
Insight to Functional Conformation and Noncovalent Interactions of Protein-Protein Assembly Using MALDI Mass Spectrometry



Abstract
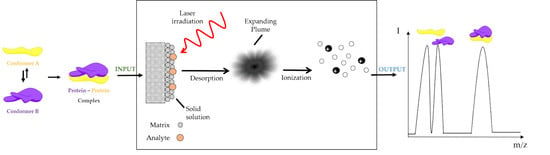
1. Introduction
2. Functional Conformations and Noncovalent Interactions
3. Types of Protein and Peptide Noncovalent Self-Assembly and Their Biological Function
3.1. Peptide Self-Assembly
3.2. Protein Self- and Non-Self-Assembly
4. Techniques to Understand Protein Assembly Interactions and Conformations
5. MALDI Mass Spectrometry Analysis as an Approach to Unravel Interactions in Protein Assembly Guided by Molecular Conformation
5.1. Limited Proteolysis MALDI-MS
5.2. HDX-MALDI-MS
5.3. Protein Derivatization
5.4. MALDI Native Mass Spectrometry
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Karas, M.; Bachmann, D.; Bahr, U.; Hillenkamp, F. Matrix-assisted ultraviolet laser desorption of non-volatile compounds. Int. J. Mass Spectrom. Ion Process. 1987, 78, 53–68. [Google Scholar] [CrossRef]
- Hillenkamp, F.; Karas, M. The MALDI Process and Method. In Maldi Ms; John Wiley & Sons: Weinheim, Germany; pp. 1–28.
- Josip, L. Introducing Proteomics: From Concepts to Sample Separation, Mass Spectrometry and Data Analysis; Wiley-Blackwell: West Sussex, UK, 2014. [Google Scholar]
- Noor, Z.; Ahn, S.B.; Baker, M.S.; Ranganathan, S.; Mohamedali, A. Mass spectrometry–based protein identification in proteomics—A review. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Bolbach, G. Matrix-Assisted Laser Desorption/Ionization Analysis of Non-Covalent Complexes: Fundamentals and Applications. Curr. Pharm. Des. 2005, 11, 2535–2557. [Google Scholar] [CrossRef]
- Kokkinidis, M.; Glykos, N.M.; Fadouloglou, V.E. Chapter 7—Protein Flexibility and Enzymatic Catalysis. In Advances in Protein Chemistry and Structural Biology; Christov, C., Karabencheva-Christova, T., Eds.; Academic Press: Cambridge, MA, USA; Volume 87, pp. 181–2018.
- Rousseau, F.; Schymkowitz, J. A systems biology perspective on protein structural dynamics and signal transduction. Curr. Opin. Struct. Biol. 2005, 15, 23–30. [Google Scholar] [CrossRef]
- Mythily, S.; Roger, W.R. Immunomodulatory Peptides from IgSF Proteins: A Review. Curr. Protein Pept. Sci. 2005, 6, 185–196. [Google Scholar]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; Wiley: Hoboken, NJ, USA, 2011; p. 1428 S. [Google Scholar]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef]
- Wilson, I.A.; Stanfield, R.L. Antibody-antigen interactions: New structures and new conformational changes. Curr. Opin. Struct. Biol. 1994, 4, 857–867. [Google Scholar] [CrossRef]
- Al Qaraghuli, M.M.; Kubiak-Ossowska, K.; Ferro, V.A.; Mulheran, P.A. Antibody-protein binding and conformational changes: Identifying allosteric signalling pathways to engineer a better effector response. Sci. Rep. 2020, 10, 13696. [Google Scholar] [CrossRef]
- Halakou, F.; Gursoy, A.; Keskin, O. Embedding Alternative Conformations of Proteins in Protein–Protein Interaction Networks. In Protein-Protein Interaction Networks: Methods and Protocols; Canzar, S., Ringeling, F.R., Eds.; Springer US: New York, NY, USA, 2020; pp. 113–124. [Google Scholar]
- Mihăşan, M.; Wormwood, K.L.; Sokolowska, I.; Roy, U.; Woods, A.G.; Darie, C.C. Mass Spectrometry- and Computational Structural Biology-Based Investigation of Proteins and Peptides. In Advancements of Mass Spectrometry in Biomedical Research; Woods, A.G., Darie, C.C., Eds.; Springer International Publishing: Cham, Germany, 2019; pp. 265–287. [Google Scholar]
- Malhotra, P.; Udgaonkar, J.B. How cooperative are protein folding and unfolding transitions? Protein Sci. 2016, 25, 1924–1941. [Google Scholar] [CrossRef]
- Chen, F.; Gülbakan, B.; Weidmann, S.; Fagerer, S.R.; Ibáñez, A.J.; Zenobi, R. Applying mass spectrometry to study non-covalent biomolecule complexes. Mass Spectrom. Rev. 2016, 35, 48–70. [Google Scholar] [CrossRef]
