
Recent Advances in Palladium-Catalyzed Isocyanide Insertions

 ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Palladium Catalysis
1.2. Isocyanides
2. Pd0-Catalyzed Isocyanide Insertions
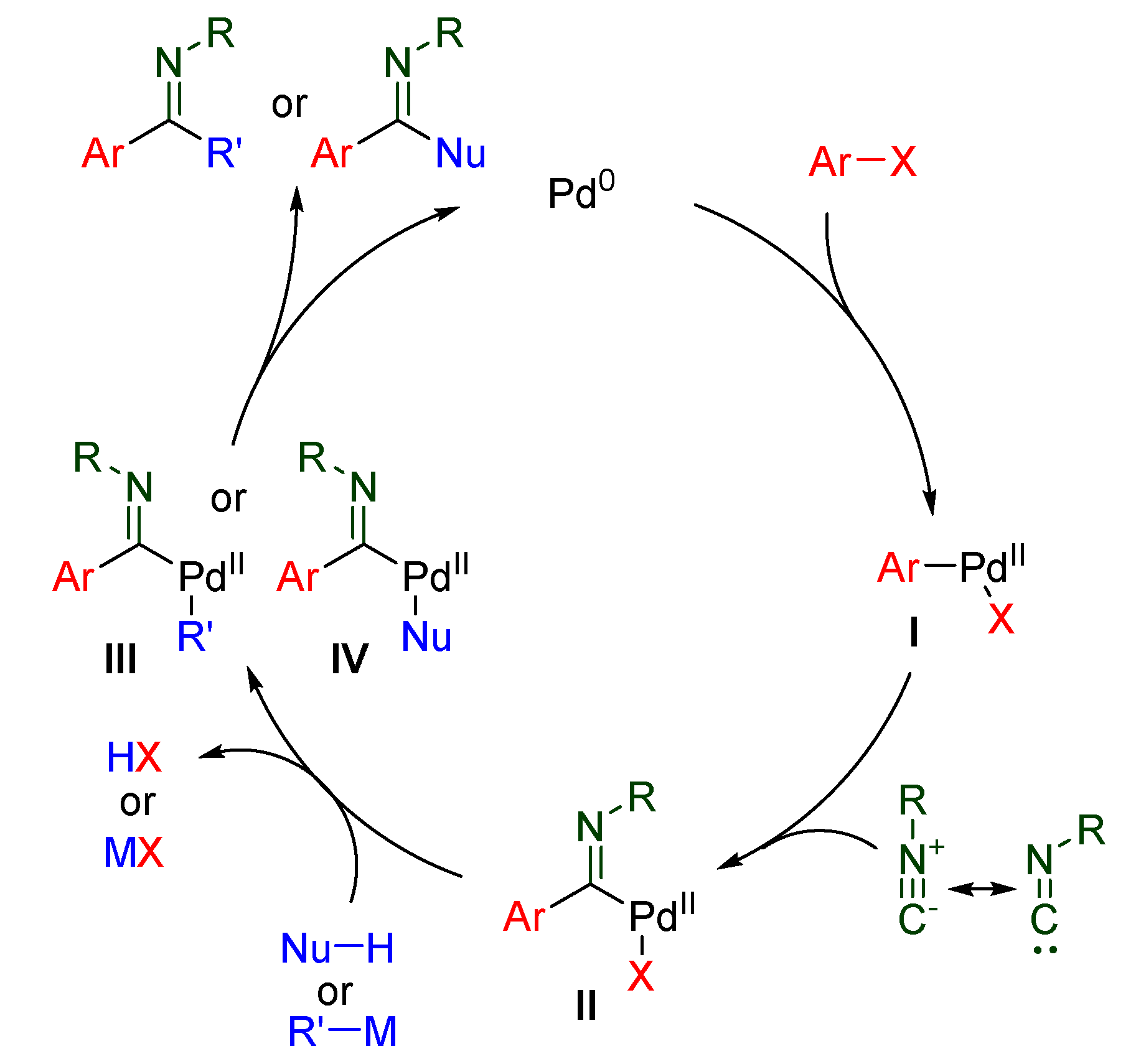
2.1. Imidoylation Initiated by Oxidative Addition of Carbohalides
2.1.1. Cross-Couplings with Organometallic Carbon Nucleophiles
2.1.2. Cross-Coupling with Other Carbon Nucleophiles
2.1.3. Cross-Couplings with Oxygen or Nitrogen Nucleophiles
2.2. Imidoylation Initiated by Oxidative Addition of Oximes
2.3. Imidoylation Initiated by Oxidative Addition of Allyl(pseudo)halides
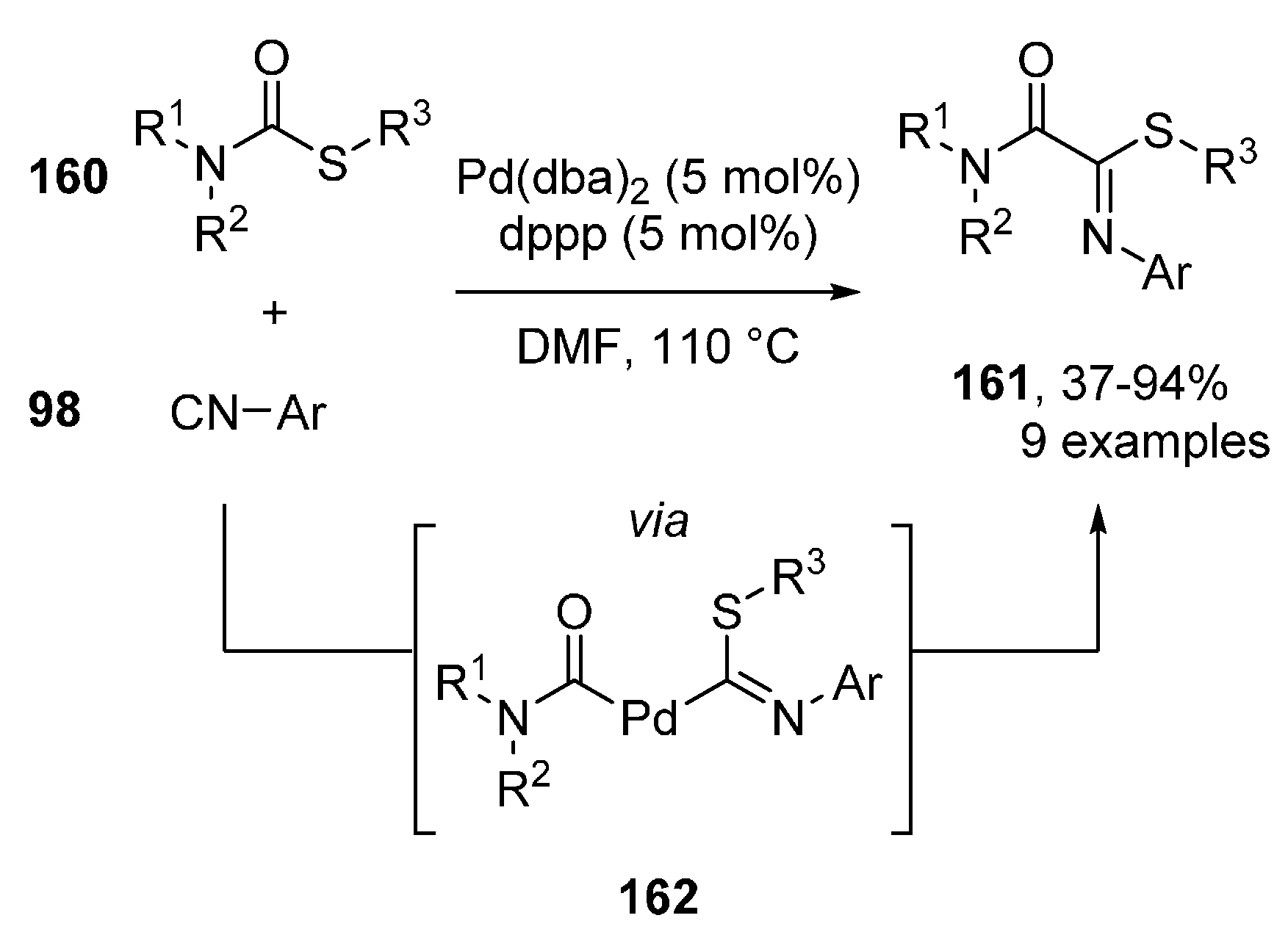
2.4. Imidoylation Initiated by Oxidative Addition of Activated Sulfur Compounds
2.5. Imidoylation of Palladium-Ligated Carbenes and Nitrenes
2.6. Miscellaneous Pd0-Catalyzed Isocyanide Insertions
3. PdII-Catalyzed Isocyanide Insertions
3.1. Introduction
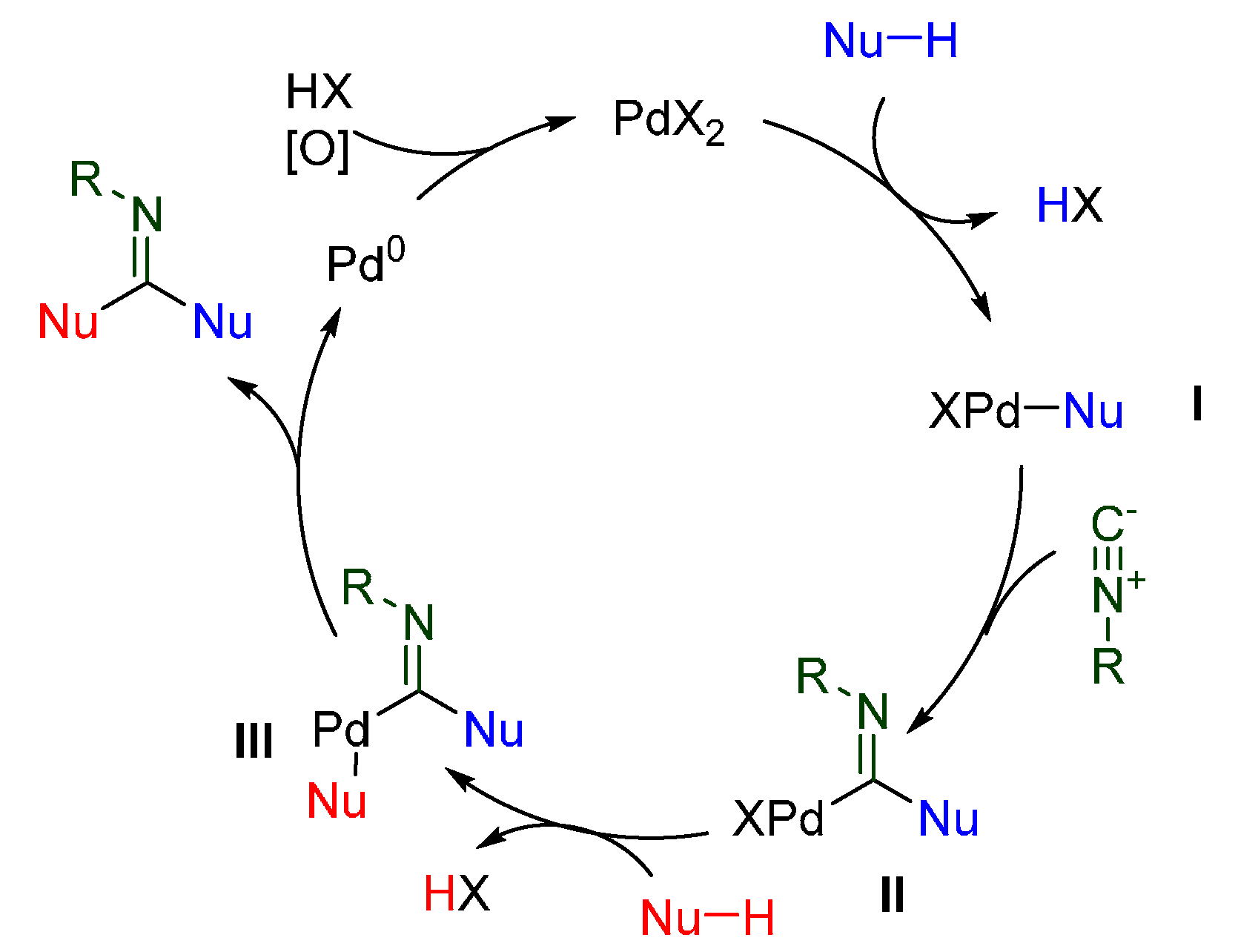
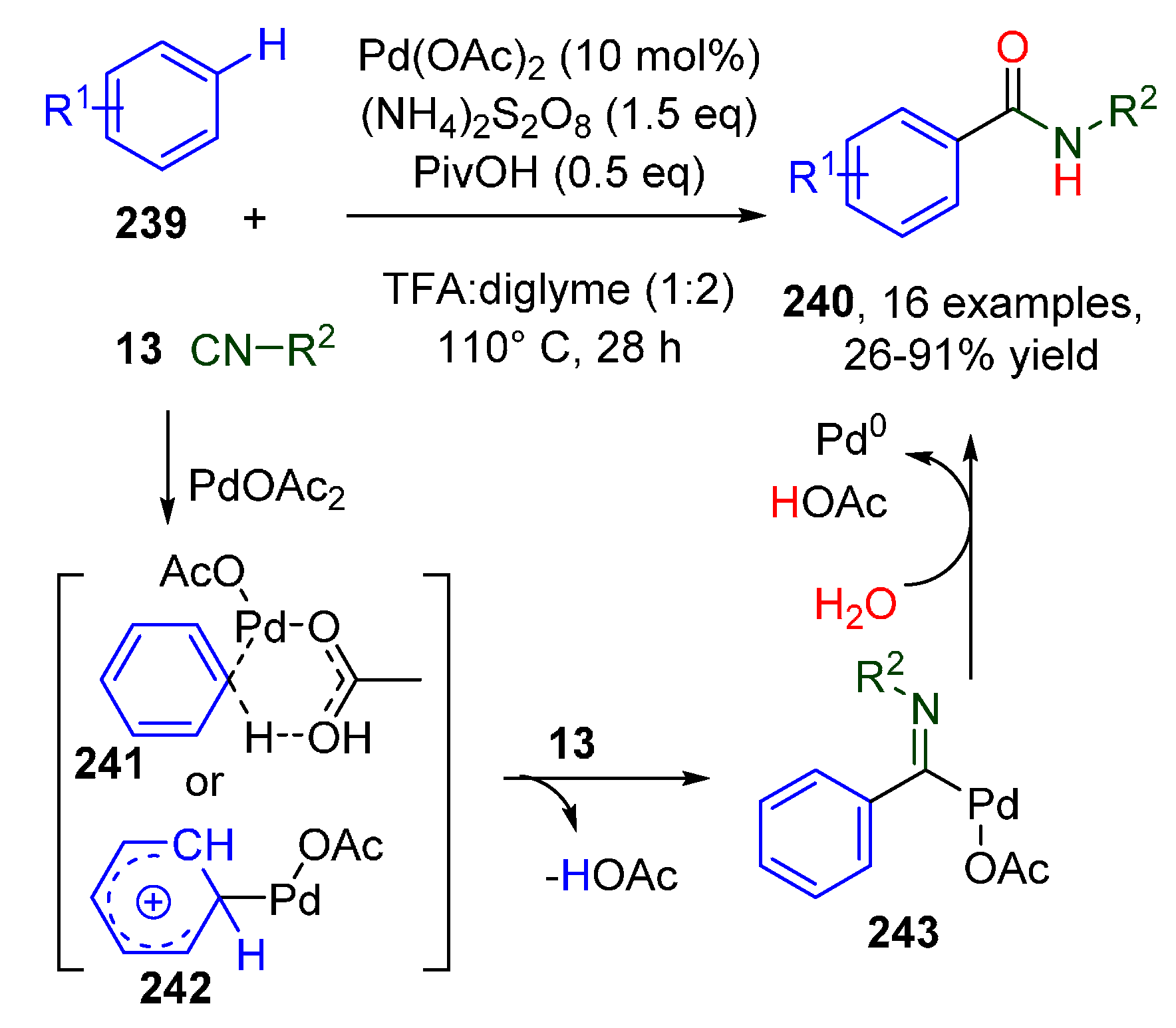
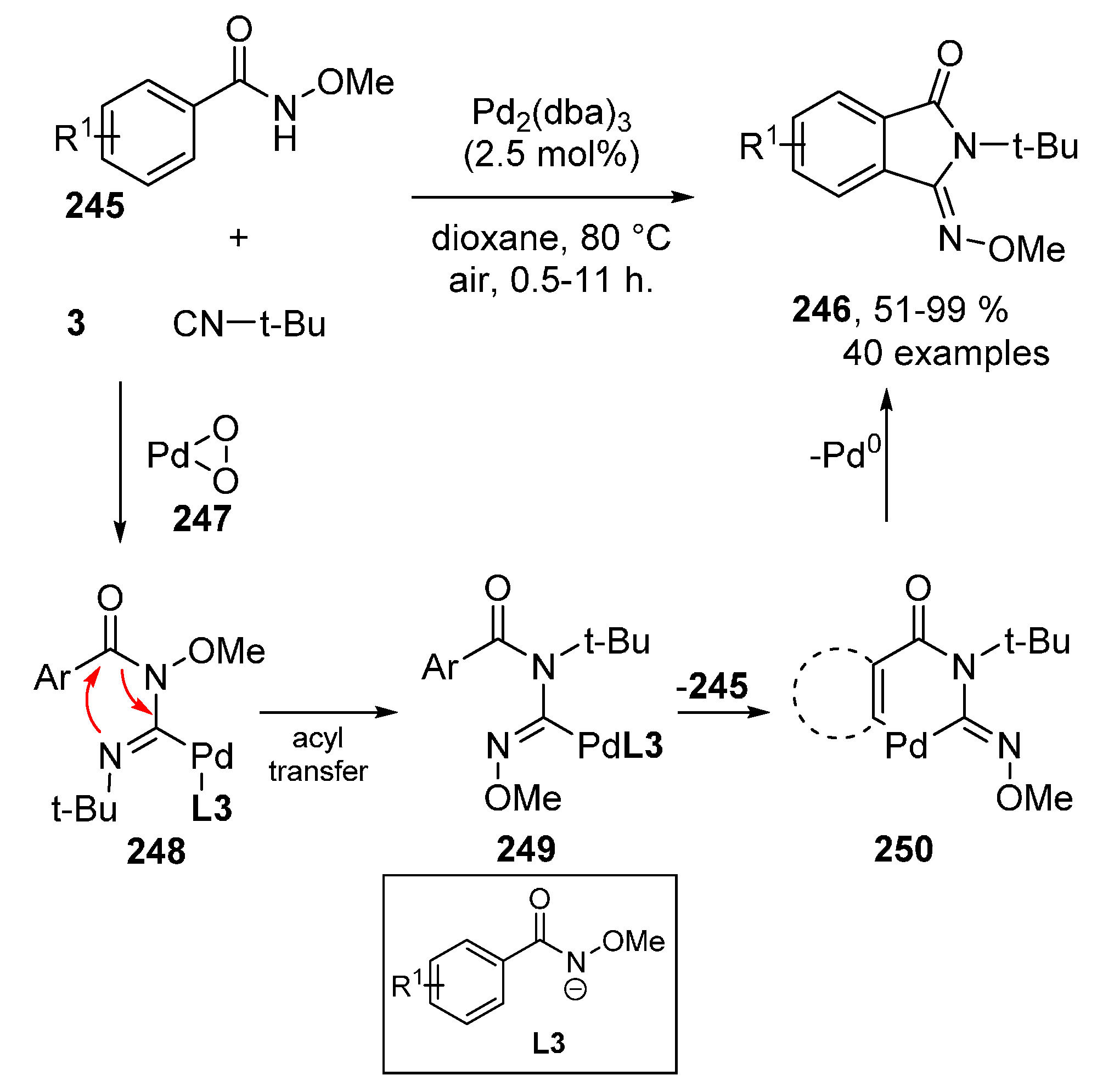
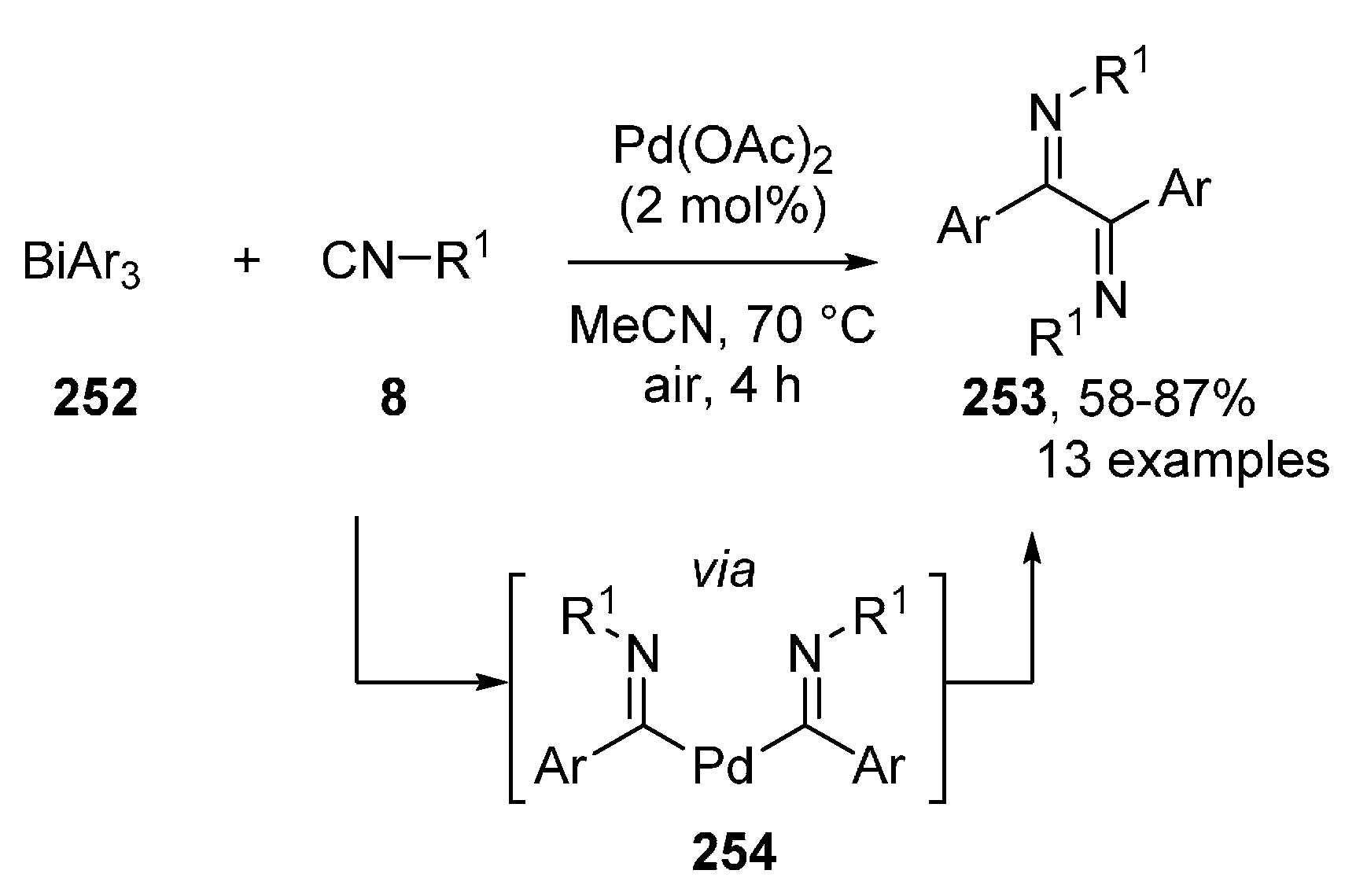
3.2. PdII-Catalyzed Intermolecular Oxidative Isocyanide Insertions
3.3. PdII-Catalyzed Intramolecular Oxidative Isocyanide Insertions
3.4. Miscellaneous PdII-Catalyzed Isocyanide Insertions
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooper, T.W.J.; Campbell, I.B.; Macdonald, S.J.F. Factors Determining the Selection of Organic Reactions by Medicinal Chemists and the Use of These Reactions in Arrays (Small Focused Libraries). Angew. Chem. Int. Ed. 2010, 49, 8082–8091. [Google Scholar] [CrossRef] [PubMed]
- Vlaar, T.; Ruijter, E.; Orru, R.V.A. Recent Advances in Palladium-Catalyzed Cascade Cyclizations. Adv. Synth. Catal. 2011, 353, 809–841. [Google Scholar] [CrossRef]
- Ugi, I.; Werner, B.; Dömling, A. The Chemistry of Isocyanides, their MultiComponent Reactions and their Libraries. Molecules 2003, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Giustiniano, M.; Basso, A.; Mercalli, V.; Massarotti, A.; Novellino, E.; Tron, G.C.; Zhu, J. To each his own: Isonitriles for all flavors. Functionalized isocyanides as valuable tools in organic synthesis. Chem. Soc. Rev. 2017, 46, 1295–1357. [Google Scholar] [CrossRef]
- Wang, Y.; Kumar, R.K.; Bi, X. Silver-catalyzed organic reactions of isocyanides. Tetrahedron Lett. 2016, 57, 5730–5741. [Google Scholar] [CrossRef]
- Lang, S. Unravelling the labyrinth of palladium-catalysed reactions involving isocyanides. Chem. Soc. Rev. 2013, 42, 4867–4880. [Google Scholar] [CrossRef]
- Vlaar, T.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Palladium-Catalyzed Migratory Insertion of Isocyanides: An Emerging Platform in Cross-Coupling Chemistry. Angew. Chem. Int. Ed. 2013, 52, 7084–7097. [Google Scholar] [CrossRef]
- Collet, J.W.; Roose, T.R.; Ruijter, E.; Maes, B.U.W.; Orru, R.V.A. Base Metal Catalyzed Isocyanide Insertions. Angew. Chem. Int. Ed. 2020, 59, 540–558. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H.; Beller, M. Palladium-catalyzed carbonylative coupling reactions between Ar-X and carbon nucleophiles. Chem. Soc. Rev. 2011, 40, 4986–5009. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H.; Beller, M. Synthesis of Heterocycles via Palladium-Catalyzed Carbonylations. Chem. Rev. 2013, 113, 1–35. [Google Scholar] [CrossRef]
- Schwartz, E.; Koepf, M.; Kitto, H.J.; Nolte, R.J.M.; Rowan, A.E. Helical poly(isocyanides): Past, present and future. Polym. Chem. 2011, 2, 33–47. [Google Scholar] [CrossRef]
- Millich, F. Polymerization of isocyanides. Chem. Rev. 1972, 72, 101–113. [Google Scholar] [CrossRef]
- Pomarico, S.K.; Lye, D.S.; Elacqua, E.; Weck, M. Synthesis of sheet-coil-helix and coil-sheet-helix triblock copolymers by combining ROMP with palladium-mediated isocyanide polymerization. Polym. Chem. 2018, 9, 5655–5659. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Jiang, Z.; Liu, F.; Liu, F.; Zhu, Y.; Liang, Y.; Wu, Z. Halogen effects on phenylethynyl palladium(II) complexes for living polymerization of isocyanides: A combined experimental and computational investigation. Sci. China Chem. 2019, 62, 491–499. [Google Scholar] [CrossRef]
- Huang, J.; Shen, L.; Zou, H.; Liu, N. Enantiomer-selective Living Polymerization of rac-Phenyl Isocyanide Using Chiral Palladium Catalyst. Chin. J. Polym. Sci. 2018, 36, 799–804. [Google Scholar] [CrossRef]
- Chen, J.-L.; Yang, L.; Wang, Q.; Jiang, Z.-Q.; Liu, N.; Yin, J.; Ding, Y.; Wu, Z.-Q. Helix-Sense-Selective and Enantiomer-Selective Living Polymerization of Phenyl Isocyanide Induced by Reusable Chiral Lactide Using Achiral Palladium Initiator. Macromolecules 2015, 48, 7737–7746. [Google Scholar] [CrossRef]
- Xue, Y.-X.; Chen, J.-L.; Jiang, Z.-Q.; Yu, Z.; Liu, N.; Yin, J.; Zhu, Y.-Y.; Wu, Z.-Q. Living polymerization of arylisocyanide initiated by the phenylethynyl palladium(ii) complex. Polym. Chem. 2014, 5, 6435–6438. [Google Scholar] [CrossRef]
