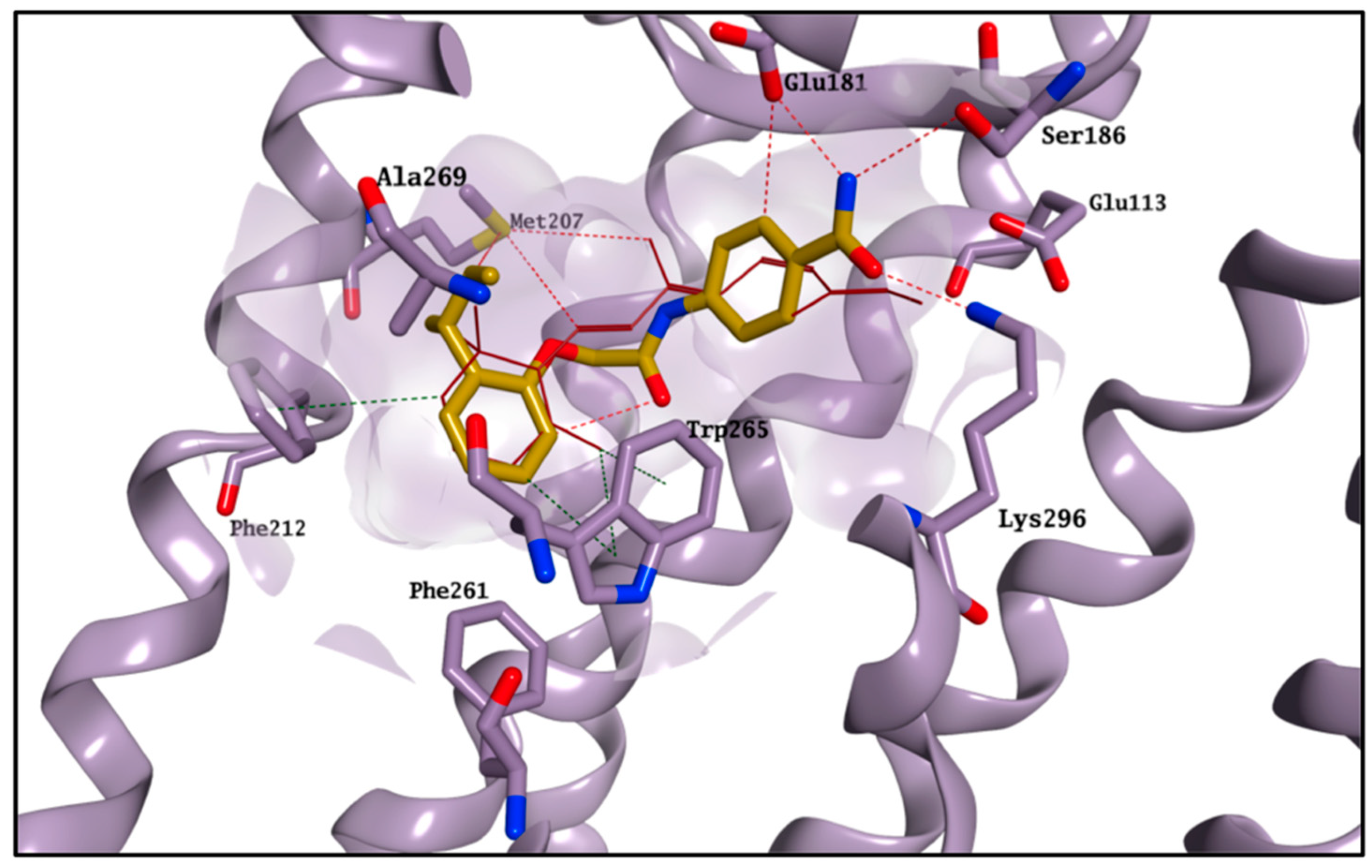
5 is shown in
Figure 4. The molecule perfectly overlaps with the cocrystallised 11-
cis-retinal, placing its
sec-butylbenzene portion in the hydrophobic area and forming H-bonds with Glu181, Ser186 and Lys296. The occupation of the chromophore pocket could reflect on the ability of
5 to act as potential chaperone for rhodopsin folding in the absence of its endogenous ligand.
2.2. Competitive Binding Assay
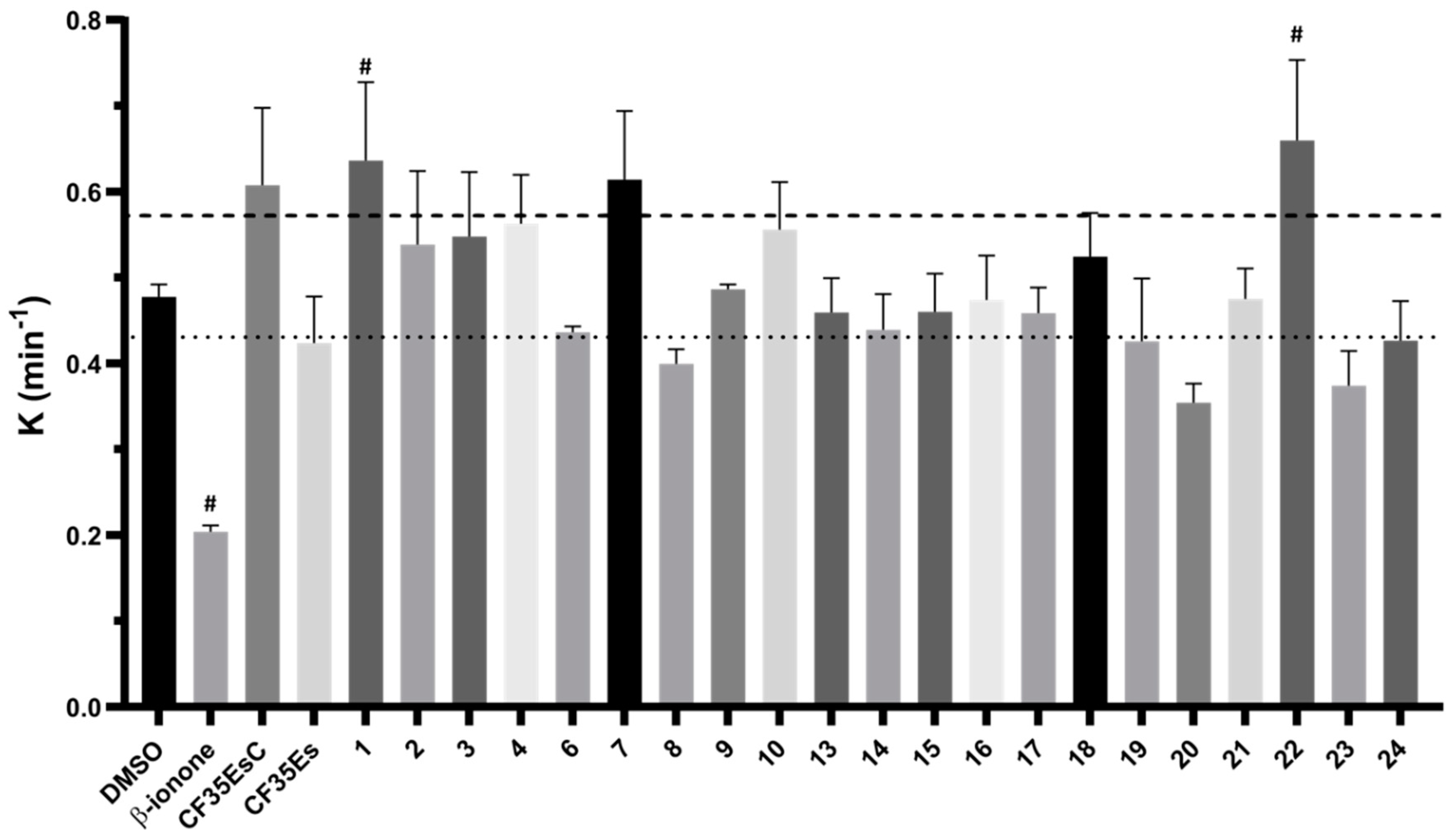
The 24 compounds selected in silico were evaluated for their ability to bind rhodopsin, by monitoring the regeneration rate of bovine isorhodopsin through time-dependent UV–Vis spectroscopy. As previously reported, the addition of 9-
cis-retinal to rhodopsin, a chromophore with similar photoactivation properties but more stable than 11-
cis-retinal, resulted in a time-dependent increase in optical density at 485 nm [
1,
17]. A compound able to compete with 9-
cis-retinal for the binding to rhodopsin should reduce the rate constant (K) of the rhodopsin-9-
cis-retinal complex formation. The compounds, tested at fixed concentration (
Section 4.2.2), were preincubated for 30 min with freshly bleached isorhodopsin followed by addition of 9-
cis-retinal. The rate constant K of the complex regeneration was calculated and compared to mock preincubated samples with vehicle only (DMSO, K = 0.48 ± 0.01 min
−1, t
1/2 = 1.52 ± 0.1 min, mean ± SEM).
β-ionone,
CF35EsC and
CF35Es were used as references.
CF35EsC and
CF35Es were synthesised as reported in the (
Supplementary Materials Figure S1). As expected, β-ionone significantly reduced the regeneration kinetic (K = 0.20 ± 0.01 min
−1, t
1/2 = 3.52 ± 0.3 min), confirming its ability to occupy the chromophore binding site (
Figure 5 and
Table S1).