- Bennett, M.J.; Eisenberg, D. The Evolving Role of 3D Domain Swapping in Proteins. Structure 2004, 12, 1339–1341. [Google Scholar] [CrossRef]
- Wodak, S.J.; Malevanets, A.; MacKinnon, S.S. The Landscape of Intertwined Associations in Homooligomeric Proteins. Biophys. J. 2015, 109, 1087–1100. [Google Scholar] [CrossRef]
- Springael, J.-Y.; Urizar, E.; Costagliola, S.; Vassart, G.; Parmentier, M. Allosteric properties of G protein-coupled receptor oligomers. Pharm. Ther. 2007, 115, 410–418. [Google Scholar] [CrossRef]
- Cui, Q.; Karplus, M. Allostery and cooperativity revisited. Protein Sci. 2008, 17, 1295–1307. [Google Scholar] [CrossRef]
- Guo, J.; Zhou, H.-X. Protein Allostery and Conformational Dynamics. Chem. Rev. 2016, 116, 6503–6515. [Google Scholar] [CrossRef]
- Cooper, A.; Dryden, D.T.F. Allostery without conformational change. Eur. Biophys. J. 1984, 11, 103–109. [Google Scholar] [CrossRef]
- Stefan, M.I.; Le Novère, N. Cooperative Binding. Plos Comp. Biol. 2013, 9, e1003106. [Google Scholar] [CrossRef]
- Goh, C.-S.; Milburn, D.; Gerstein, M. Conformational changes associated with protein–protein interactions. Curr. Opin. Struct. Biol. 2004, 14, 104–109. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Structural Symmetry and Protein Function. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 105–153. [Google Scholar] [CrossRef]
- Jaffe, E.K. Morpheeins—A new structural paradigm for allosteric regulation. Trends Biochem. Sci. 2005, 30, 490–497. [Google Scholar] [CrossRef]
- Gabizon, R.; Friedler, A. Allosteric modulation of protein oligomerization: An emerging approach to drug design. Front. Chem. 2014, 2, 9. [Google Scholar] [CrossRef]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Peselis, A.; Gao, A.; Serganov, A. Cooperativity, allostery and synergism in ligand binding to riboswitches. Biochimie 2015, 117, 100–109. [Google Scholar] [CrossRef]
- Mulder, A.; Huskens, J.; Reinhoudt, D.N. Multivalency in supramolecular chemistry and nanofabrication. Org. Biomol. Chem. 2004, 2, 3409–3424. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-T.; Chen, Y.-C.; Liou, H.-H.; Chao, C.-Y. Structural basis for cooperative oxygen binding and bracelet-assisted assembly of Lumbricus terrestris hemoglobin. Sci. Rep. 2015, 5, 9494. [Google Scholar] [CrossRef] [PubMed]
- Koshland, D.E.; Némethy, G.; Filmer, D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits*. Biochemistry 1966, 5, 365–385. [Google Scholar] [CrossRef] [PubMed]
- Cooperman, B.S. Allosteric Regulation. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 71–74. [Google Scholar]
- Huang, Y.-M.M.; Chen, W.; Potter, M.J.; Chang, C.-E.A. Insights from Free-Energy Calculations: Protein Conformational Equilibrium, Driving Forces, and Ligand-Binding Modes. Biophys. J. 2012, 103, 342–351. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef]
- Yang, Q.-F.; Tang, C. On the necessity of an integrative approach to understand protein structural dynamics. J. Zhejiang Univ. Sci. B 2019, 20, 496–502. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal Mode Analysis of Biomolecular Structures: Functional Mechanisms of Membrane Proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef]
- Tang, C.; Schwieters, C.D.; Clore, G.M. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 2007, 449, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J. Intermolecular and Surface Forces, 2nd ed.; Academic Press: London, UK, 1991. [Google Scholar]
- Cook, E.C.; Creamer, T.P. Influence of electrostatic forces on the association kinetics and conformational ensemble of an intrinsically disordered protein. Proteins 2020, n/a. [Google Scholar] [CrossRef]
