An Updated Review on the Synthesis and Antibacterial Activity of Molecular Hybrids and Conjugates Bearing Imidazole Moiety


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Heterocyclic Conjugates and Hybrids Bearing Nitroimidazole Moiety
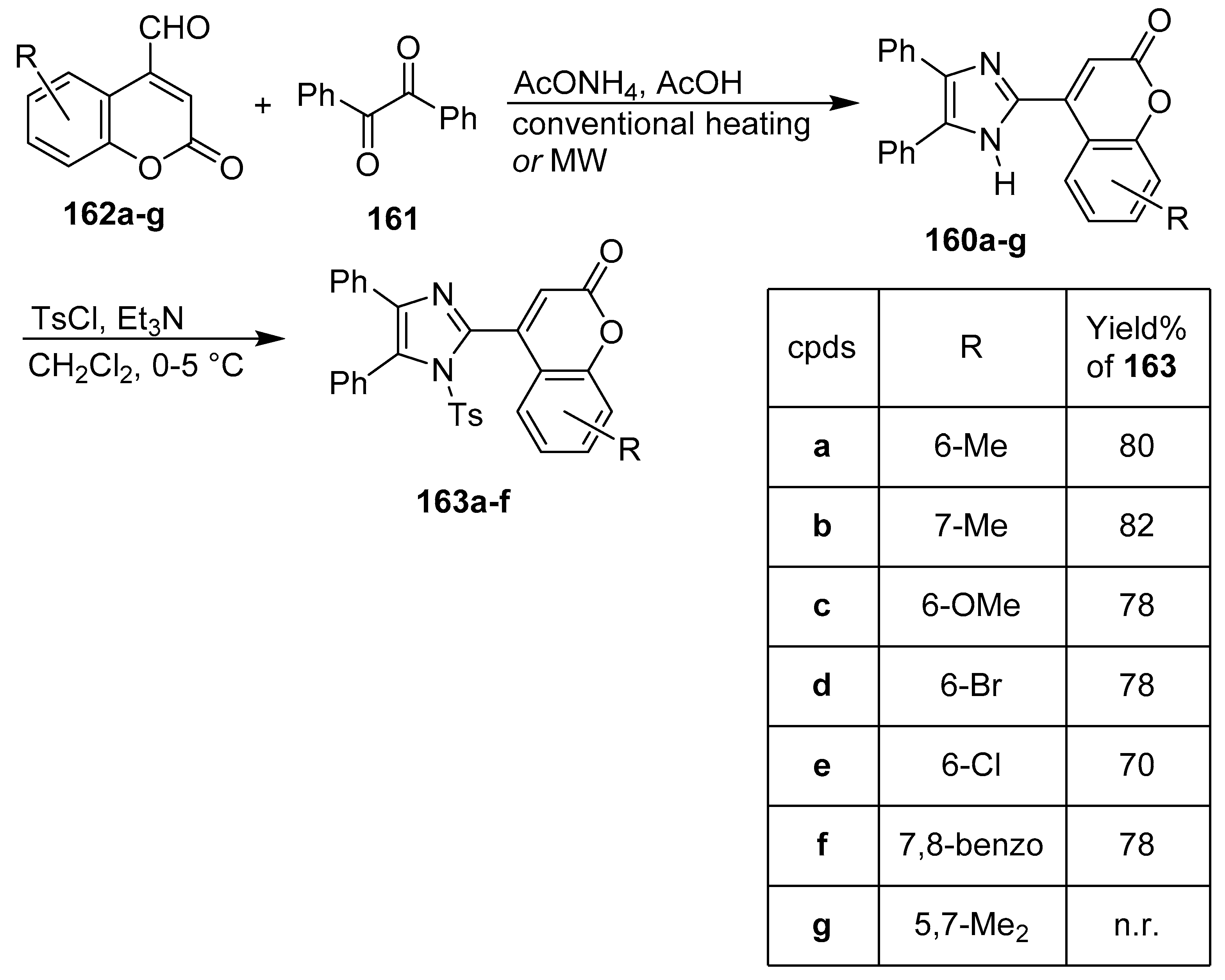
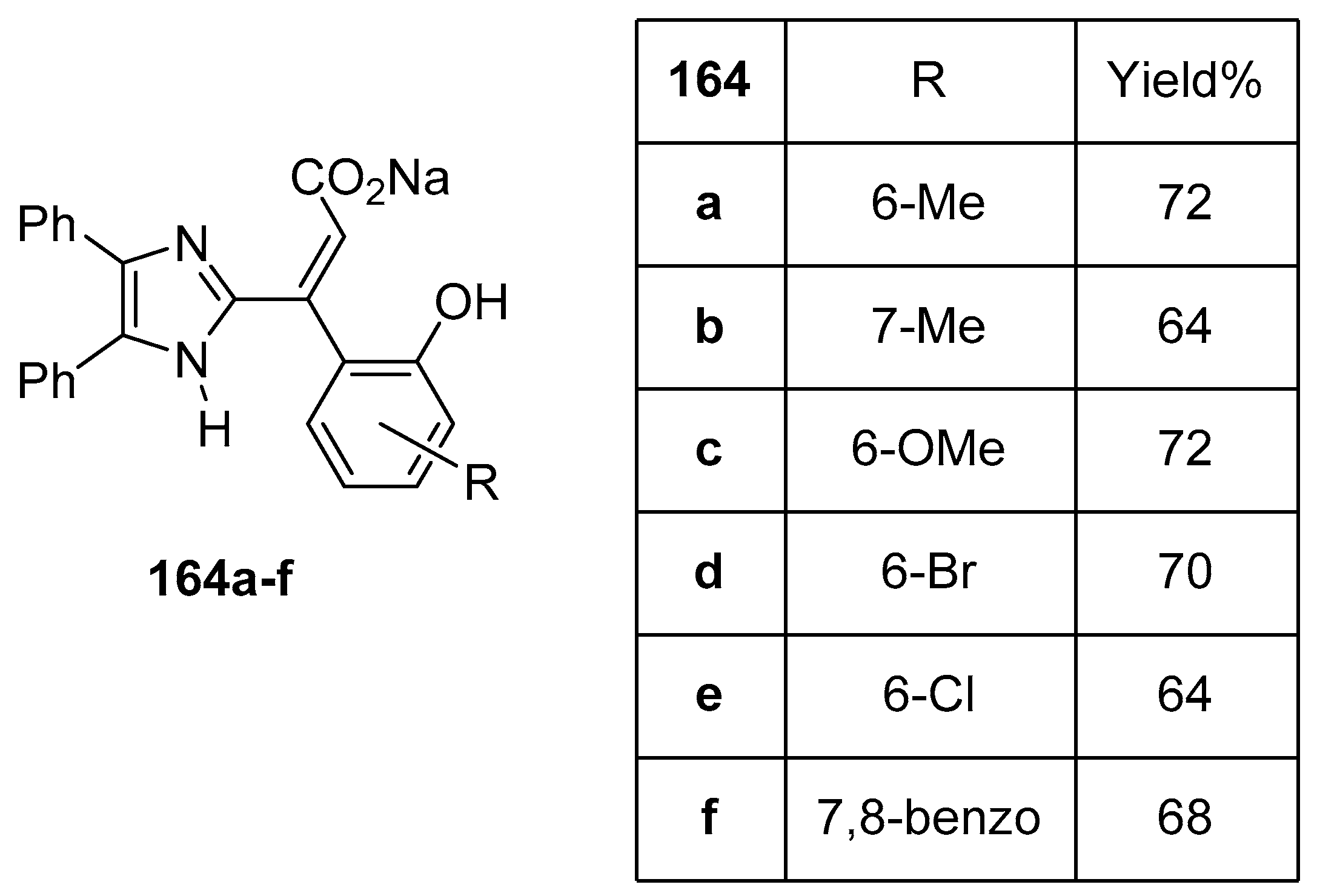
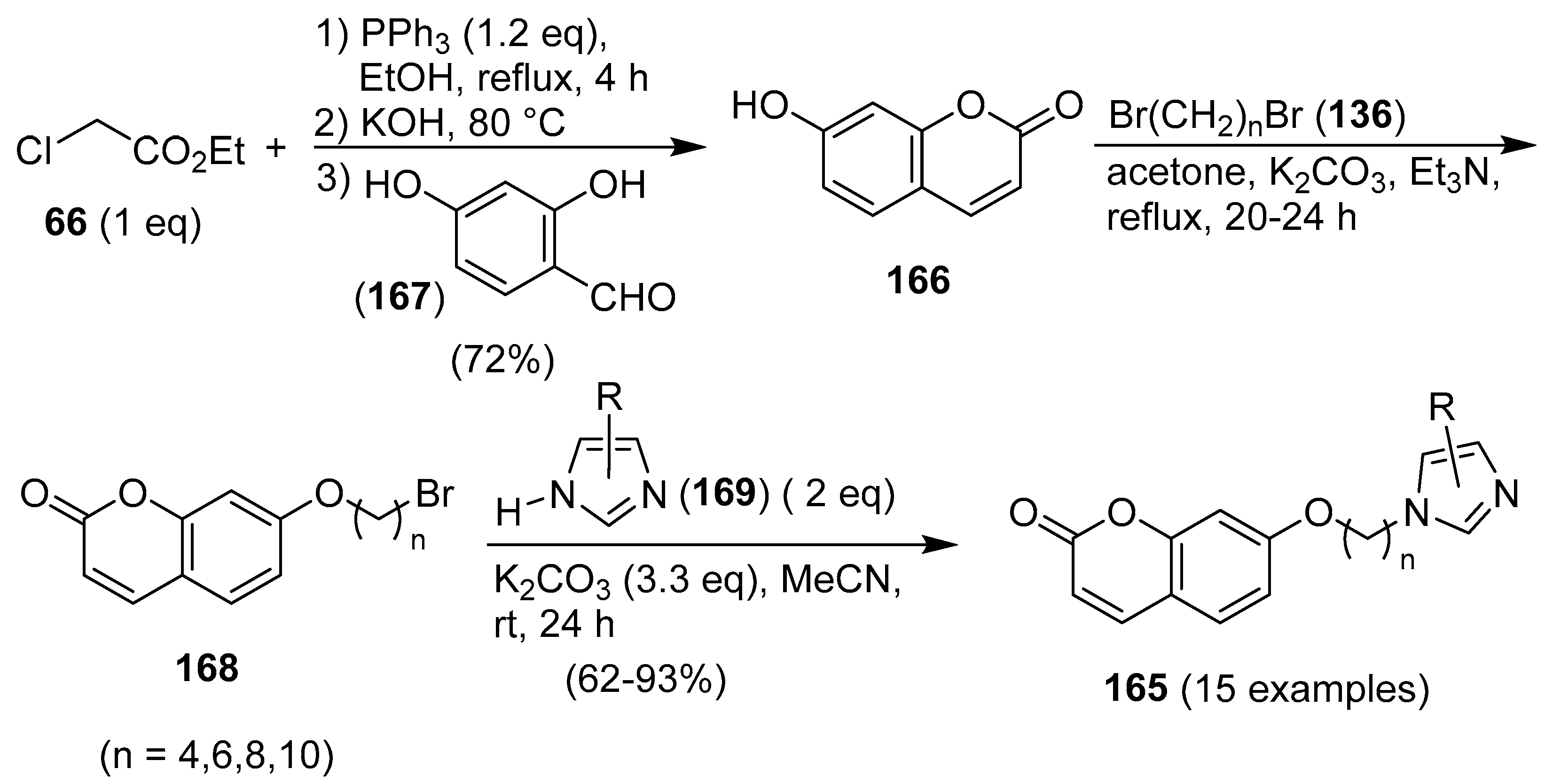