Both standards,
CF35EsC and
CF35Es, have been previously reported as being able to induce proper trafficking of P23H rhodopsin from ER to the cell surface, but their direct binding to the chromophore active site has only been speculated and not directly confirmed [
8]. Our data suggest that while
CF35Es, to some extent, competed for the occupation of the chromophore site, as shown by the 10% decrease of K value (K = 0.42 ± 0.05 min
−1) compared to DMSO,
CF35EsC possessed an unexpected ability to increase the K value by over 20% (K = 0.61 ± 0.09 min
−1). Combining our data with what was previously reported [
8],
CF35EsC could act as an allosteric modulator of rhodopsin, either by facilitating the retinal access to the binding site enhancing and speeding up the formation of the opsin-9-
cis-retinal complex, or by stabilising the already formed complex. Previous studies have reported β-ionone as an enhancer of the catalytic activity of different visual pigments, in which the chromophore-binding site was already occupied, potentially acting as an allosteric modulator [
18]. Ortega et al. described a series of flavonoids as potential allosteric modulators able to enhance opsin stability by modulating its conformation [
19]. In a similar way,
CF35EsC could still occupy the binding site of retinal, but the ability of facilitating/stabilising the rhodopsin-9-
cis-retinal complex formation appears to be predominant. In general, compounds able to stabilise rhodopsin (stabilisers) and prevent its degradation, in the absence of 11-
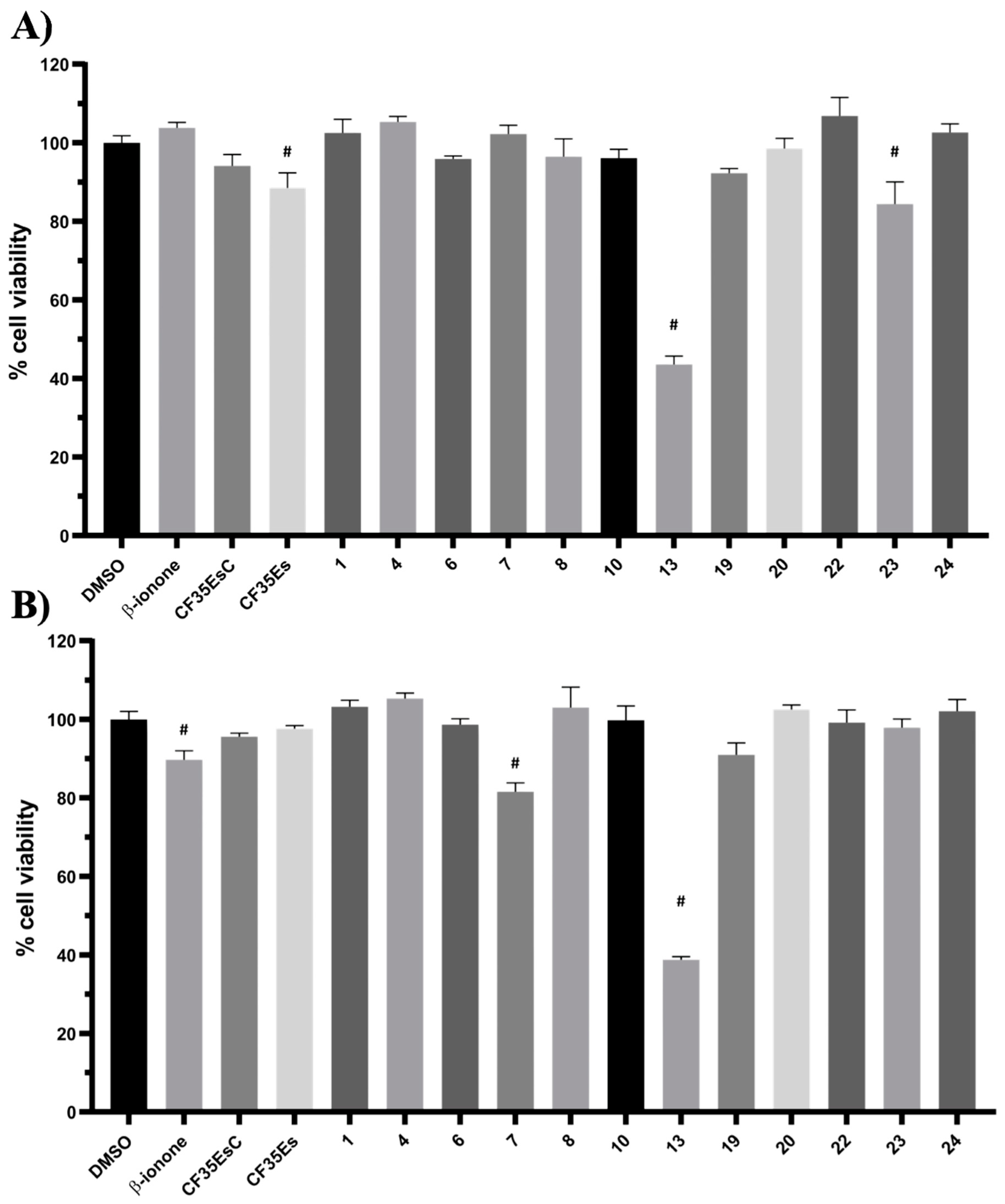
cis-retinal (LCA) or in the presence of aberrant mutations that impair its folding and structural stability (RP), could provide a novel therapeutic mechanism to further explore, in addition to the desired chaperone function by occupation of the main chromophore binding site. Of the 24 molecules tested, four compounds (
6: K = 0.43 ± 0.01 min
−1;
8: K = 0.40 ± 0.02 min
−1;
20: K = 0.35 ± 0.02 min
−1;
23: K = 0.37 ± 0.04 min
−1) exhibited a promising decrease by 10% (or over) of the rate constant K, showing the ability to bind the chromophore pocket and compete with 9-
cis-retinal. In particular,
20 presented the best activity profile reducing K by 20%. Interestingly, five molecules (
1: K = 0.64 ± 0.09 min
−1;
4: K = 0.56 ± 0.06 min
−1;
7: K = 0.61 ± 0.08 min
−1;
10: K = 0.56 ± 0.06 min
−1;
22: K = 0.66 ± 0.09 min
−1) presented the same behaviour found for
CF35EsC increasing the K value by 10–20%, potentially possessing the ability to stabilise and enhance the opsin-9-
cis-retinal complex formation.
No pan-assay interference compounds (PAINS) were found on the molecules presenting activity after checking their chemical structure in two different web servers [
20,
21], providing at this stage two potential different classes of compounds with therapeutic potential for RP and LCA: molecules, which compete for the binding to the chromophore pocket, which could act as chemical chaperones, and molecules, which seem able to facilitate/stabilise the opsin-9-
cis-retinal complex, which could act as opsin stabilisers by binding to a different site. Both types of molecules were further explored both in silico and in vitro, as detailed below.
2.3. Molecular Modelling Studies on the Chromophore Binding Pocket
In order to further explore the binding of our hit molecules to opsin, 100 ns molecular dynamic (MD) simulations were performed on the rhodopsin structure, both free and in complex with the cocrystallised 11-
cis-retinal, using the Desmond software package [