- Kaltashov, I.A.; Bobst, C.E.; Abzalimov, R.R. Mass spectrometry-based methods to study protein architecture and dynamics. Protein Sci. 2013, 22, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Edwards-Gayle, C.J.C.; Hamley, I.W. Self-assembly of bioactive peptides, peptide conjugates, and peptide mimetic materials. Org. Biomol. Chem. 2017, 15, 5867–5876. [Google Scholar] [CrossRef] [PubMed]
- Stendahl, J.C.; Rao, M.S.; Guler, M.O.; Stupp, S.I. Intermolecular Forces in the Self-Assembly of Peptide Amphiphile Nanofibers. Adv. Funct. Mater. 2006, 16, 499–508. [Google Scholar] [CrossRef]
- Hutchinson, J.A.; Burholt, S.; Hamley, I.W. Peptide hormones and lipopeptides: From self-assembly to therapeutic applications. J. Pept. Sci. 2017, 23, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Deng, L.; Wang, Y.; Lu, J.R.; Xu, H. Different nanostructures caused by competition of intra- and inter-β-sheet interactions in hierarchical self-assembly of short peptides. J. Colloid Interface Sci. 2016, 464, 219–228. [Google Scholar] [CrossRef]
- Cui, H.; Webber, M.J.; Stupp, S.I. Self-assembly of peptide amphiphiles: From molecules to nanostructures to biomaterials. Pept. Sci. 2010, 94, 1–18. [Google Scholar] [CrossRef]
- Kirkham, S.; Castelletto, V.; Hamley, I.W.; Inoue, K.; Rambo, R.; Reza, M.; Ruokolainen, J. Self-Assembly of the Cyclic Lipopeptide Daptomycin: Spherical Micelle Formation Does Not Depend on the Presence of Calcium Chloride. ChemPhysChem 2016, 17, 2118–2122. [Google Scholar] [CrossRef]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef]
- Hamley, I.W. Lipopeptides: From self-assembly to bioactivity. Chem. Commun. 2015, 51, 8574–8583. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Das, S.; Nandi, A.K. A review on recent advances in polymer and peptide hydrogels. Soft Matter 2020, 16, 1404–1454. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Castelletto, V.; Jones, R.R.; Connon, C.J.; Hamley, I.W. Hydrogelation of self-assembling RGD-based peptides. Soft Matter 2011, 7, 1326–1333. [Google Scholar] [CrossRef]
- Marianayagam, N.J.; Sunde, M.; Matthews, J.M. The power of two: Protein dimerization in biology. Trends Biochem. Sci. 2004, 29, 618–625. [Google Scholar] [CrossRef]
- Singh, H.; Ashley, R.H. Redox Regulation of CLIC1 by Cysteine Residues Associated with the Putative Channel Pore. Biophys. J. 2006, 90, 1628–1638. [Google Scholar] [CrossRef]
- Frain, K.M.; Robinson, C.; van Dijl, J.M. Transport of Folded Proteins by the Tat System. Protein J. 2019, 38, 377–388. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. How and why do GPCRs dimerize? Trends Pharm. Sci. 2008, 29, 234–240. [Google Scholar] [CrossRef]
- Lee, M.J.; Yaffe, M.B. Protein Regulation in Signal Transduction. Cold Spring Harb. Perspect. Biol. 2016, 8, a005918. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Lu, B.; Evans, R.; Gutkind, J.S. Signals and Receptors. Cold Spring Harb. Perspect. Biol. 2016, 8, a005900. [Google Scholar] [CrossRef]
- Ishigai, M.; Langridge, J.I.; Bordoli, R.S.; Gaskell, S.J. Noncovalent associations of glutathione S-transferase and ligands: A study using electrospray quadrupole/time-of-flight mass spectrometry. J. Am. Soc. Mass Spectrom. 2000, 11, 606–614. [Google Scholar] [CrossRef][Green Version]
- van Hateren, A.; Bailey, A.; Elliott, T. Recent advances in Major Histocompatibility Complex (MHC) class I antigen presentation: Plastic MHC molecules and TAPBPR-mediated quality control. F1000Research 2017, 6, 158. [Google Scholar] [CrossRef] [PubMed]
- Chapter 8—The T Cell Receptor: Proteins and Genes. In Primer to the Immune Response (Second Edition); Mak, T.W., Saunders, M.E., Jett, B.D., Eds.; Academic Cell: Boston, MA, USA, 2014; pp. 181–196. [Google Scholar]
- Reverberi, R.; Reverberi, L. Factors affecting the antigen-antibody reaction. Blood Transfus. 2007, 5, 227–240. [Google Scholar] [PubMed]