- Xue, Y.-X.; Zhu, Y.-Y.; Gao, L.-M.; He, X.-Y.; Liu, N.; Zhang, W.-Y.; Yin, J.; Ding, Y.; Zhou, H.; Wu, Z.-Q. Air-Stable (Phenylbuta-1,3-diynyl)palladium(II) Complexes: Highly Active Initiators for Living Polymerization of Isocyanides. J. Am. Chem. Soc. 2014, 136, 4706–4713. [Google Scholar] [CrossRef]
- Masanori, K.; Toshimi, O.; Hiroyuki, T.; Hiroshi, S.; Toshihiko, M. Palladium Catalyzed Iminocarbonylation of Bromobenzene with Isocyanide and Organotin Compounds. Chem. Lett. 1986, 15, 1197–1200. [Google Scholar]
- Ishiyama, T.; Oh-e, T.; Miyaura, N.; Suzuki, A. Palladium-catalyzed iminocarbonylative cross-coupling reaction between haloarenes, t-BuNC, and 9-alkyl-9-BBN derivatives. Synthesis of alkyl aryl ketones. Tetrahedron Lett. 1992, 33, 4465–4468. [Google Scholar] [CrossRef]
- Chen, Z.; Duan, H.; Jiang, X.; Wang, J.; Zhu, Y.; Yang, S. Palladium-Catalyzed Synthesis of Biaryl Ketones via tert-Butyl Isocyanide Insertion. Synlett 2014, 25, 1425–1430. [Google Scholar] [CrossRef]
- Dechert-Schmitt, A.-M.; Garnsey, M.R.; Wisniewska, H.M.; Murray, J.I.; Lee, T.; Kung, D.W.; Sach, N.; Blackmond, D.G. Highly Modular Synthesis of 1,2-Diketones via Multicomponent Coupling Reactions of Isocyanides as CO Equivalents. ACS Catal. 2019, 9, 4508–4515. [Google Scholar] [CrossRef]
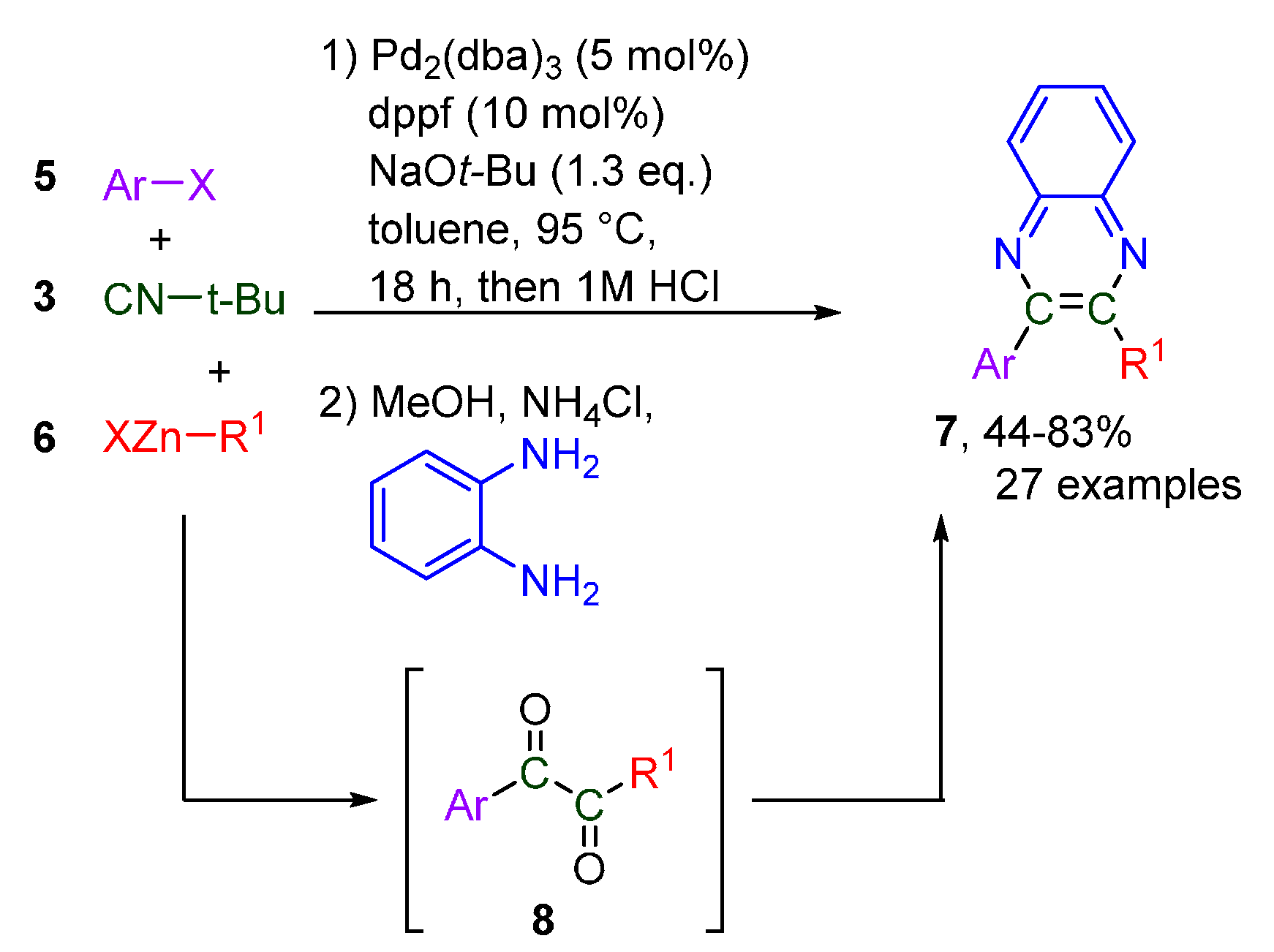
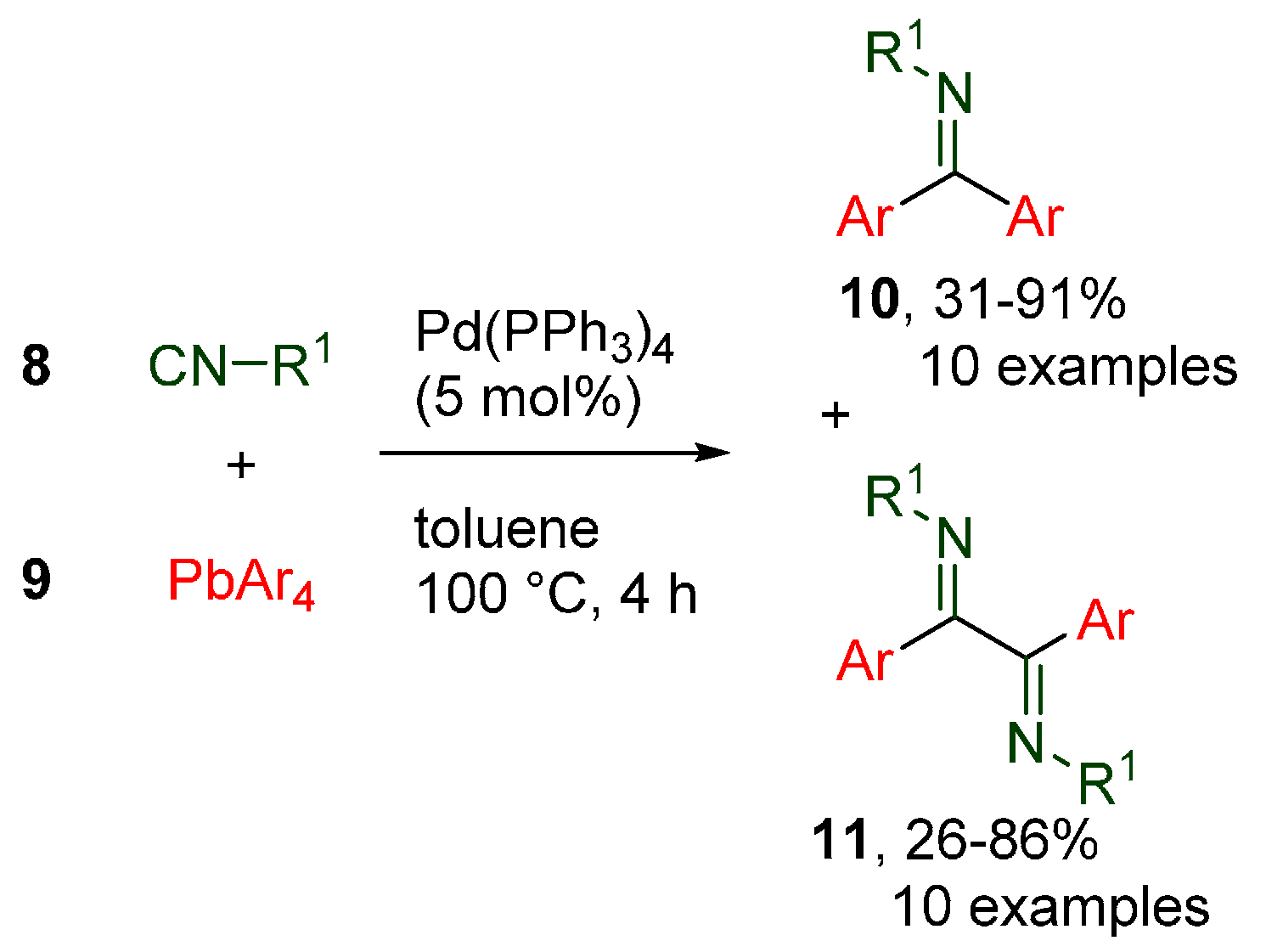
- Tran, C.C.; Kawaguchi, S.-I.; Kobiki, Y.; Matsubara, H.; Tran, D.P.; Kodama, S.; Nomoto, A.; Ogawa, A. Palladium-Catalyzed Diarylation of Isocyanides with Tetraarylleads for the Selective Synthesis of Imines and α-Diimines. J. Org. Chem. 2019, 84, 11741–11751. [Google Scholar] [CrossRef]
- Tang, T.; Fei, X.-D.; Ge, Z.-Y.; Chen, Z.; Zhu, Y.-M.; Ji, S.-J. Palladium-Catalyzed Carbonylative Sonogashira Coupling of Aryl Bromides via tert-Butyl Isocyanide Insertion. J. Org. Chem. 2013, 78, 3170–3175. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.W.; Ackermans, K.; Lambregts, J.; Maes, B.U.W.; Orru, R.V.A.; Ruijter, E. Modular Three-Component Synthesis of 4-Aminoquinolines via an Imidoylative Sonogashira/Cyclization Cascade. J. Org. Chem. 2018, 83, 854–861. [Google Scholar] [CrossRef]
- Zhang, F.-L.; Chen, Z.-B.; Liu, K.; Yuan, Q.; Jiang, Q.; Zhu, Y.-M. Carbonylative Synthesis of Thiochromenones via Palladium-Catalyzed tert-Butyl Isocyanide Insertion. Synlett 2018, 29, 621–626. [Google Scholar]
- Duan, H.; Chen, Z.; Han, L.; Feng, Y.; Zhu, Y.; Yang, S. Palladium-catalyzed chemoselective synthesis of indane-1,3-dione derivatives via tert-butyl isocyanide insertion. Org. Biomol. Chem. 2015, 13, 6782–6788. [Google Scholar] [CrossRef] [PubMed]
- Fei, X.-D.; Ge, Z.-Y.; Tang, T.; Zhu, Y.-M.; Ji, S.-J. Palladium-Catalyzed Synthesis of Isocoumarins and Phthalides via tert-Butyl Isocyanide Insertion. J. Org. Chem. 2012, 77, 10321–10328. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Duan, H.-Q.; Jiang, X.; Zhu, Y.-M.; Ji, S.-J.; Yang, S.-L. Palladium-Catalyzed Domino Synthesis of 4-Amino-3-acyl-2-naphthols via Isocyanide Chemoselective Insertion. J. Org. Chem. 2015, 80, 8183–8188. [Google Scholar] [CrossRef]
- Gu, Z.-Y.; Zhu, T.-H.; Cao, J.-J.; Xu, X.-P.; Wang, S.-Y.; Ji, S.-J. Palladium-Catalyzed Cascade Reactions of Isocyanides with Enaminones: Synthesis of 4-Aminoquinoline Derivatives. ACS Catal. 2014, 4, 49–52. [Google Scholar] [CrossRef]
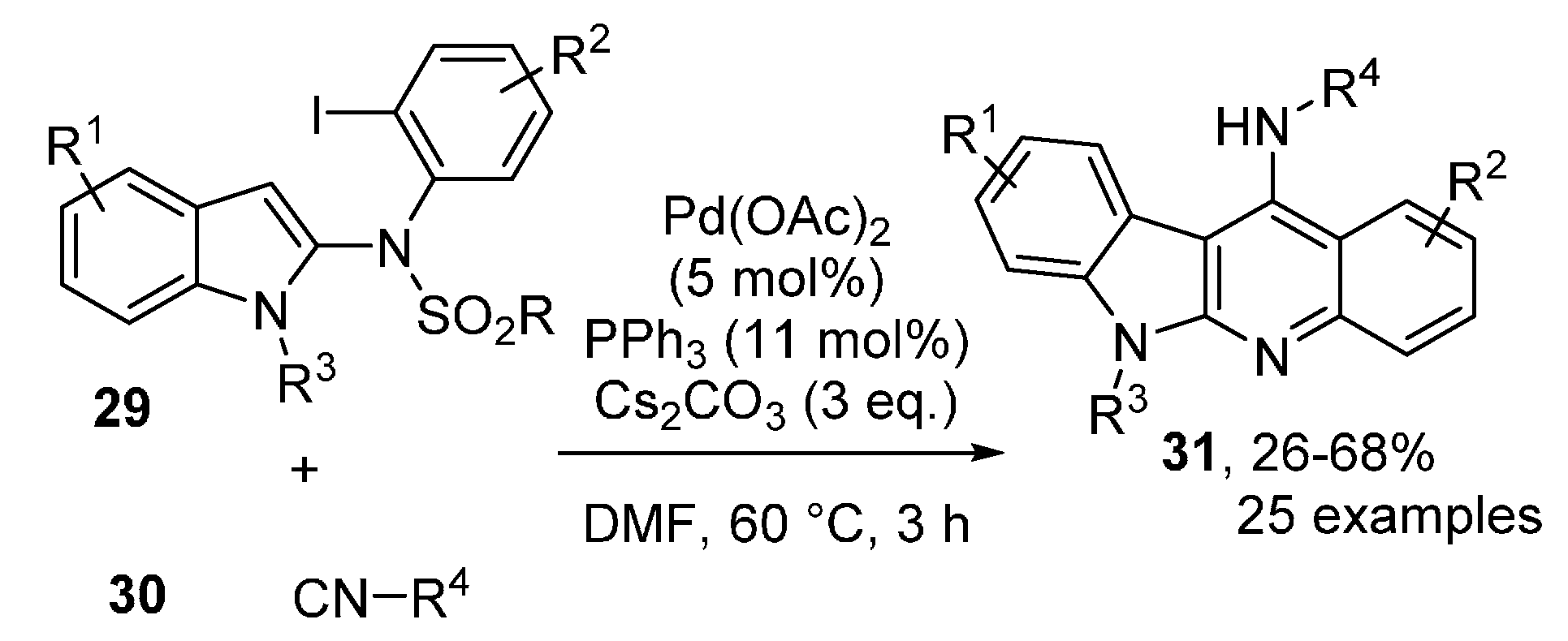
- Prasad, B.; Nallapati, S.B.; Kolli, S.K.; Sharma, A.K.; Yellanki, S.; Medisetti, R.; Kulkarni, P.; Sripelly, S.; Mukkanti, K.; Pal, M. Pd-catalyzed isocyanide insertion/nucleophilic attack by indole C-3/desulfonylation in the same pot: A direct access to indoloquinolines of pharmacological interest. RSC Adv. 2015, 5, 62966–62970. [Google Scholar] [CrossRef]
- Nallapati, S.B.; Prasad, B.; Sreenivas, B.Y.; Sunke, R.; Poornachandra, Y.; Kumar, C.G.; Sridhar, B.; Shivashankar, S.; Mukkanti, K.; Pal, M. Apparent Carbon Monoxide Insertion via Double Isocyanide Incorporation during Palladium-Catalyzed Construction of Indoloquinoline Ring in a Single Pot: Synthesis of New Cytotoxic Agents. Adv. Synth. Catal. 2016, 358, 3387–3393. [Google Scholar] [CrossRef]
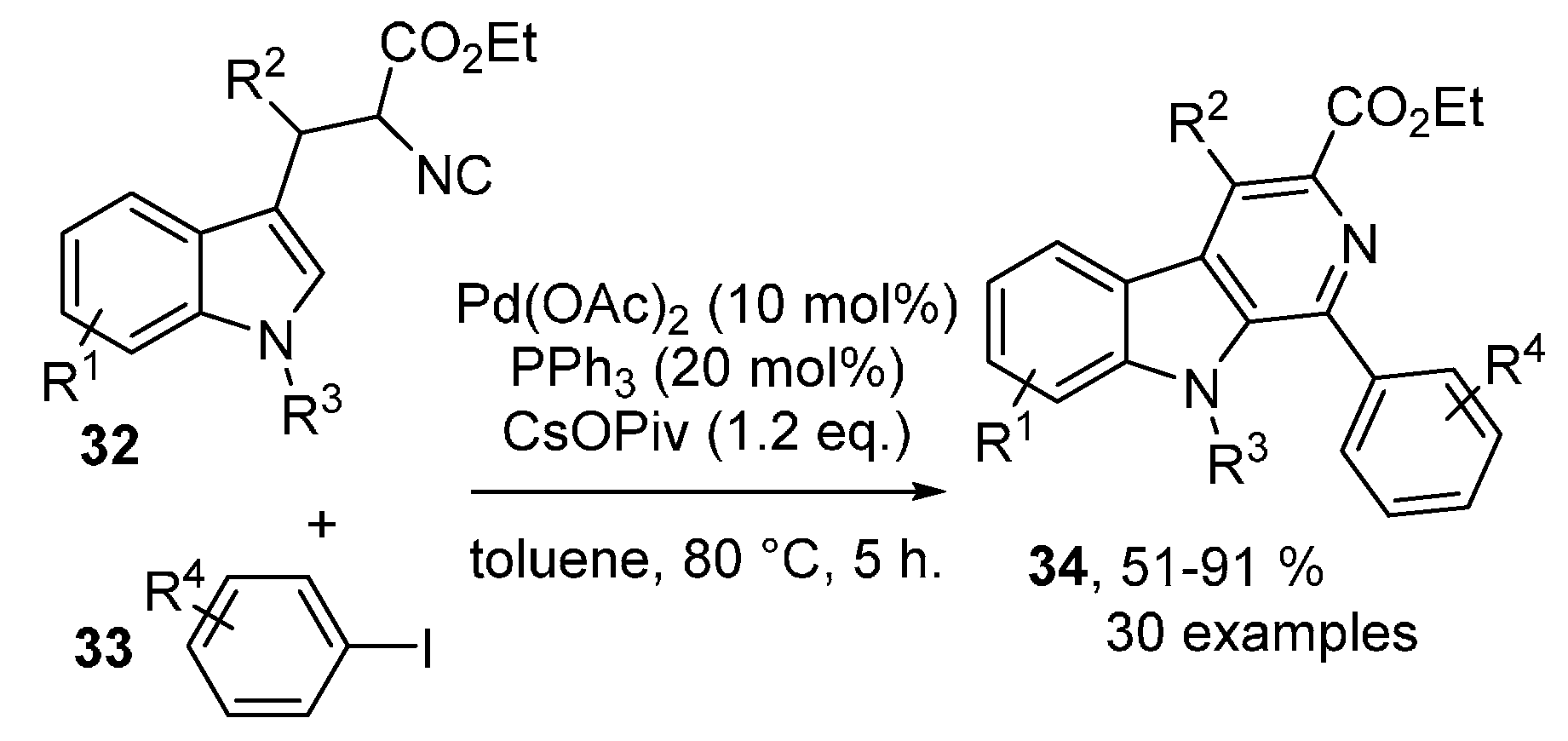
- Tang, S.; Wang, J.; Xiong, Z.; Xie, Z.; Li, D.; Huang, J.; Zhu, Q. Palladium-Catalyzed Imidoylative Cyclization of Tryptophan-Derived Isocyanides: Access to β-Carbolines. Org. Lett. 2017, 19, 5577–5580. [Google Scholar] [CrossRef] [PubMed]
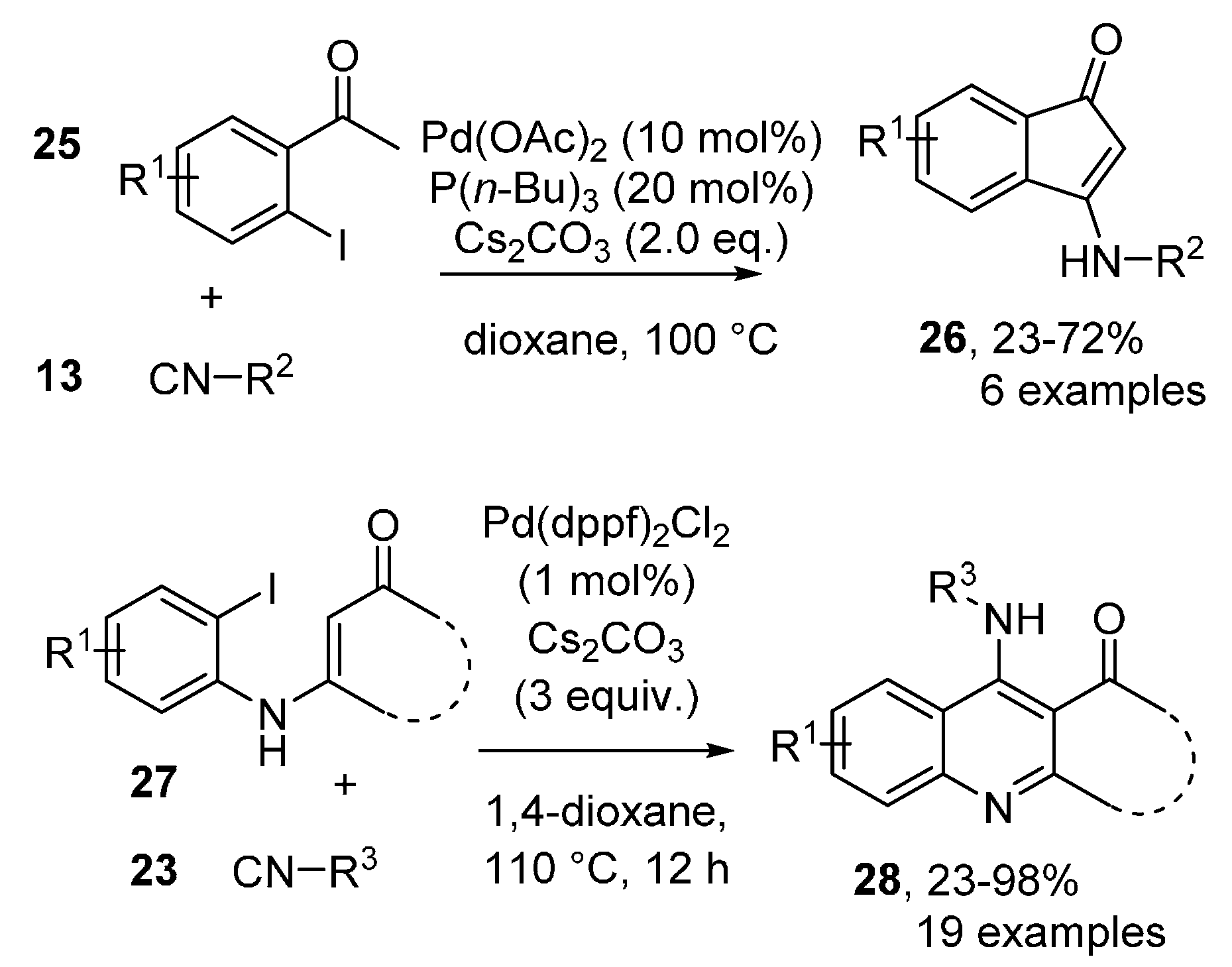
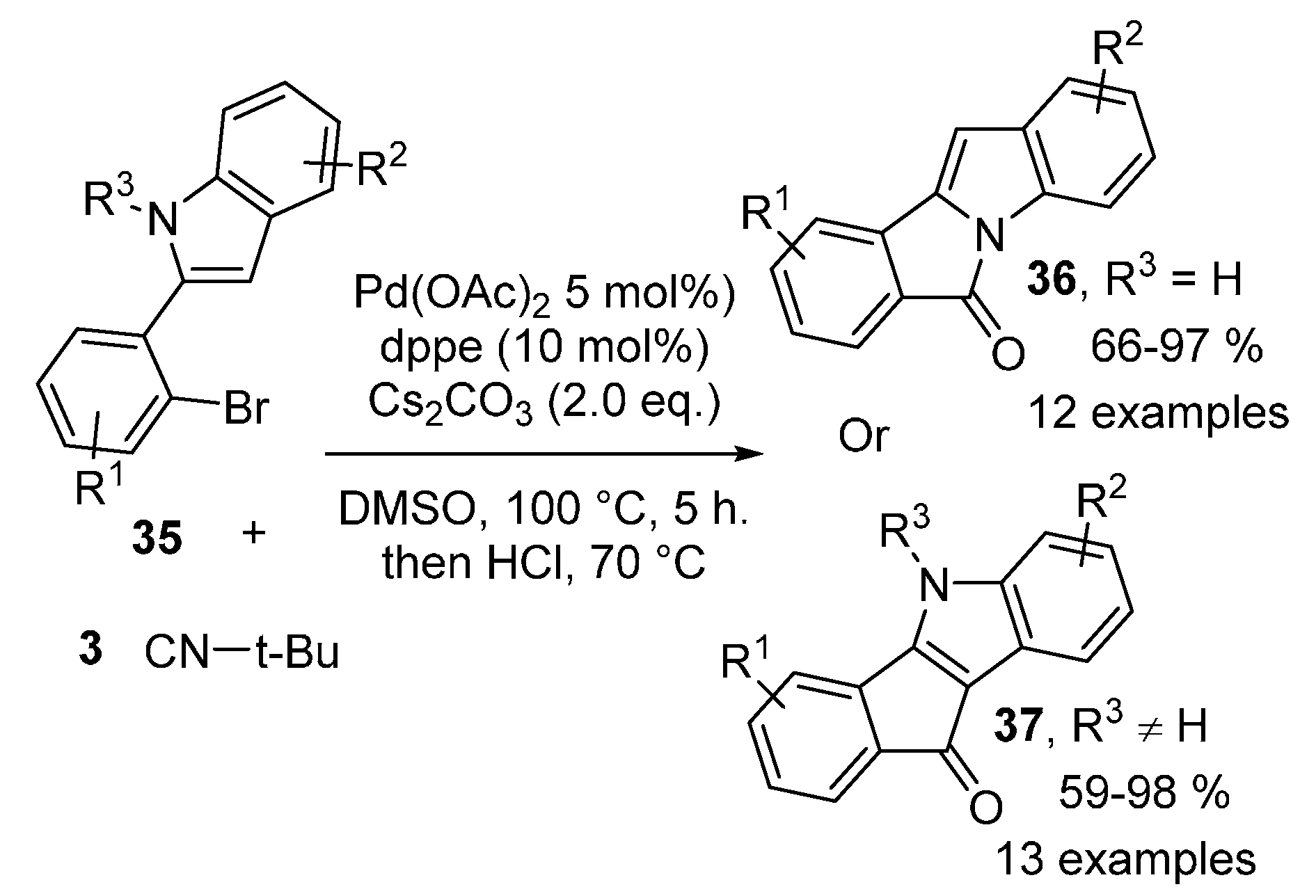
- Tang, T.; Jiang, X.; Wang, J.-M.; Sun, Y.-X.; Zhu, Y.-M. Divergent synthesis of 6H-isoindolo[2,1-a]indol-6-ones and indenoindolones: An investigation of Pd-catalyzed isocyanide insertion. Tetrahedron 2014, 70, 2999–3004. [Google Scholar] [CrossRef]