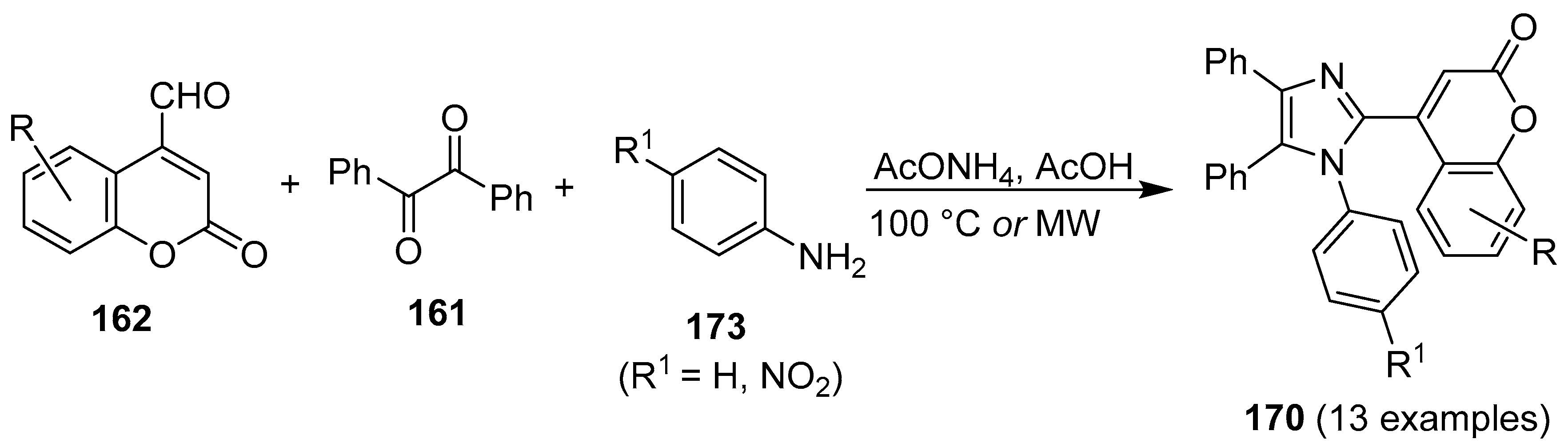
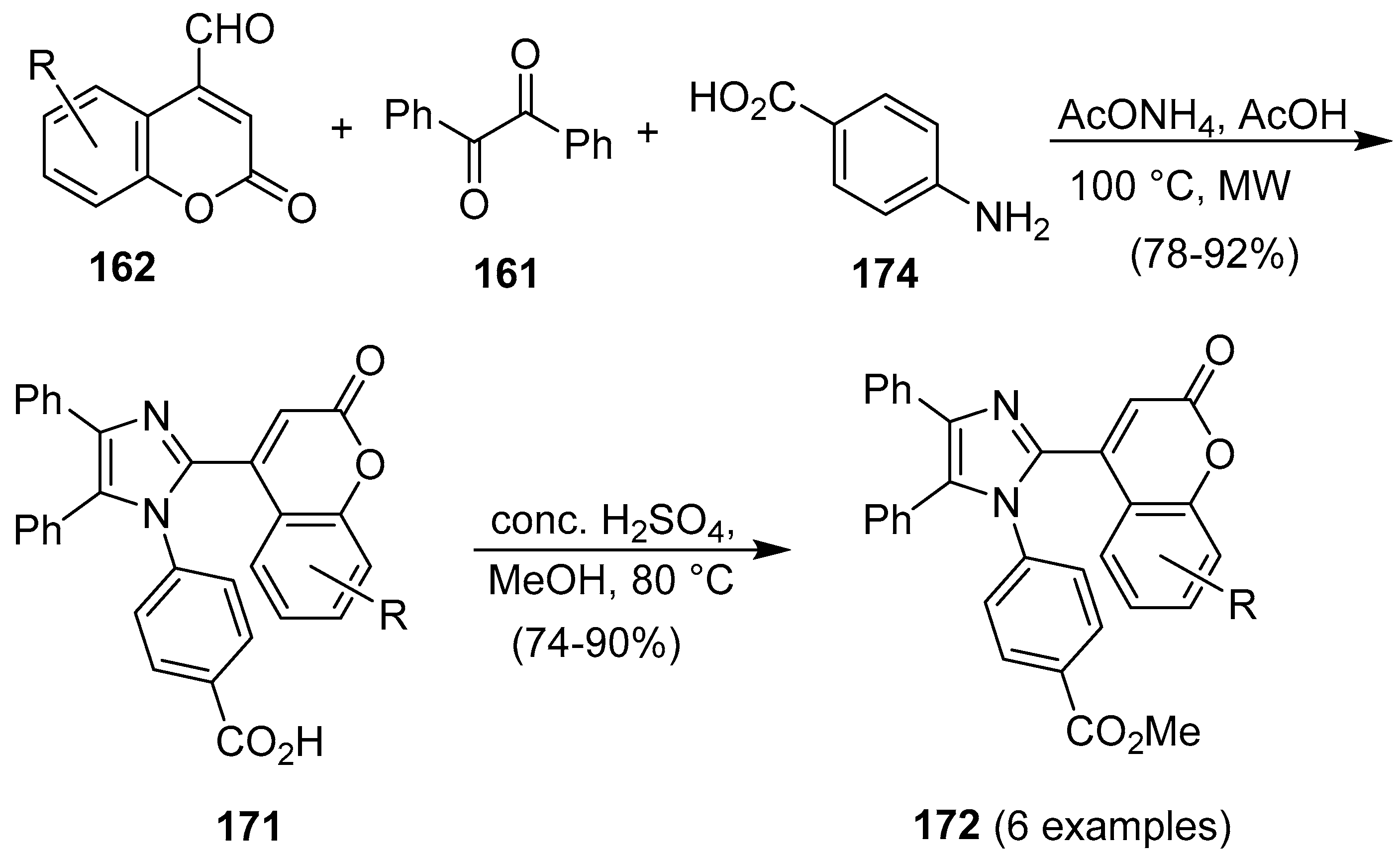
3. Coumarin/Imidazole Hybrids and Conjugates