22,
23]. All simulations were run in triplicate. Overall, after an initial 40 ns of equilibration, the presence of 11-
cis-retinal seemed to confer a higher stability to rhodopsin, with the simulation system converging around a fixed RMSD value, as shown by the small C-alpha RMSD variation (
Figure S2). On the contrary, the ligand-free opsin was not able to reach stability after 40 ns and the RMSD value was still growing toward the end of the simulation (
Figure S2). Interestingly, this result seemed in line with the findings that 11-
cis-retinal is required to enhance rhodopsin intrinsic stability and it is an indication of the reliability of the simulation system used [
1,
17,
19]. In order to validate the binding mode suggested by the docking program, and to find a rational discrimination between active and inactive molecules, a series of 100 ns MD simulations were also carried out on selected compounds (
6,
8,
9,
13,
17,
20,
21,
22,
23,
CF35EsC and
CF35Es). The compounds’ relative binding free energies (ΔG
binding) were then calculated using the Prime/MM-GBSA calculation method (
Table 1) [
24].
All the protein–ligand systems, with the exception of the ones with compounds
9,
21 and
22, reached stability after 40 ns, in line with the 11-
cis-retinal-rhodospin complex, and therefore only the remaining 60 ns of the simulations were considered in our analysis (
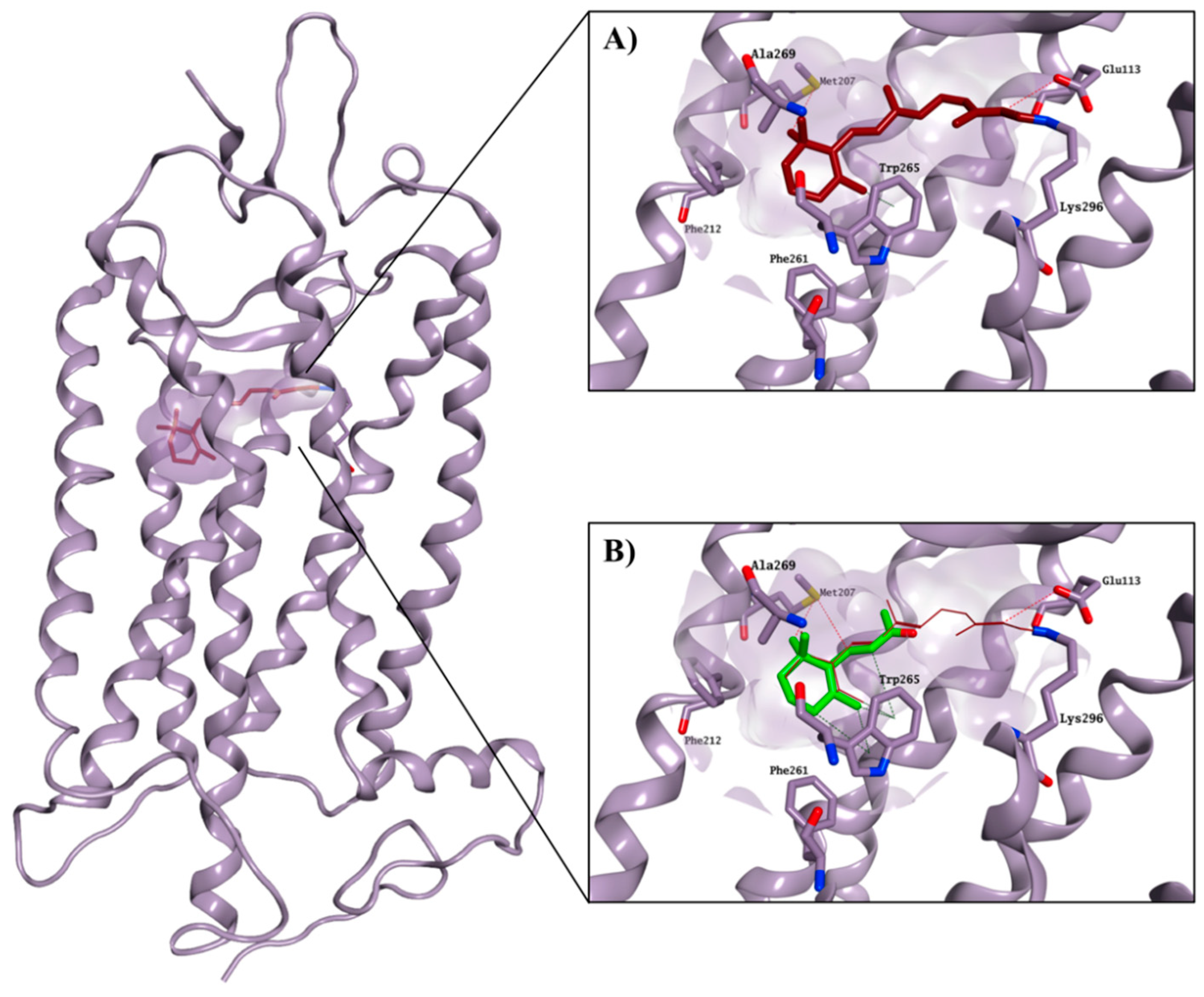
Figures S3 and S4). In general, the four active molecules (
6,
8,
20 and
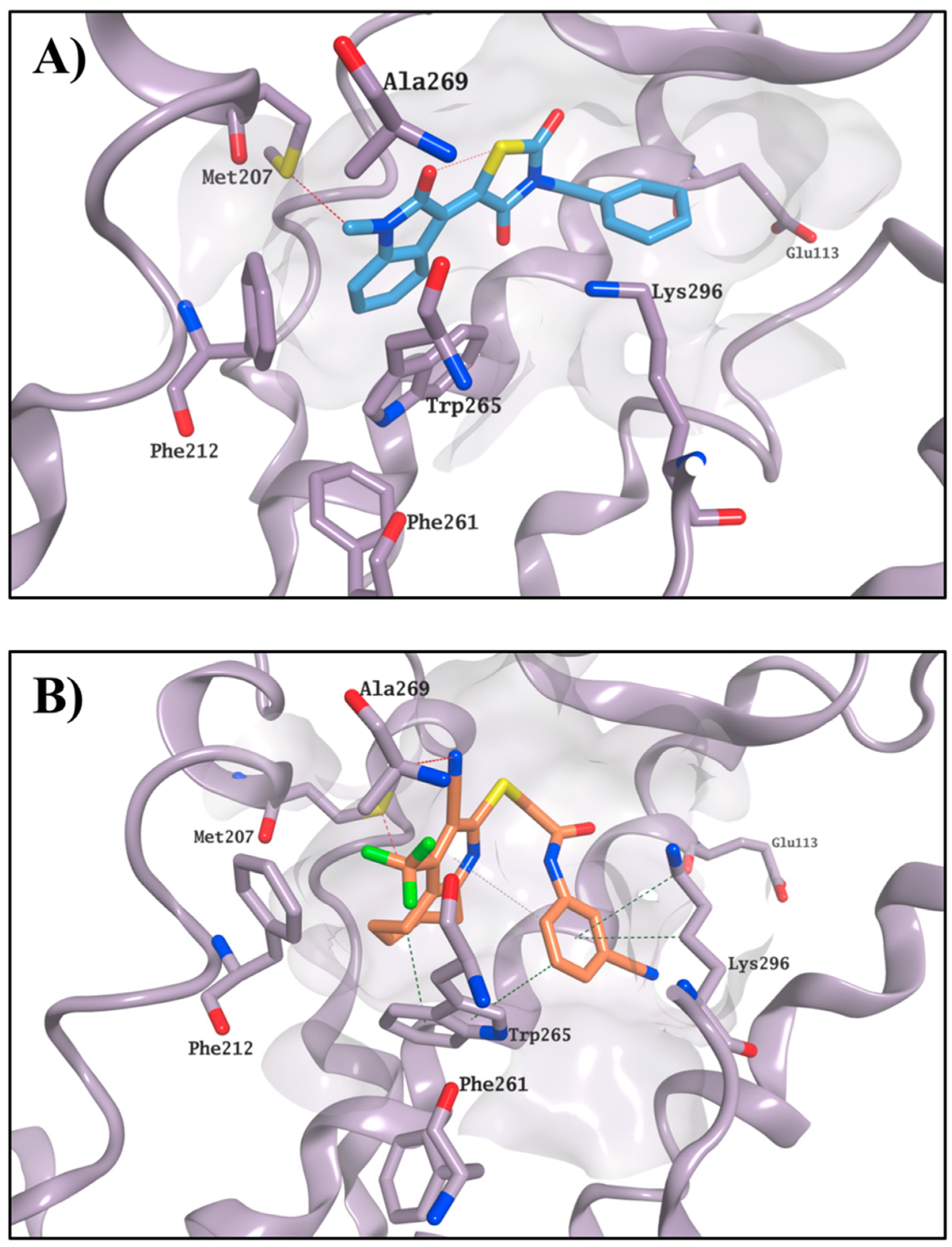
23) tended to optimise their occupation of the active site, maintaining a stable position during the entire MD simulation. In particular, the compounds seemed to adjust their orientation toward the hydrophobic portion of the binding site, creating hydrophobic contacts with the surrounding residues (i.e., Met207, Phe212 and Trp265), which were maintained for the entire simulation. The stable occupation of this area could confer to these four molecules their ability to compete for the chromophore-binding pocket. Moreover, the ΔG
binding values obtained further confirmed their potential to interact with the active site and appeared to be in line with the competitive bidding assay results. The best ΔG
binding was found for compound
20, in line with this compound’s lowest observed K value. Binding of
20 and
23 is shown in
Figure 6 as an example.
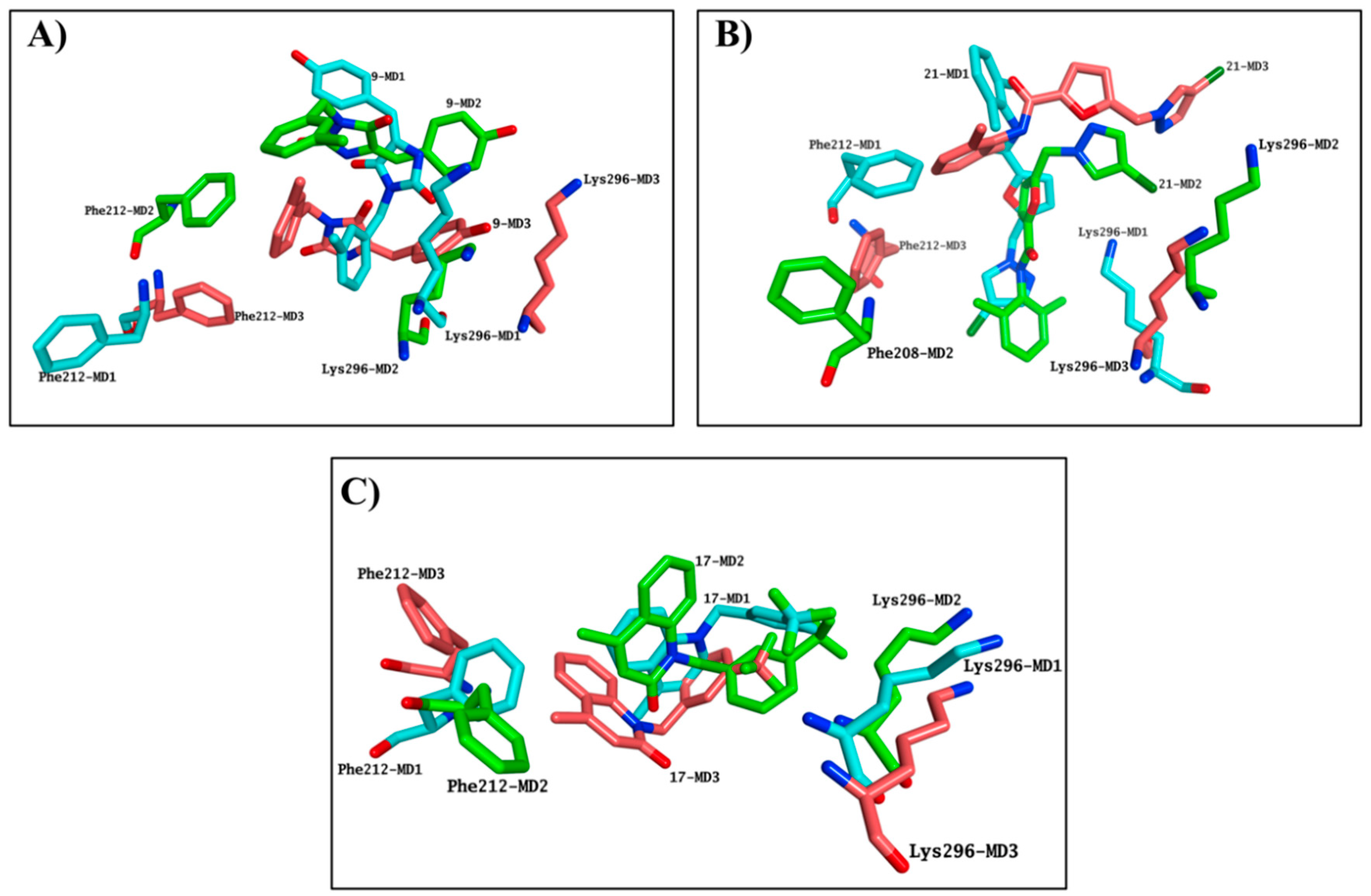
Simulation systems for
9 and
21 were not able to equilibrate during the entire MD, indicating that these molecules were not likely to bind the chromophore pocket. Both molecules presented highly variable results in terms of occupation of the binding site in each simulation performed, further confirming the inability of
9 and
21 to consistently occupy the chromophore pocket, in line with the negative results obtained in the competitive binding assay (
Figure 7A,B). Although the
17-rhodhopsin complex does reach stability after 40 ns, as with the active molecules, variable binding modes were obtained, indicating that
17 is also not likely to compete for binding to the active site (
Figure 7C).
The MD results predict that
13 should bind to the chromophore active site, with its ΔG
binding suggesting it should provide a reduction of the K value similar to
6. This inconsistency between the competitive binding assay and the molecular modelling prediction could be caused by the simultaneous presence of both potential effects detected on the competitive binding assay, which could affect the final read out of the assay itself.