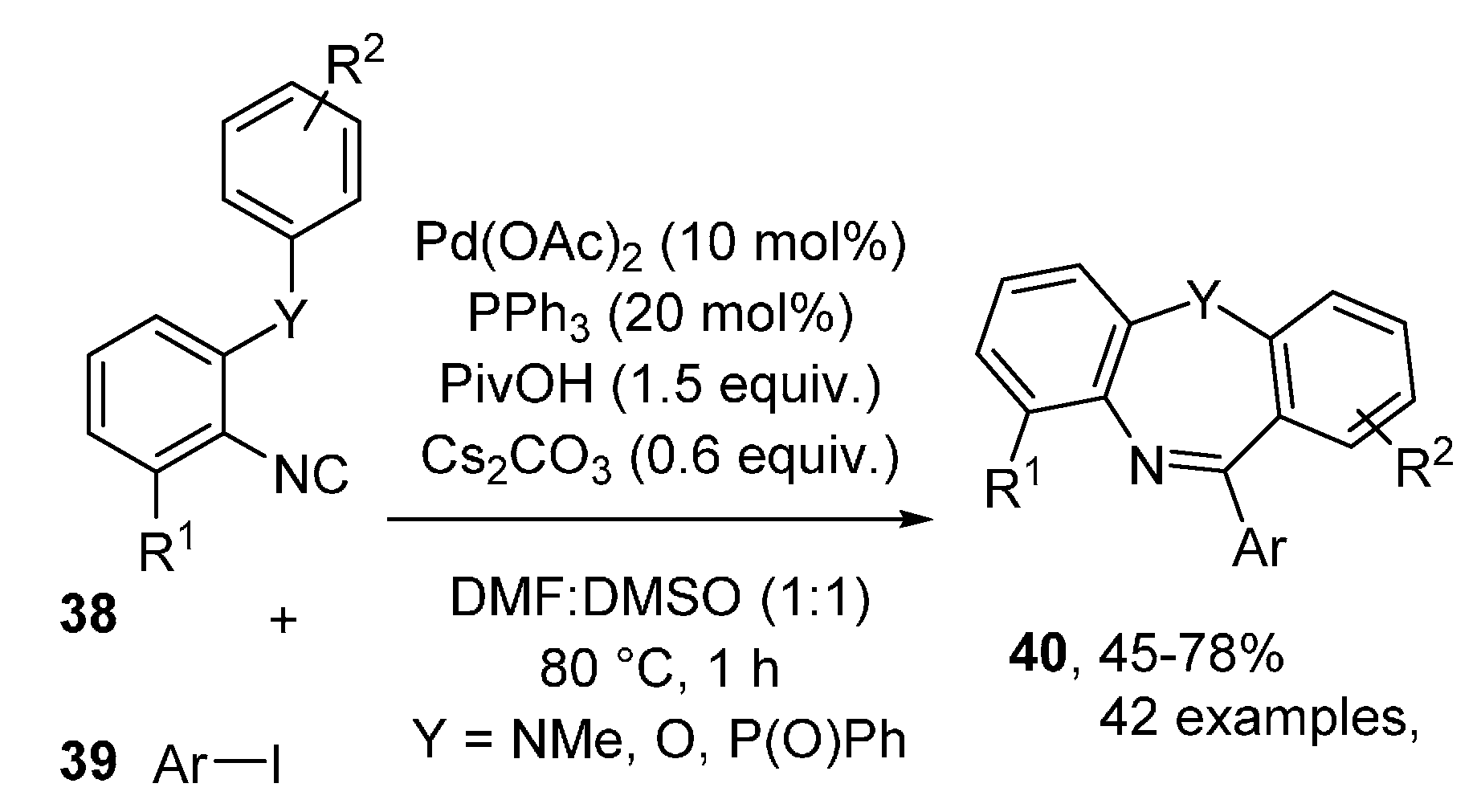
- Hu, W.; Teng, F.; Hu, H.; Luo, S.; Zhu, Q. Pd-Catalyzed C(sp2)-H Imidoylative Annulation: A General Approach To Construct Dibenzoox(di)azepines. J. Org. Chem. 2019, 84, 6524–6535. [Google Scholar] [CrossRef]
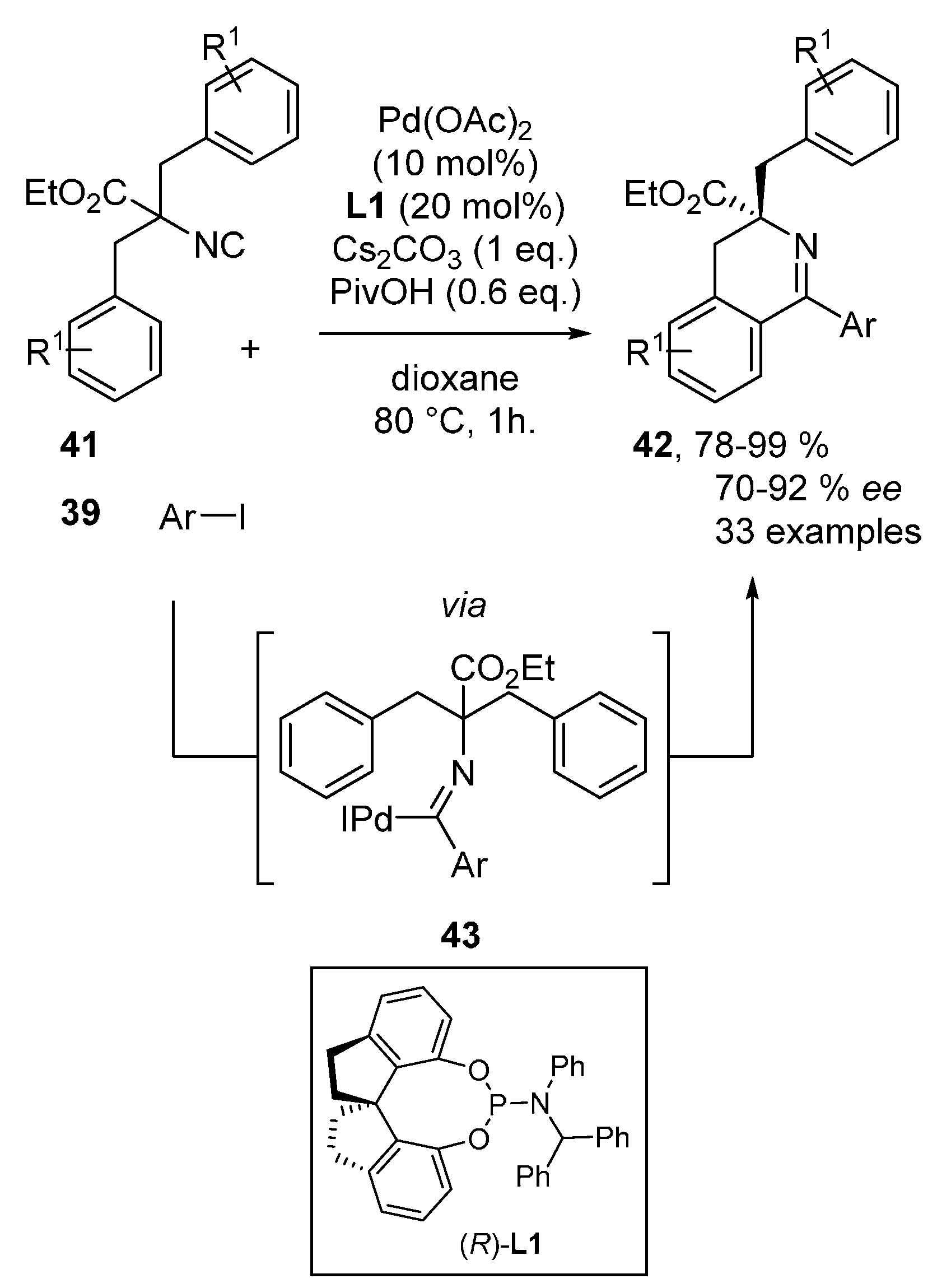
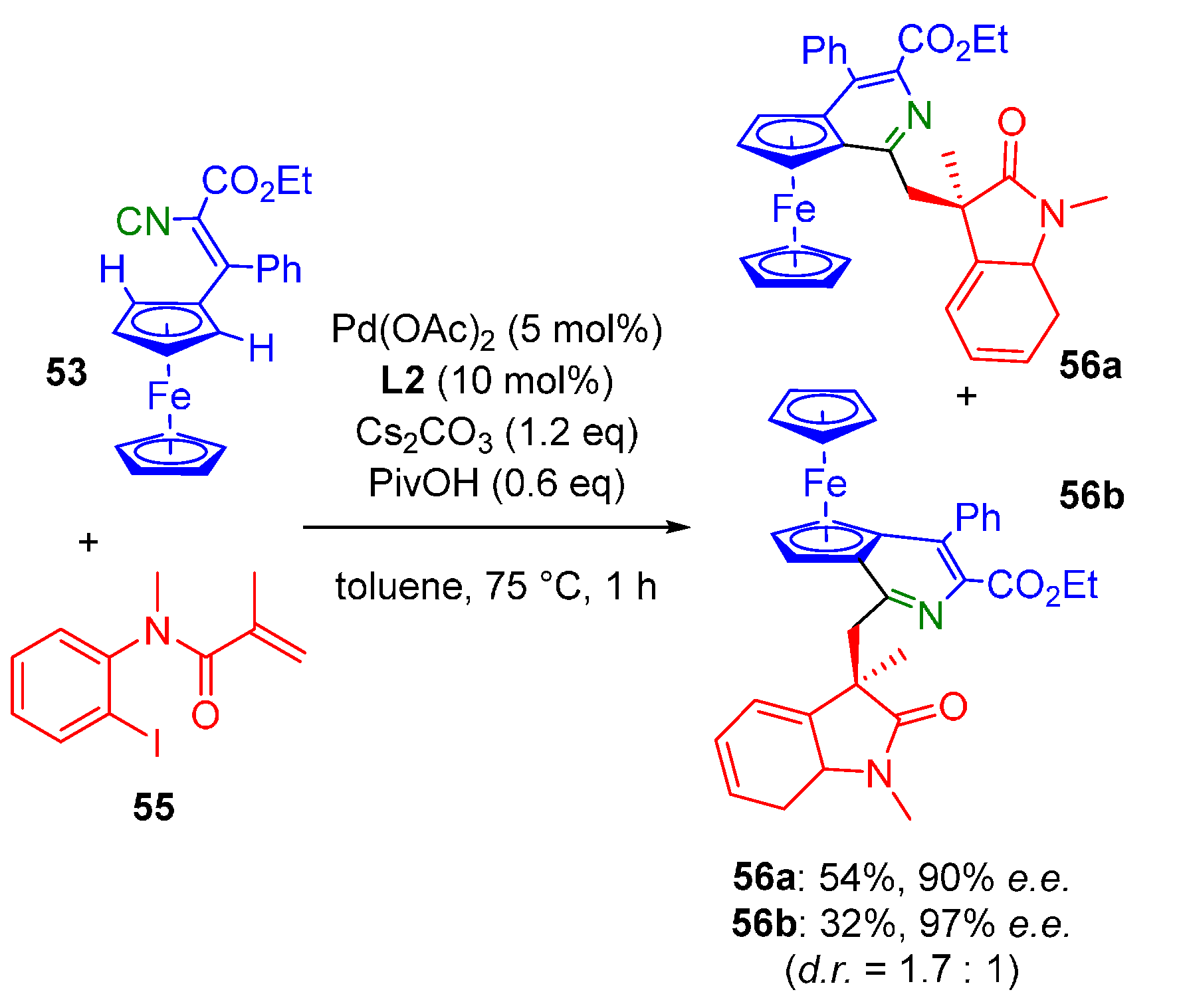
- Wang, J.; Gao, D.-W.; Huang, J.; Tang, S.; Xiong, Z.; Hu, H.; You, S.-L.; Zhu, Q. Palladium-Catalyzed Enantioselective C(sp2)-H Imidoylation by Desymmetrization. ACS Catal. 2017, 7, 3832–3836. [Google Scholar] [CrossRef]
- Zhang, B.; Studer, A. Recent advances in the synthesis of nitrogen heterocycles via radical cascade reactions using isonitriles as radical acceptors. Chem. Soc. Rev. 2015, 44, 3505–3521. [Google Scholar] [CrossRef]
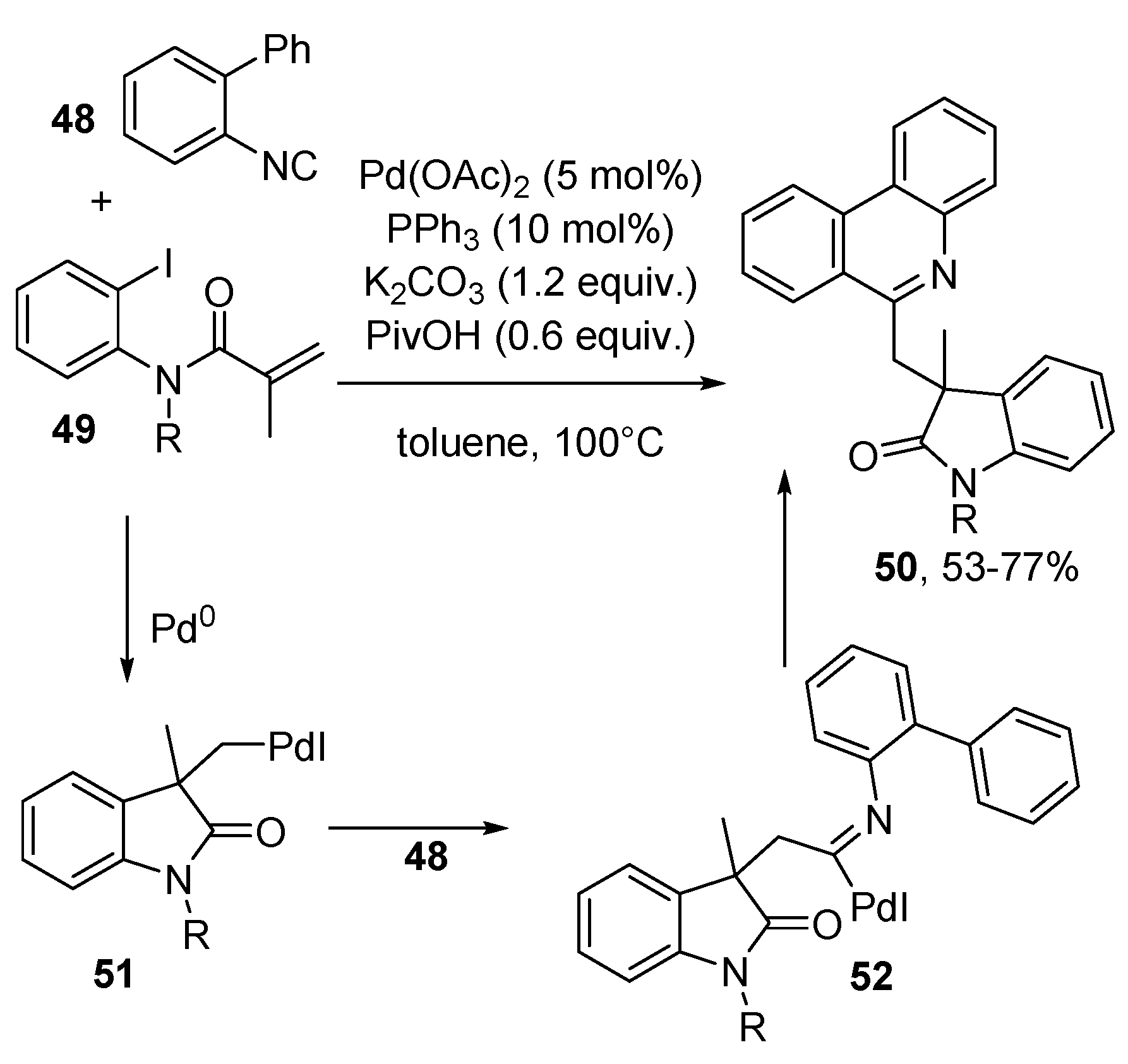
- Li, J.; He, Y.; Luo, S.; Lei, J.; Wang, J.; Xie, Z.; Zhu, Q. Palladium-Catalyzed Intramolecular C(sp2)-H Imidoylation for the Synthesis of Six-Membered N-Heterocycles. J. Org. Chem. 2015, 80, 2223–2230. [Google Scholar] [CrossRef]
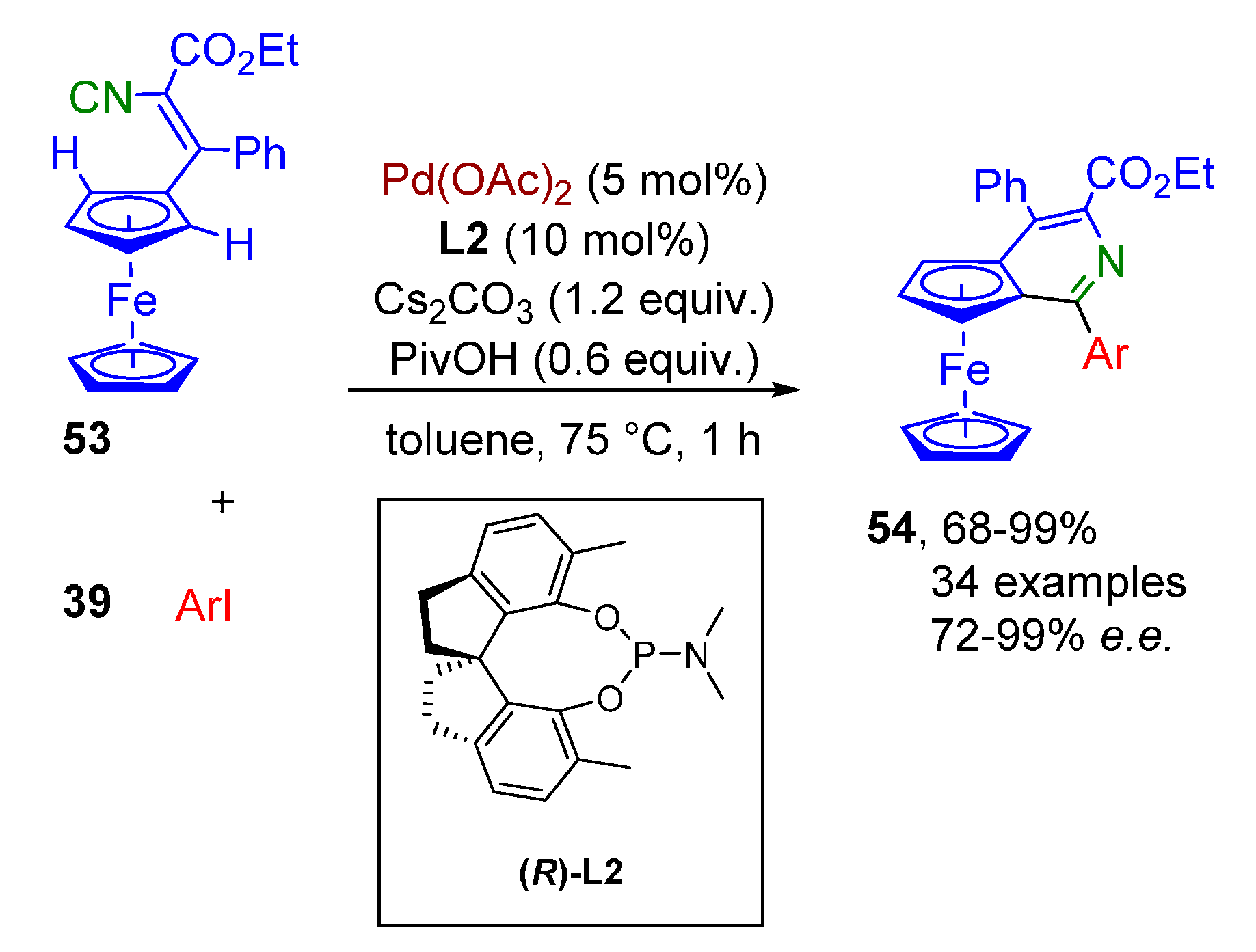
- Luo, S.; Xiong, Z.; Lu, Y.; Zhu, Q. Enantioselective Synthesis of Planar Chiral Pyridoferrocenes via Palladium-Catalyzed Imidoylative Cyclization Reactions. Org. Lett. 2018, 20, 1837–1840. [Google Scholar] [CrossRef]
- Perego, L.A.; Fleurat-Lessard, P.; El Kaïm, L.; Ciofini, I.; Grimaud, L. Multiple Roles of Isocyanides in Palladium-Catalyzed Imidoylative Couplings: A Mechanistic Study. Chem. Eur. J. 2016, 22, 15491–15500. [Google Scholar] [CrossRef]
- Liang, Y.; Ren, Y.; Jia, J.; Wu, H.-S. Mechanistic investigation of palladium-catalyzed amidation of aryl halides. J. Mol. Model. 2016, 22, 53. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, Y.; Wang, Z.; Orru, R.V.A.; Maes, B.U.W.; Wu, X.-F. Palladium-Catalyzed Construction of Amidines from Arylboronic Acids under Oxidative Conditions. Chem. Eur. J. 2016, 22, 7743–7746. [Google Scholar] [CrossRef] [PubMed]
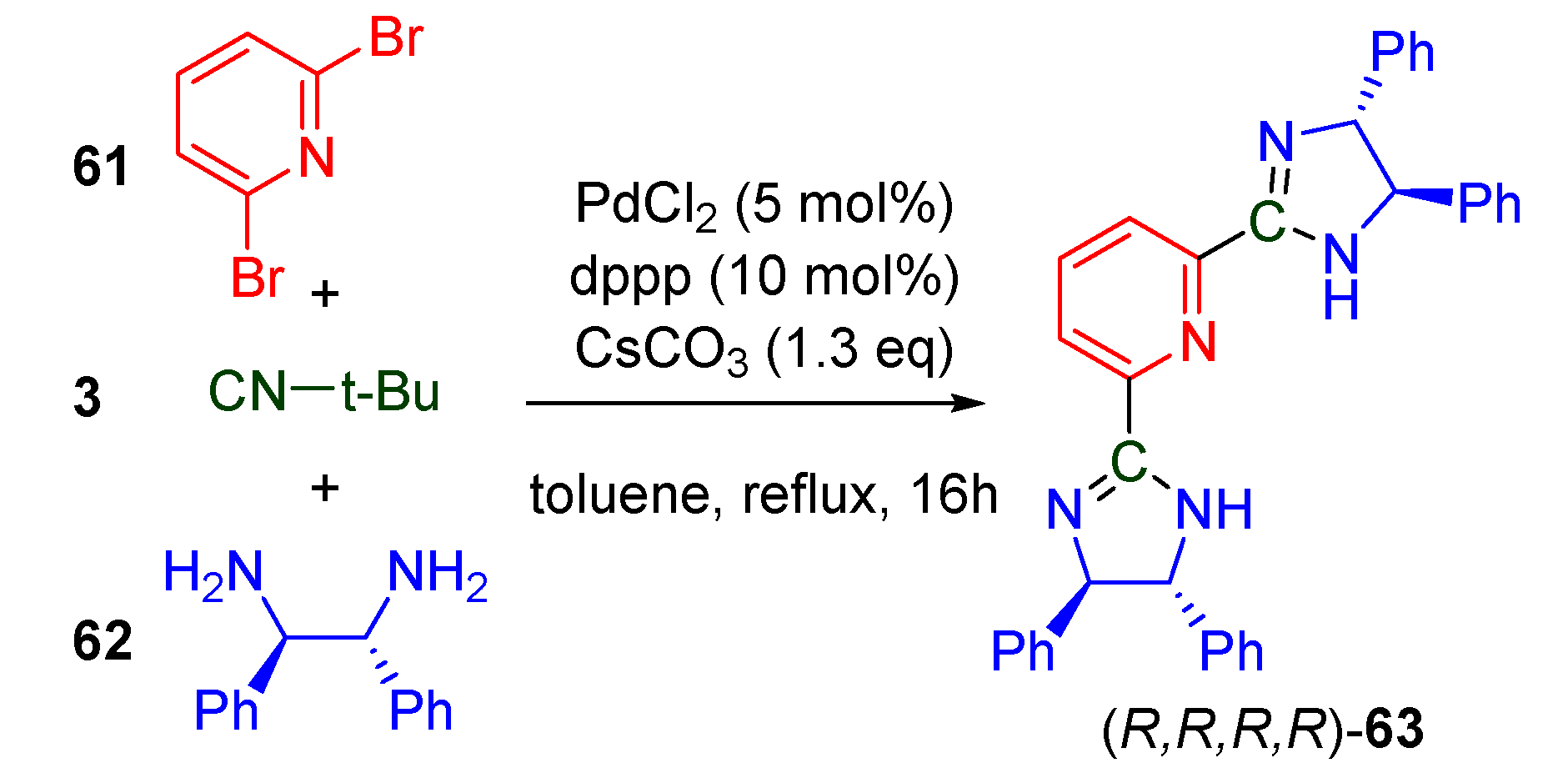
- Geden, J.V.; Pancholi, A.K.; Shipman, M. Palladium-Catalyzed Multicomponent Synthesis of 2-Aryl-2-imidazolines from Aryl Halides and Diamines. J. Org. Chem. 2013, 78, 4158–4164. [Google Scholar] [CrossRef]
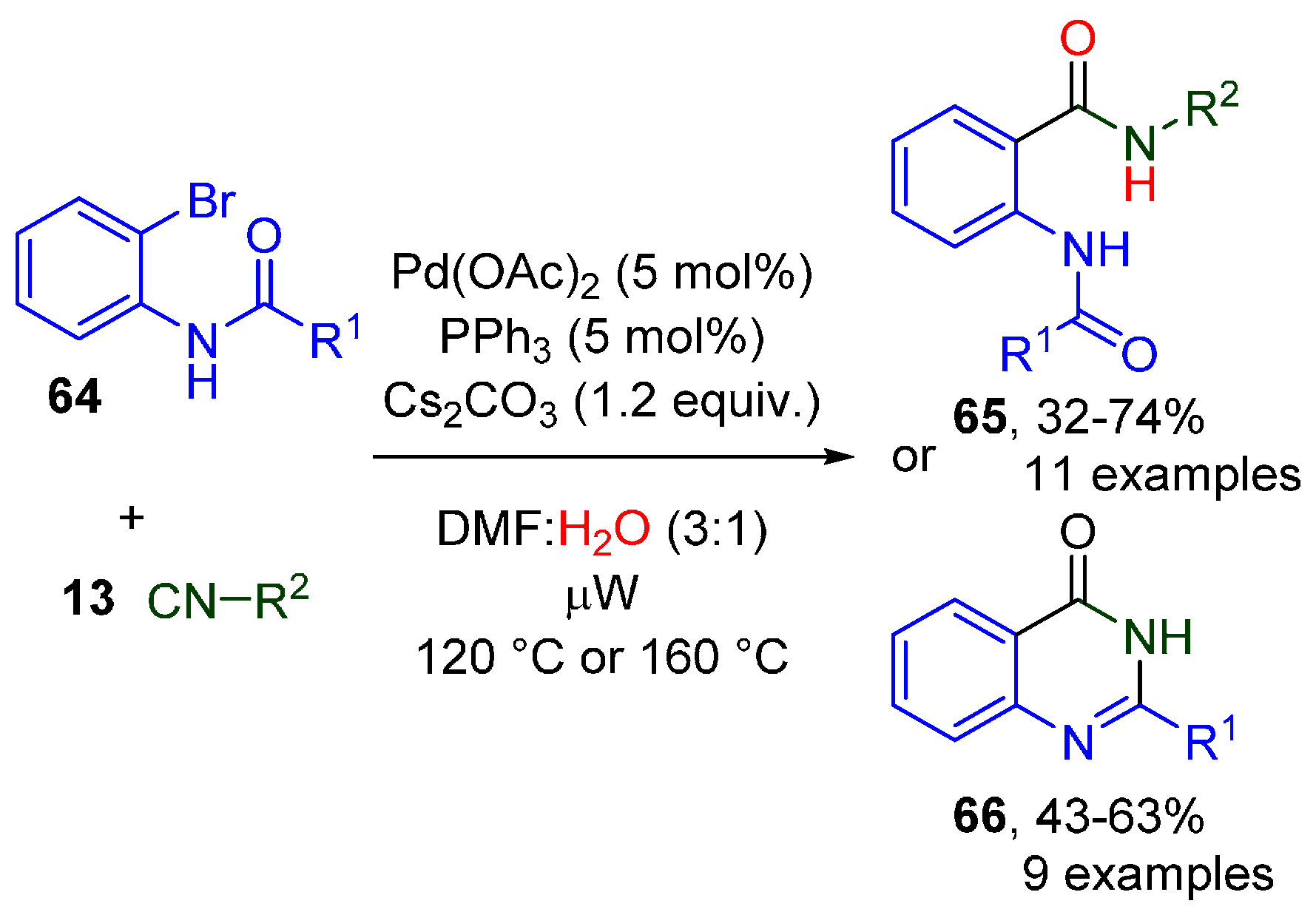
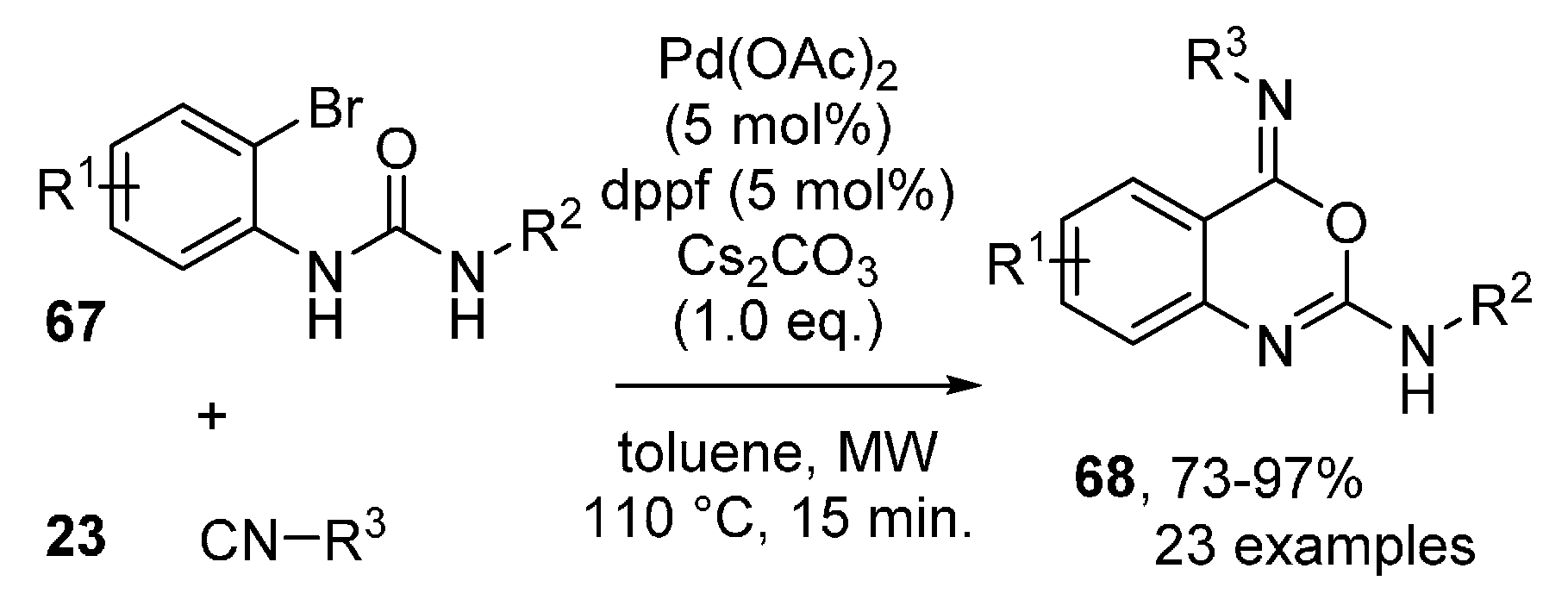
- Khan, I.; Singh, J.; Khan, I.; Dutt, S.; Khan, S.; Tyagi, V. Temperature-controlled synthesis of N-acyl anthranilamides and quinazoline-4-ones via Pd-catalysed cascade consisting of isocyanide insertion. ARKIVOC 2019, 2019, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Pathare, R.S.; Sharma, S.; Elagandhula, S.; Saini, V.; Sawant, D.M.; Yadav, M.; Sharon, A.; Khan, S.; Pardasani, R.T. Application of Isocyanides as Amide Surrogates in the Synthesis of Diverse Isoindolin-1-one Derivatives by a Palladium-Catalyzed Tandem Carboxamidation/Hydroamidation Reaction. Eur. J. Org. Chem. 2016, 2016, 5579–5587. [Google Scholar] [CrossRef]
- Shiri, M.; Ranjbar, M.; Yasaei, Z.; Zamanian, F.; Notash, B. Palladium-catalyzed tandem reaction of 2-chloroquinoline-3-carbaldehydes and isocyanides. Org. Biomol. Chem. 2017, 15, 10073–10081. [Google Scholar] [CrossRef]
- Pandey, G.; Batra, S. Microwave-assisted palladium-catalysed isonitrile insertion in 2-bromophenylureas for efficient synthesis of 4-substituted imino 4H-benzo[d][1,3]oxazin-2-amines. RSC Adv. 2015, 5, 28875–28878. [Google Scholar] [CrossRef]
- Sharma, S.; Jain, A. Ligand-free palladium assisted insertion of isocyanides to urea derivatives for cascade synthesis of phenylamino-substituted quinazolinones. Tetrahedron Lett. 2014, 55, 6051–6054. [Google Scholar] [CrossRef]
- Pandey, G.; Bhowmik, S.; Batra, S. Synthesis of 4-substituted imino-4H-benzo[d][1,3] thiazin-2-amines via palladium-catalysed isocyanide insertion in 2-bromophenylthioureas. RSC Adv. 2014, 4, 41433–41436. [Google Scholar] [CrossRef]
- Wang, J.-M.; Jiang, X.; Zhang, Y.; Zhu, Y.-M.; Shen, J.-K. Palladium-catalyzed synthesis of 4H-benzo[d][1,3]oxazin-4-ones and N-(2-cyanophenyl)benzamides via tert-butyl isocyanide insertion. Tetrahedron Lett. 2015, 56, 2349–2354. [Google Scholar] [CrossRef]
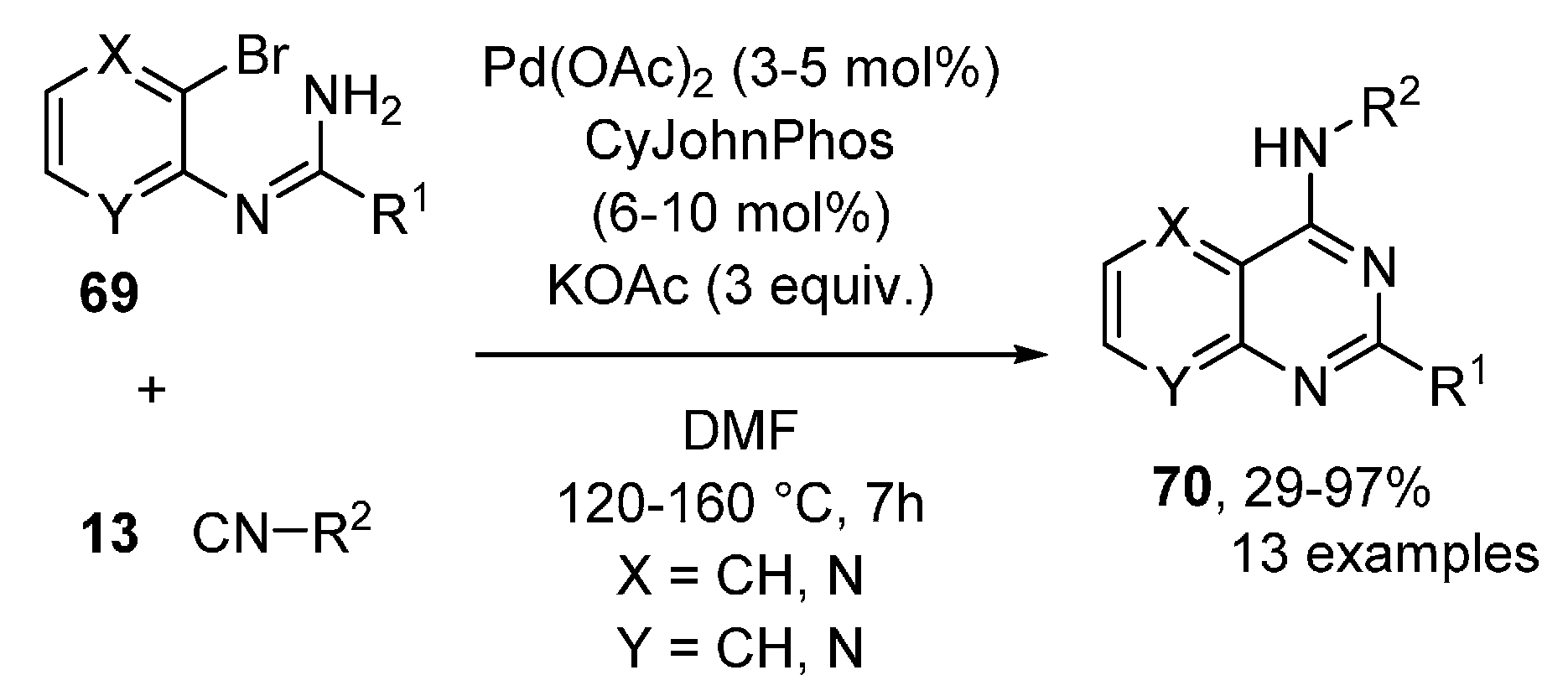
- Estévez, V.; Van Baelen, G.; Lentferink, B.H.; Vlaar, T.; Janssen, E.; Maes, B.U.W.; Orru, R.V.A.; Ruijter, E. Synthesis of Pyridopyrimidines by Palladium-Catalyzed Isocyanide Insertion. ACS Catal. 2014, 4, 40–43. [Google Scholar] [CrossRef]
- Fan, X.-Y.; Jiang, X.; Zhang, Y.; Chen, Z.-B.; Zhu, Y.-M. Palladium-catalyzed one-pot synthesis of diazoles via tert-butyl isocyanide insertion. Org. Biomol. Chem. 2015, 13, 10402–10408. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Hu, L.; Gui, W. Facile synthesis of 2-amino-3-bromoquinolines by palladium-catalyzed isocyanide insertion and cyclization of gem-dibromovinylanilines. RSC Adv. 2014, 4, 13850–13853. [Google Scholar] [CrossRef]