- Hornby, J.A.T.; Codreanu, S.G.; Armstrong, R.N.; Dirr, H.W. Molecular Recognition at the Dimer Interface of a Class Mu Glutathione Transferase: Role of a Hydrophobic Interaction Motif in Dimer Stability and Protein Function. Biochemistry 2002, 41, 14238–14247. [Google Scholar] [CrossRef] [PubMed]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef]
- Pelletier, H.; Kraut, J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science 1992, 258, 1748. [Google Scholar] [CrossRef]
- Drenth, J. Principles of protein X-ray crystallography; Springer: New York, NY, USA, 2007. [Google Scholar]
- Garbuzynskiy, S.O.; Melnik, B.S.; Lobanov, M.Y.; Finkelstein, A.V.; Galzitskaya, O.V. Comparison of X-ray and NMR structures: Is there a systematic difference in residue contacts between X-ray- and NMR-resolved protein structures? Proteins Struct. Funct. Bioinf. 2005, 60, 139–147. [Google Scholar] [CrossRef]
- Billeter, M. Comparison of protein structures determined by NMR in solution and by X-ray diffraction in single crystals. Q. Rev. Biophys. 1992, 25, 325–377. [Google Scholar] [CrossRef]
- Wuthrich, K. NMR of Proteins and Nucleic Acids. 1986; John Wiley & Sons: Michigan, MI, USA, 2001; Volume 19, p. 19. [Google Scholar]
- Berova, N.; Nakanishi, K.; Woody, R.W. Circular Dichroism: Principles and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2000. [Google Scholar]
- Daly, S.; Rosu, F.; Gabelica, V. Mass-resolved electronic circular dichroism ion spectroscopy. Science 2020, 368, 1465–1468. [Google Scholar] [CrossRef]
- Adrian, M.; Dubochet, J.; Lepault, J.; McDowall, A.W. Cryo-electron microscopy of viruses. Nature 1984, 308, 32–36. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid Fibrils of the HET-s(218–289) Prion Form a β Solenoid with a Triangular Hydrophobic Core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Gui, X.; Zhou, H.; Gu, J.; Li, Y.; Liu, X.; Zhao, M.; Li, D.; Li, X.; Liu, C. Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation. Nat. Struct. Mol. Biol. 2018, 25, 341–346. [Google Scholar] [CrossRef]
- LeBlanc, S.J.; Kulkarni, P.; Weninger, K.R. Single molecule FRET: A powerful tool to study intrinsically disordered proteins. Biomolecules 2018, 8, 140. [Google Scholar] [CrossRef]
- Pierce, M.M.; Raman, C.S.; Nall, B.T. Isothermal Titration Calorimetry of Protein–Protein Interactions. Methods 1999, 19, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.; Prenner, E. Differential scanning calorimetry: An invaluable tool for a detailed thermodynamic characterization of macromolecules and their interactions. J. Pharm. Bioallied Sci. 2011, 3, 39–59. [Google Scholar] [PubMed]
- Douzi, B. Protein–Protein Interactions: Surface Plasmon Resonance. In Bacterial Protein Secretion Systems: Methods and Protocols; Journet, L., Cascales, E., Eds.; Springer: New York, NY, USA, 2017; pp. 257–275. [Google Scholar]
- Lambrughi, M.; Tiberti, M.; Allega, M.F.; Sora, V.; Nygaard, M.; Toth, A.; Salamanca Viloria, J.; Bignon, E.; Papaleo, E. Analyzing Biomolecular Ensembles. In Biomolecular Simulations: Methods and Protocols; Bonomi, M., Camilloni, C., Eds.; Springer: New York, NY, USA, 2019; pp. 415–451. [Google Scholar]
- McLafferty, F. Tandem mass spectrometry. Science 1981, 214, 280–287. [Google Scholar] [CrossRef]
- Loo, J.A. Electrospray ionization mass spectrometry: A technology for studying noncovalent macromolecular complexes. Int. J. Mass Spectrom. 2000, 200, 175–186. [Google Scholar] [CrossRef]
- Daniel, J.M.; Friess, S.D.; Rajagopalan, S.; Wendt, S.; Zenobi, R. Quantitative determination of noncovalent binding interactions using soft ionization mass spectrometry. Int. J. Mass Spectrom. 2002, 216, 1–27. [Google Scholar] [CrossRef]
- Mitra, G. Emerging Role of Mass Spectrometry-Based Structural Proteomics in Elucidating Intrinsic Disorder in Proteins. Proteomics 2020, 2000011. [Google Scholar] [CrossRef]