13 may still occupy the main chromophore binding area, as predicted by the MD simulations, but it may also be able to stabilise the formation of the rhodopsin-9-
cis-retinal complex, thus affecting the outcome of the competitive binding assay. The failure of the simulation system to reach equilibration in all the three experiments for
22, associated with the highest increase for the K value, suggests that this compound is not likely to compete for the chromophore active site, and it may act purely as a stabiliser of the rhodopsin-9-
cis-retinal complex. According to the calculated ΔG
binding, carboxylic acid derivative
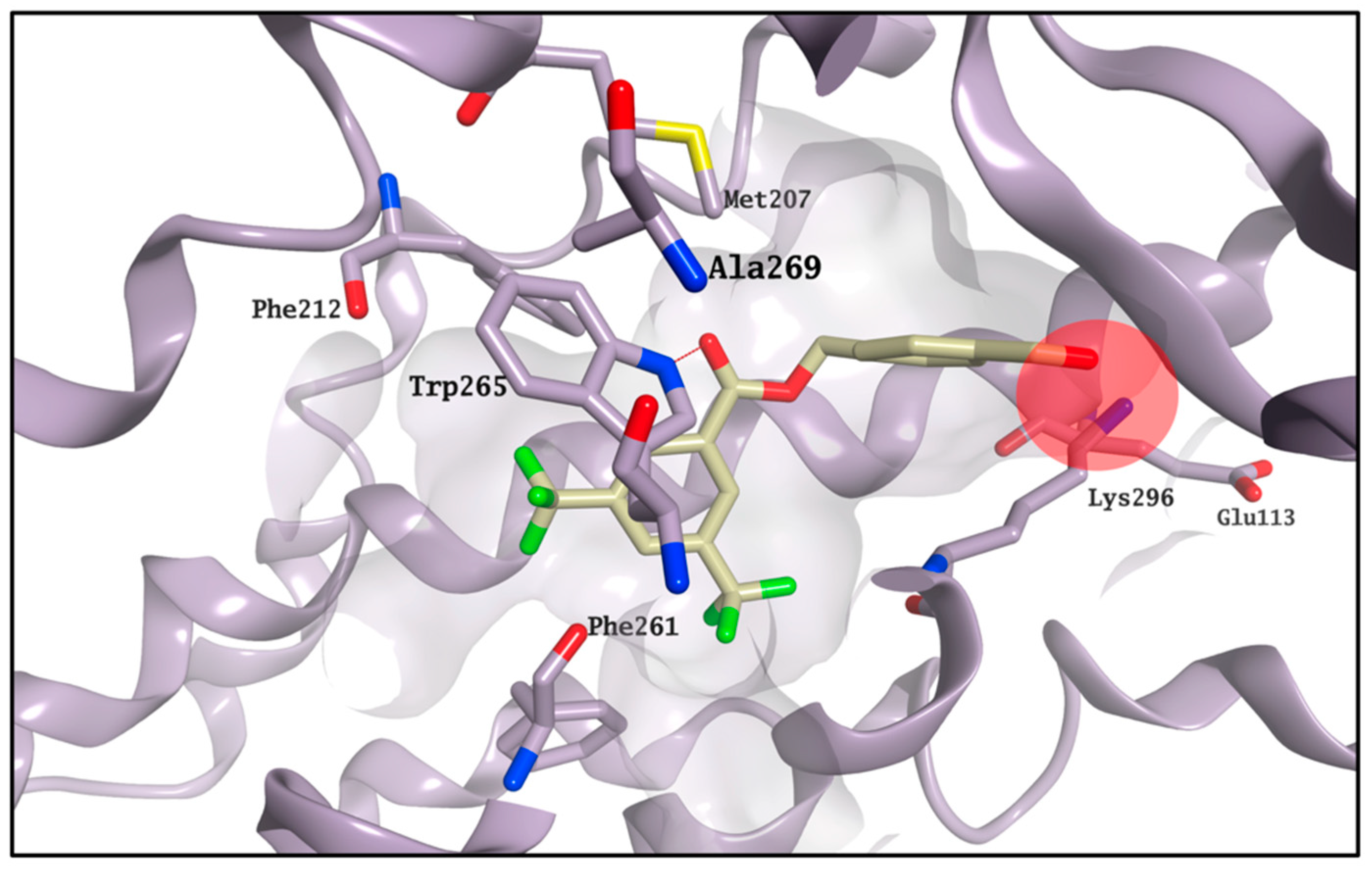
CF35EsC seems to possess a limited ability to bind the chromophore site, in line with the competitive binding assay results. On the other hand, the aldehyde moiety on
CF35Es could form a transient Schiff base with rhodopsin (ΔG
binding calculation cannot predict covalent interactions), as also confirmed by the vicinity of this group to Lys296 during the MD simulation (
Figure 8), potentially making this compound much more stable in the binding site than
CF35EsC, thus potentially explaining its ability to reduce the K value.
In order to find possible common structural features among the compounds able to bind the chromophore pocket, a pure ligand-based approach was performed. This approach uses a molecular field points-based similarity method to generate a series of low-energy conformations for each compound [
25,
26]. The molecular field points define the shape, electrostatic and hydrophobic properties of a molecule and their spatial distribution. The lowest energy conformation and its associated 3D electrostatic-hydrophobic and shape properties for
20 and
23, the compounds showing the best activity profile, were then generated. These electrostatic and hydrophobic properties of the two conformations obtained were then compared and used to derive a pharmacophore model to identify common motifs between the two active molecules [
26,
27].
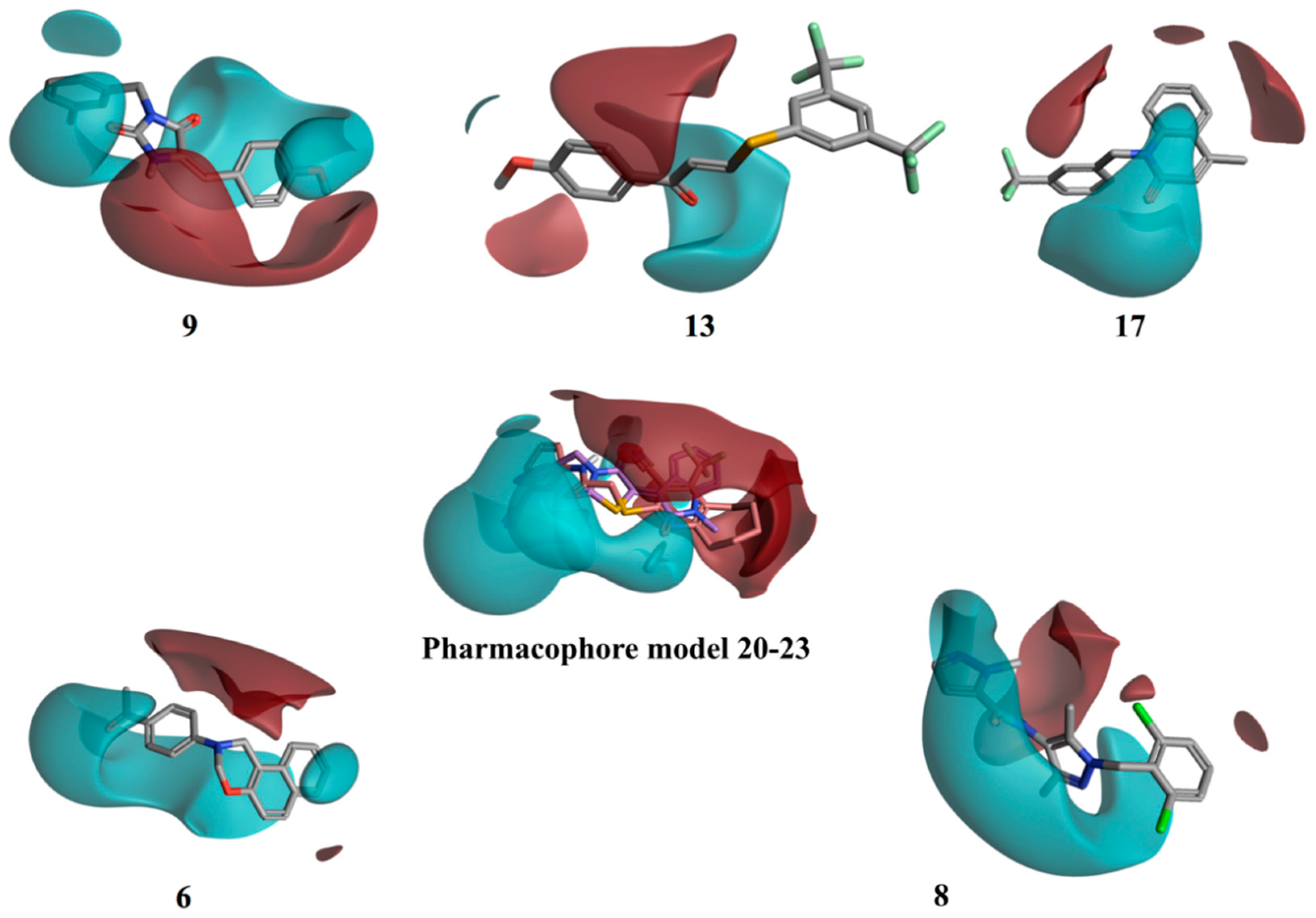
Figure 9 shows the resulting pharmacophore model. Two well distinct and separated regions can be identified: a positive electrostatic region, in red, and a negative electrostatic region, in cyan. The other compounds were then aligned with the identified pharmacophore, using a field-based alignment approach, to identify potential differences between active and inactive molecules. Interestingly, the best alignment results obtained for the inactive/weakly active compounds present a quite different distribution of the two electrostatic regions in comparison with the active pharmacophore query. The separation between the positive and the negative region was less distinct, with the distribution of the negative filed area remarkably less wide (
13 and
17), or in a completely different orientation (
9), for the inactive compounds (
Figure 9). Compounds
6 and
8 are characterised by a wide negative electrostatic region oriented correctly, matching the pharmacophore model for the active molecules (
Figure 9).
A further calculation of the protein–ligand electrostatic complementarity for some of the active molecules, comparing the protein and ligand electrostatic potential (ESP) values, revealed the presence of a positive ESP surface on the binding pocket that could facilitate the binding of a ligand possessing a large negative ESP [
26,
28,
29].