- Hu, L.; Gui, W.; Liu, Z.; Jiang, B. Synthesis of 3-aryl-2-aminoquinolines: Palladium-catalyzed cascade reactions of gem-dibromovinylanilines with tert-butyl isocyanide and arylboronic acids. RSC Adv. 2014, 4, 38258–38262. [Google Scholar] [CrossRef]
- Salehi, P.; Shiri, M. Palladium-Catalyzed Regioselective Synthesis of 3-(Hetero)Arylpropynamides from gem-Dibromoalkenes and Isocyanides. Adv. Synth. Catal. 2019, 361, 118–125. [Google Scholar] [CrossRef]
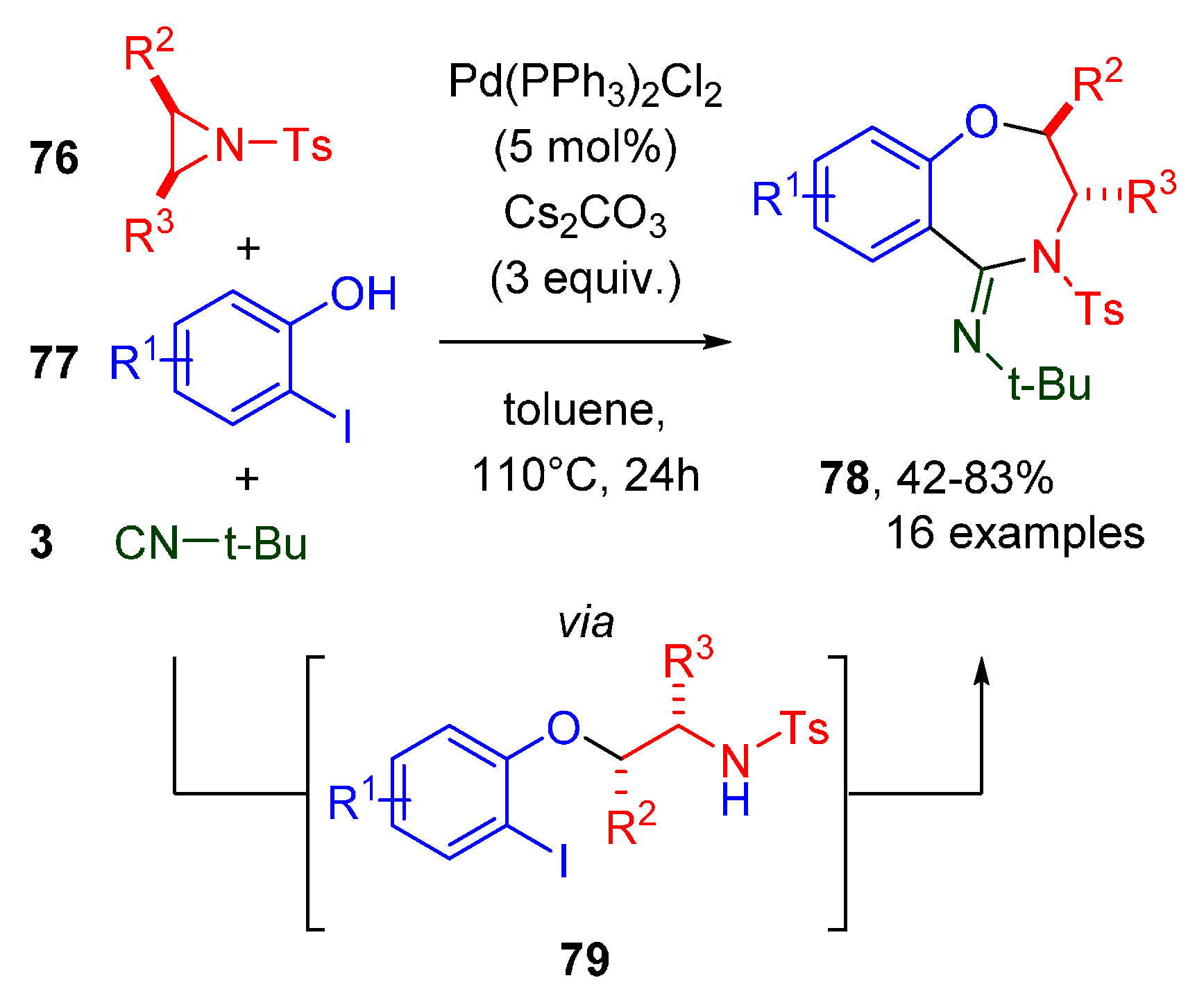
- Ji, F.; Lv, M.-F.; Yi, W.-B.; Cai, C. Synthesis of 1,4-Benzoxazepine Derivatives via a Novel Domino Aziridine Ring-Opening and Isocyanide-Insertion Reaction. Adv. Synth. Catal. 2013, 355, 3401–3406. [Google Scholar] [CrossRef]
- Pan, Y.-Y.; Wu, Y.-N.; Chen, Z.-Z.; Hao, W.-J.; Li, G.; Tu, S.-J.; Jiang, B. Synthesis of 3-Iminoindol-2-amines and Cyclic Enaminones via Palladium-Catalyzed Isocyanide Insertion-Cyclization. J. Org. Chem. 2015, 80, 5764–5770. [Google Scholar] [CrossRef]
- Pálinkás, N.; Kollár, L.; Kégl, T. Palladium-Catalyzed Synthesis of Amidines via tert-Butyl isocyanide Insertion. ACS Omega 2018, 3, 16118–16126. [Google Scholar] [CrossRef]
- Mamboury, M.; Wang, Q.; Zhu, J. α-Oxo-Ketenimines from Isocyanides and α-Haloketones: Synthesis and Divergent Reactivity. Chem. Eur. J. 2017, 23, 12744–12748. [Google Scholar] [CrossRef]
- Ren, Z.-L.; He, P.; Lu, W.-T.; Sun, M.; Ding, M.-W. Synthesis of iminoisoindolinones via a cascade of the three-component Ugi reaction, palladium catalyzed isocyanide insertion, hydroxylation and an unexpected rearrangement reaction. Org. Biomol. Chem. 2018, 16, 6322–6331. [Google Scholar] [CrossRef]
- Wang, B.; He, D.; Ren, B.; Yao, T. Synthesis of imides via palladium-catalyzed three-component coupling of aryl halides, isocyanides and carboxylic acids. Chem. Commun. 2020, 56, 900–903. [Google Scholar] [CrossRef]
- Ji, F.; Lv, M.-F.; Yi, W.-B.; Cai, C. A Practical, Ligand-Free, Palladium-Catalyzed Isocyanide Insertion Reaction for the Construction of Novel Ring-Fused Quinazolinones. Synthesis 2013, 45, 1965–1974. [Google Scholar]
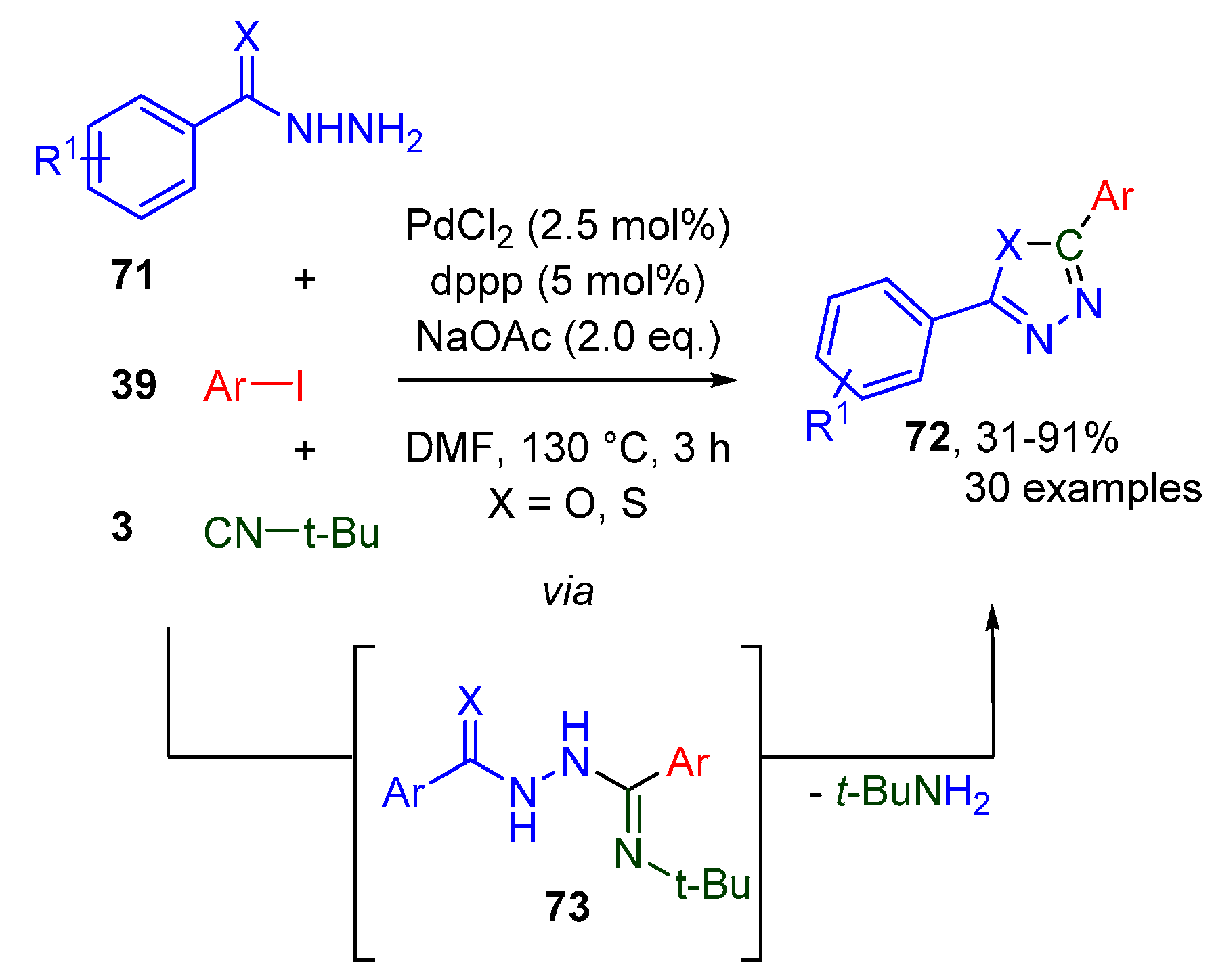
- Ukhin, L.Y.; Kuz’mina, L.G.; Gribanov, T.N.; Belousova, L.V.; Orlova, Z.I. Anthranilic acid hydrazide in the synthesis of fused polycyclic compounds with quinazoline moieties. Russ. Chem. Bull. 2009, 57, 2340. [Google Scholar] [CrossRef]
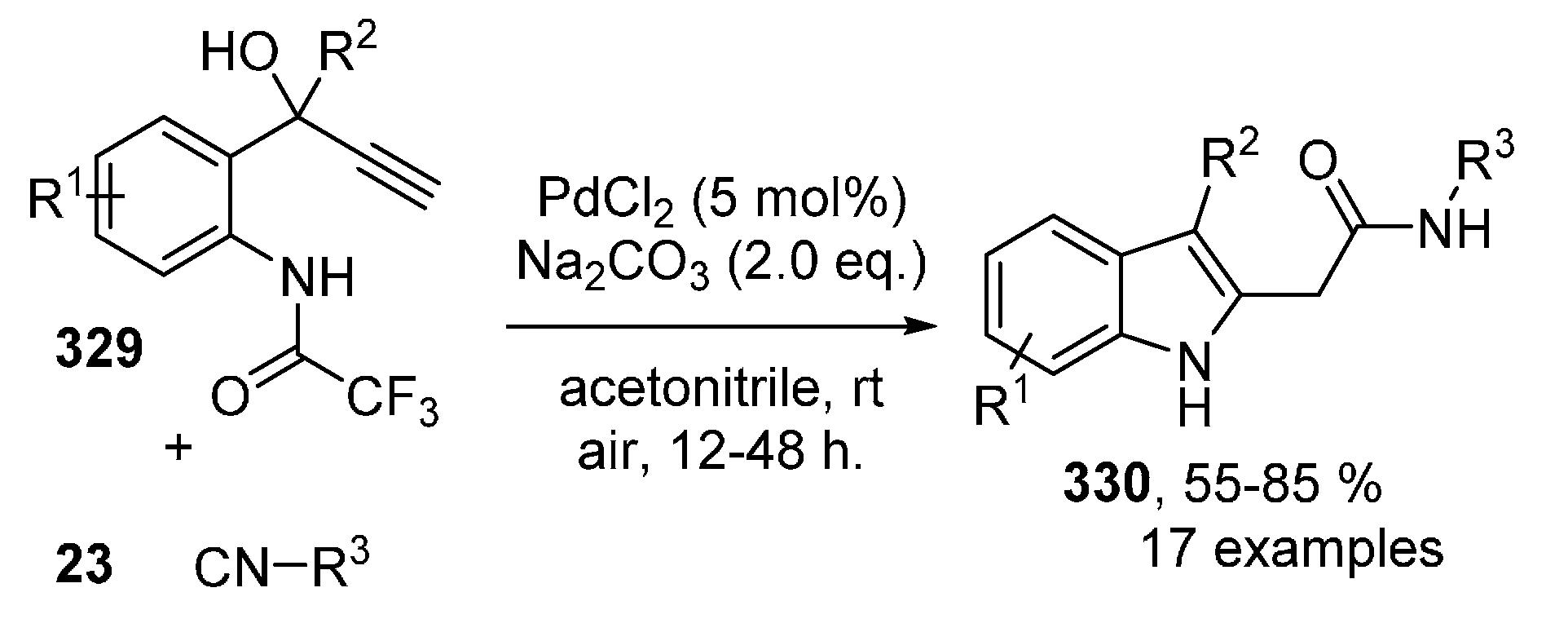
- Nanjo, T.; Yamamoto, S.; Tsukano, C.; Takemoto, Y. Synthesis of 3-Acyl-2-arylindole via Palladium-catalyzed Isocyanide Insertion and Oxypalladation of Alkyne. Org. Lett. 2013, 15, 3754–3757. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, S.; Huang, J.; Mao, T.; Zhu, Q. Palladium-Catalyzed Imidoylative Cyclization of α-Isocyanoacetamides: Efficient Access to C2-Diversified Oxazoles. Chem. Eur. J. 2014, 20, 11220–11224. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luo, S.; Li, J.; Zhu, Q. A room-temperature synthesis of 2,2′-bisoxazoles through palladium-catalyzed oxidative coupling of α-isocyanoacetamides. Org. Chem. Front. 2014, 1, 1285–1288. [Google Scholar] [CrossRef]
- Senadi, G.C.; Lu, T.-Y.; Dhandabani, G.K.; Wang, J.-J. Palladium-Catalyzed Double-Isocyanide Insertion via Oxidative N-O Cleavage of Acetyl Oximes: Syntheses of 2H-Pyrrol-2-imines. Org. Lett. 2017, 19, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Senadi, G.C.; Guo, B.-C.; Chang, Y.-C.; Hu, W.-P.; Wang, J.-J. Synthesis of Fused-Pyrazines via Palladium-Catalyzed Double Benzyl Isocyanide Insertion and Cross-Dehydrogenative Coupling. Adv. Synth. Catal. 2018, 360, 491–501. [Google Scholar] [CrossRef]
- Xiong, Z.; Cai, P.; Mei, Y.; Wang, J. Access to 1-amino-3,4-dihydroisoquinolines via palladium-catalyzed C-H bond aminoimidoylation reaction from functionalized isocyanides. RSC Adv. 2019, 9, 42072–42076. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Wang, J.; Wang, Y.; Luo, S.; Zhu, Q. Palladium-catalyzed C(sp2)-H aminoimidoylation of isocyano-containing arenes: Synthesis of amino substituted N-heterocycles. Org. Chem. Front. 2017, 4, 1768–1771. [Google Scholar] [CrossRef]
- Nanjo, T.; Tsukano, C.; Takemoto, Y. Synthesis of 3,3-Disubstituted 2-Aminoindolenines by Palladium-Catalyzed Allylic Amidination with Isocyanide. Synlett 2014, 25, 1473–1477. [Google Scholar] [CrossRef]
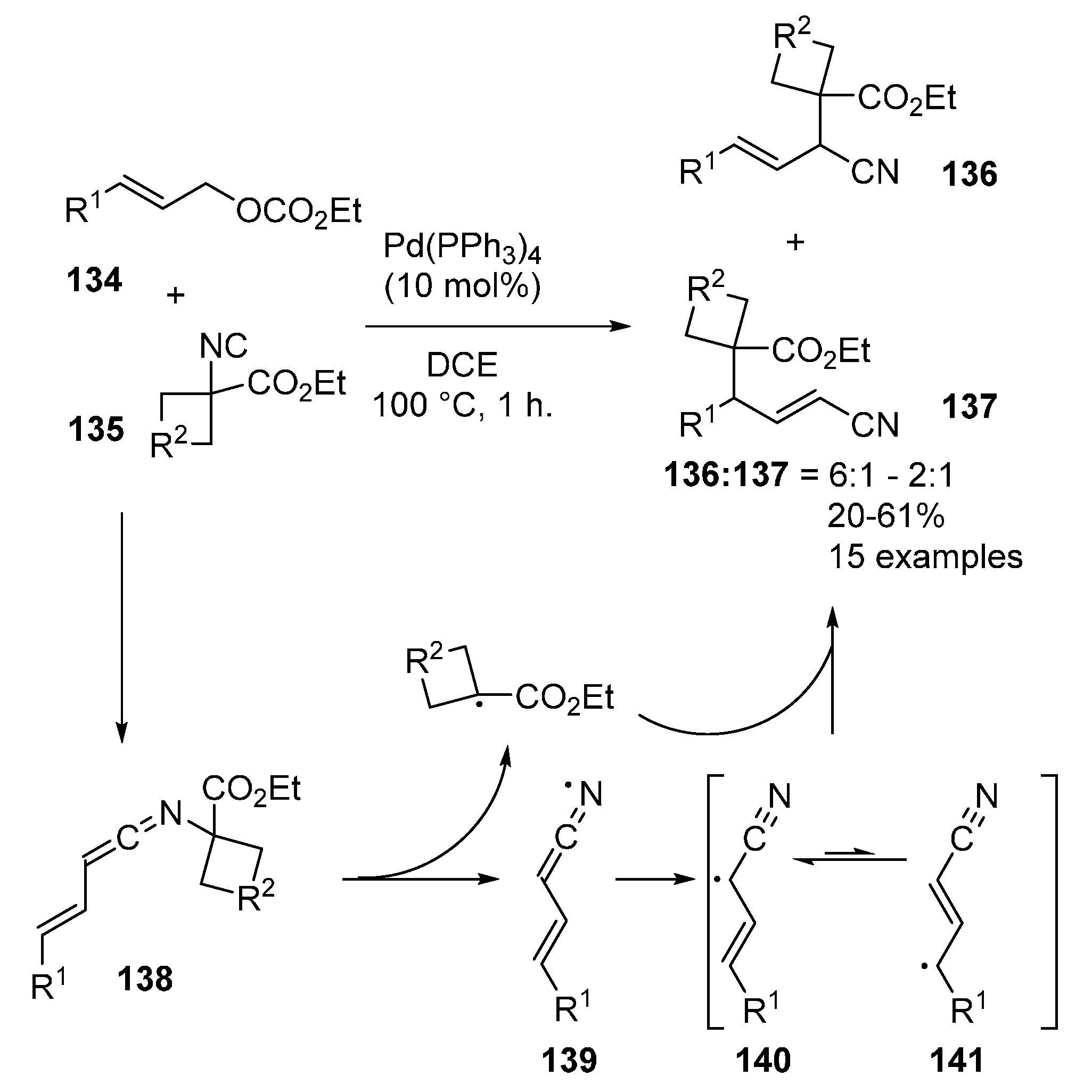
- Qiu, G.; Mamboury, M.; Wang, Q.; Zhu, J. Ketenimines from Isocyanides and Allyl Carbonates: Palladium-Catalyzed Synthesis of β,γ-Unsaturated Amides and Tetrazoles. Angew. Chem. Int. Ed. 2016, 55, 15377–15381. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Sornay, C.; Savary, D.; Zheng, S.-C.; Wang, Q.; Zhu, J. From isonitrile to nitrile via ketenimine intermediate: Palladium-catalyzed 1,1-carbocyanation of allyl carbonate by α-isocyanoacetate. Tetrahedron 2018, 74, 6966–6971. [Google Scholar] [CrossRef]