- Boeri Erba, E.; Signor, L.; Petosa, C. Exploring the structure and dynamics of macromolecular complexes by native mass spectrometry. J. Proteom. 2020, 222, 103799. [Google Scholar] [CrossRef]
- Simon, R.P.; Winter, M.; Kleiner, C.; Ries, R.; Schnapp, G.; Heimann, A.; Li, J.; Zuvela-jelaska, L.; Bretschneider, T.; Luippold, A.H.; et al. MALDI-TOF Mass Spectrometry-Based High-Throughput Screening for Inhibitors of the Cytosolic DNA Sensor cGAS. SAGE 2019, 25. [Google Scholar] [CrossRef] [PubMed]
- Israr, M.Z.; Bernieh, D.; Salzano, A.; Cassambai, S.; Yazaki, Y.; Suzuki, T. Matrix-assisted laser desorption ionisation (MALDI) mass spectrometry (MS): Basics and clinical applications. Clin. Chem. Lab. Med. (Cclm) 2020, 58, 883. [Google Scholar] [CrossRef] [PubMed]
- Weidmann, S.; Zenobi, R. High-Mass MALDI-MS Using Ion Conversion Dynode Detectors: Influence of the Conversion Voltage on Sensitivity and Spectral Quality. J. Am. Soc. Mass Spectrom. 2014, 25, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-C.; Lai, Y.-H.; Ou, Y.-M.; Chang, H.-T.; Wang, Y.-S. Critical factors determining the quantification capability of matrix-assisted laser desorption/ionization—time-of-flight mass spectrometry. Philos. Trans. R. Soc. A: Math. Phys. Eng. Sci. 2016, 374, 20150371. [Google Scholar] [CrossRef]
- Pepelnjak, M.; de Souza, N.; Picotti, P. Detecting Protein–Small Molecule Interactions Using Limited Proteolysis–Mass Spectrometry (LiP-MS). Trends Biochem. Sci. 2020. [Google Scholar] [CrossRef]
- Uversky, V.; Longhi, S. Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 3. [Google Scholar]
- Cohen, S.L.; Ferré-D’Amaré, A.R.; Burley, S.K.; Chait, B.T. Probing the solution structure of the DNA-binding protein Max by a combination of proteolysis and mass spectrometry. Protein Sci. 1995, 4, 1088–1099. [Google Scholar] [CrossRef]
- Tomer, K.B.; Trojak, S.J.; Parker, C.E. Structural Studies of Protein-Protein Interactions Using Proteolytic Footprinting and MALDI/MS. In New Methods for the Study of Biomolecular Complexes; Ens, W., Standing, K.G., Chernushevich, I.V., Eds.; Springer: Dordrecht, The Netherlands, 1998; pp. 59–65. [Google Scholar]
- Kriwacki, R.W.; Wu, J.; Siuzdak, G.; Wright, P.E. Probing Protein/Protein Interactions with Mass Spectrometry and Isotopic Labeling: Analysis of the p21/Cdk2 Complex. JACS 1996, 118, 5320–5321. [Google Scholar] [CrossRef]
- Kriwacki, R.W.; Wu, J.; Tennant, L.; Wright, P.E.; Siuzdak, G. Probing protein structure using biochemical and biophysical methods: Proteolysis, matrix-assisted laser desorption/ionization mass spectrometry, high-performance liquid chromatography and size-exclusion chromatography of p21Waf1/Cip1/Sdi1. J. Chromatogr. A 1997, 777, 23–30. [Google Scholar] [CrossRef]
- Siuzdak, G. Probing viruses with mass spectrometry. J. Mass Spectrom. 1998, 33, 203–211. [Google Scholar] [CrossRef]
- Zlotnick, A.; Reddy, V.S.; Dasgupta, R.; Schneemann, A.; Ray, W.J.; Rueckert, R.R.; Johnson, J.E. Capsid assembly in a family of animal viruses primes an autoproteolytic maturation that depends on a single aspartic acid residue. J. Biol. Chem. 1994, 269, 13680–13684. [Google Scholar]
- Zhao, Y.; Xiao, K. A Mass Spectrometry-Based Structural Assay for Activation-Dependent Conformational Changes in β-Arrestins. In Beta-Arrestins; Humana Press: New York, NY, USA, 2019; pp. 293–308. [Google Scholar]
- Xiao, K.; Shenoy, S.K.; Nobles, K.; Lefkowitz, R.J. Activation-dependent conformational changes in {beta}-arrestin 2. J. Biol. Chem. 2004, 279, 55744–55753. [Google Scholar] [CrossRef]
- Shao, J.; Irwin, A.; Hartson, S.D.; Matts, R.L. Functional Dissection of Cdc37: Characterization of Domain Structure and Amino Acid Residues Critical for Protein Kinase Binding. Biochemistry 2003, 42, 12577–12588. [Google Scholar] [CrossRef] [PubMed]