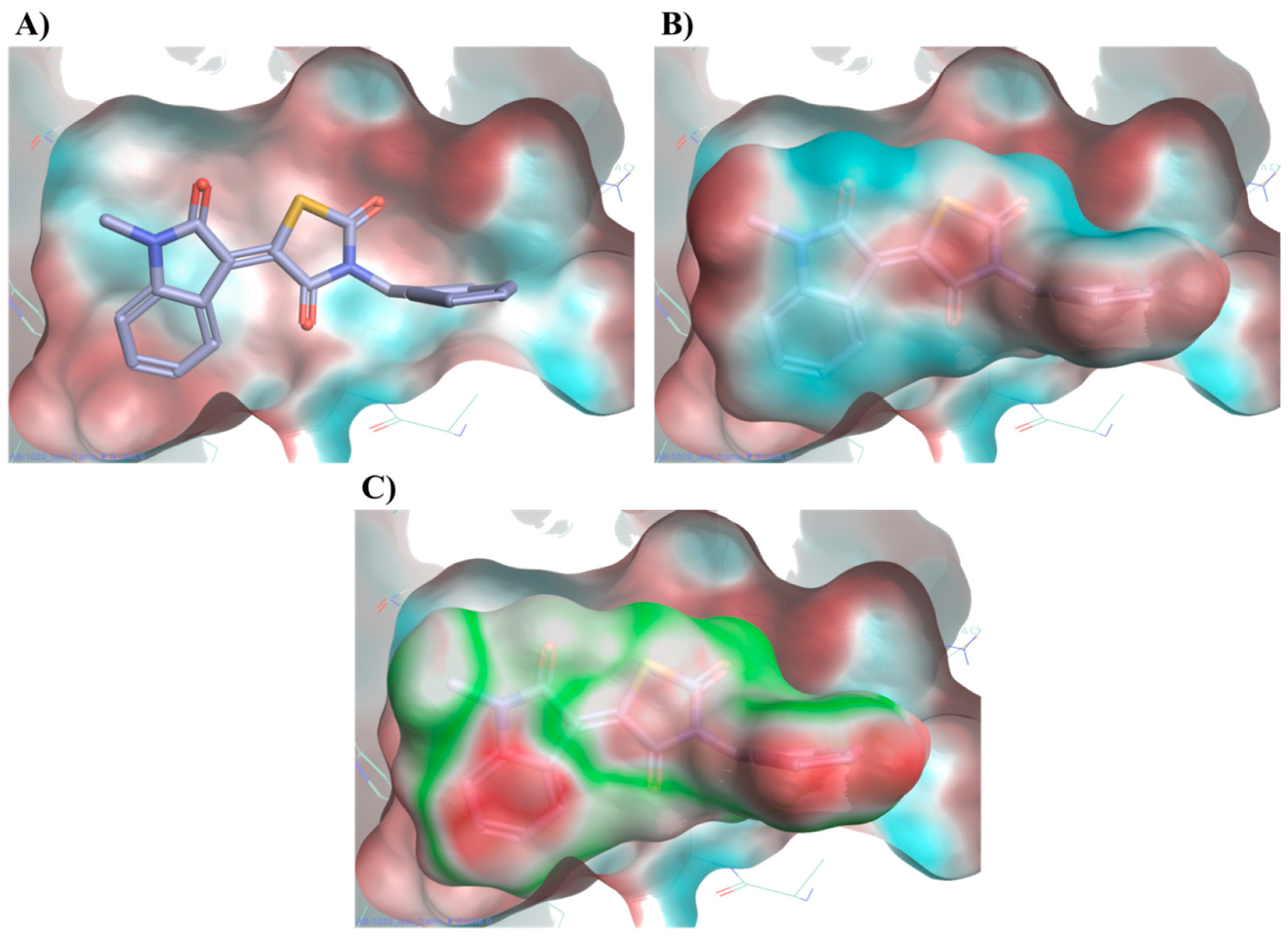
Figure 10 displays compound
20 placing its large negative electrostatic area, having as the centre the thiazolidinedione ring, in correspondence to this positive portion of the protein, showing a high electrostatic complementary with the binding pocket. According to these findings, the presence of a well-defined negative electrostatic region on the compound appears to be an essential feature, and its electrostatic complementarity with the positive portion of binding site a critical factor, for the ligand–protein interaction.
2.4. Molecular Modelling Studies to Investigate the Observed Stabilisation Effect of the Rhodopsin-9-cis-Retinal Complex
According to our competitive binding assay, some of the selected molecules increase the rate constant K for the formation of the rhodopsin-9-
cis-retinal complex, and could therefore act as allosteric modulators/stabilisers of rhodopsin structure, either by facilitating the retinal access to its main binding site, or by stabilising its interaction with this site. Different alternative binding pockets on the opsin structure have been previously suggested, but none of them has been directly confirmed through cocrystallisation with any active molecule so far [
19,
30]. Although the exact mechanism behind the ability of β-ionone to increase the catalytic activity of different visual pigments without interacting with the chromophore-binding site is still not known [
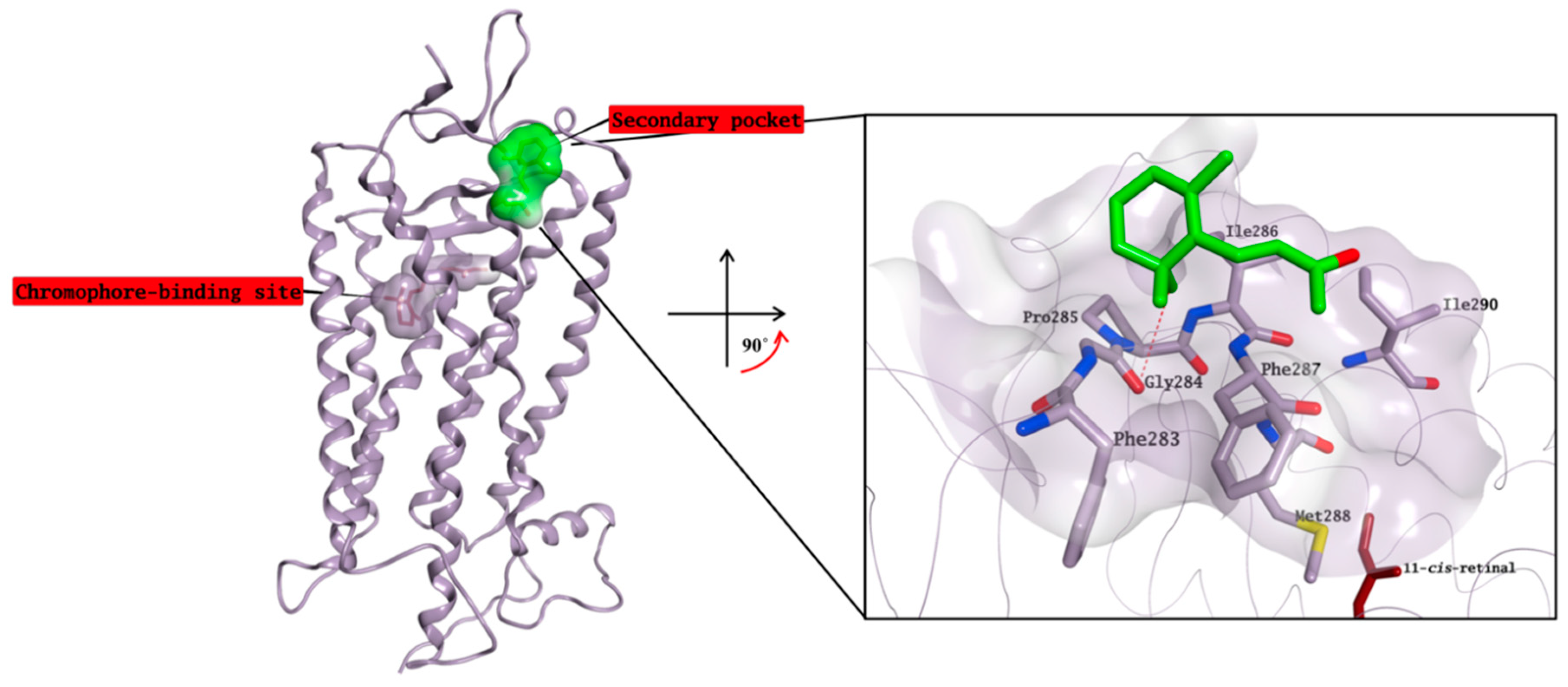
18], a crystal structure in complex with rhodopsin in which the chromophore-binding pocket is already occupied by 11-cis-retinal has been resolved by Makino et al. [
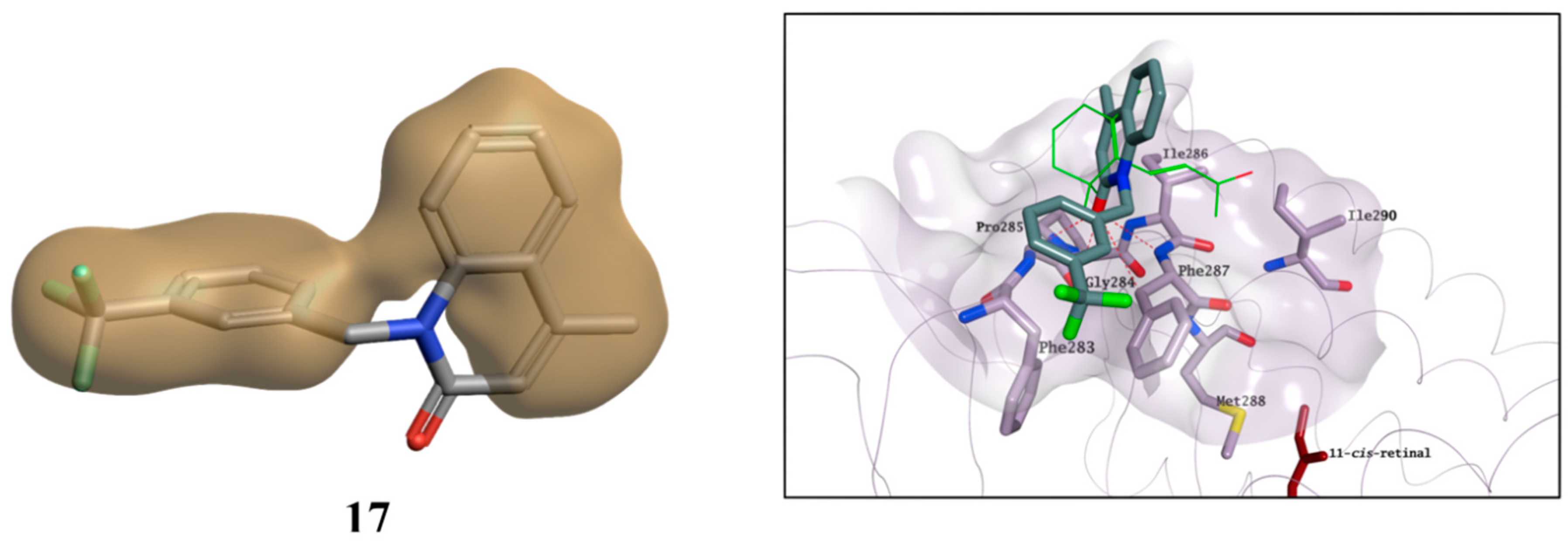
13]. In this structure, β-ionone is bound to a small, surface-exposed and highly hydrophobic pocket formed by Phe283, Gly284, Pro285, Ile286, Phe287, Met288 and Ile290, mainly forming hydrophobic interactions with the surrounding residues (
Figure 11). The pocket is not distant from the primary chromophore-binding site, and it is present in both bovine opsin-11-cis-retinal complex and in ligand-free opsin crystal structures [
9,
31]. Furthermore, Behnen et al., studied the effect of different mutations in weakening the network of native links that confers stability to opsin, and some of these mutations weaken the network of interactions mainly in an area that is in close proximity to the β-ionone binding pocket [
32]. Binding of β-ionone or other small molecules in this pocket could have a stabilising effect on this network, enhancing the intrinsic stability of native opsin.