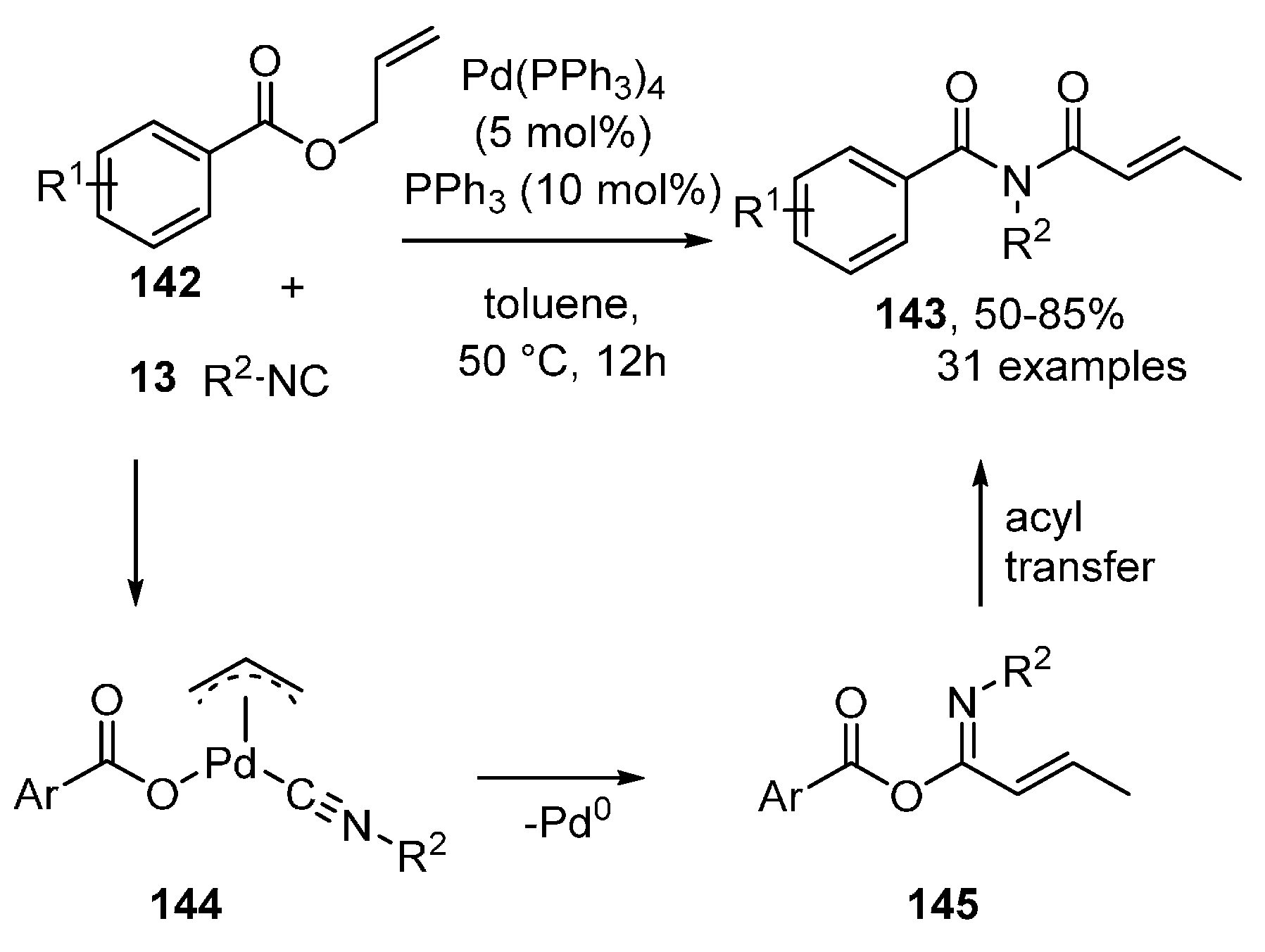
- Chen, S.; Wei, W.-X.; Wang, J.; Xia, Y.; Shen, Y.; Wu, X.-X.; Jing, H.; Liang, Y.-M. Palladium-Catalyzed Isocyanide Insertion with Allylic Esters: Synthesis of N-(But-2-enoyl)-N-(tert-butyl)benzamide Derivatives via Intramolecular Acyl Transfer Termination. Adv. Synth. Catal. 2017, 359, 3538–3544. [Google Scholar] [CrossRef]
- Canovese, L.; Chessa, G.; Visentin, F. A study on the insertion of isocyanides into palladium allyl bond: Effect of their nature on the strategic steps of the process. Inorg. Chim. Acta 2010, 363, 3426–3431. [Google Scholar] [CrossRef]
- Canovese, L.; Visentin, F.; Santo, C.; Levi, C. Migratory Insertion of Allyl Groups across the Pd-C Bond in Palladium(II) Isocyanide Complexes. The Fundamental Interplay between Temperature and Allyl Hapticity. Organometallics 2009, 28, 6762–6770. [Google Scholar] [CrossRef]
- Chuc, L.T.N.; Chen, C.-S.; Lo, W.-S.; Shen, P.-C.; Hsuan, Y.-C.; Tsai, H.-H.G.; Shieh, F.-K.; Hou, D.-R. Long-Range Olefin Isomerization Catalyzed by Palladium(0) Nanoparticles. ACS Omega 2017, 2, 698–711. [Google Scholar] [CrossRef]
- Qiu, G.; Wang, Q.; Zhu, J. Palladium-Catalyzed Three-Component Reaction of Propargyl Carbonates, Isocyanides, and Alcohols or Water: Switchable Synthesis of Pyrroles and Its Bicyclic Analogues. Org. Lett. 2017, 19, 270–273. [Google Scholar] [CrossRef]
- Cheng, H.-G.; Chen, H.; Liu, Y.; Zhou, Q. The Liebeskind-Srogl Cross-Coupling Reaction and its Synthetic Applications. Asian J. Org. Chem. 2018, 7, 490–508. [Google Scholar] [CrossRef] [Green Version]
- Prokopcová, H.; Kappe, C.O. The Liebeskind-Srogl C—C Cross-Coupling Reaction. Angew. Chem. Int. Ed. 2009, 48, 2276–2286. [Google Scholar] [CrossRef]
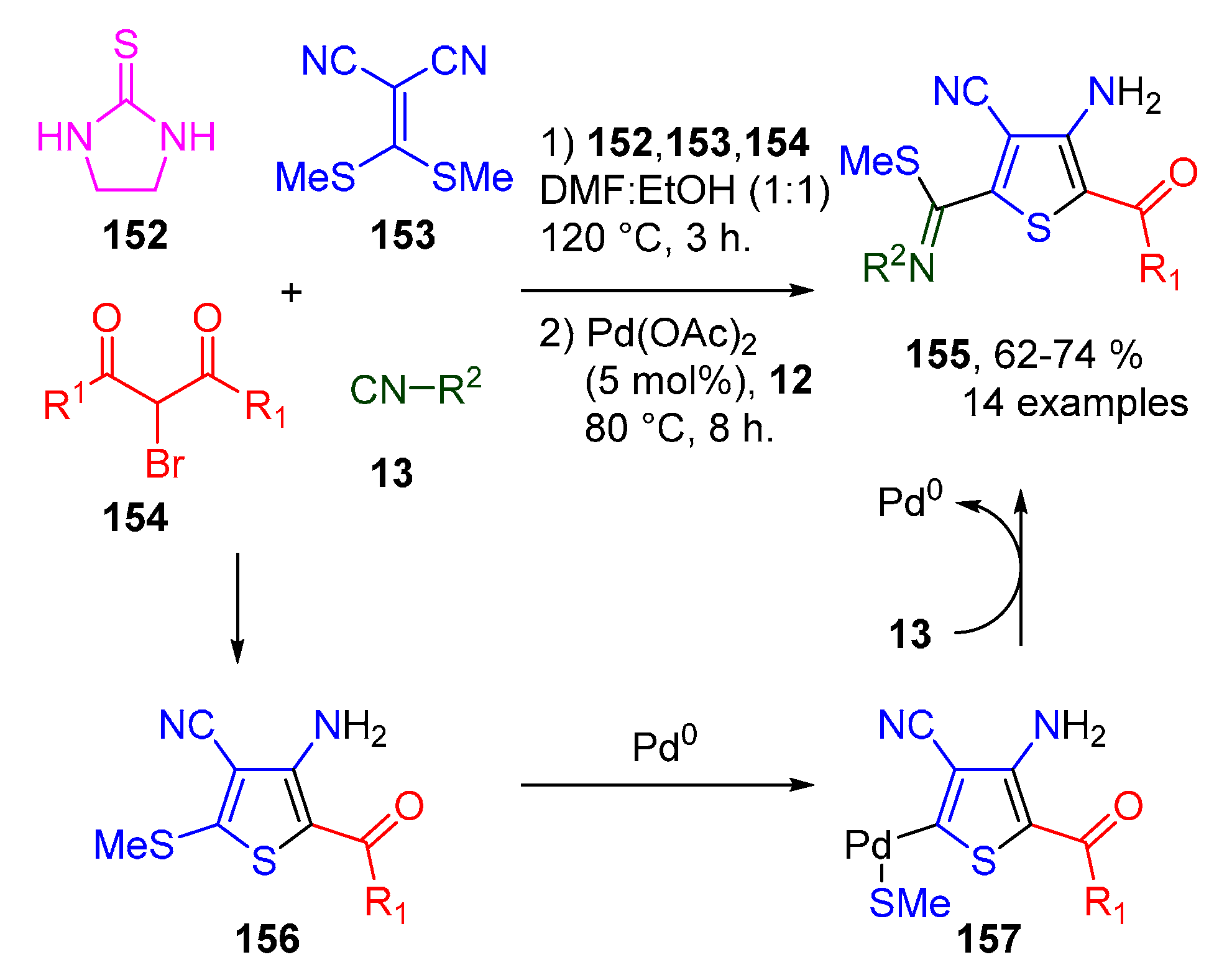
- Vahabi, A.H.; Alizadeh, A.; Khavasi, H.R.; Bazgir, A. Palladium-Catalyzed Migratory Insertion of Isocyanides into C(thiophene)-SMe Bonds: Access to Atom-Transfer Reactions. Eur. J. Org. Chem. 2017, 2017, 5347–5356. [Google Scholar] [CrossRef]
- Alizadeh, A.; Vahabi, A.H.; Bazgir, A.; Khavasi, H.R.; Zhu, Z.; Ng, S.W. Highly mild approach towards synthesis of tetrasubstituted thiophenes by an organic salt afforded by cyclic thioureas and ketene dithioacetals. RSC Adv. 2015, 5, 85028–85034. [Google Scholar] [CrossRef]
- Otsuka, S.; Nogi, K.; Yorimitsu, H. Palladium-Catalyzed Insertion of Isocyanides into the C-S Bonds of Heteroaryl Sulfides. Angew. Chem. Int. Ed. 2018, 57, 6653–6657. [Google Scholar] [CrossRef] [PubMed]
- Shiro, D.; Fujiwara, S.-i.; Tsuda, S.; Iwasaki, T.; Kuniyasu, H.; Kambe, N. Palladium-catalyzed Insertion Reactions of Isocyanides into Thiocarbamates and Selenocarbamates. Chem. Lett. 2015, 44, 465–467. [Google Scholar] [CrossRef]
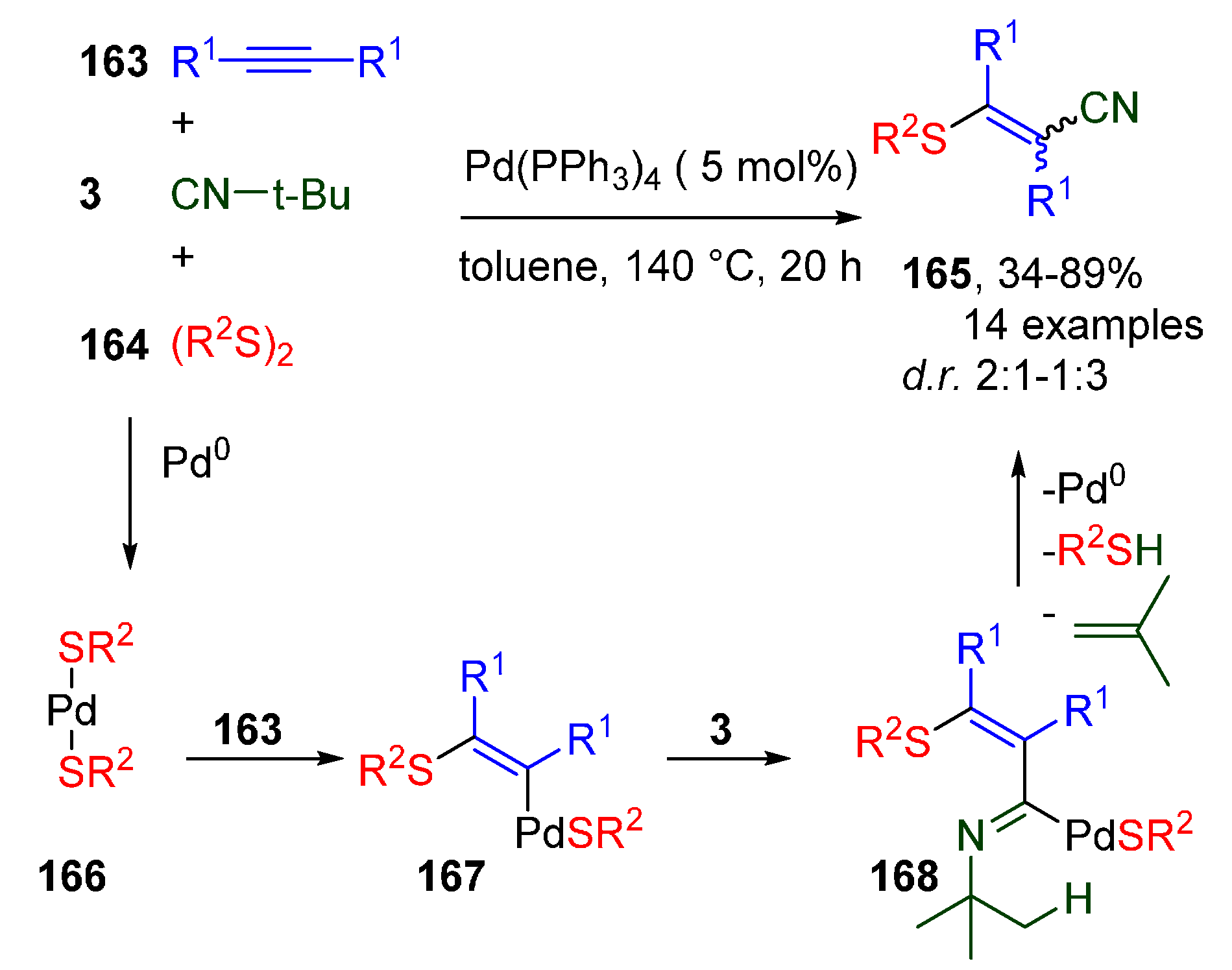
- Higashimae, S.; Kurata, D.; Kawaguchi, S.-I.; Kodama, S.; Sonoda, M.; Nomoto, A.; Ogawa, A. Palladium-Catalyzed Cyanothiolation of Internal Alkynes Using Organic Disulfides and tert-Butyl Isocyanide. J. Org. Chem. 2018, 83, 5267–5273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Ding, K.; Cai, Q. Palladium-Catalyzed Amidation of N-Tosylhydrazones with Isocyanides. Chem. Eur. J. 2011, 17, 12268–12271. [Google Scholar] [CrossRef]
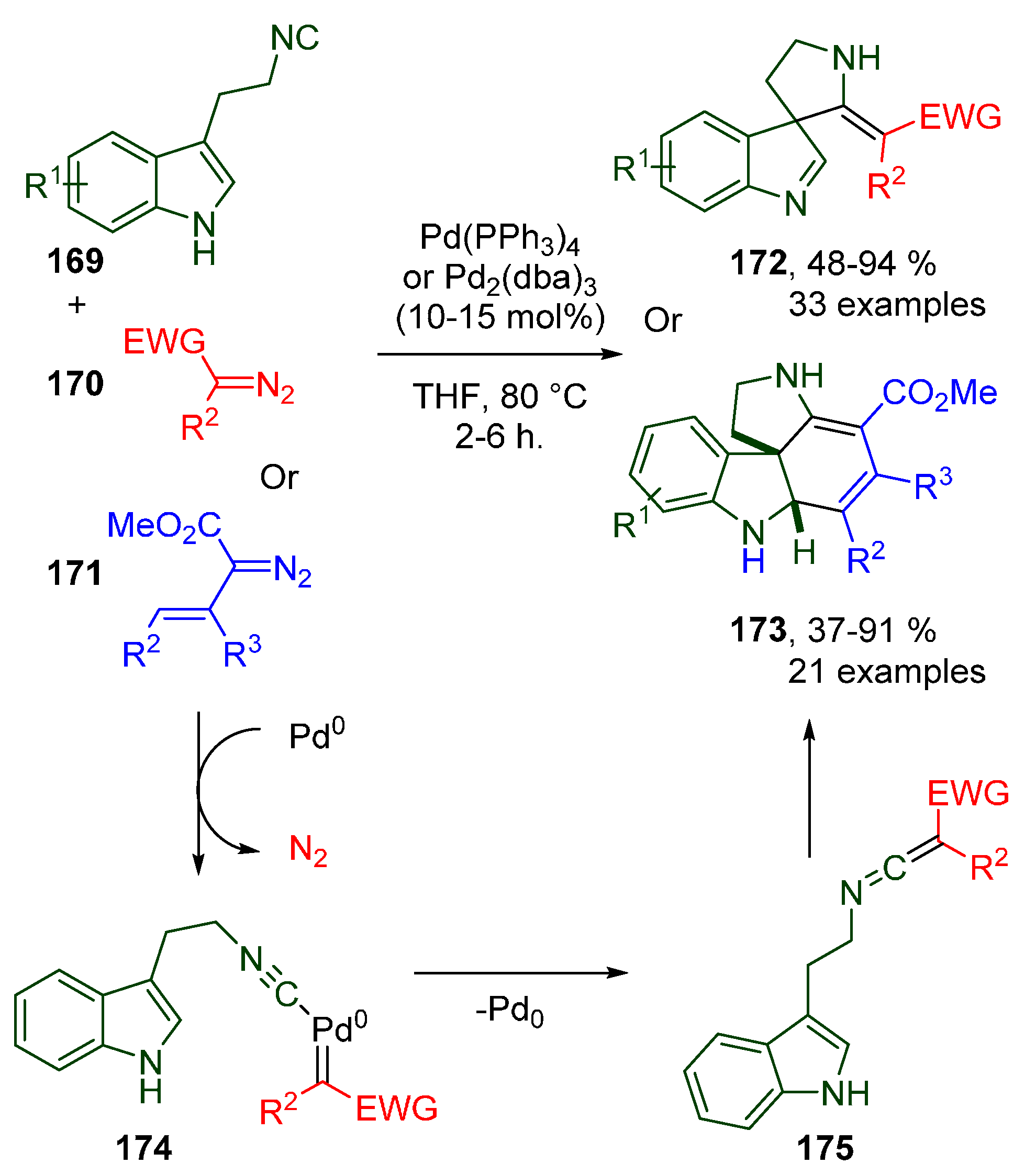
- Chen, G.-S.; Chen, S.-J.; Luo, J.; Mao, X.-Y.; Chan, A.S.-C.; Sun, R.W.-Y.; Liu, Y.-L. Tandem Cross-Coupling/Spirocyclization/Mannich-Type Reactions of 3-(2-Isocyanoethyl)indoles with Diazo Compounds toward Polycyclic Spiroindolines. Angew. Chem. Int. Ed. 2020, 59, 614–621. [Google Scholar] [CrossRef]
- Huang, J.; Li, F.; Cui, L.; Su, S.; Jia, X.; Li, J. Palladium-catalyzed cascade reactions of enynones and isocyanides: Access towards functionalized ketenimine and its application. Chem. Commun. 2020, 56, 4555–4558. [Google Scholar] [CrossRef]
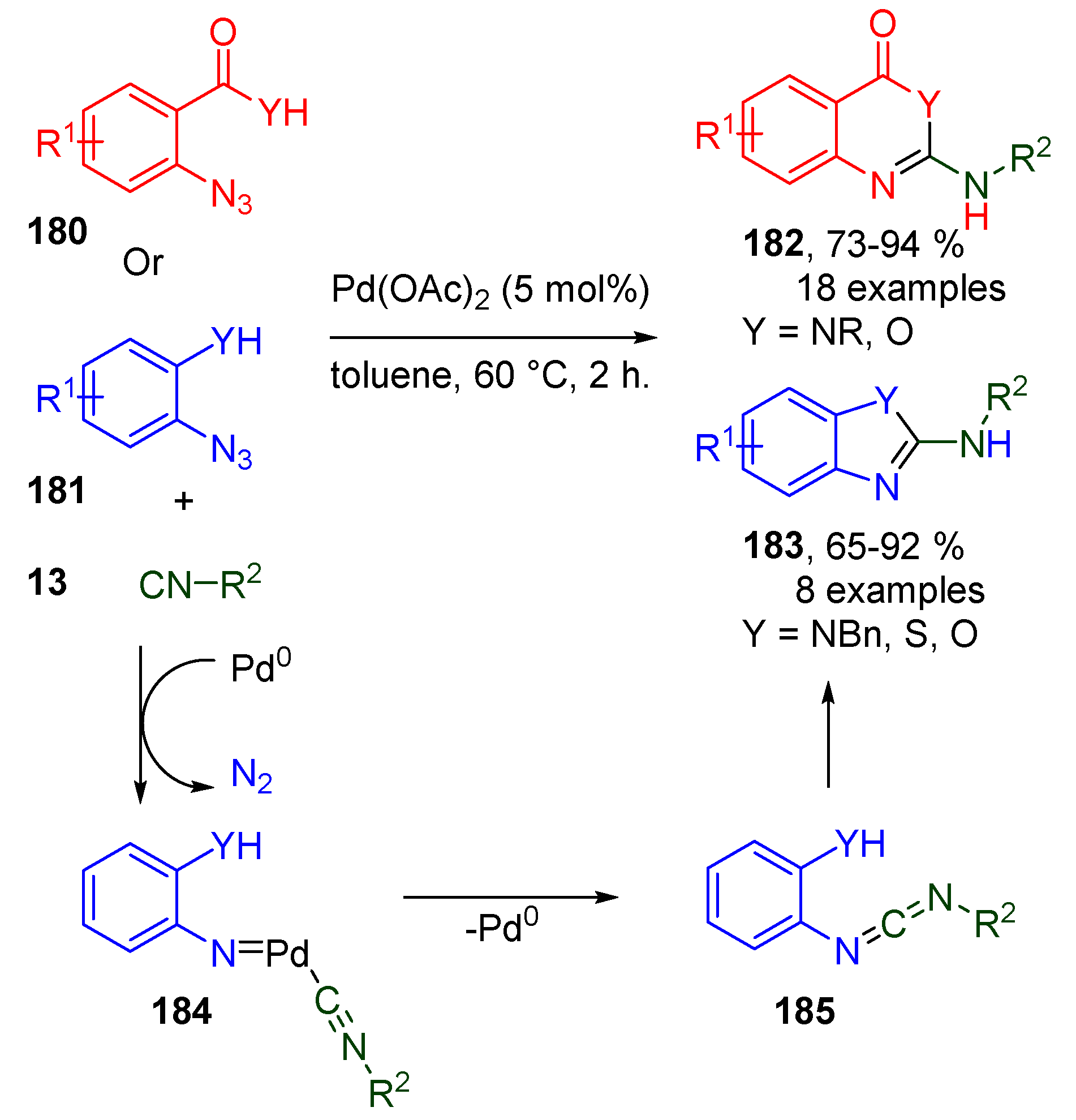
- Ansari, A.J.; Pathare, R.S.; Maurya, A.K.; Agnihotri, V.K.; Khan, S.; Roy, T.K.; Sawant, D.M.; Pardasani, R.T. Synthesis of Diverse Nitrogen Heterocycles via Palladium-Catalyzed Tandem Azide-Isocyanide Cross-Coupling/Cyclization: Mechanistic Insight using Experimental and Theoretical Studies. Adv. Synth. Catal. 2018, 360, 290–297. [Google Scholar] [CrossRef]
- Pathare, R.S.; Maurya, A.K.; Kumari, A.; Agnihotri, V.K.; Verma, V.P.; Sawant, D.M. Synthesis of quinazoline-3-oxides via a Pd(ii) catalyzed azide-isocyanide coupling/cyclocondensation reaction. Org. Biomol. Chem. 2019, 17, 363–368. [Google Scholar] [CrossRef]
- Ren, Z.-L.; Kong, H.-H.; Lu, W.-T.; Sun, M.; Ding, M.-W. One-pot synthesis of quinazolin-4(3H)-ones and fused quinazolinones by a palladium-catalyzed domino process. Tetrahedron 2018, 74, 184–193. [Google Scholar] [CrossRef]