- Dhungana, S.; Williams, J.G.; Fessler, M.B.; Tomer, K.B. Epitope Mapping by Proteolysis of Antigen–Antibody Complexes. In Epitope Mapping Protocols: Second Edition; Schutkowski, M., Reineke, U., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 87–101. [Google Scholar]
- Chalmers, M.J.; Busby, S.A.; Pascal, B.D.; West, G.M.; Griffin, P.R. Differential hydrogen/deuterium exchange mass spectrometry analysis of protein–ligand interactions. Expert Rev. Proteom. 2011, 8, 43–59. [Google Scholar] [CrossRef] [PubMed]
- Chapter 2 Hydrogen Exchange. Advances in Heterocyclic Chemistry Volume 47; Katritzky, A.R., Taylor, R., Eds.; Academic Press: San Diego, CA, USA, 1990; Volume 47, pp. 7–37. [Google Scholar]
- Persson, F.; Halle, B. How amide hydrogens exchange in native proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10383–10388. [Google Scholar] [CrossRef]
- Weis, D.D. Hydrogen Exchange Mass Spectrometry of Proteins: Fundamentals, Methods, and Applications; John Wiley & Sons: West Sussex, UK, 2016. [Google Scholar]
- Englander, S.W. Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Soc. Mass Spectrom. 2006, 17, 1481–1489. [Google Scholar] [CrossRef]
- Beckett, D. Hydrogen–Deuterium Exchange Study of an Allosteric Energy Cycle. In Allostery: Methods and Protocols; Fenton, A.W., Ed.; Springer: New York, NY, USA, 2012; pp. 261–278. [Google Scholar]
- Hodge, E.A.; Benhaim, M.A.; Lee, K.K. Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 2020, 29, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Mandell, J.G.; Falick, A.M.; Komives, E.A. Identification of protein–protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. USA 1998, 95, 14705–14710. [Google Scholar] [CrossRef]
- Mandell, J.G.; Falick, A.M.; Komives, E.A. Measurement of Amide Hydrogen Exchange by MALDI-TOF Mass Spectrometry. Anal. Chem. 1998, 70, 3987–3995. [Google Scholar] [CrossRef]
- Narayanan, S.; Mitra, G.; Muralidharan, M.; Mathew, B.; Mandal, A.K. Protein Structure–Function Correlation in Living Human Red Blood Cells Probed by Isotope Exchange-based Mass Spectrometry. Anal. Chem. 2015, 87, 11812–11818. [Google Scholar] [CrossRef]
- Woofter, R.T.; Maurer, M.C. Role of calcium in the conformational dynamics of factor XIII activation examined by hydrogen–deuterium exchange coupled with MALDI-TOF MS. Arch. Biochem. Biophys. 2011, 512, 87–95. [Google Scholar] [CrossRef]
- McDonald, C.; Li, L. Limited proteolysis combined with isotope labeling and quantitative LC-MALDI MS for monitoring protein conformational changes: A study on calcium-binding sites of cardiac Troponin C. Anal. Chim. Acta 2005, 534, 3–10. [Google Scholar] [CrossRef]
- Rand, K.D.; Bache, N.; Nedertoft, M.M.; Jørgensen, T.J.D. Spatially Resolved Protein Hydrogen Exchange Measured by Matrix-Assisted Laser Desorption Ionization In-Source Decay. Anal. Chem. 2011, 83, 8859–8862. [Google Scholar] [CrossRef] [PubMed]
- Pirrone, G.F.; Wang, H.; Canfield, N.; Chin, A.S.; Rhodes, T.A.; Makarov, A.A. Use of MALDI-MS Combined with Differential Hydrogen–Deuterium Exchange for Semiautomated Protein Global Conformational Screening. Anal. Chem. 2017, 89, 8351–8357. [Google Scholar] [CrossRef] [PubMed]
- Alexovič, M.; Urban, P.L.; Tabani, H.; Sabo, J. Recent advances in robotic protein sample preparation for clinical analysis and other biomedical applications. Clin. Chim. Acta 2020, 507, 104–116. [Google Scholar] [CrossRef]
- Kabaria, S.R.; Mangion, I.; Makarov, A.A.; Pirrone, G.F. Use of MALDI-MS with solid-state hydrogen deuterium exchange for semi-automated assessment of peptide and protein physical stability in lyophilized solids. Anal. Chim. Acta 2019, 1054, 114–121. [Google Scholar] [CrossRef]