The identified compounds able to increase K for the formation of the opsin-9-
cis-retinal complex could likely interact with the same secondary pocket, thus facilitating/stabilising the formation of the opsin-11-
cis-retinal complex. Molecular docking studies show that all compounds able to increase K (
1,
4,
7,
10,
13 and
22)
, on top of the two reference molecules included in our assay (
CF35EsC and
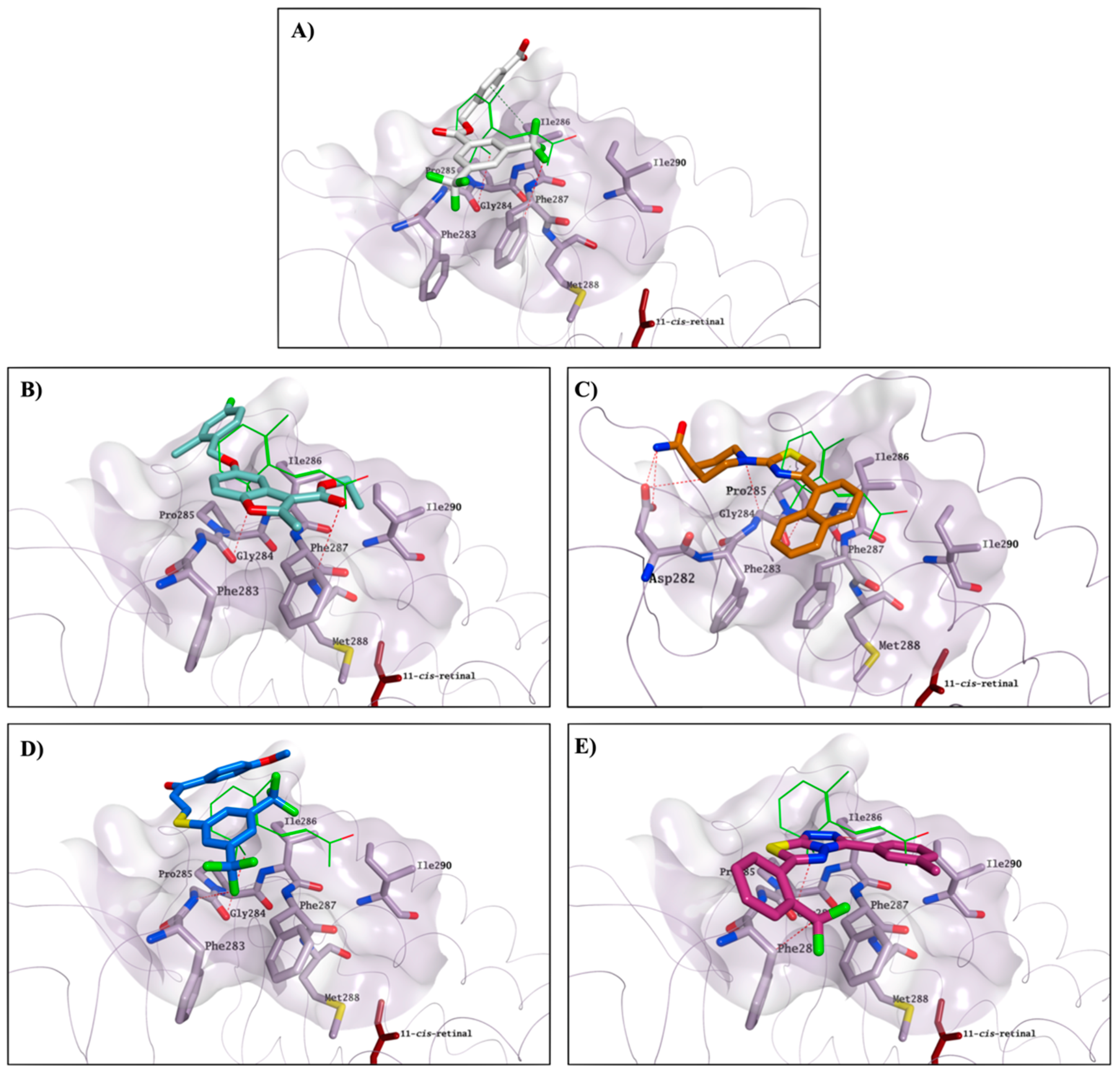
CF35Es), have the potential to interact with this secondary pocket, placing one hydrophobic portion of their structure inside the binding cavity, similarly to the binding of the β-ionone cyclohexenyl ring found in the crystal structure. Predicted binding of
CF35EsC,
4,
7,
13 and
22 to this secondary site is shown in
Figure 12. Compound
7 is also establishing multiple H-bonds with Asp282, which can further stabilise the molecule on the binding site.
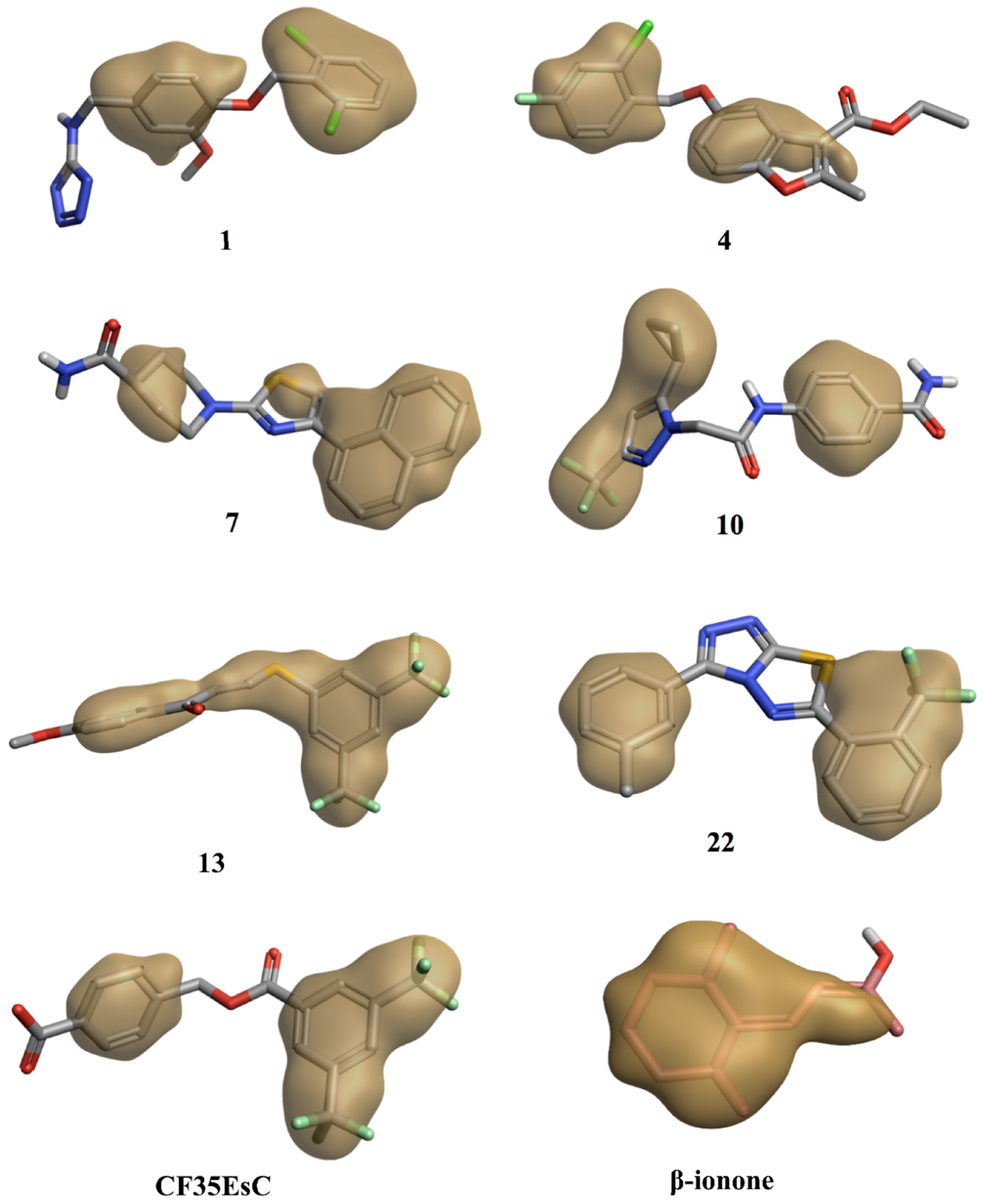
Interestingly, the chemical structure of all these compounds is characterised by either an extended planar hydrophobic region, or by two hydrophobic areas with a spatial orientation, which is relatively coplanar (
Figure 13). Combination of hydrophobicity and coplanarity may be an essential feature to correctly occupy this shallow hydrophobic pocket, and to act as potential allosteric modulators of rhodopsin. Although
17 is characterised by two hydrophobic areas, the lack of coplanarity between them does not allow the molecule to correctly interact with the pocket, in line with its inability to affect K found in the competitive binding assay (
Figure 14).