- Pathare, R.S.; Ansari, A.J.; Verma, S.; Maurya, A.; Maurya, A.K.; Agnihotri, V.K.; Sharon, A.; Pardasani, R.T.; Sawant, D.M. Sequential Pd(0)/Fe(III) Catalyzed Azide-Isocyanide Coupling/Cyclization Reaction: One-Pot Synthesis of Aminotetrazoles. J. Org. Chem. 2018, 83, 9530–9537. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.J.; Joshi, G.; Yadav, U.P.; Maurya, A.K.; Agnihotri, V.K.; Kalra, S.; Kumar, R.; Singh, S.; Sawant, D.M. Exploration of Pd-catalysed four-component tandem reaction for one-pot assembly of pyrazolo[1,5-c]quinazolines as potential EGFR inhibitors. Bioorg. Chem. 2019, 93, 103314. [Google Scholar] [CrossRef]
- Sawant, D.M.; Sharma, S.; Pathare, R.S.; Joshi, G.; Kalra, S.; Sukanya, S.; Maurya, A.K.; Metre, R.K.; Agnihotri, V.K.; Khan, S.; et al. Relay tricyclic Pd(ii)/Ag(i) catalysis: Design of a four-component reaction driven by nitrene-transfer on isocyanide yields inhibitors of EGFR. Chem. Commun. 2018, 54, 11530–11533. [Google Scholar] [CrossRef] [PubMed]
- Mampuys, P.; Neumann, H.; Sergeyev, S.; Orru, R.V.A.; Jiao, H.; Spannenberg, A.; Maes, B.U.W.; Beller, M. Combining Isocyanides with Carbon Dioxide in Palladium-Catalyzed Heterocycle Synthesis: N3-Substituted Quinazoline-2,4(1H,3H)-diones via a Three-Component Reaction. ACS Catal. 2017, 7, 5549–5556. [Google Scholar] [CrossRef]
- Xu, P.; Wang, F.; Wei, T.-Q.; Yin, L.; Wang, S.-Y.; Ji, S.-J. Palladium-Catalyzed Incorporation of Two C1 Building Blocks: The Reaction of Atmospheric CO2 and Isocyanides with 2-Iodoanilines Leading to the Synthesis of Quinazoline-2,4(1H,3H)-diones. Org. Lett. 2017, 19, 4484–4487. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Khatun, R.; Dolai, M.; Haque Biswas, I.; Haque, N.; Sengupta, M.; Islam, M.S.; Islam, S.M. Catalytic formation of N3-substituted quinazoline-2,4(1H,3H)-diones by Pd(ii)EN@GO composite and its mechanistic investigations through DFT calculations. New J. Chem. 2020, 44, 141–151. [Google Scholar] [CrossRef]
- Chen, Z.-B.; Zhang, Y.; Yuan, Q.; Zhang, F.-L.; Zhu, Y.-M.; Shen, J.-K. Palladium-Catalyzed Synthesis of α-Iminonitriles from Aryl Halides via Isocyanide Double Insertion Reaction. J. Org. Chem. 2016, 81, 1610–1616. [Google Scholar] [CrossRef]
- Ali, W.; Dahiya, A.; Patel, B.K. Cascade Synthesis of Dihydrobenzofurans and Aurones via Palladium-Catalyzed Isocyanides Insertion into 2-Halophenoxy Acrylates. Adv. Synth. Catal. 2018, 360, 1232–1239. [Google Scholar] [CrossRef]
- Kong, W.; Wang, Q.; Zhu, J. Synthesis of Diversely Functionalized Oxindoles Enabled by Migratory Insertion of Isocyanide to a Transient σ-Alkylpalladium(II) Complex. Angew. Chem. Int. Ed. 2016, 55, 9714–9718. [Google Scholar] [CrossRef]
- Chen, X.; Qiu, G.; Liu, R.; Chen, D.; Chen, Z. Divergent oriented synthesis (DOS) of aza-heterocyclic amides through palladium-catalyzed ketenimination of 2-iodo-N-(propa-1,2-dien-1-yl)anilines. Org. Chem. Front. 2020, 7, 890–895. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Xiong, Z.; Zhong, L.; Ding, S.; Li, L.; Zhao, H.; Chen, C.; Shang, Y. Palladium-catalysed dearomative aryl/cycloimidoylation of indoles. Chem. Commun. 2020, 56, 3249–3252. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Tang, S.; Zhu, Q. Intramolecular Imidoylative Heck Reaction: Synthesis of Cyclic Ketoimines from Functionalized Isocyanide. Org. Lett. 2016, 18, 3074–3077. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, J.-M.; Zhang, Y.; Chen, Z.; Zhu, Y.-M.; Ji, S.-J. Palladium-Catalyzed Formylation of Aryl Halides with tert-Butyl Isocyanide. Org. Lett. 2014, 16, 3492–3495. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, X.; Wang, J.-M.; Chen, J.-L.; Zhu, Y.-M. Palladium-catalyzed synthesis of aldehydes from aryl halides and tert-butyl isocyanide using formate salts as hydride donors. RSC Advances 2015, 5, 17060–17063. [Google Scholar] [CrossRef]
- Hu, W.; Li, M.; Jiang, G.; Wu, W.; Jiang, H. Synthesis of 2,3-Difunctionalized Benzofuran Derivatives through Palladium-Catalyzed Double Isocyanide Insertion Reaction. Org. Lett. 2018, 20, 3500–3503. [Google Scholar] [CrossRef]
- Chang, J.; Liu, B.; Yang, Y.; Wang, M. Pd-Catalyzed C-S Activation/Isocyanide Insertion/Hydrogenation Enables a Selective Aerobic Oxidation/Cyclization. Org. Lett. 2016, 18, 3984–3987. [Google Scholar] [CrossRef]
- Sterckx, H.; Morel, B.; Maes, B.U.W. Catalytic Aerobic Oxidation of C(sp3)-H Bonds. Angew. Chem. Int. Ed. 2019, 58, 7946–7970. [Google Scholar] [CrossRef]
- Teong, S.P.; Li, X.; Zhang, Y. Hydrogen peroxide as an oxidant in biomass-to-chemical processes of industrial interest. Green Chem. 2019, 21, 5753–5780. [Google Scholar] [CrossRef]
- Khalaj, M.; Taherkhani, M.; Mousavi-Safavi, S.M.; Akbari, J. Synthesis of Benzamide Derivatives by the Reaction of Arenes and Isocyanides through a C–H Bond Activation Strategy. Synlett 2018, 29, 94–98. [Google Scholar] [CrossRef]