4. Furanchalcone/Imidazole Hybrids
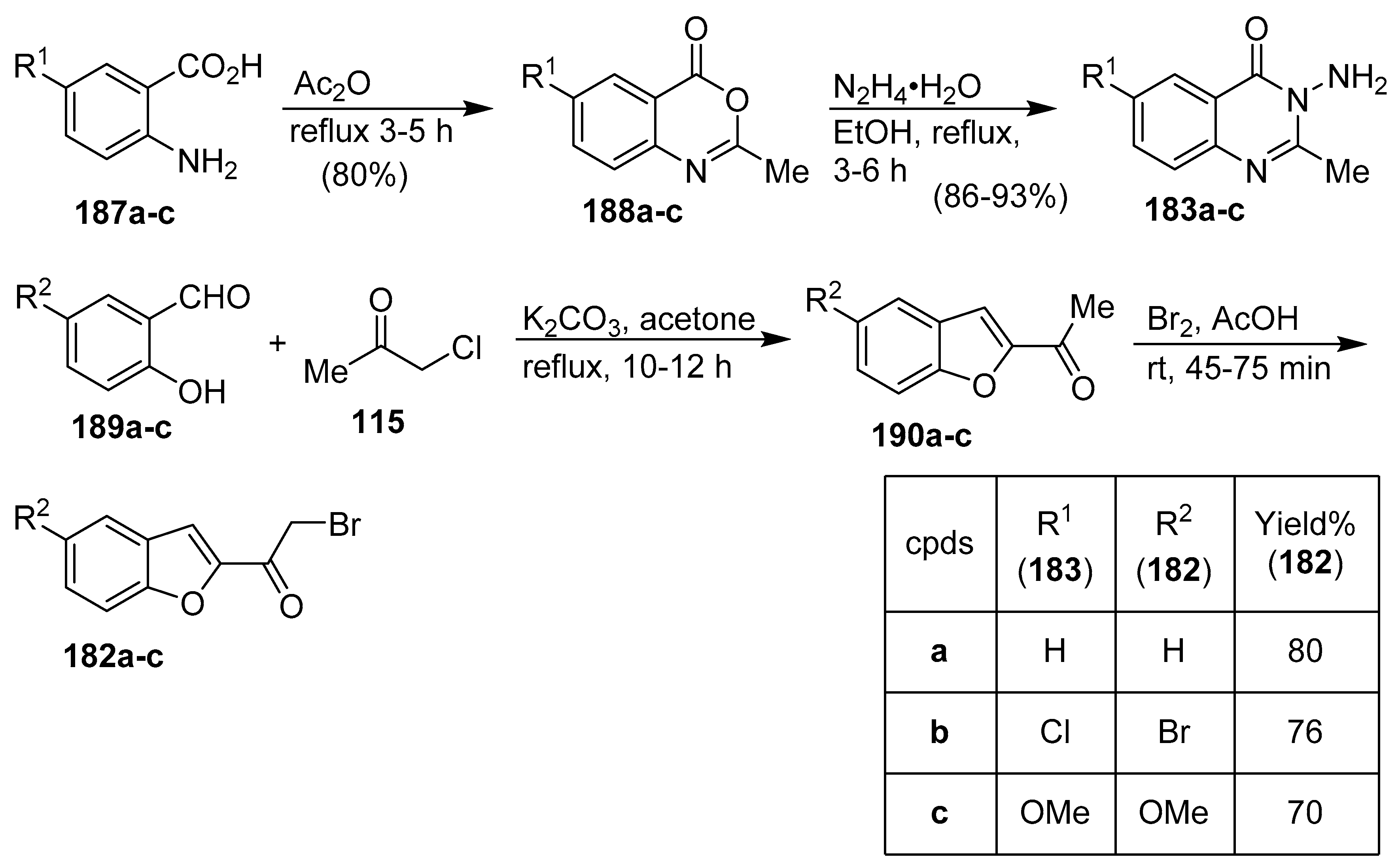
5. Hybrids Based on