- Reddy, N.C.; Kumar, M.; Molla, R.; Rai, V. Chemical methods for modification of proteins. Org. Biomol. Chem. 2020, 18, 4669–4691. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Hu, P.; MacGregor, P.; Xue, Y.; Fan, H.; Suchecki, P.; Olszewski, L.; Liu, A. Understanding the Conformational Impact of Chemical Modifications on Monoclonal Antibodies with Diverse Sequence Variation Using Hydrogen/Deuterium Exchange Mass Spectrometry and Structural Modeling. Anal. Chem. 2014, 86, 3468–3475. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Gerber, S.; Heuser, K.; Korkhov, V.M.; Lizak, C.; Mireku, S.; Locher, K.P.; Zenobi, R. High-Mass Matrix-Assisted Laser Desorption Ionization-Mass Spectrometry of Integral Membrane Proteins and Their Complexes. Anal. Chem. 2013, 85, 3483–3488. [Google Scholar] [CrossRef]
- Chavez, J.D.; Mohr, J.P.; Mathay, M.; Zhong, X.; Keller, A.; Bruce, J.E. Systems structural biology measurements by in vivo cross-linking with mass spectrometry. Nat. Protoc. 2019, 14, 2318–2343. [Google Scholar] [CrossRef]
- Iacobucci, C.; Götze, M.; Sinz, A. Cross-linking/mass spectrometry to get a closer view on protein interaction networks. Curr. Opin. Biotechnol. 2020, 63, 48–53. [Google Scholar] [CrossRef]
- Chang, Z.; Kuchar, J.; Hausinger, R.P. Chemical cross-linking and mass spectrometric identification of sites of interaction for UreD, UreF, and urease. J. Biol. Chem. 2004, 279, 15305–15313. [Google Scholar] [CrossRef]
- Nguyen-Huynh, N.-T.; Sharov, G.; Potel, C.; Fichter, P.; Trowitzsch, S.; Berger, I.; Lamour, V.; Schultz, P.; Potier, N.; Leize-Wagner, E. Chemical cross-linking and mass spectrometry to determine the subunit interaction network in a recombinant human SAGA HAT subcomplex. Protein Sci. 2015, 24, 1232–1246. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: San Diego, CA, USA, 2013. [Google Scholar]
- Wong, S.S. Chemistry of Protein Conjugation and Cross-Linking; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Müller, M.Q.; Zeiser, J.J.; Dreiocker, F.; Pich, A.; Schäfer, M.; Sinz, A. A universal matrix-assisted laser desorption/ionization cleavable cross-linker for protein structure analysis. Rapid Commun. Mass Spectrom. 2011, 25, 155–161. [Google Scholar] [CrossRef]
- Mädler, S.; Barylyuk, K.; Boeri Erba, E.; Nieckarz, R.J.; Zenobi, R. Compelling Advantages of Negative Ion Mode Detection in High-Mass MALDI-MS for Homomeric Protein Complexes. J. Am. Soc. Mass Spectrom. 2012, 23, 213–224. [Google Scholar] [CrossRef][Green Version]
- Köhler, M.; Neff, C.; Perez, C.; Brunner, C.; Pardon, E.; Steyaert, J.; Schneider, G.; Locher, K.P.; Zenobi, R. Binding Specificities of Nanobody•Membrane Protein Complexes Obtained from Chemical Cross-Linking and High-Mass MALDI Mass Spectrometry. Anal. Chem. 2018, 90, 5306–5313. [Google Scholar] [CrossRef]
- Krauth, F.; Ihling, C.H.; Rüttinger, H.H.; Sinz, A. Heterobifunctional isotope-labeled amine-reactive photo-cross-linker for structural investigation of proteins by matrix-assisted laser desorption/ionization tandem time-of-flight and electrospray ionization LTQ-Orbitrap mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Sinz, A.; Kalkhof, S.; Ihling, C. Mapping protein interfaces by a trifunctional cross-linker combined with MALDI-TOF and ESI-FTICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2005, 16, 1921–1931. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ptáčková, R.; Ječmen, T.; Novák, P.; Hudeček, J.; Stiborová, M.; Šulc, M. The application of an emerging technique for protein-protein interaction interface mapping: The combination of photo-initiated cross-linking protein nanoprobes with mass spectrometry. Int. J. Mol. Sci. 2014, 15, 9224–9241. [Google Scholar] [CrossRef]
- Li, G.; Ma, F.; Cao, Q.; Zheng, Z.; DeLaney, K.; Liu, R.; Li, L. Nanosecond photochemically promoted click chemistry for enhanced neuropeptide visualization and rapid protein labeling. Nat. Commun. 2019, 10, 4697. [Google Scholar] [CrossRef]
- Petrotchenko, E.V.; Xiao, K.; Cable, J.; Chen, Y.; Dokholyan, N.V.; Borchers, C.H. BiPS, a Photocleavable, Isotopically Coded, Fluorescent Cross-linker for Structural Proteomics. Mol. Cell. Proteom. 2009, 8, 273–286. [Google Scholar] [CrossRef]