- Blondiaux, E.; Bomon, J.; Smoleń, M.; Kaval, N.; Lemière, F.; Sergeyev, S.; Diels, L.; Sels, B.; Maes, B.U.W. Bio-based Aromatic Amines from Lignin-Derived Monomers. ACS Sustain. Chem. Eng. 2019, 7, 6906–6916. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Xu, H.; Kong, W.-J.; Shang, M.; Dai, H.-X.; Yu, J.-Q. Overcoming the limitations of directed C-H functionalizations of heterocycles. Nature 2014, 515, 389–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tereniak, S.J.; Stahl, S.S. Mechanistic Basis for Efficient, Site-Selective, Aerobic Catalytic Turnover in Pd-Catalyzed C-H Imidoylation of Heterocycle-Containing Molecules. J. Am. Chem. Soc. 2017, 139, 14533–14541. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-J.; Wang, R.; Gao, J.; Wang, Y.-Y.; Luo, D.-H.; Wu, Y.-C. Bismuth(III) Bromide-Catalysed Substitution of Benzyl Alcohols with Arylsulfonylmethyl Isocyanides: An Unexpected Access to Sulfinates. Adv. Synth. Catal. 2015, 357, 1393–1397. [Google Scholar] [CrossRef]
- He, Y.; Wang, Y.-C.; Hu, K.; Xu, X.-L.; Wang, H.-S.; Pan, Y.-M. Palladium-Catalyzed Synthesis of 5-Iminopyrrolones through Isocyanide Double Insertion Reaction with Terminal Alkynes and Water. J. Org. Chem. 2016, 81, 11813–11818. [Google Scholar] [CrossRef]
- Hu, W.; Zheng, J.; Li, J.; Liu, B.; Wu, W.; Liu, H.; Jiang, H. Assembly of Polysubstituted Maleimides via Palladium-Catalyzed Cyclization Reaction of Alkynes with Isocyanides. J. Org. Chem. 2016, 81, 12451–12458. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, W.; Huang, Y.; Zhu, J.; Hu, Q.; Wu, W.; Jiang, H. Palladium-Catalyzed Synthesis of 1H-Indenes and Phthalimides via Isocyanide Insertion. Org. Lett. 2017, 19, 5818–5821. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Y.; Liang, X.; Huang, B.; Wang, H.; Pan, Y.-M. Palladium-Catalyzed Three-Component Reaction: A Novel Method for the Synthesis of N-Acyl Propiolamides. Org. Lett. 2018, 20, 7117–7120. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zou, M.; Wang, J.; Tan, Q.; Liu, B.; Xu, B. Palladium-Catalyzed Multicomponent Reaction of Alkynes, Carboxylic Acids, and Isocyanides: A Direct Approach to Captodative Olefins. Org. Lett. 2019, 21, 1593–1597. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, J.; Liang, D.; Zhu, Q. Synthesis of 1H-Indole-3-carboxamidines through a Palladium-Catalyzed Three-Component Reaction Involving Isocyanide Insertion as a Key Step. Adv. Synth. Catal. 2013, 355, 3290–3294. [Google Scholar] [CrossRef]
- Li, P.-X.; Meng, X.-J.; Yang, L.; Tang, H.-T.; Pan, Y.-M. Synthesis of Multisubstituted Guanidines through Palladium-Catalyzed Insertion of Isonitriles. Synlett 2018, 29, 2326–2330. [Google Scholar]
- Vlaar, T.; Cioc, R.C.; Mampuys, P.; Maes, B.U.W.; Orru, R.V.A.; Ruijter, E. Sustainable Synthesis of Diverse Privileged Heterocycles by Palladium-Catalyzed Aerobic Oxidative Isocyanide Insertion. Angew. Chem. Int. Ed. 2012, 51, 13058–13061. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Qiu, X.; Wu, J. Generation of 4-Cyanoisoquinolines and 4-Amidylisoquinolines via a Palladium-Catalyzed Cyanative Reaction of 2-Alkynylbenzaldimines with Isocyanides. Adv. Synth. Catal. 2013, 355, 3205–3209. [Google Scholar] [CrossRef]
- Qiu, G.; Qiu, X.; Liu, J.; Wu, J. Switchable Synthesis of 3-Cyanoindoles and 3-Amidylindoles via a Palladium-Catalyzed Reaction of N,N-Dimethyl-2-alkynylaniline with Isocyanide. Adv. Synth. Catal. 2013, 355, 2441–2446. [Google Scholar] [CrossRef]
- Lu, F.; Chen, Z.; Li, Z.; Wang, X.; Peng, X.; Li, C.; Fang, L.; Liu, D.; Gao, M.; Lei, A. Palladium/Copper-Catalyzed Oxidative Coupling of Arylboronic Acids with Isocyanides: Selective Routes to Amides and Diaryl Ketones. Org. Lett. 2017, 19, 3954–3957. [Google Scholar] [CrossRef]
- Chen, Z.-B.; Liu, K.; Zhang, F.-L.; Yuan, Q.; Zhu, Y.-M. Palladium-catalyzed oxidative coupling of arylboronic acid with isocyanide to form aromatic carboxylic acids. Org. Biomol. Chem. 2017, 15, 8078–8083. [Google Scholar] [CrossRef]
- Tao, S.-W.; Zhou, J.-Y.; Liu, R.-Q.; Zhu, Y.-M. One-Pot Synthesis of N-Imidoyl-(1H)-indoles via Palladium-Catalyzed Oxidative Insertion Domino Reaction with Isocyanide and Arylboronic Acid. J. Org. Chem. 2019, 84, 8121–8130. [Google Scholar] [CrossRef]
- Vlaar, T.; Orru, R.V.A.; Maes, B.U.W.; Ruijter, E. Palladium-Catalyzed Synthesis of 2-Aminobenzoxazinones by Aerobic Oxidative Coupling of Anthranilic Acids and Isocyanides. J. Org. Chem. 2013, 78, 10469–10475. [Google Scholar] [CrossRef]
- Ji, F.; Lv, M.-F.; Yi, W.-B.; Cai, C. One-pot synthesis of 2-amino-4(3H)-quinazolinones via ring-opening of isatoic anhydride and palladium-catalyzed oxidative isocyanide-insertion. Org. Biomol. Chem. 2014, 12, 5766–5772. [Google Scholar] [CrossRef]