Benzofuran, Quinazolinone, and Imidazolium Moieties
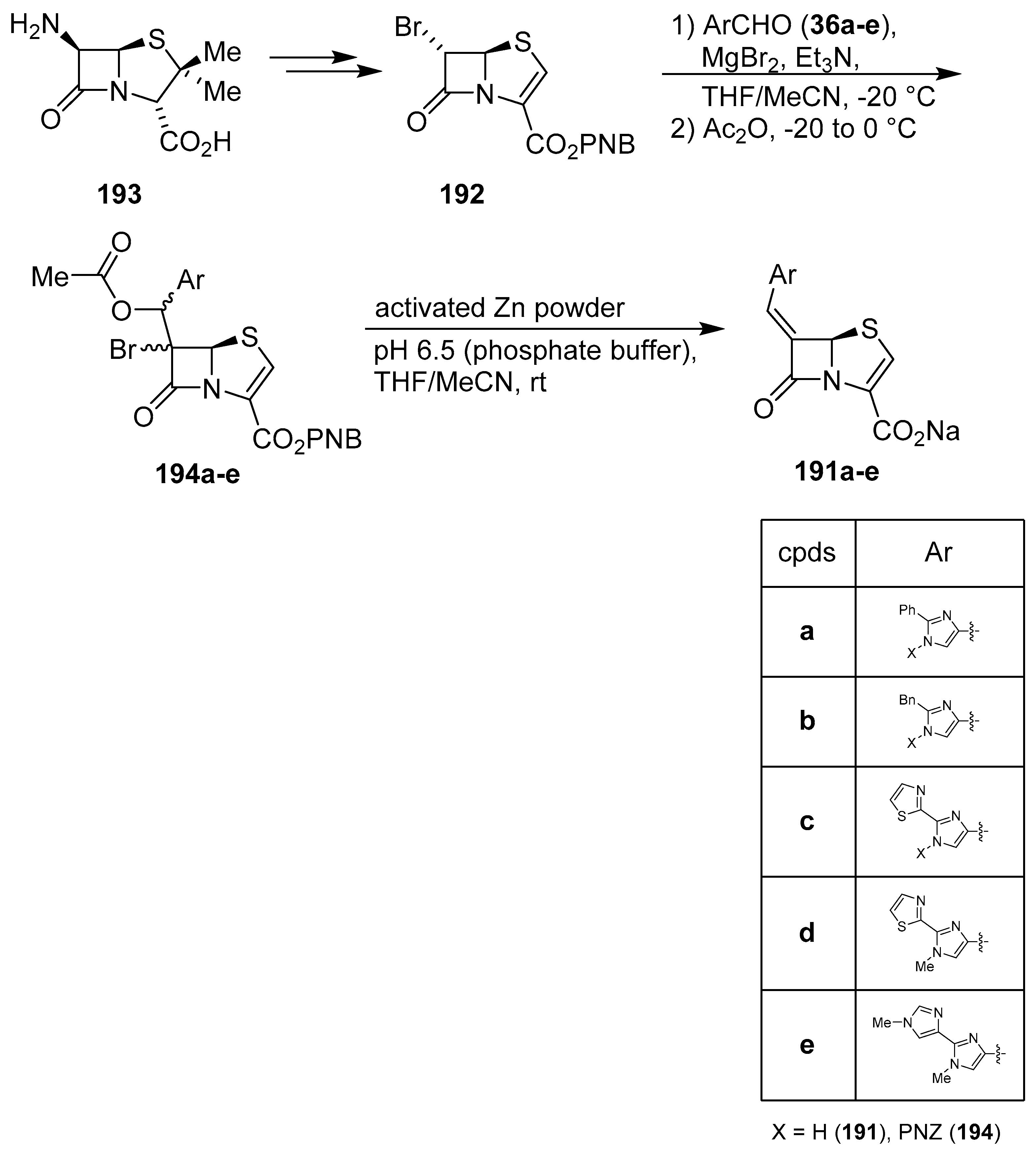
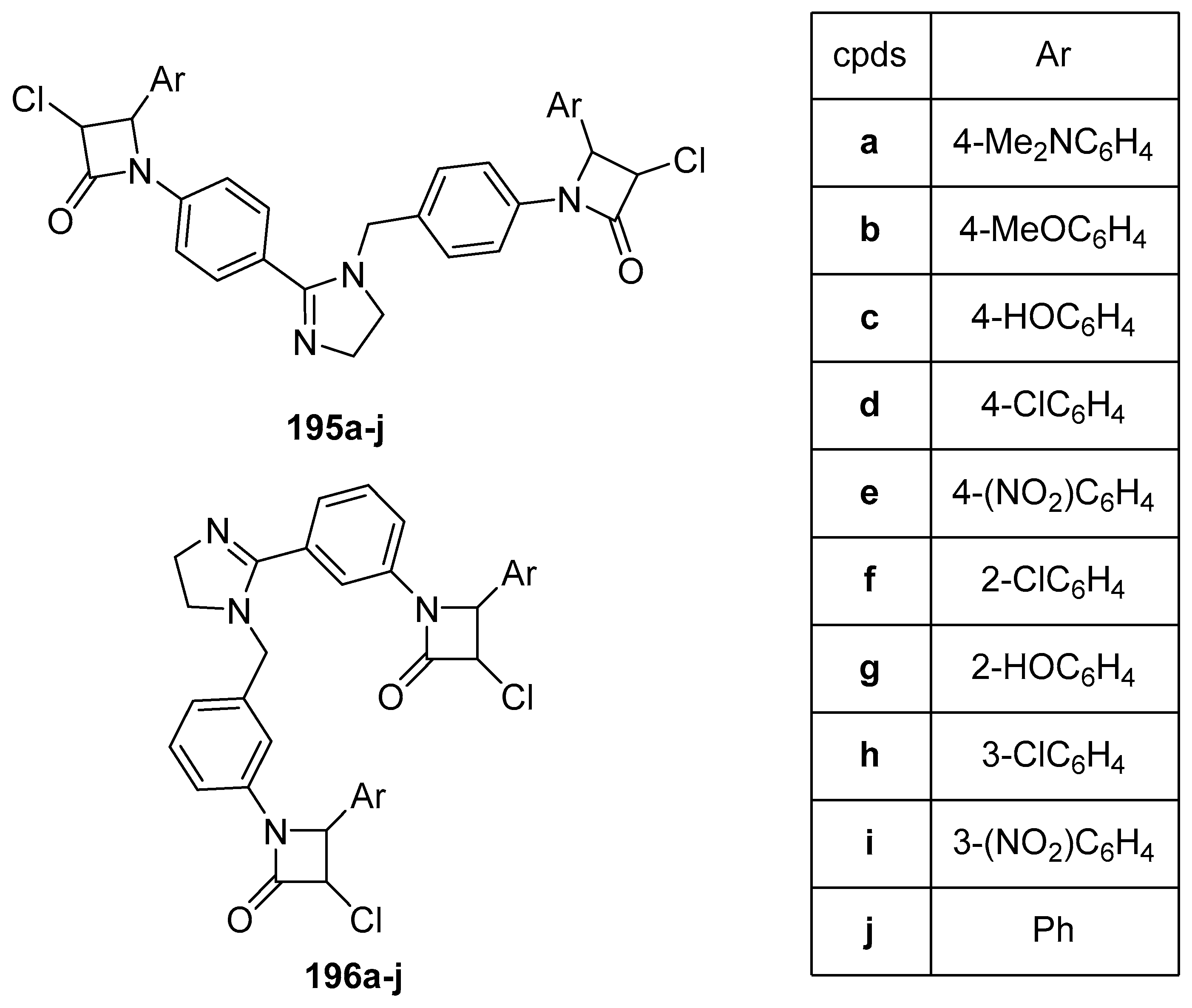