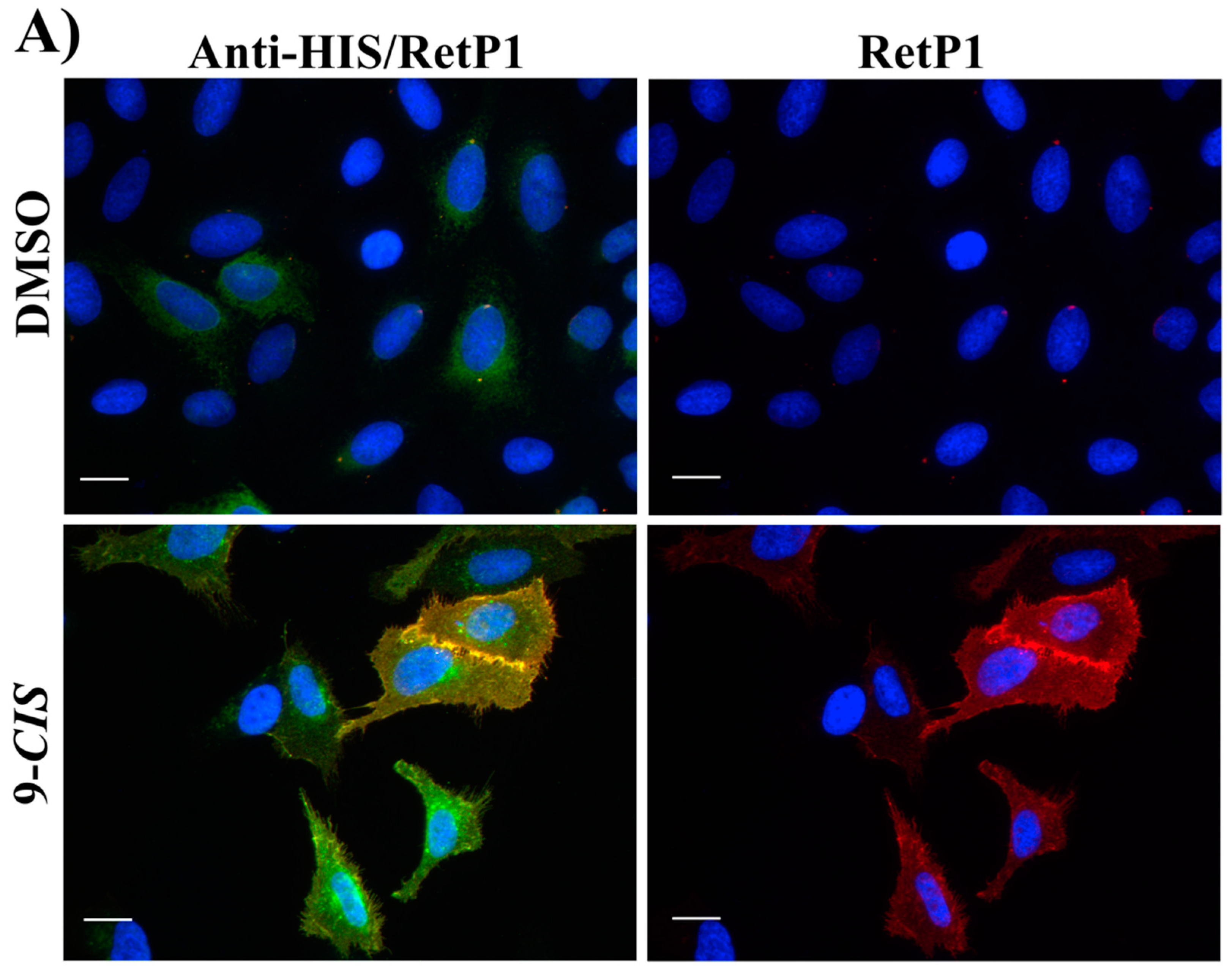
2.6. Immunofluorescence Microscopy
In this experiment, the localisation of P23H human rhodopsin His-tag (hRHO P23H His-Tag) was evaluated by immunostaining under both cell membrane non-permeabilised and membrane-permeabilised conditions. Rhodopsin trafficked to the cell membrane was detected with an anti-rhodopsin antibody (RetP1, which recognise an extracellular epitope) under non-permeabilised conditions. The total expressed rhodopsin was then detected with an anti-His-tag antibody after the cell membrane was permeabilised. In U2OS cells transiently transfected with hRHO WT His-tag, anti-rhodopsin staining (red) showed proper trafficking of rhodopsin with homogeneous distribution on the cell membrane in the presence of 9-
cis-retinal that was minimally affected by the absence of 9-
cis-retinal, as expected for wild-type human rhodopsin in this assay (
Figure S5). This is in line with previous findings [
34] and suggests that a small tag (His-tag) at the C-terminus of wild-type rhodopsin does not affect its correct trafficking to the membrane. Additionally, in accordance with a previous study [
34], in the established cell line, P23H mutant rhodopsin failed to show proper trafficking and a homogeneous cell surface distribution, which was substantially rescued when cells were incubated with 9-