- Fang, T.; Tan, Q.; Ding, Z.; Liu, B.; Xu, B. Pd-Catalyzed Oxidative Annulation of Hydrazides with Isocyanides: Synthesis of 2-Amino-1,3,4-oxadiazoles. Org. Lett. 2014, 16, 2342–2345. [Google Scholar] [CrossRef]
- Nagao, Y.; Hirata, T.; Goto, S.; Sano, S.; Kakehi, A.; Iizuka, K.; Shiro, M. Intramolecular Nonbonded S···O Interaction Recognized in (Acylimino)thiadiazoline Derivatives as Angiotensin II Receptor Antagonists and Related Compounds. J. Am. Chem. Soc. 1998, 120, 3104–3110. [Google Scholar] [CrossRef]
- Zheng, Q.; Ding, Q.; Wang, C.; Chen, W.; Peng, Y. Synthesis of 2-aminoquinolines via palladium-catalyzed intermolecular oxidative cyclization of 2-vinylanilines with isocyanides. Tetrahedron 2016, 72, 952–958. [Google Scholar] [CrossRef]
- Vidyacharan, S.; Murugan, A.; Sharada, D.S. C(sp2)-H Functionalization of 2H-Indazoles at C3-Position via Palladium(II)-Catalyzed Isocyanide Insertion Strategy Leading to Diverse Heterocycles. J. Org. Chem. 2016, 81, 2837–2848. [Google Scholar] [CrossRef]
- Vlaar, T.; Maes, B.U.W.; Ruijter, E.; Orru, R.V.A. Synthesis of 4-aminoquinolines by aerobic oxidative palladium-catalyzed double C-H activation and isocyanide insertion. Chem. Heterocycl. Compd. 2013, 49, 902–908. [Google Scholar] [CrossRef]
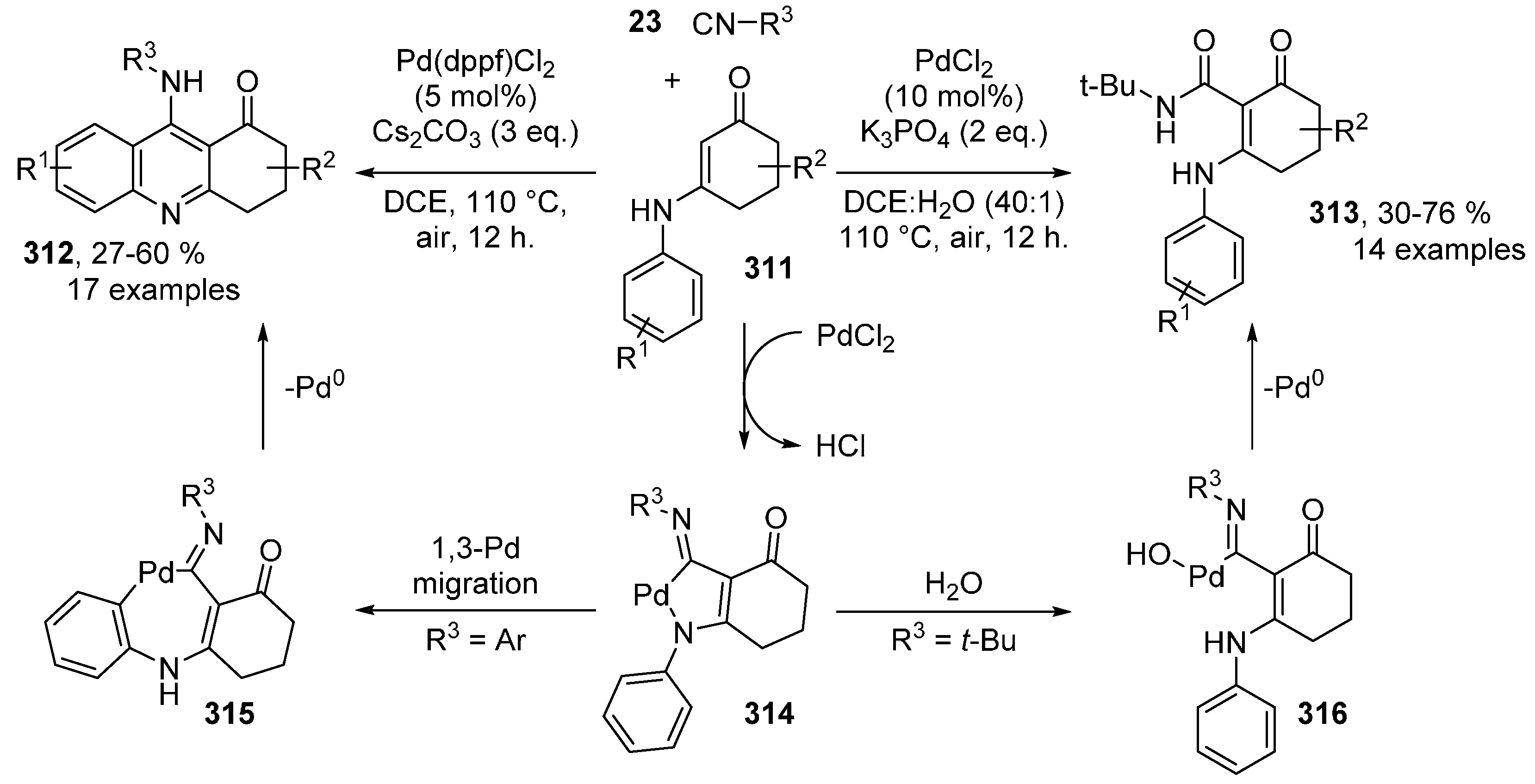
- Gu, Z.-Y.; Wang, X.; Cao, J.-J.; Wang, S.-Y.; Ji, S.-J. Chemoselective Pd-Catalyzed Isocyanide Insertion Reaction of Enaminones by C-H Functionalization: Hydrolysis or Cyclization through 1,3-Palladium Migration. Eur. J. Org. Chem. 2015, 2015, 4699–4709. [Google Scholar] [CrossRef]
- Xiong, Z.; Liang, D.; Luo, S. Palladium-catalyzed β-selective C(sp2)-H carboxamidation of enamides by isocyanide insertion: Synthesis of N-acyl enamine amides. Org. Chem. Front. 2017, 4, 1103–1106. [Google Scholar] [CrossRef]
- Chen, D.; Shan, Y.; Li, J.; You, J.; Sun, X.; Qiu, G. External Reductant-Free Palladium-Catalyzed Reductive Insertion of Isocyanide: Synthesis of Polysubstituted Pyrroles and Its Applications as a Cysteine Probe. Org. Lett. 2019, 21, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
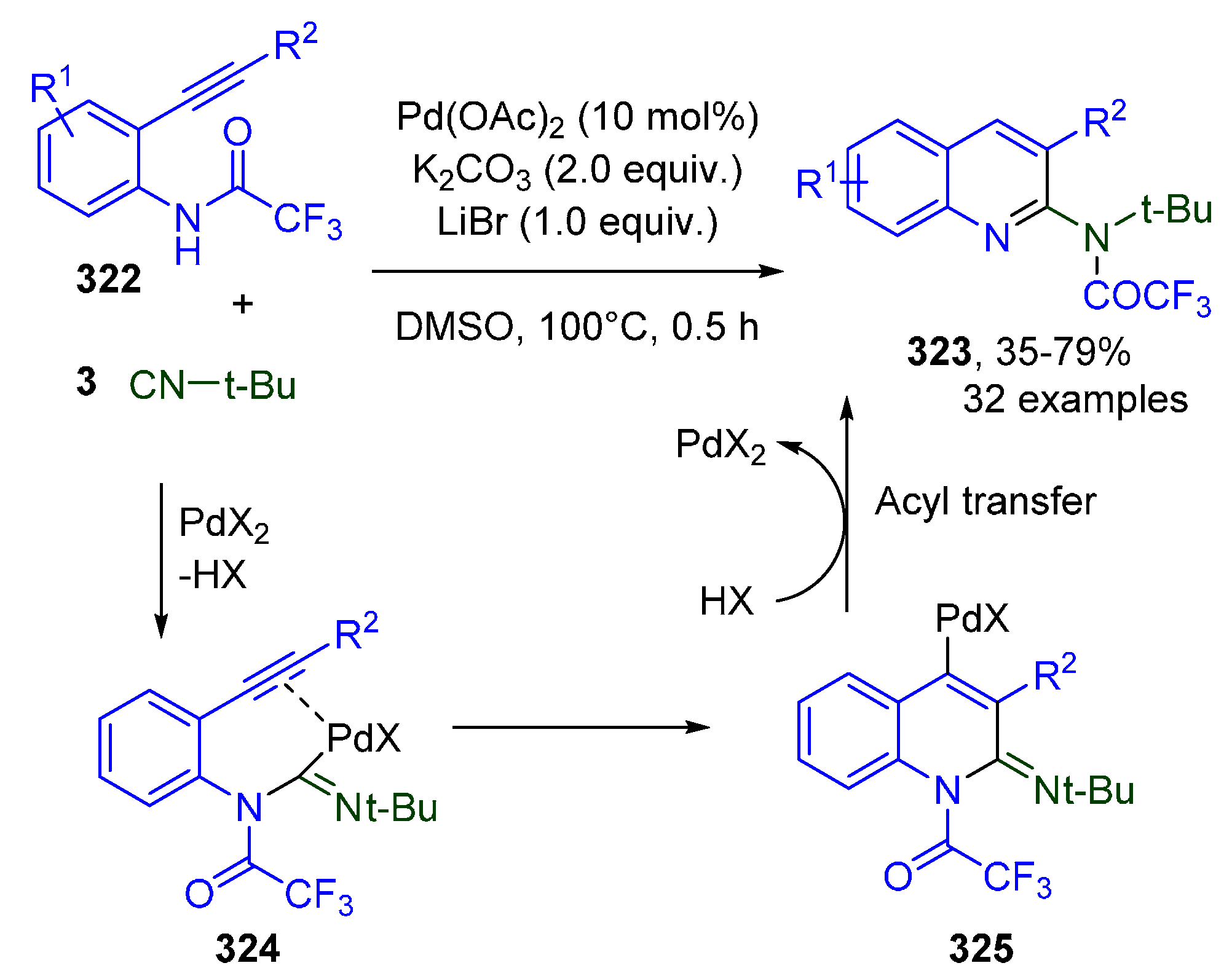
- Li, M.; Zheng, J.; Hu, W.; Li, C.; Li, J.; Fang, S.; Jiang, H.; Wu, W. Palladium-Catalyzed Cyclization of N-Acyl-o-alkynylanilines with Isocyanides Involving a 1,3-Acyl Migration: Rapid Access to Functionalized 2-Aminoquinolines. Org. Lett. 2018, 20, 7245–7248. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, M.; Zheng, J.; Hu, W.; Li, C.; Jiang, H. Tandem cyclization of o-alkynylanilines with isocyanides triggered by intramolecular nucleopalladation: Access to heterocyclic fused 2-aminoquinolines. Chem. Commun. 2018, 54, 6855–6858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirupathi, N.; Hari Babu, M.; Dwivedi, V.; Kant, R.; Sridhar Reddy, M. Palladium-Catalyzed Tandem Intramolecular Oxy/Amino-Palladation/Isocyanide Insertion: Synthesis of α-Benzofuranyl/Indolylacetamides. Org. Lett. 2014, 16, 2908–2911. [Google Scholar] [CrossRef]

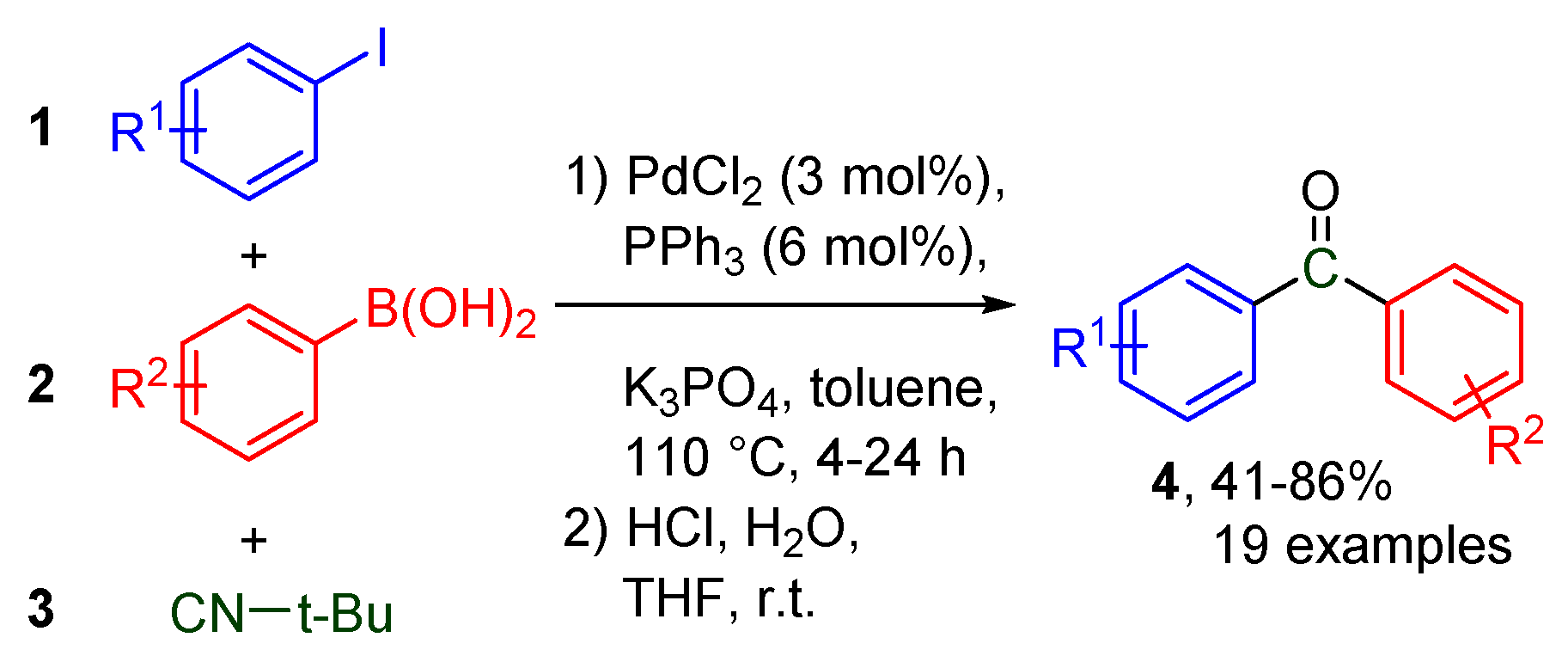




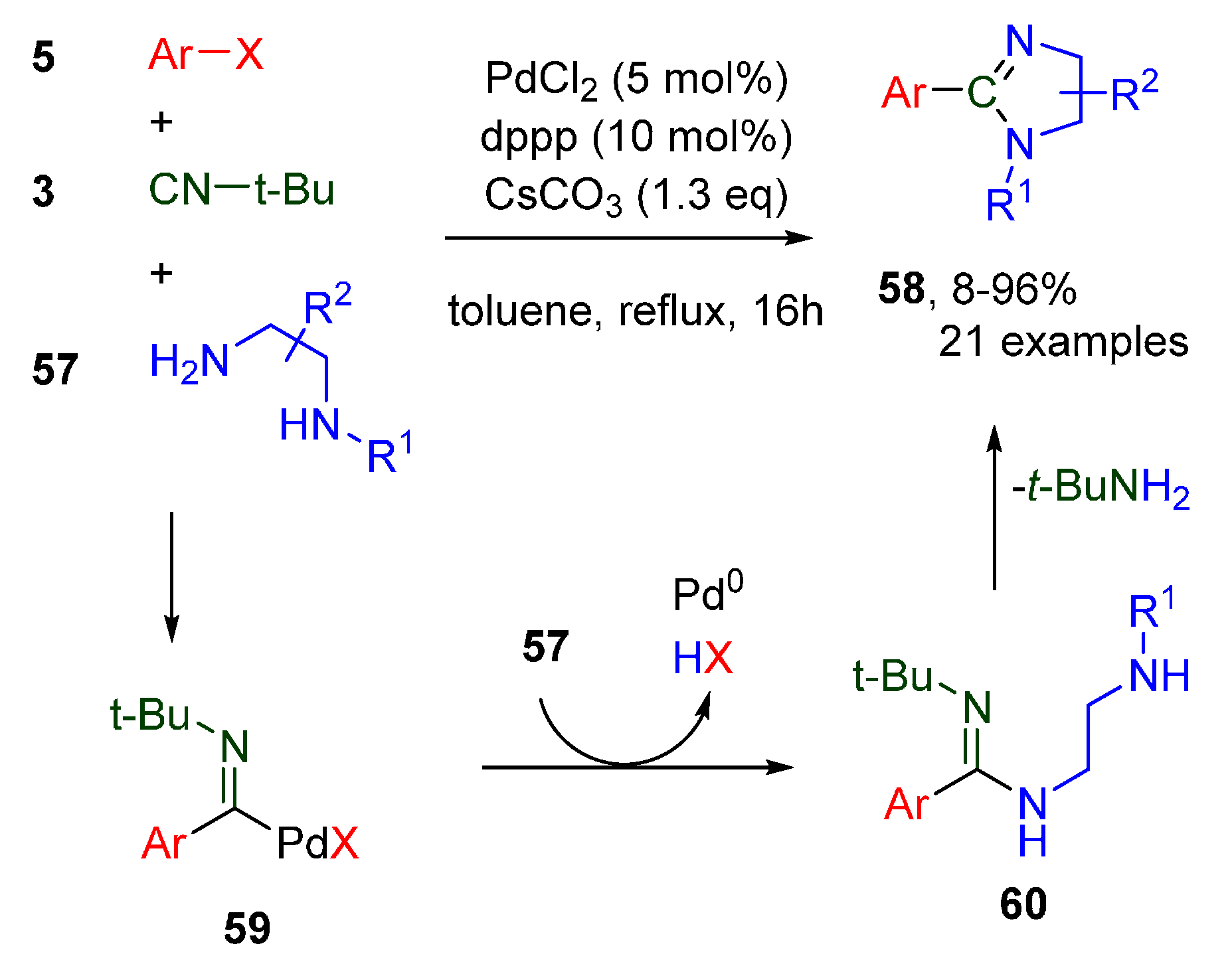






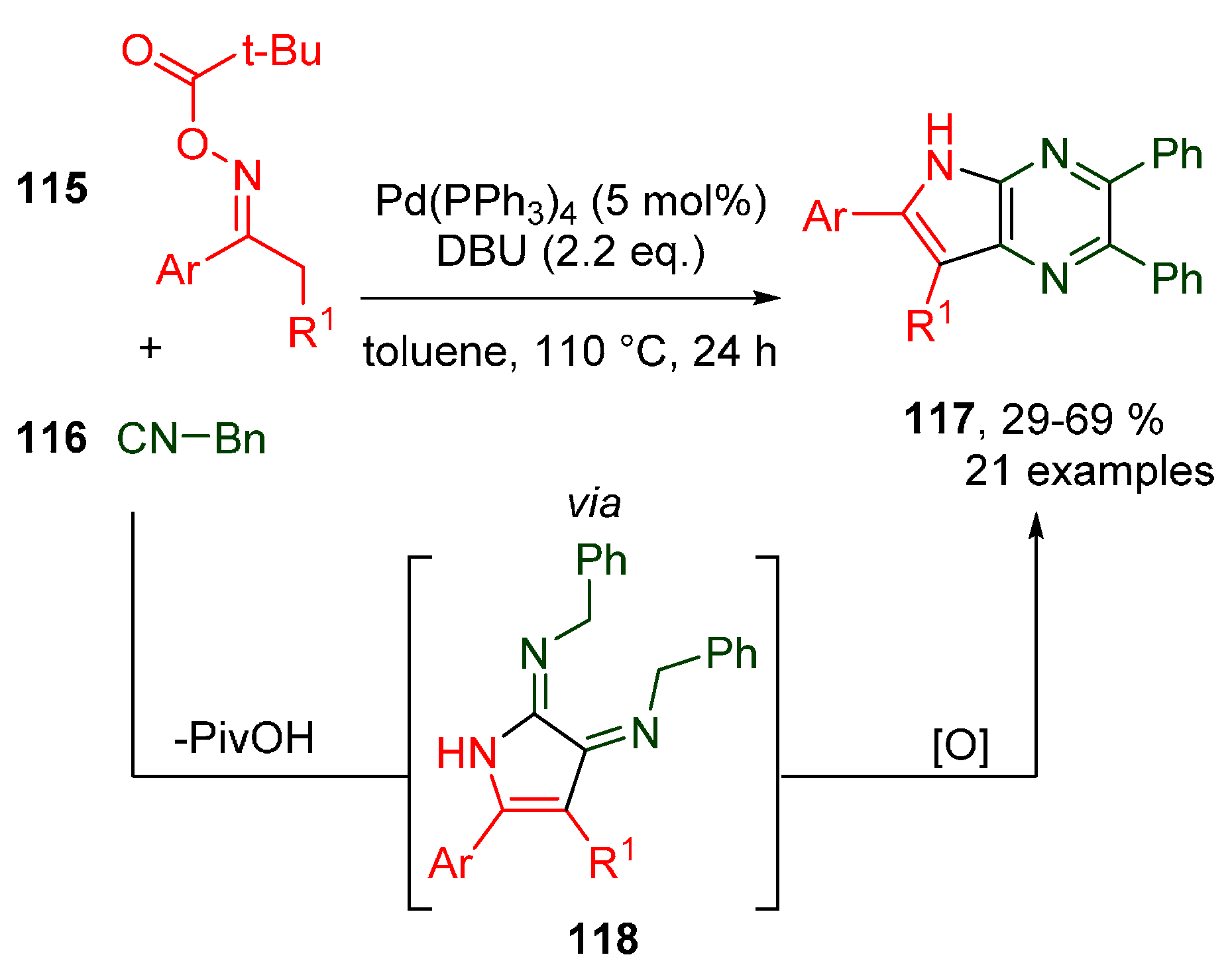

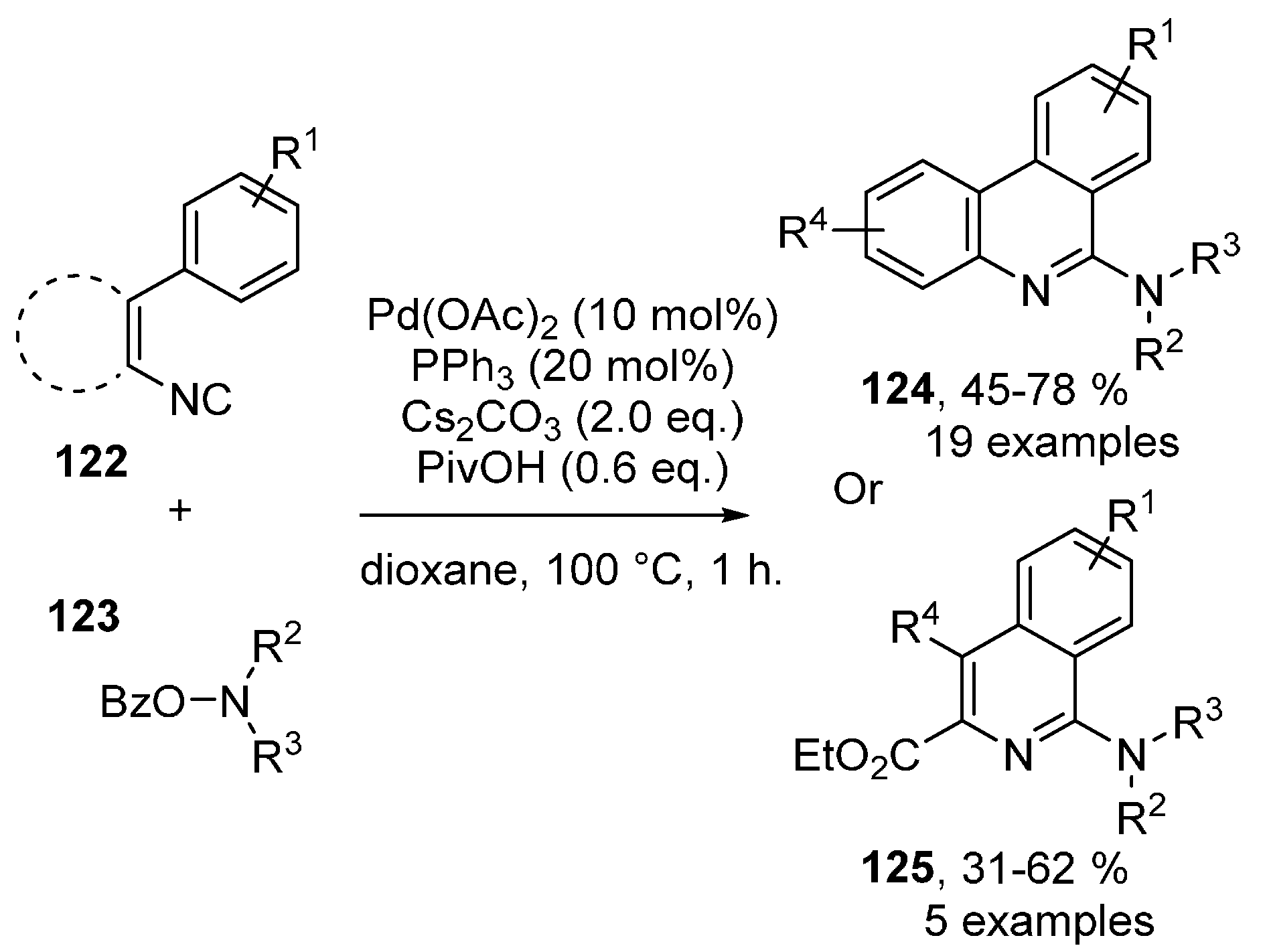
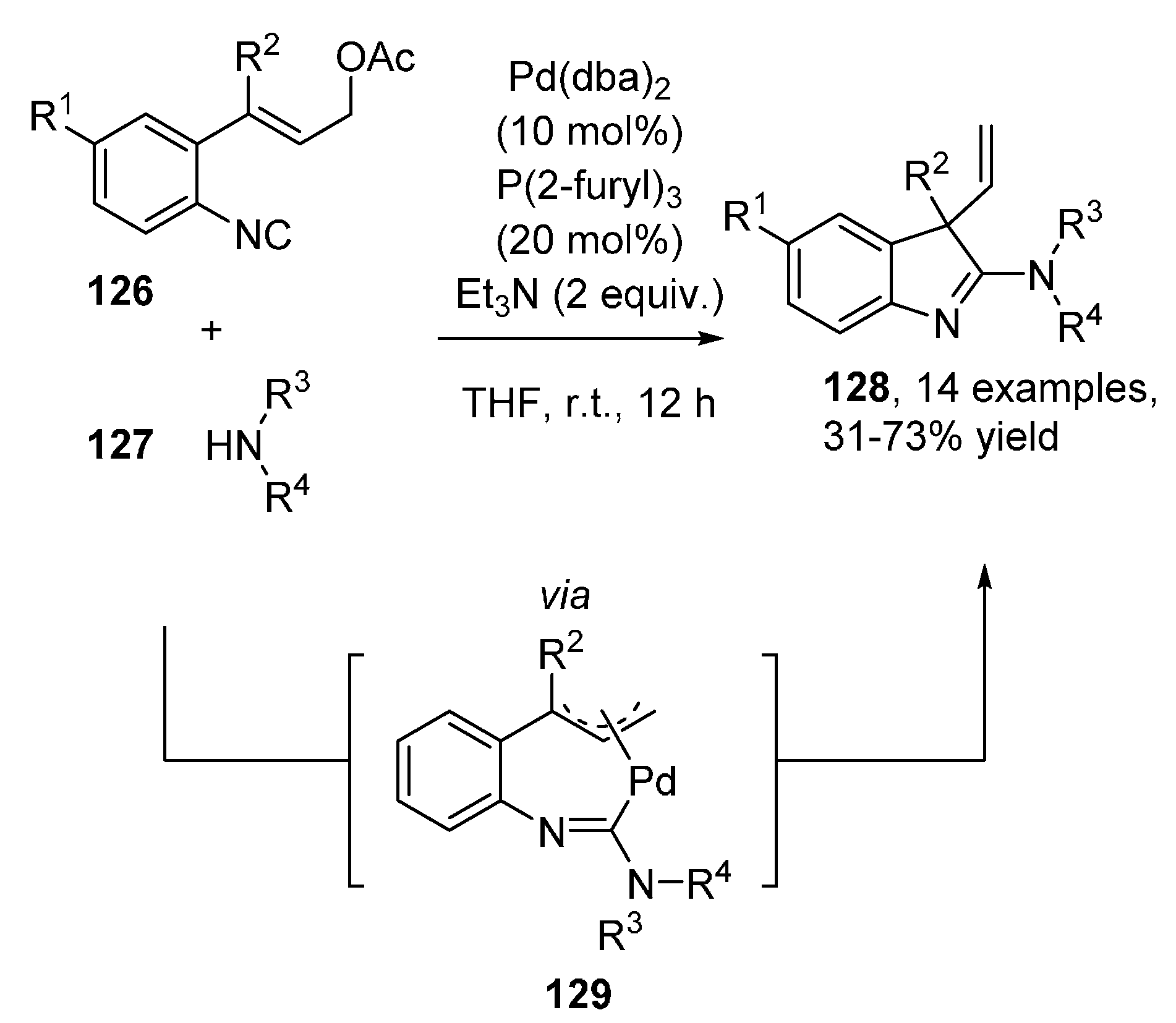
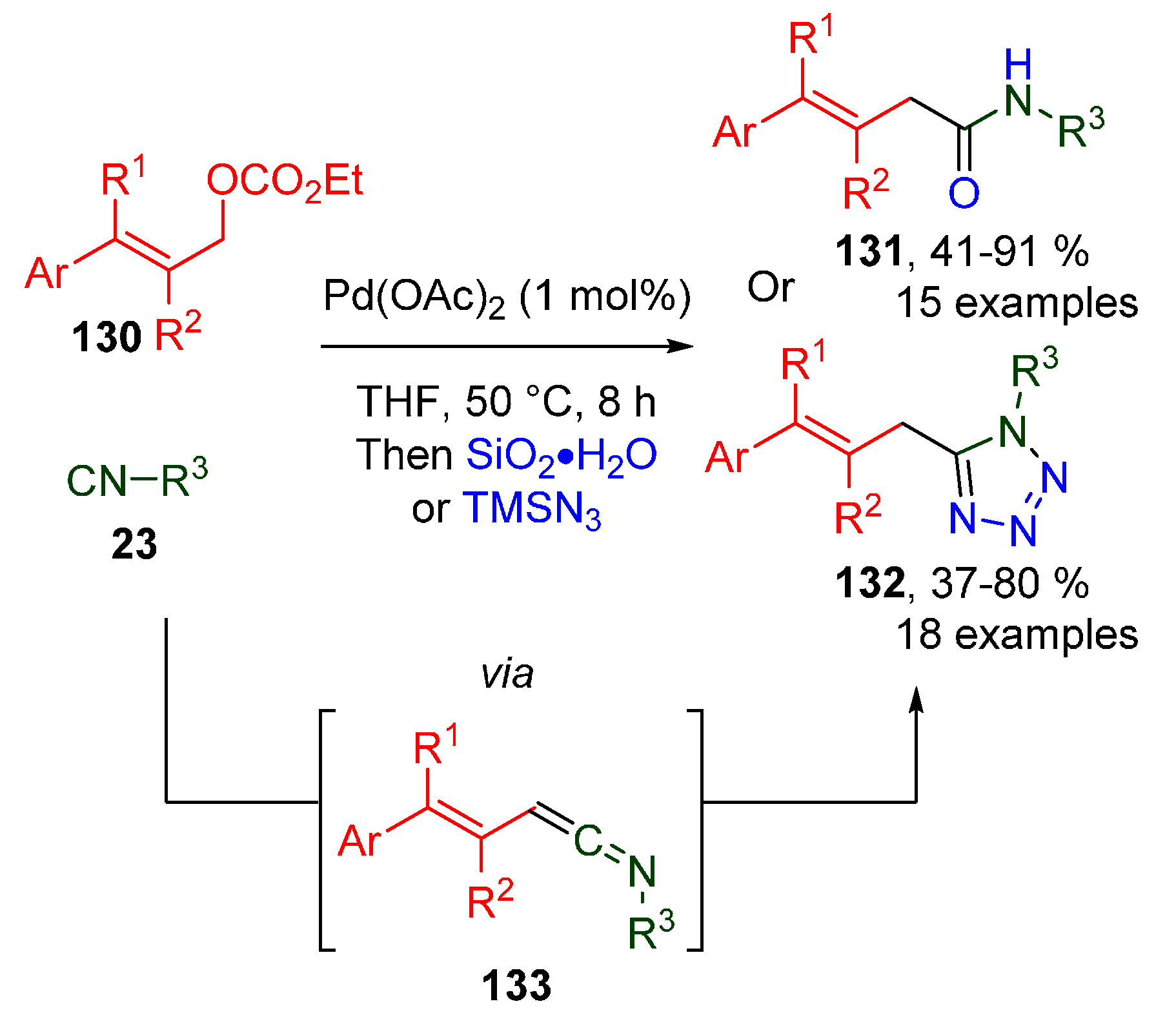

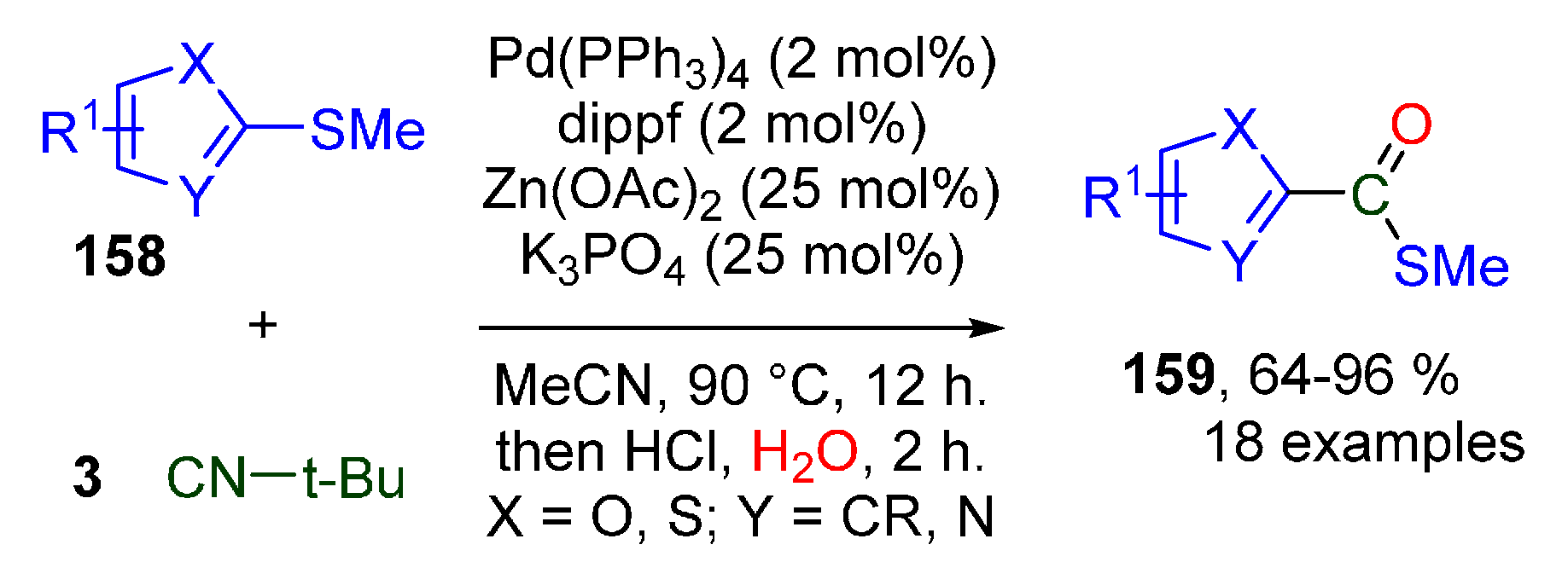





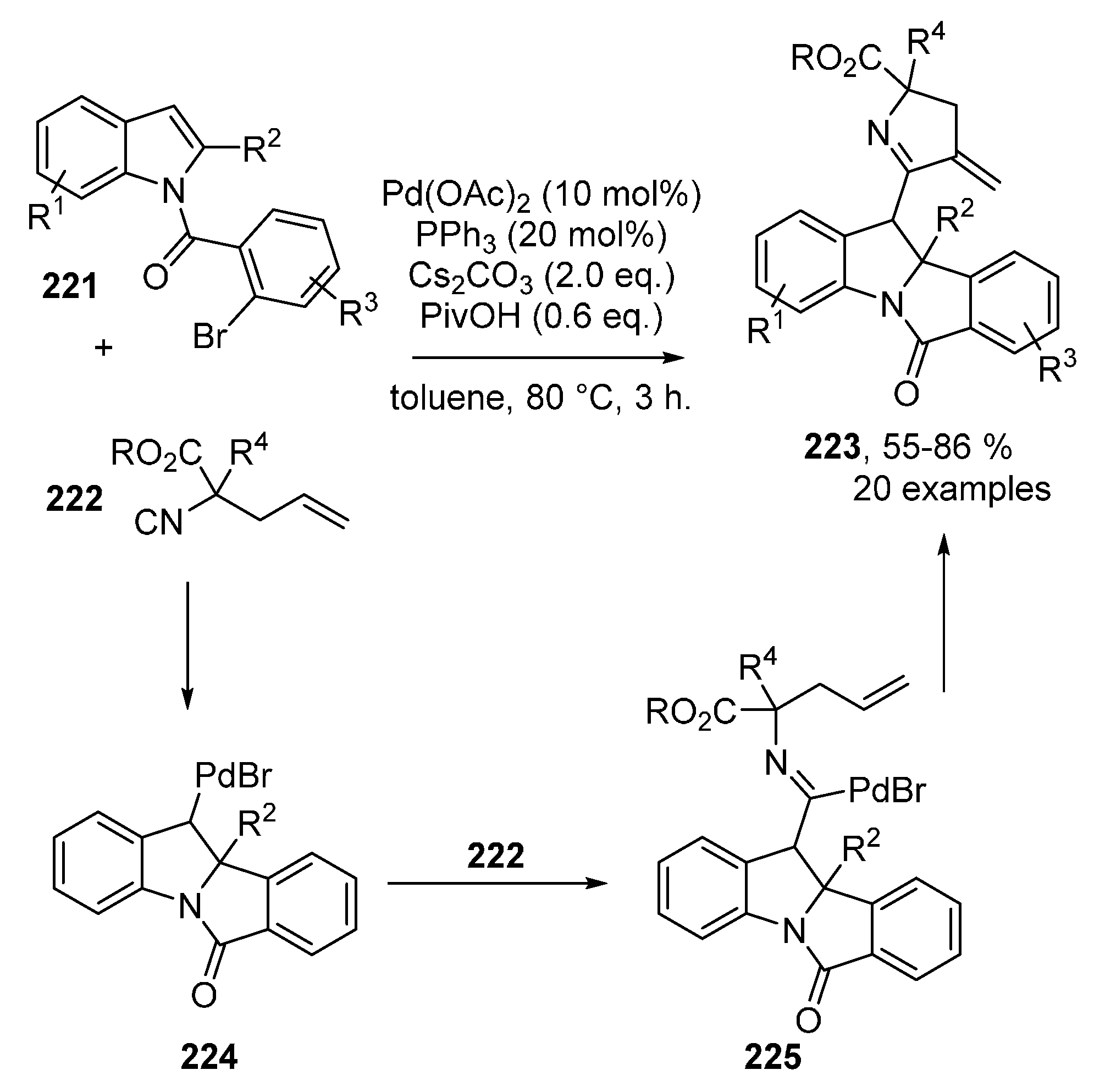
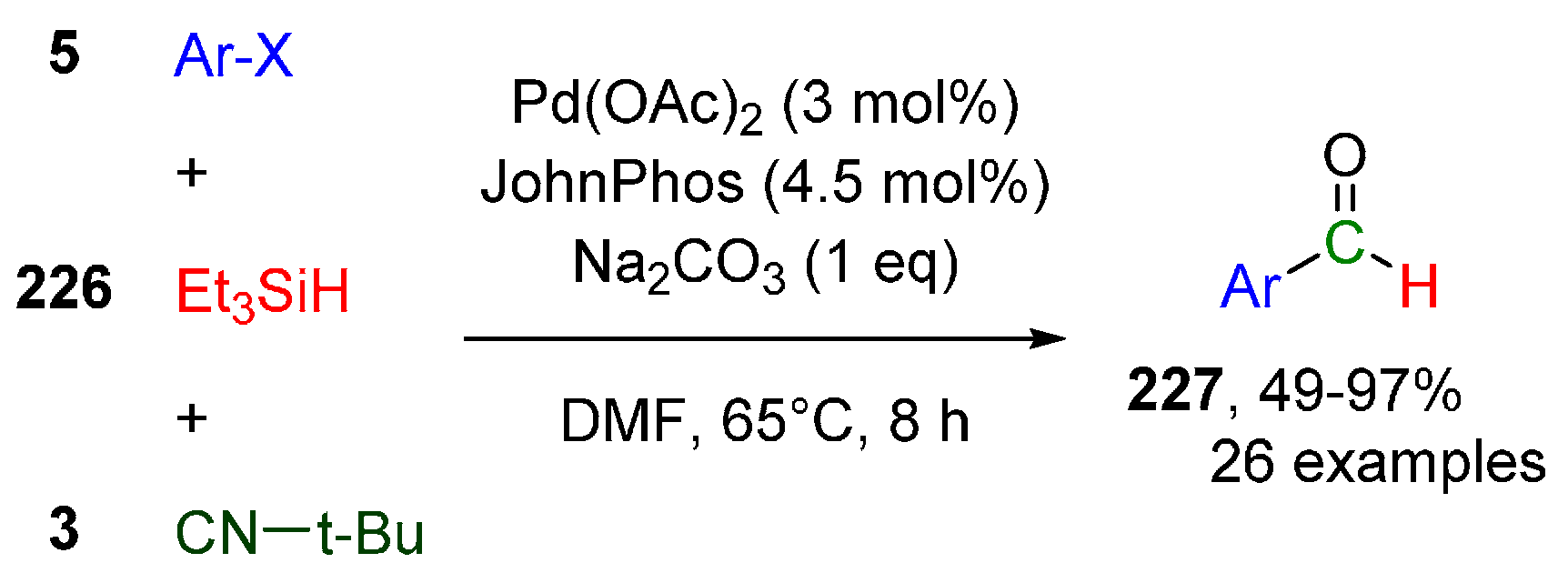
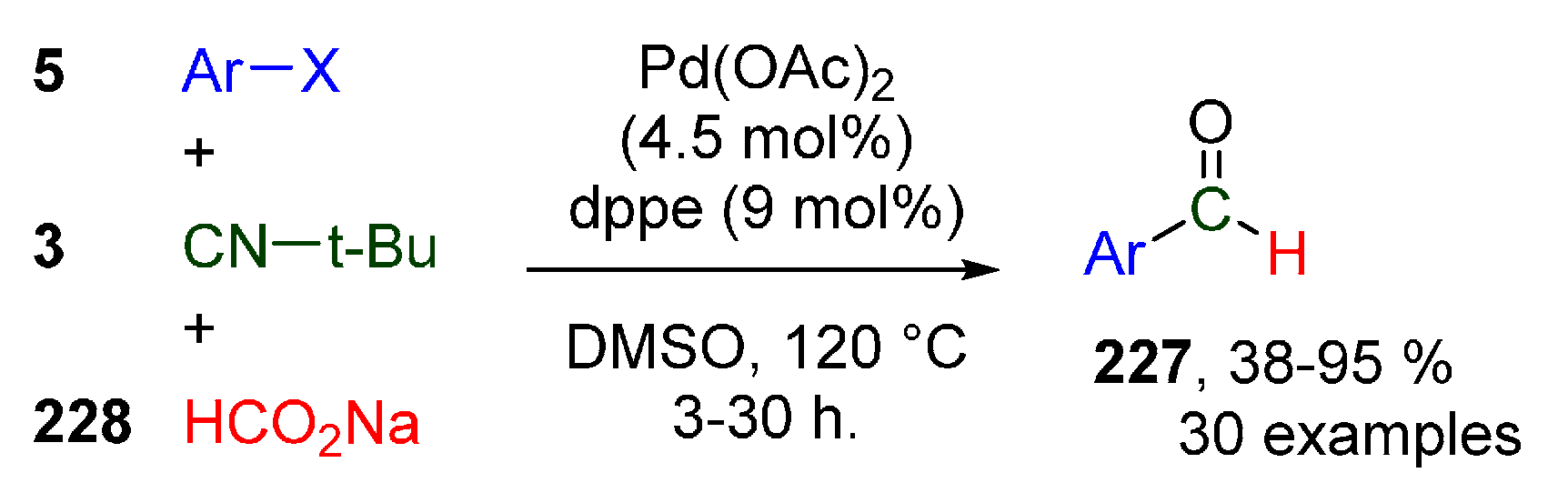
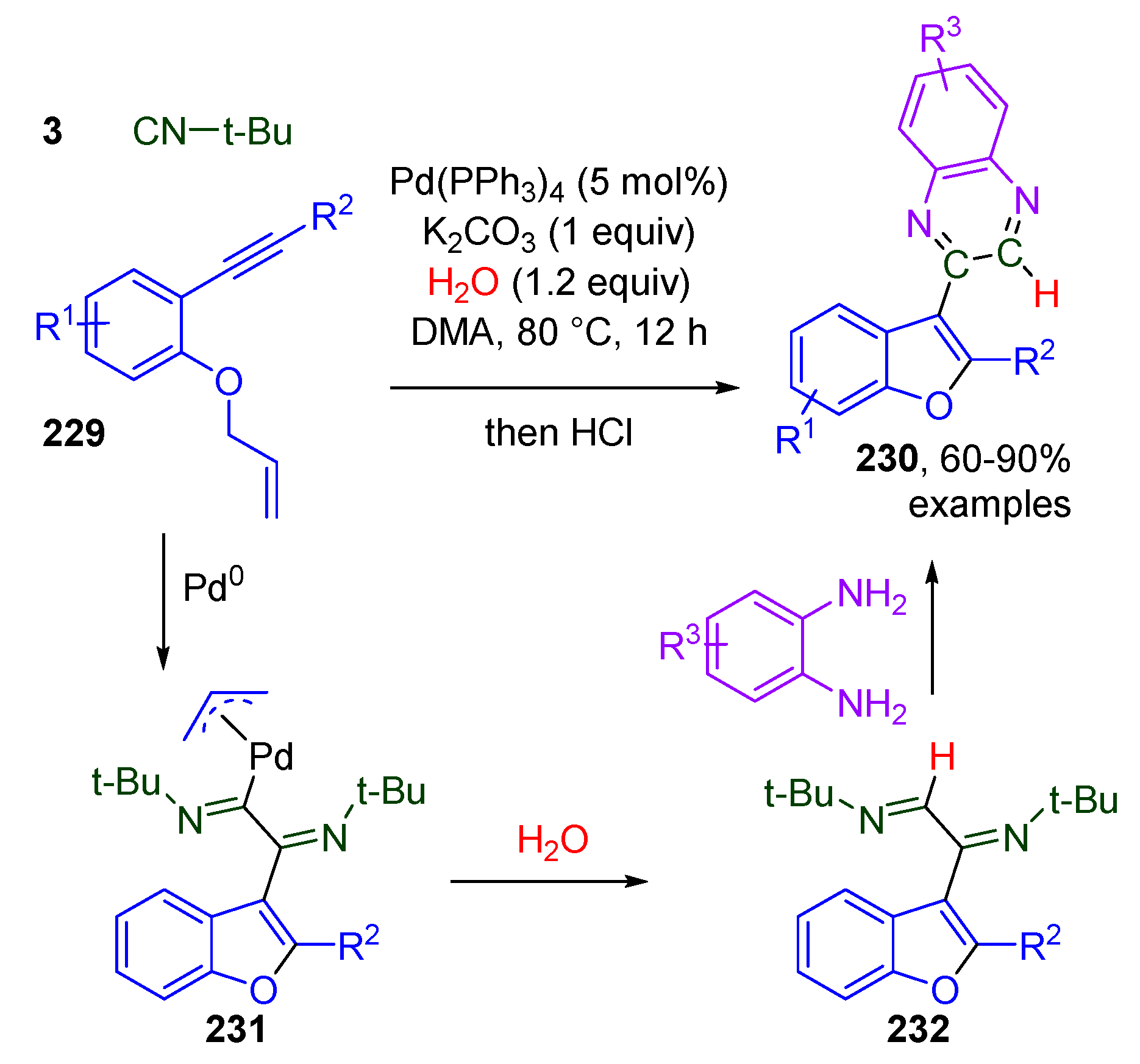

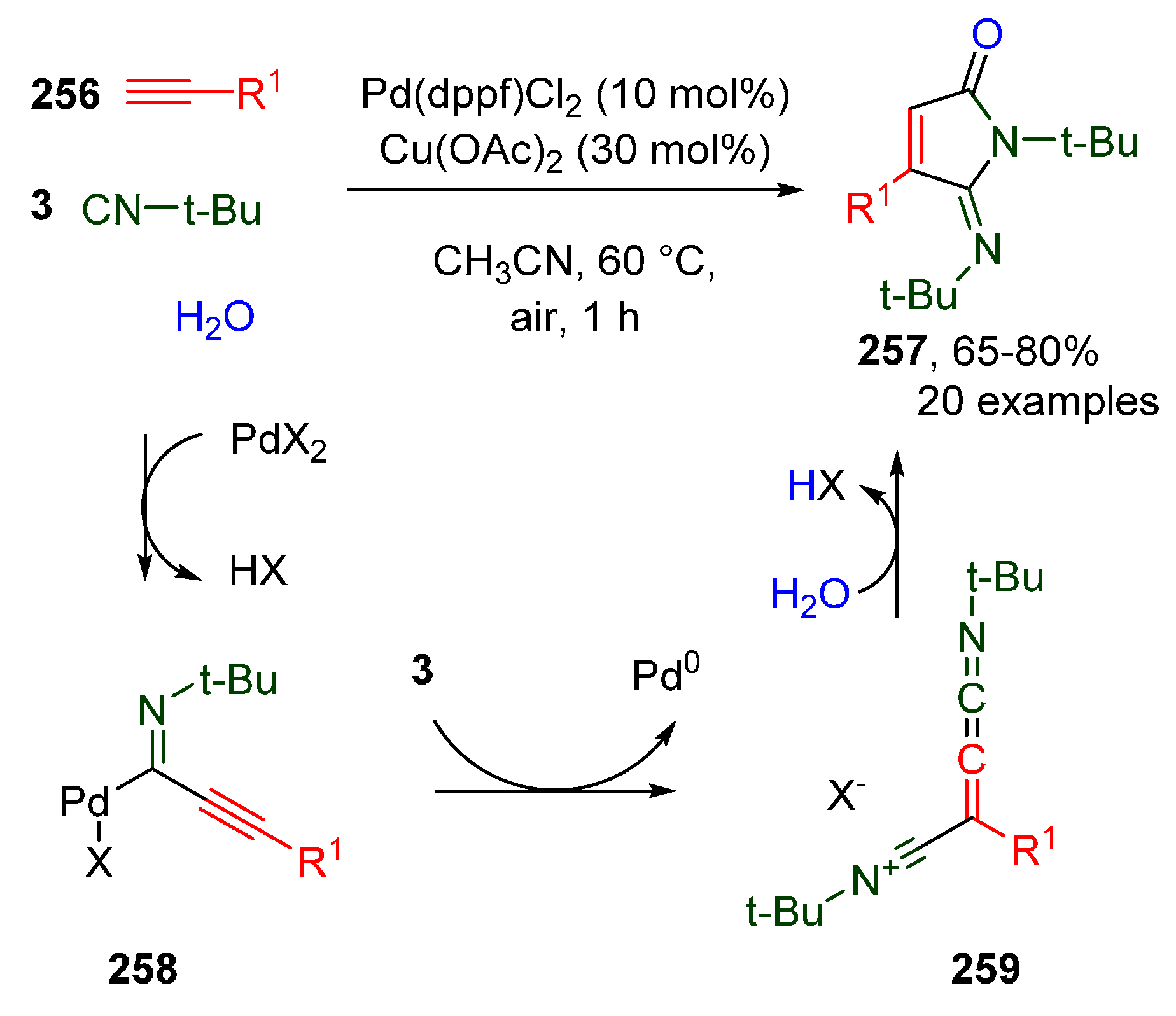
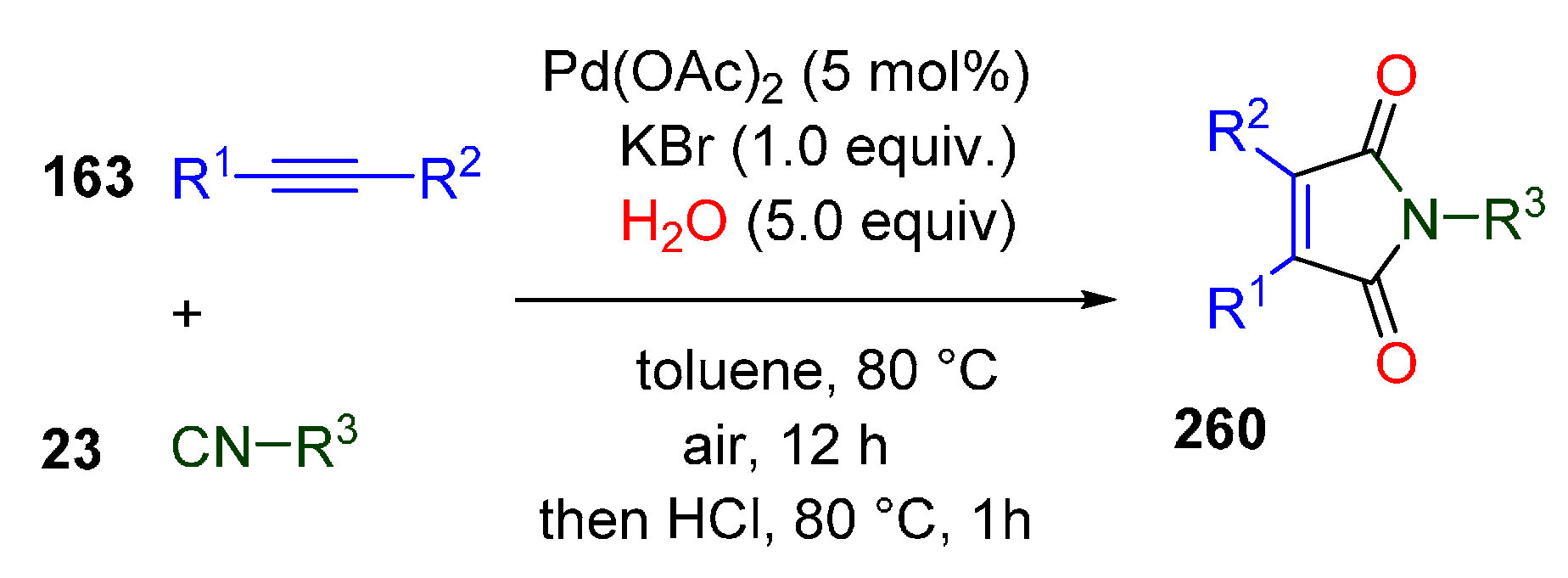

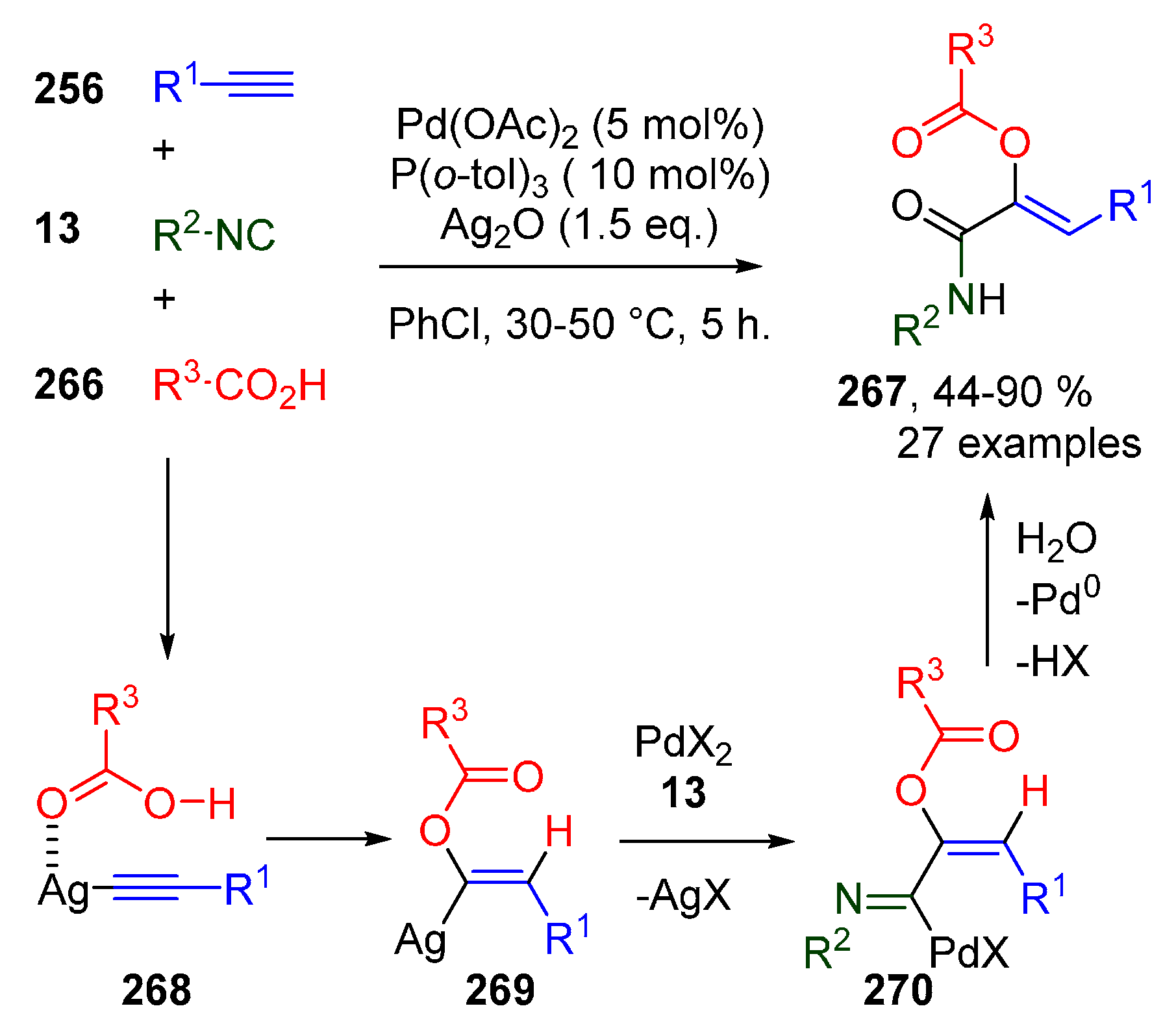
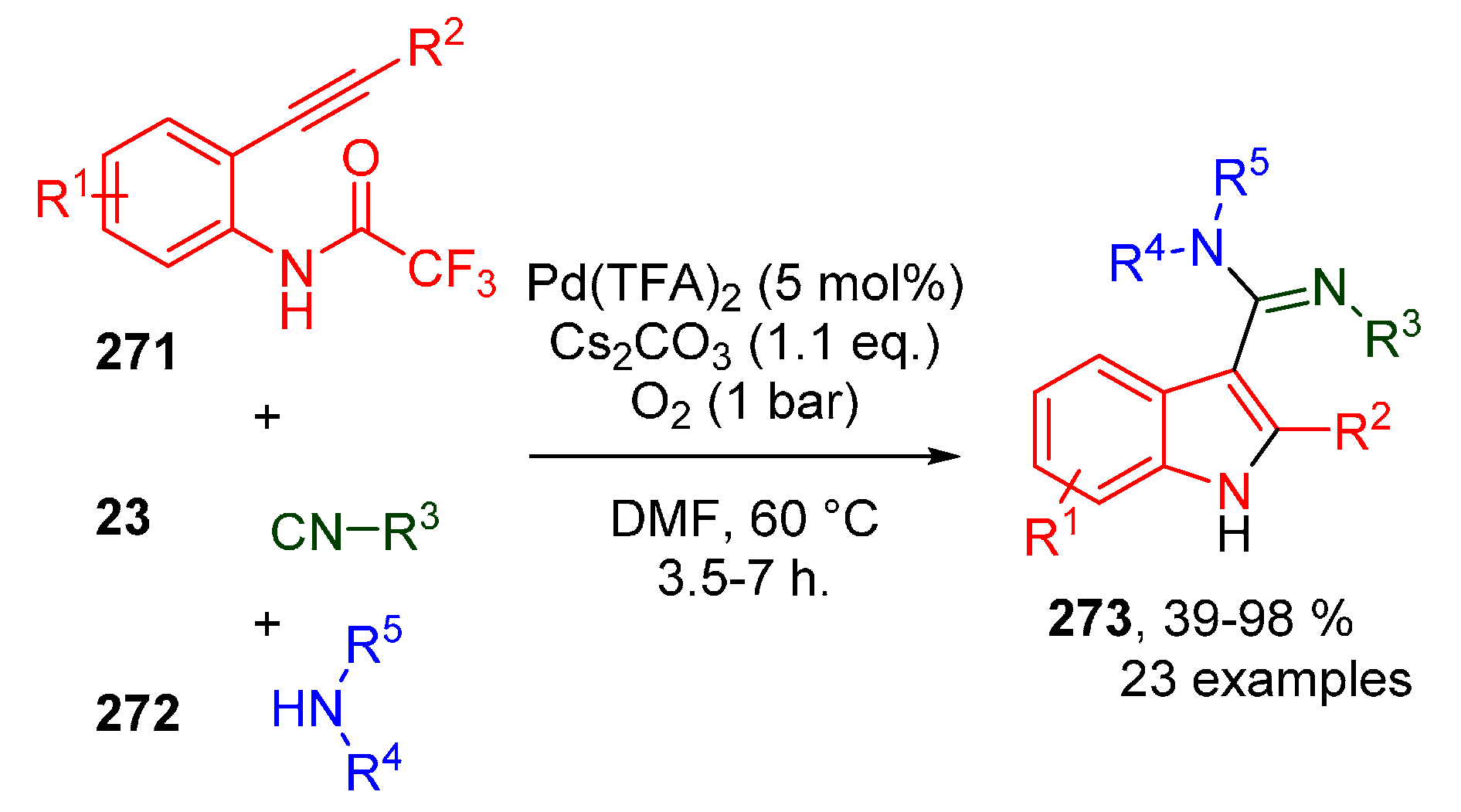
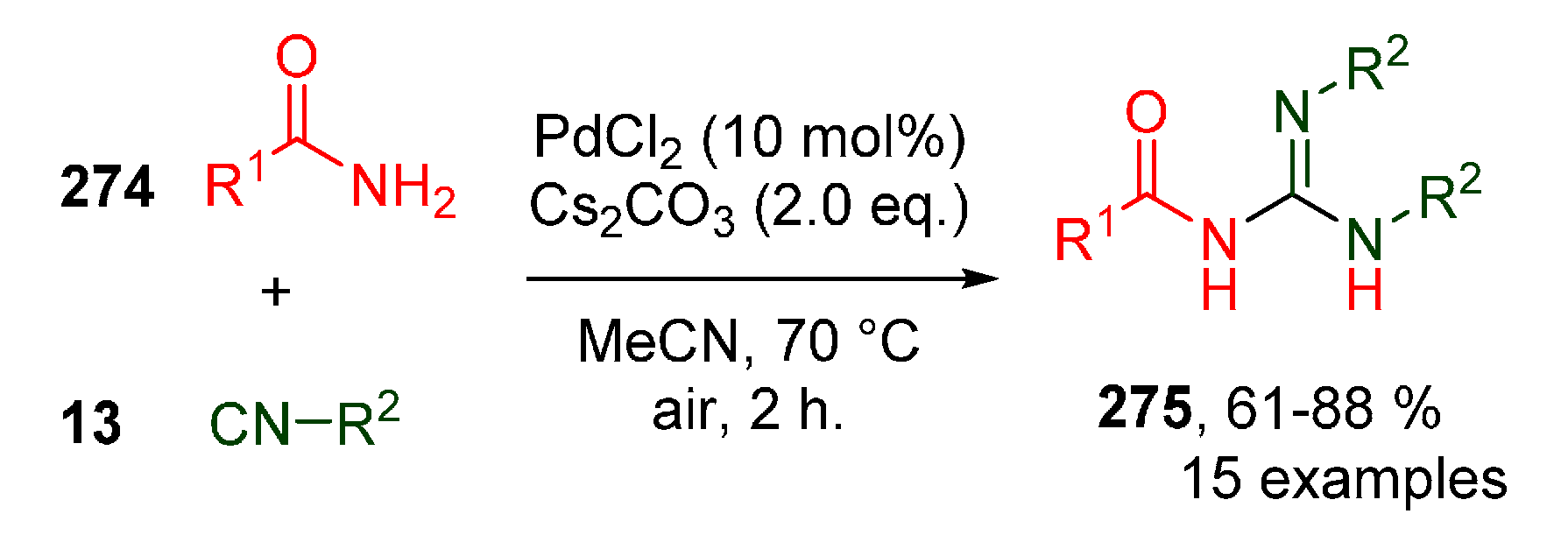
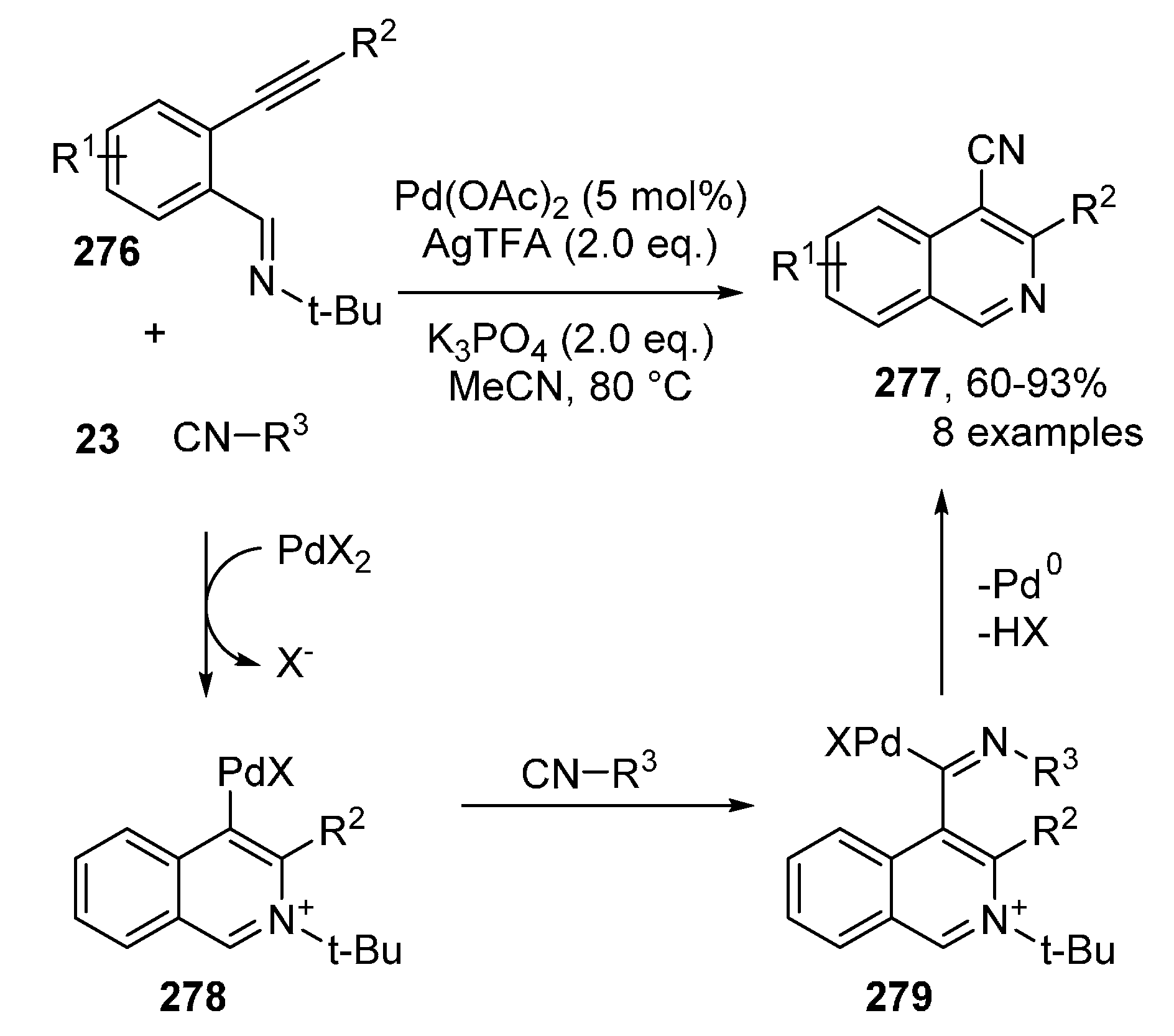
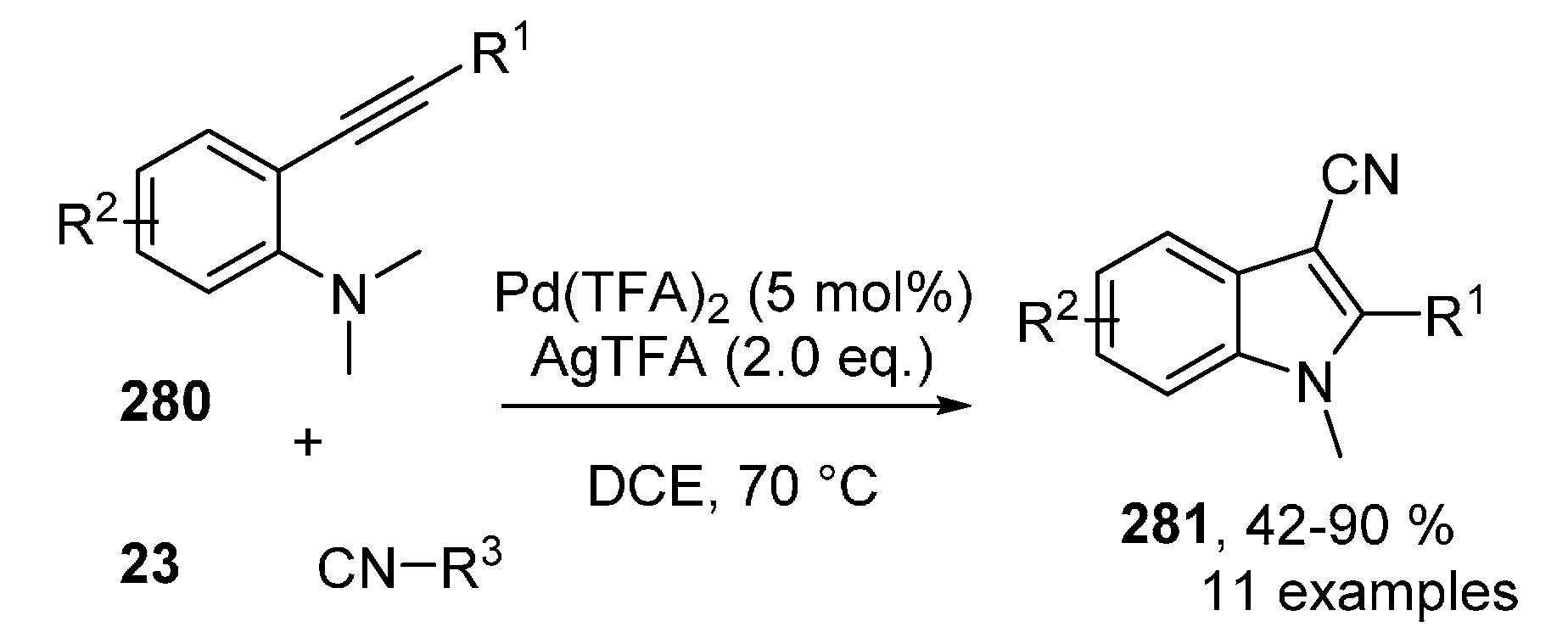
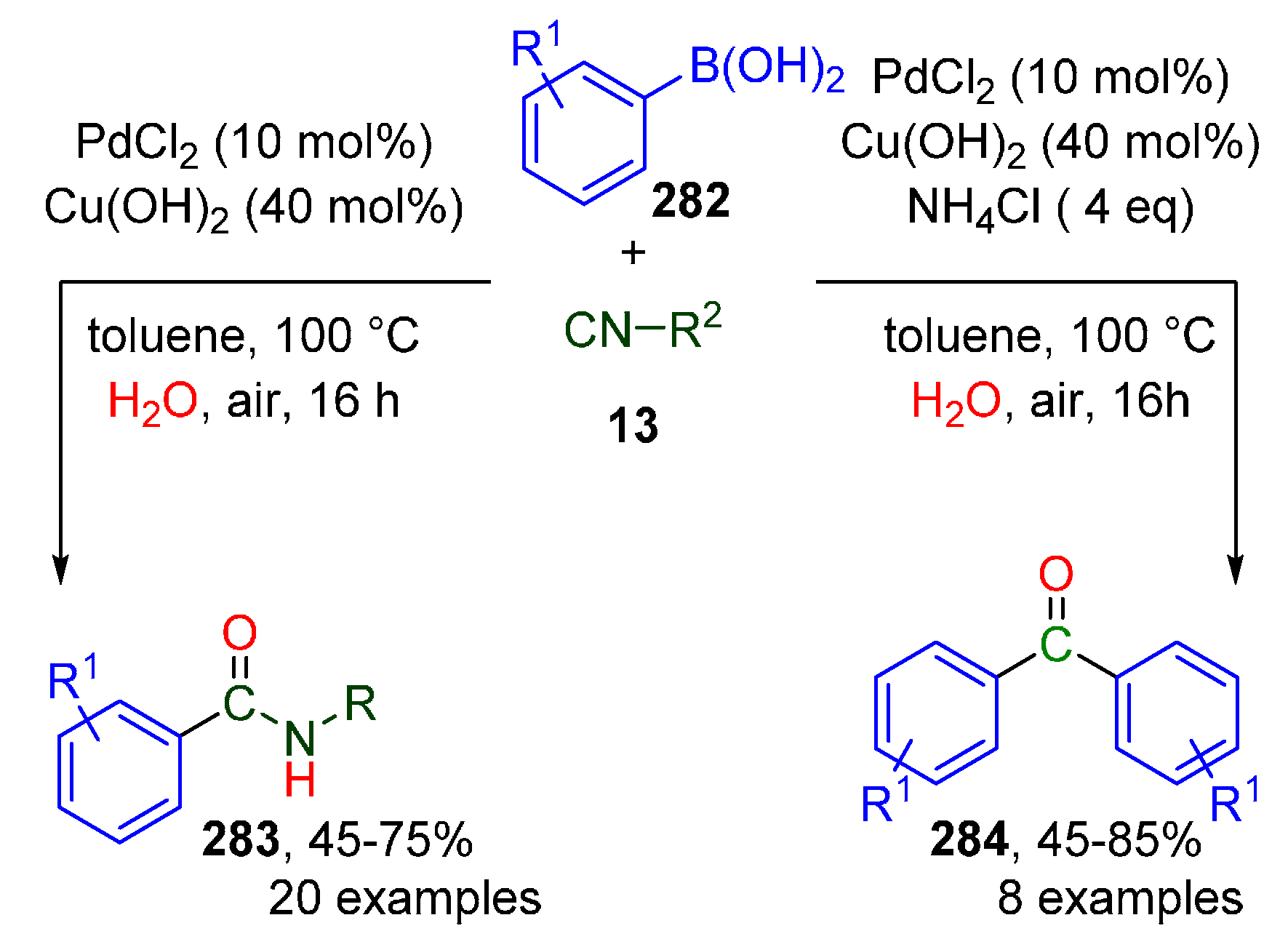

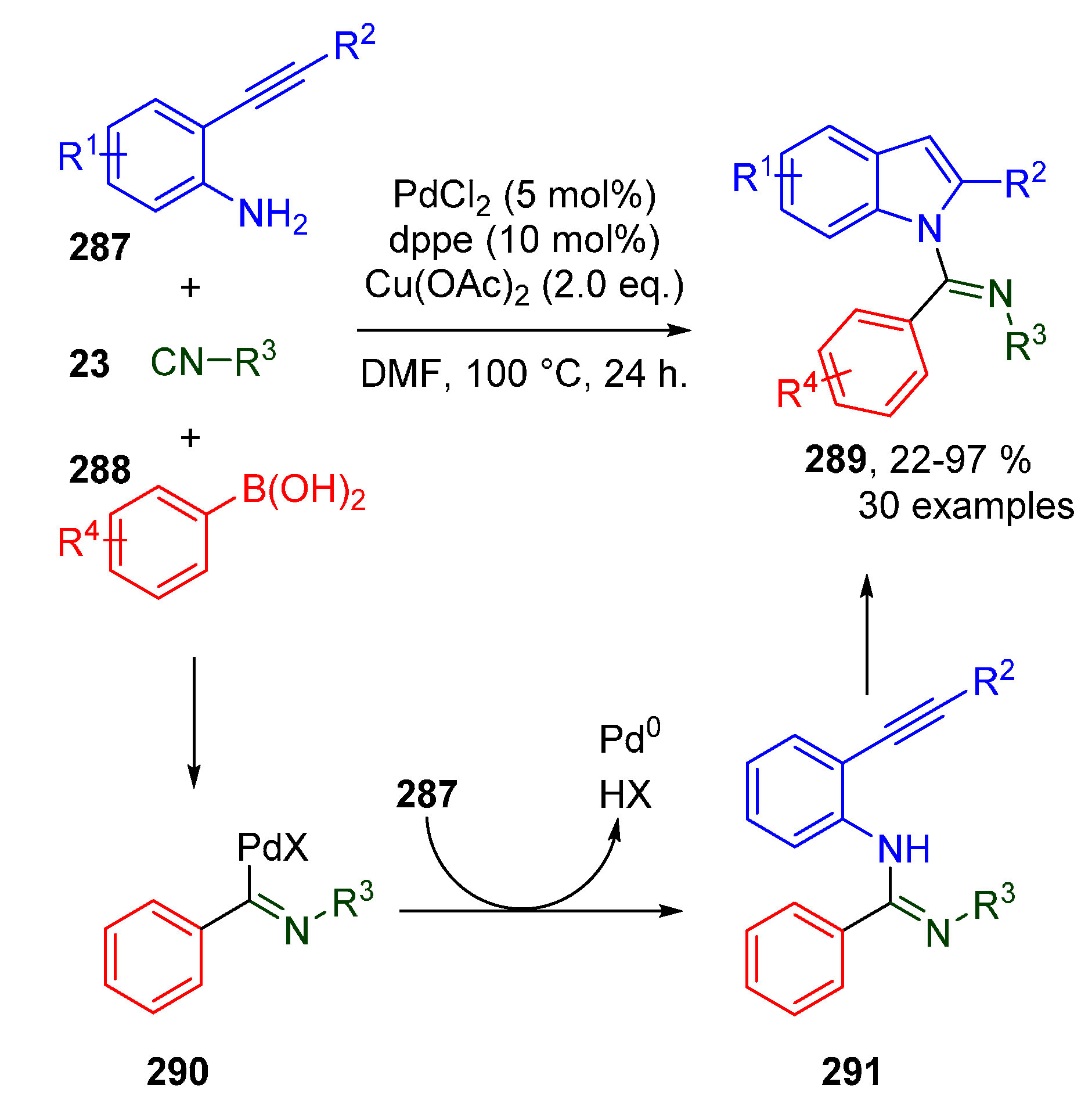
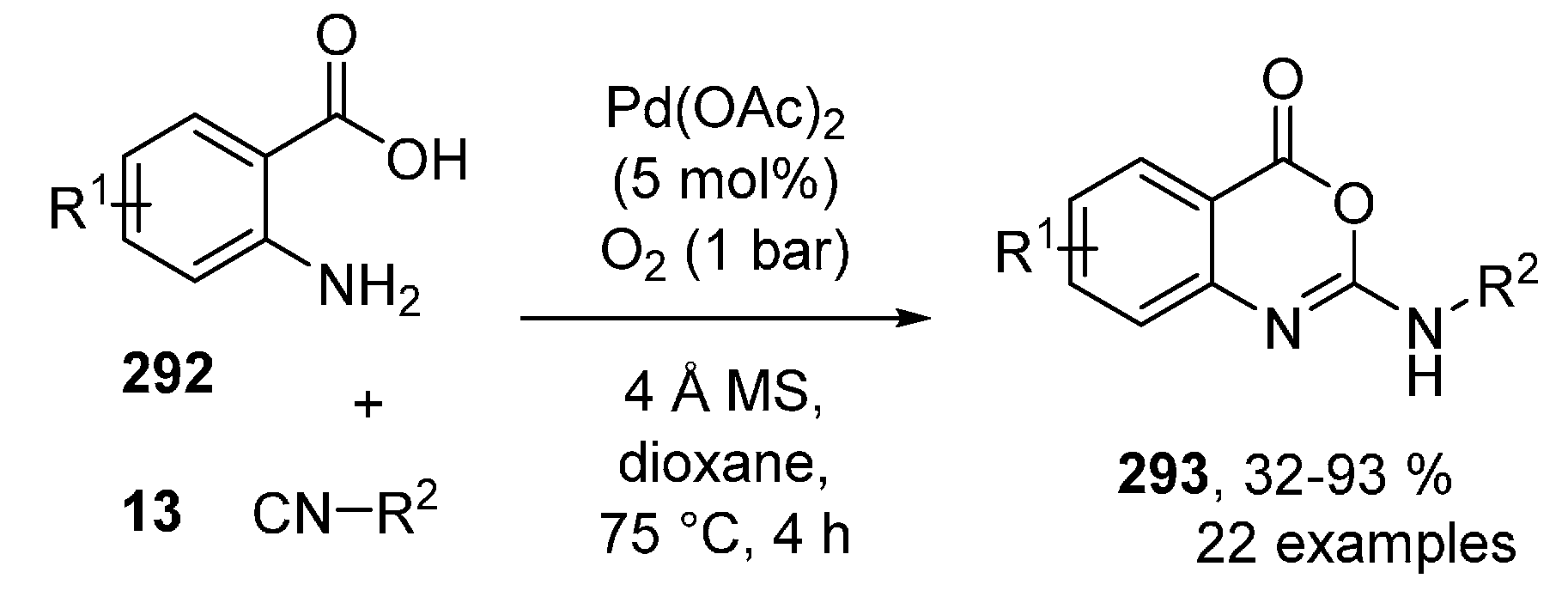

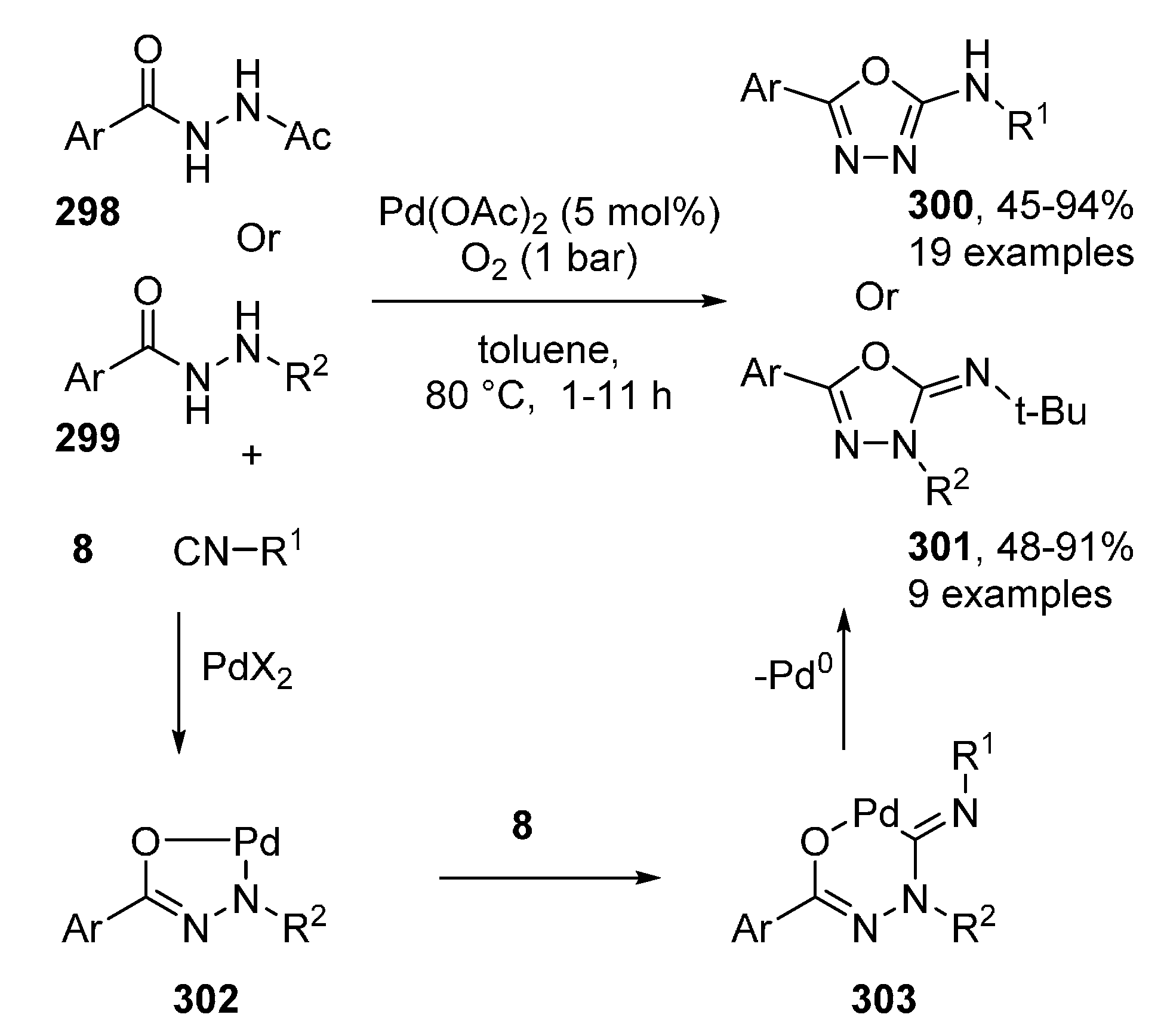


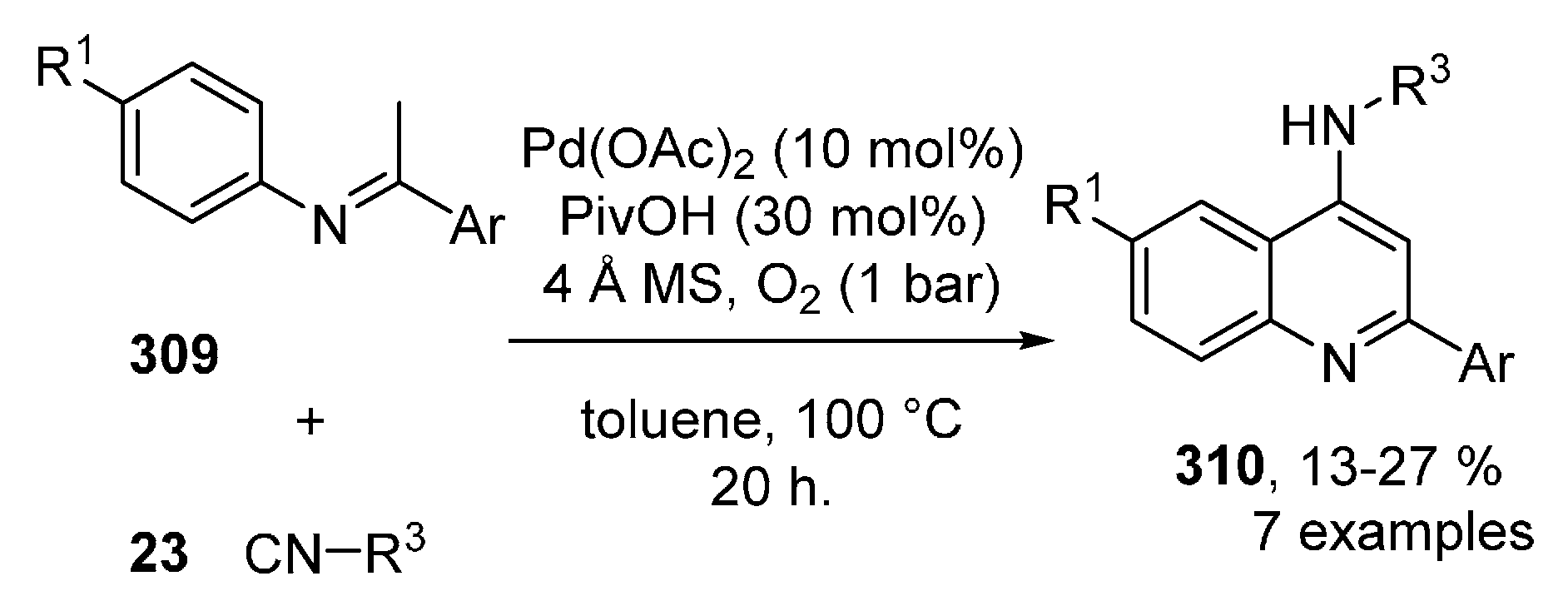


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Collet, J.W.; Roose, T.R.; Weijers, B.; Maes, B.U.W.; Ruijter, E.; Orru, R.V.A. Recent Advances in Palladium-Catalyzed Isocyanide Insertions. Molecules 2020, 25, 4906. https://doi.org/10.3390/molecules25214906
Collet JW, Roose TR, Weijers B, Maes BUW, Ruijter E, Orru RVA. Recent Advances in Palladium-Catalyzed Isocyanide Insertions. Molecules. 2020; 25(21):4906. https://doi.org/10.3390/molecules25214906
Chicago/Turabian StyleCollet, Jurriën W., Thomas R. Roose, Bram Weijers, Bert U. W. Maes, Eelco Ruijter, and Romano V. A. Orru. 2020. "Recent Advances in Palladium-Catalyzed Isocyanide Insertions" Molecules 25, no. 21: 4906. https://doi.org/10.3390/molecules25214906
APA StyleCollet, J. W., Roose, T. R., Weijers, B., Maes, B. U. W., Ruijter, E., & Orru, R. V. A. (2020). Recent Advances in Palladium-Catalyzed Isocyanide Insertions. Molecules, 25(21), 4906. https://doi.org/10.3390/molecules25214906