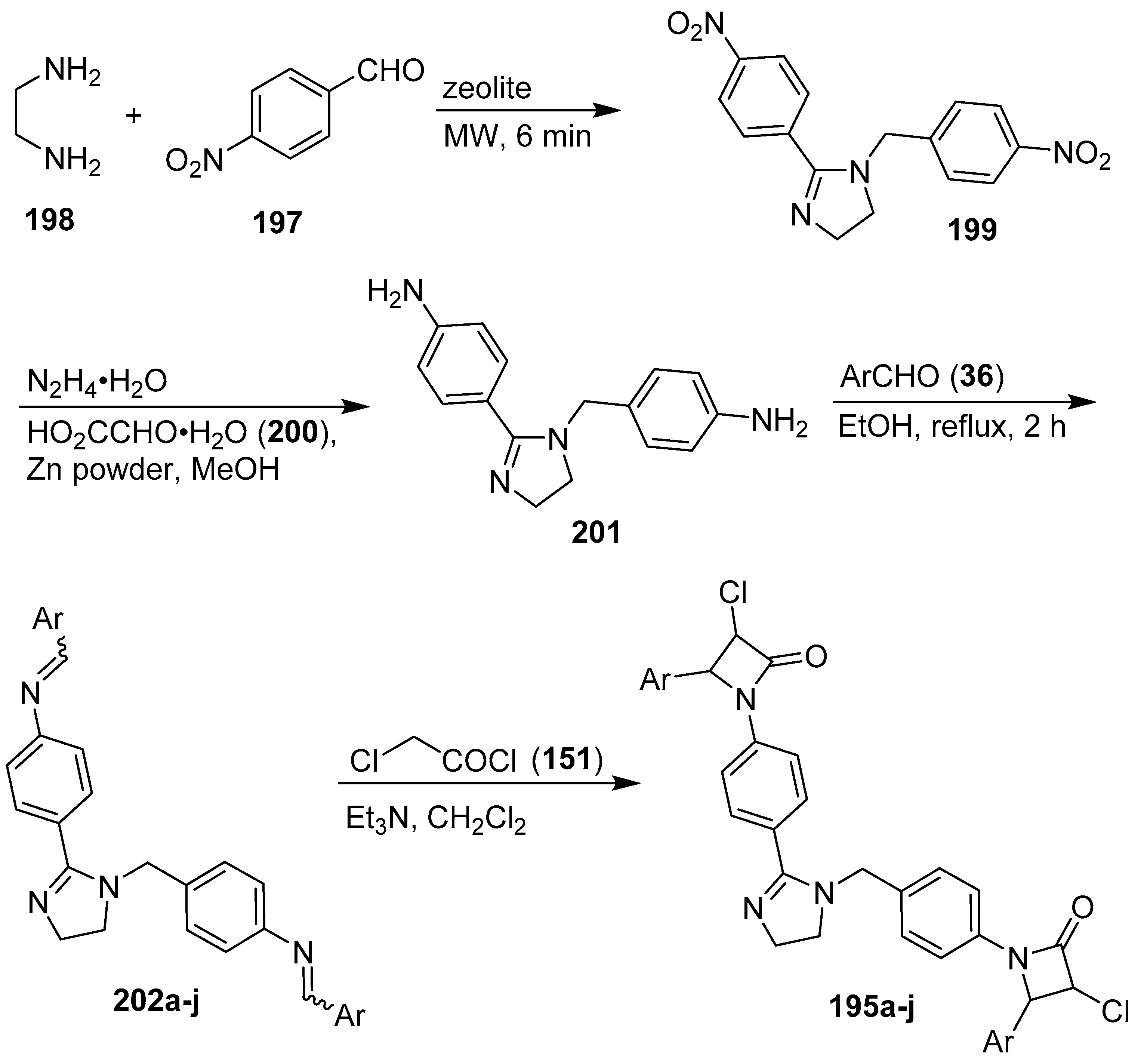
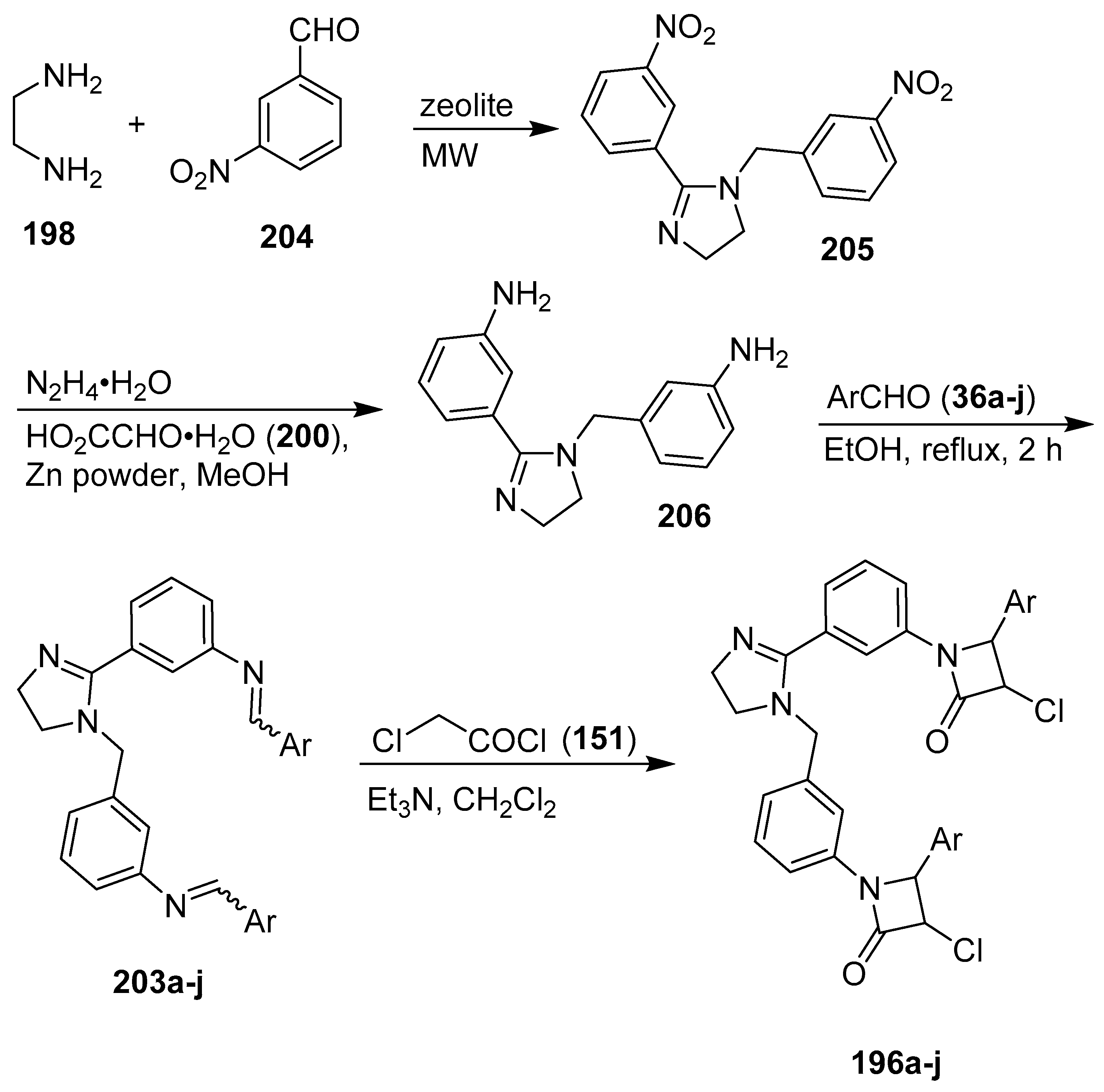
6. 1H-Imidazoles Containing