cis-retinal (
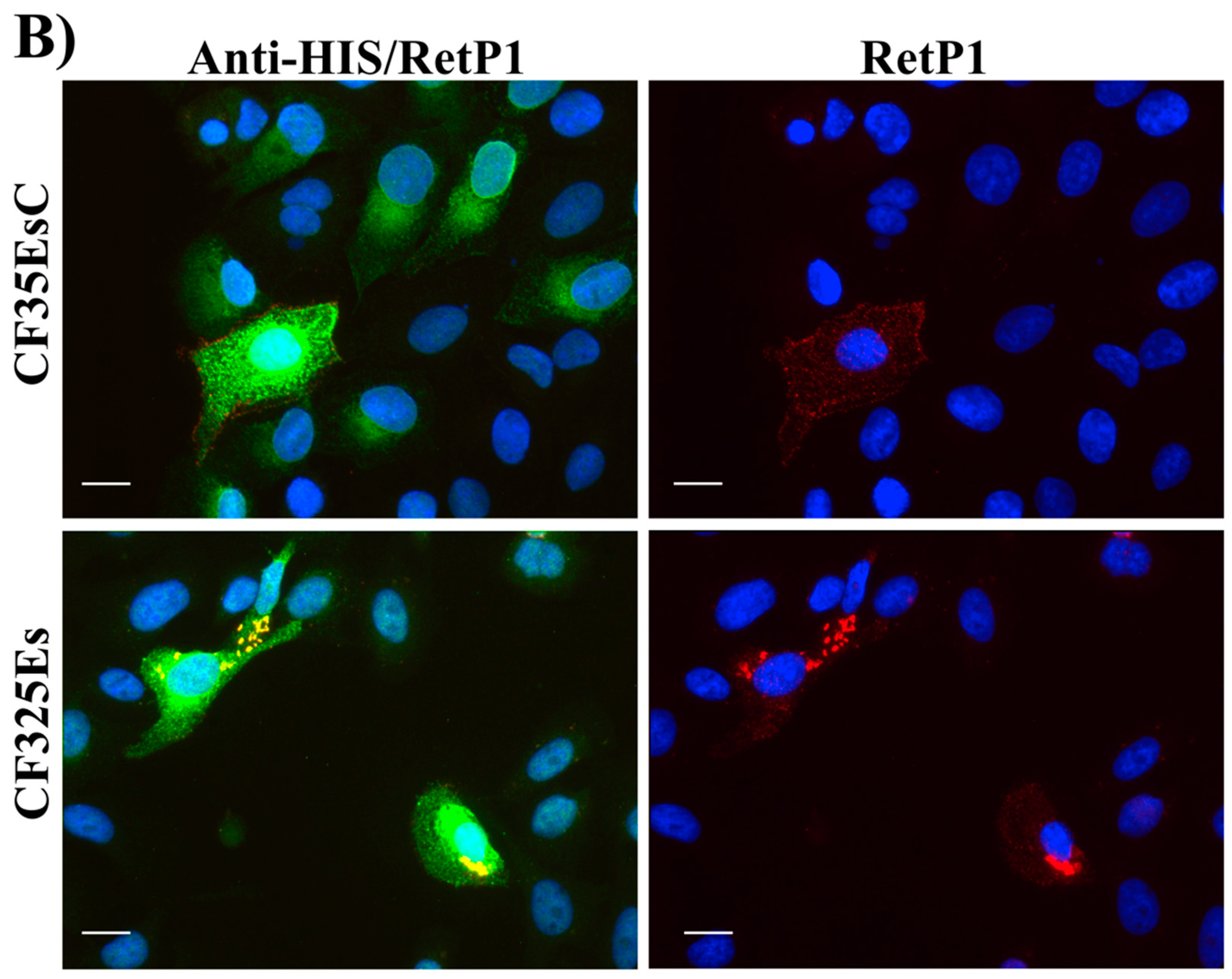
Figure 16A). Cells treated with the two standards
CF35EsC and
CF35Es also showed a mild improvement in P23H mutant opsin localisation on the membrane (
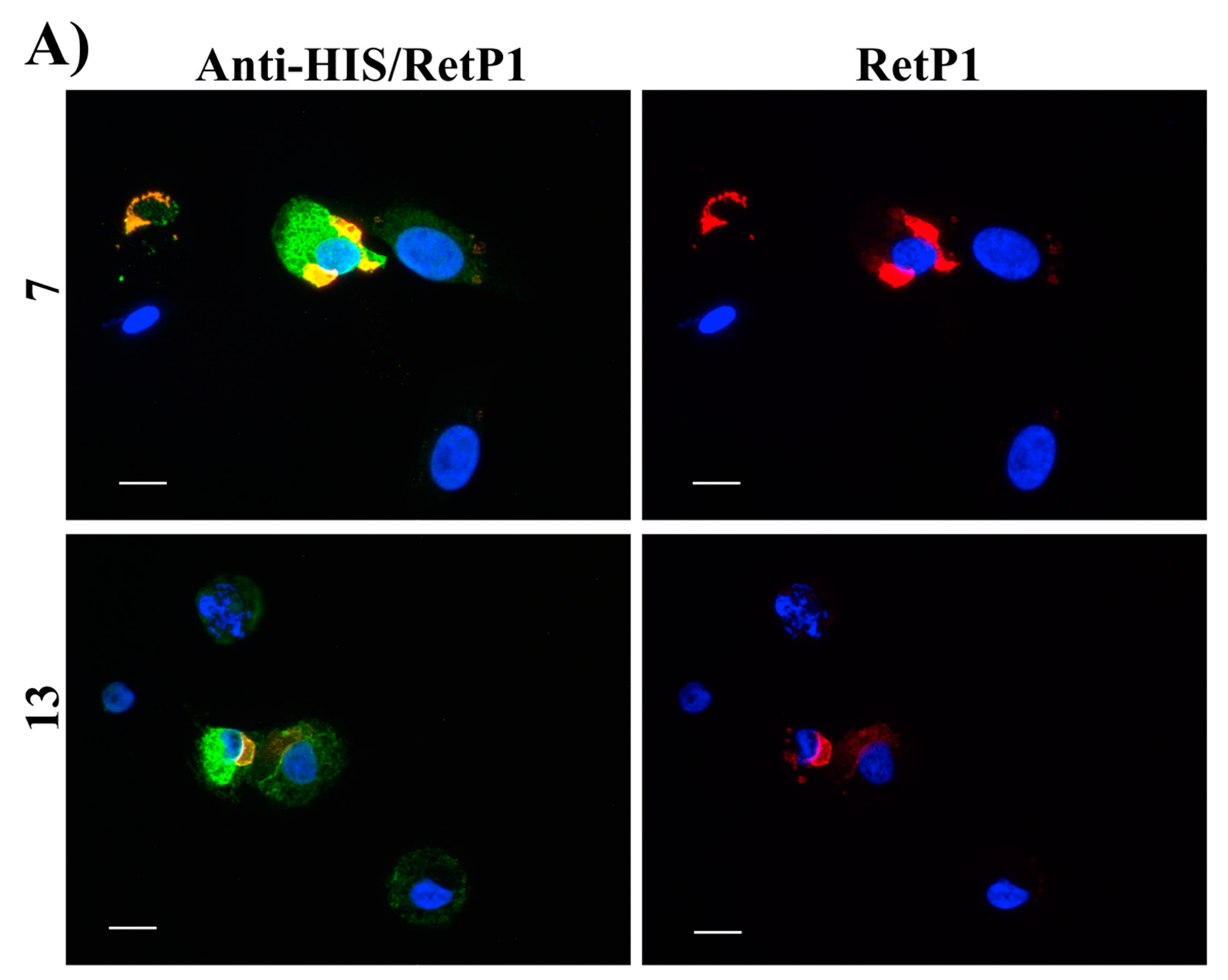
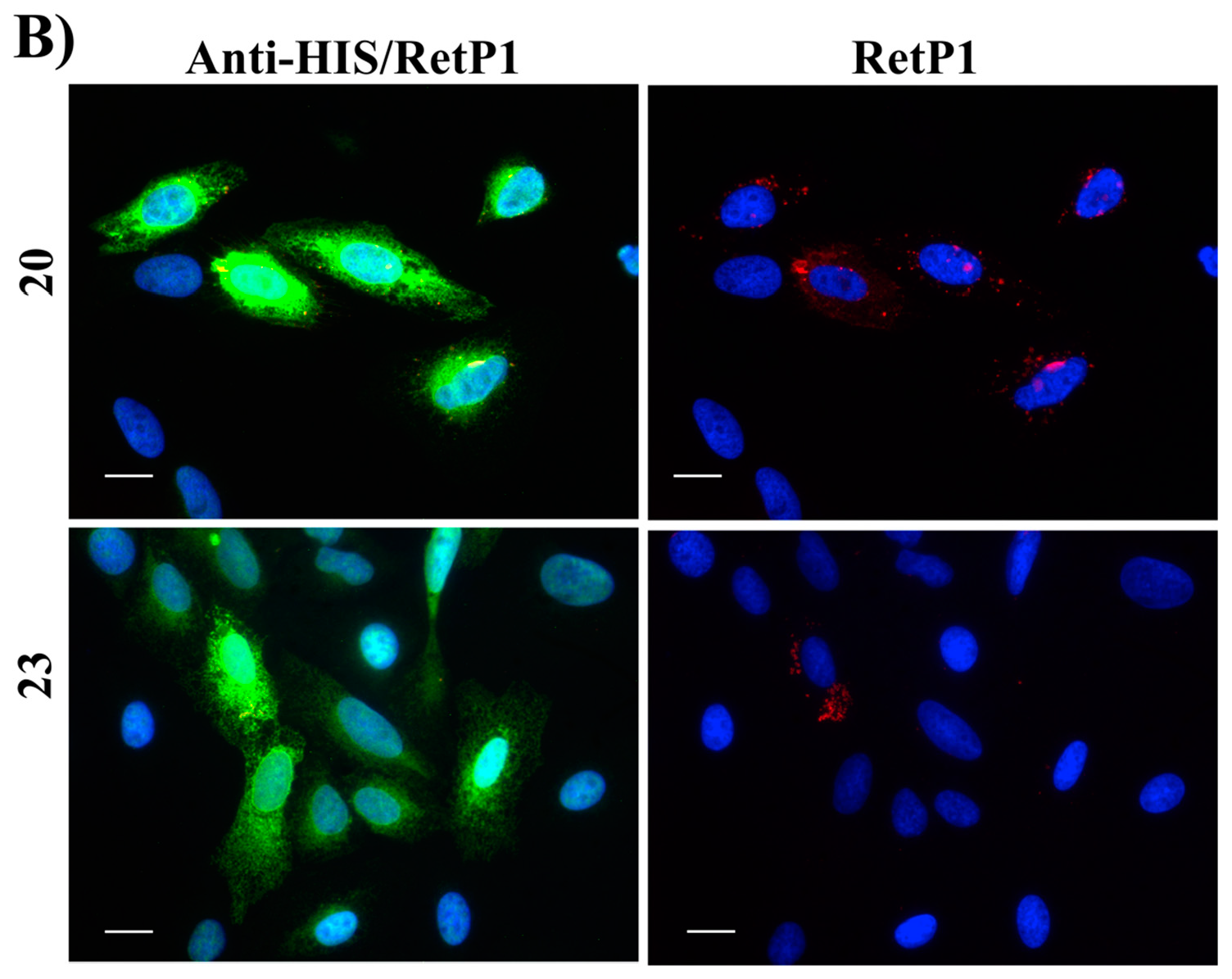
Figure 16B, red). Among our hit compounds, only
7,
13,
20 and
23 elicited a detectable increase in the trafficking of P23H mutant from the ER to the cell membrane (
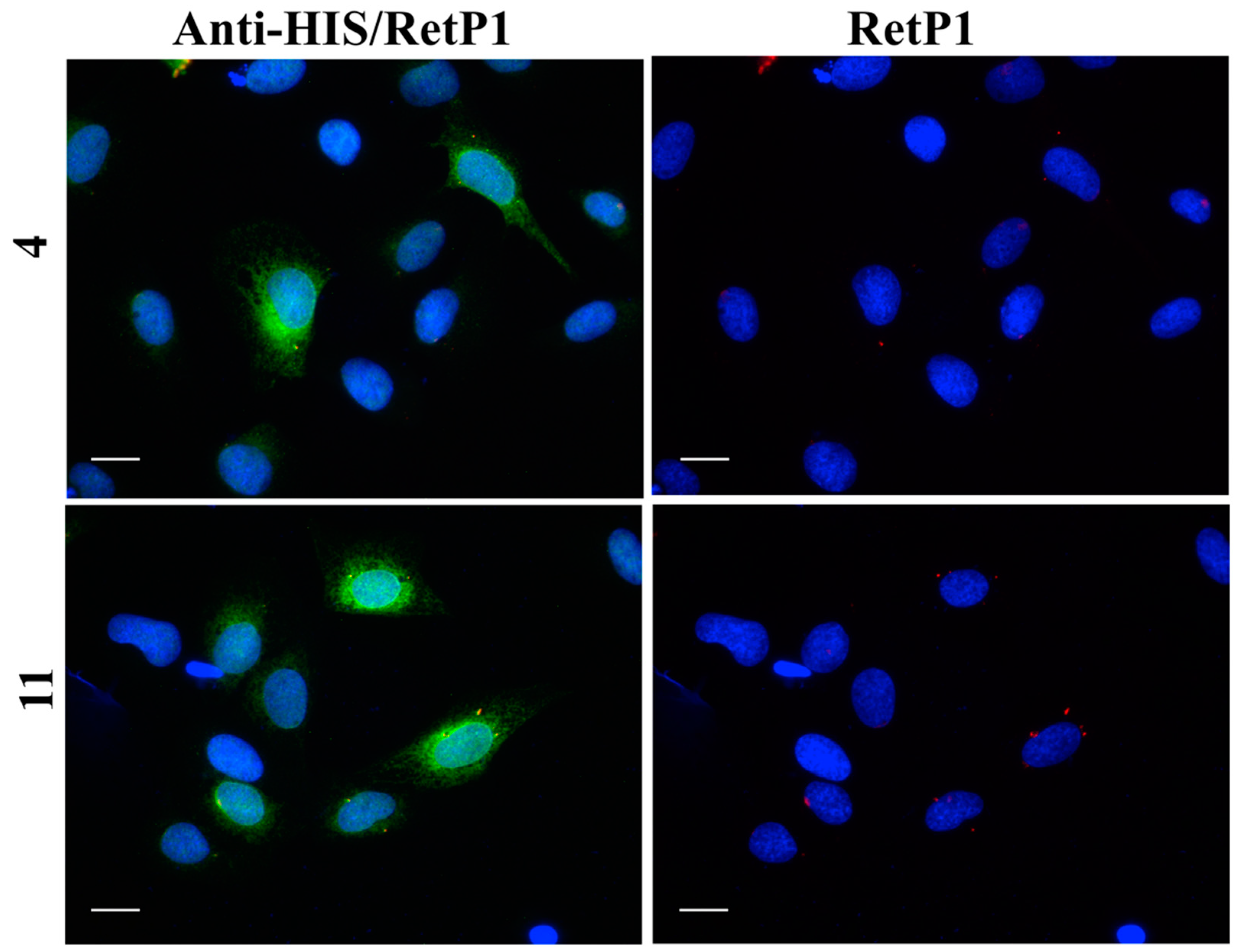
Figure 17A,B). For the remaining compounds, only reticular distribution consistent with ER retention was detected (
Figure 18). According to our molecular modelling studies,
7 and
13 are likely to act as rhodopsin
stabilisers, with a stabilisation effect of rhodopsin 3D structure by binding to a secondary site, whereas
20 and
23 are likely to act as chemical
chaperones, competing for the binding to the main chromophore pocket. In both cases, the correct folding of mutant P23H rhodopsin was partially restored, allowing its proper trafficking to the membrane. Although
13 was found to be toxic, its mild ability of increasing the trafficking of the P23H mutant can be considered a very promising starting point for further development of its molecular scaffold.


 ,
, 




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}