- Rosinke, B.; Strupat, K.; Hillenkamp, F.; Rosenbusch, J.; Dencher, N.; Krüger, U.; Galla, H.-J. Matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) of membrane proteins and non-covalent complexes. J. Mass Spectrom. 1995, 30, 1462–1468. [Google Scholar] [CrossRef]
- Cohen, S.L.; Padovan, J.C.; Chait, B.T. Mass Spectrometric Analysis of Mercury Incorporation into Proteins for X-ray Diffraction Phase Determination. Anal. Chem. 2000, 72, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, A.; Pimenova, T.; Alves, S.; Zenobi, R. Investigation of the first shot phenomenon in MALDI mass spectrometry of protein complexes. Analyst 2007, 132, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Beaufour, M.; Ginguené, D.; Le Meur, R.; Castaing, B.; Cadene, M. Liquid Native MALDI Mass Spectrometry for the Detection of Protein-Protein Complexes. J. Am. Soc. Mass Spectrom. 2018, 29, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, R.L.; Cooper, H.J. Direct Tissue Profiling of Protein Complexes: Toward Native Mass Spectrometry Imaging. Anal. Chem. 2016, 88, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Hale, O.J.; Cooper, H.J. Native Mass Spectrometry Imaging and In Situ Top-Down Identification of Intact Proteins Directly from Tissue. J. Am. Soc. Mass Spectrom. 2020. [Google Scholar] [CrossRef]
- Zavalin, A.; Todd, E.M.; Rawhouser, P.D.; Yang, J.; Norris, J.L.; Caprioli, R.M. Direct imaging of single cells and tissue at sub-cellular spatial resolution using transmission geometry MALDI MS. J. Mass Spectrom. 2012, 47, 1473–1481. [Google Scholar] [CrossRef]
- Thomen, A.; Najafinobar, N.; Penen, F.; Kay, E.; Upadhyay, P.P.; Li, X.; Phan, N.T.N.; Malmberg, P.; Klarqvist, M.; Andersson, S.; et al. Subcellular Mass Spectrometry Imaging and Absolute Quantitative Analysis across Organelles. Acs Nano 2020, 14, 4316–4325. [Google Scholar] [CrossRef]
- Meng, Y.F.; Cheng, X.L.; Wang, T.T.; Hang, W.; Li, X.P.; Nie, W.; Liu, R.; Lin, Z.; Hang, L.; Yin, Z.B.; et al. Micro-Lensed Fiber Laser Desorption Mass Spectrometry Imaging Reveals Subcellular Distribution of Drugs within Single Cells. Angew. Chem. 2020. [Google Scholar]
- Hytönen, V.P.; Wehrle-Haller, B. Protein conformation as a regulator of cell–matrix adhesion. Phys. Chem. Chem. Phys. 2014, 16, 6342–6357. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Basis of Interaction | Bond Energy (kcal/mol) |
|---|---|---|
| Covalent bond | Sharing electron pairs | 50–110 |
| Hydrogen bond | Sharing H atom | 3–7 |
| Ionic bond | The attraction of opposite charges | 3–7 |
| Hydrophobic interaction | Interaction of non-polar group | 1–2 |
| Van der Waals interaction | Interaction of electrons in the presence of polar substances | 1 |
| π-π interaction | Π-molecular orbital interaction | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giampà, M.; Sgobba, E. Insight to Functional Conformation and Noncovalent Interactions of Protein-Protein Assembly Using MALDI Mass Spectrometry. Molecules 2020, 25, 4979. https://doi.org/10.3390/molecules25214979
Giampà M, Sgobba E. Insight to Functional Conformation and Noncovalent Interactions of Protein-Protein Assembly Using MALDI Mass Spectrometry. Molecules. 2020; 25(21):4979. https://doi.org/10.3390/molecules25214979
Chicago/Turabian StyleGiampà, Marco, and Elvira Sgobba. 2020. "Insight to Functional Conformation and Noncovalent Interactions of Protein-Protein Assembly Using MALDI Mass Spectrometry" Molecules 25, no. 21: 4979. https://doi.org/10.3390/molecules25214979
APA StyleGiampà, M., & Sgobba, E. (2020). Insight to Functional Conformation and Noncovalent Interactions of Protein-Protein Assembly Using MALDI Mass Spectrometry. Molecules, 25(21), 4979. https://doi.org/10.3390/molecules25214979