Azetidin-2-one (β-lactam) Derivatives
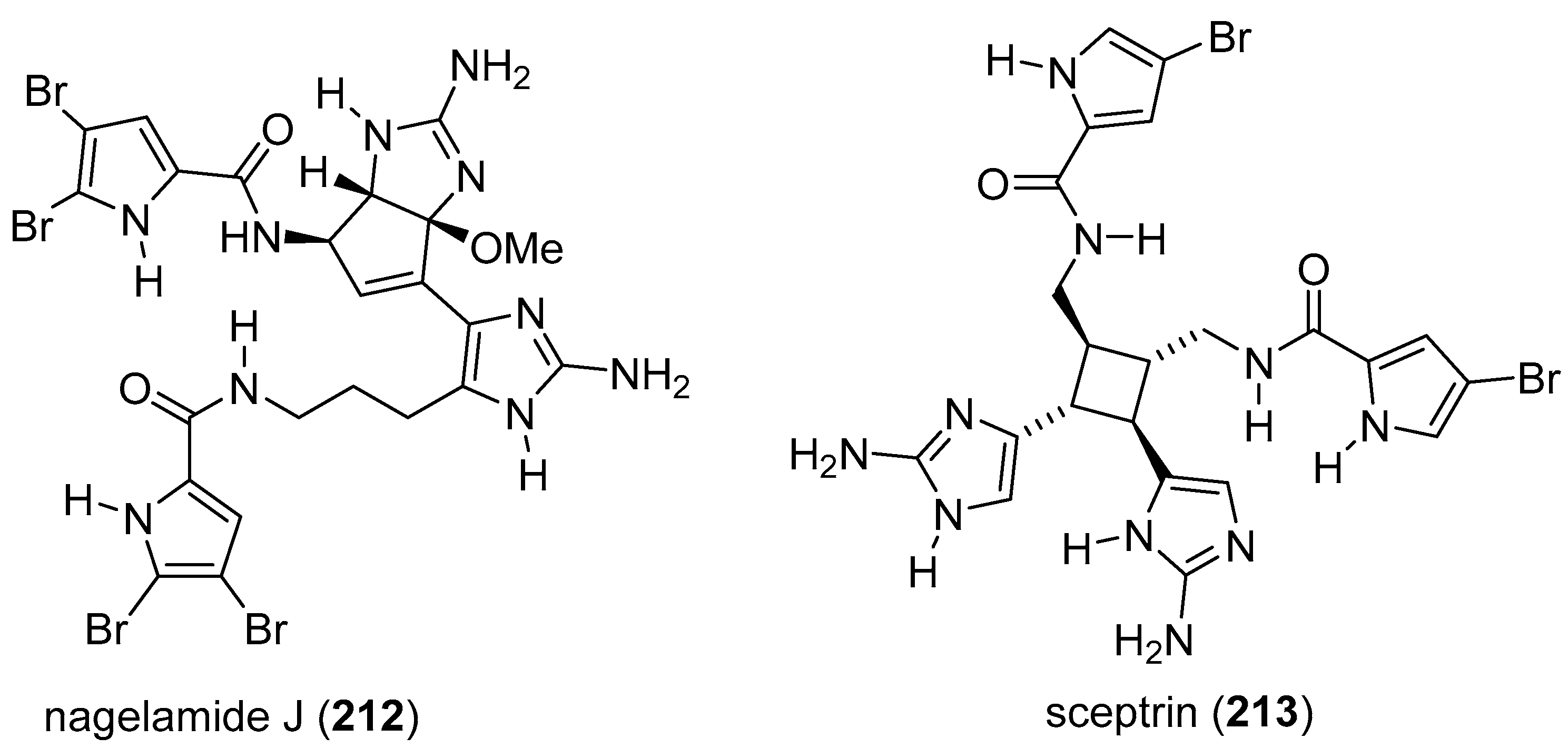
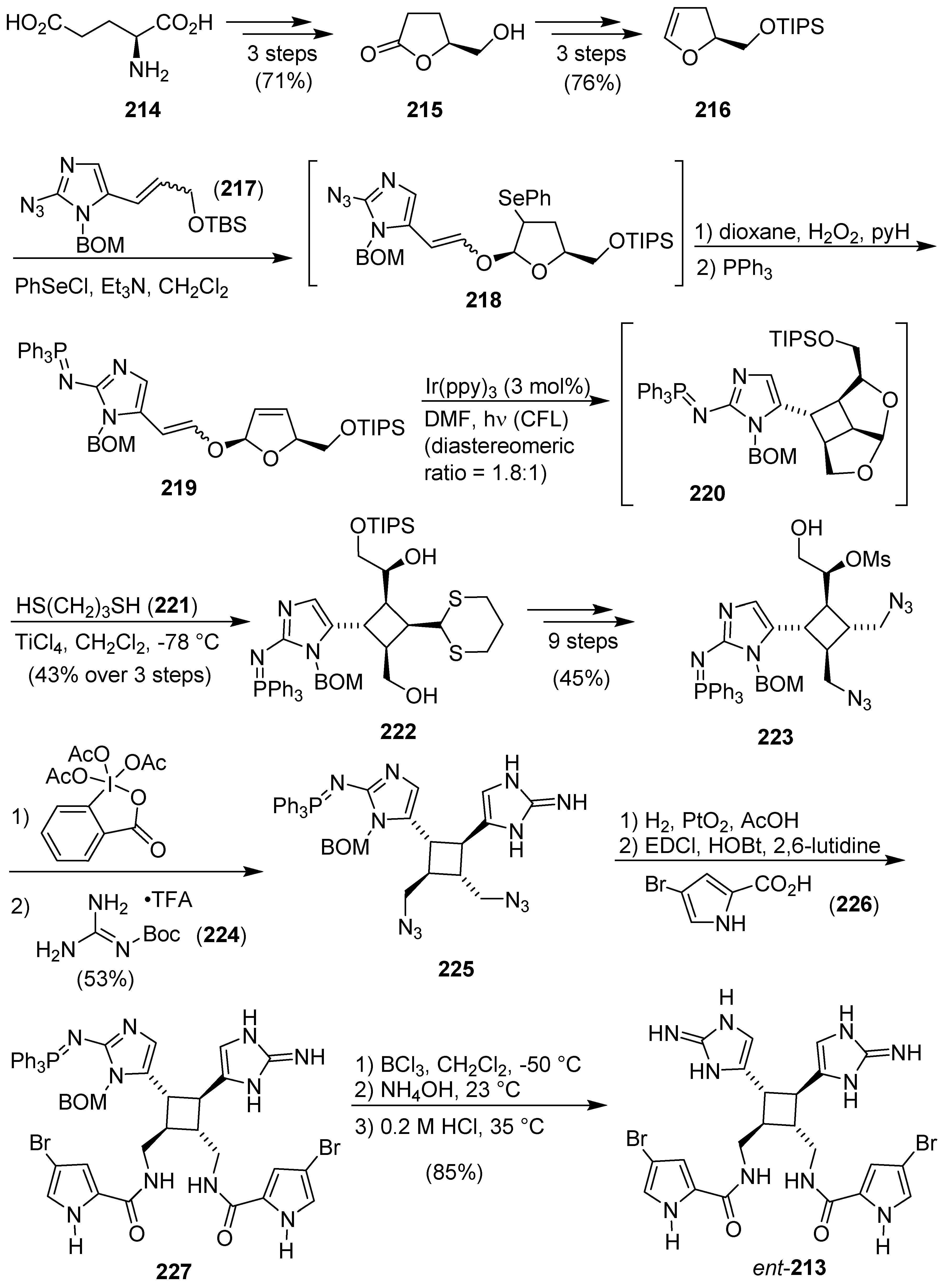
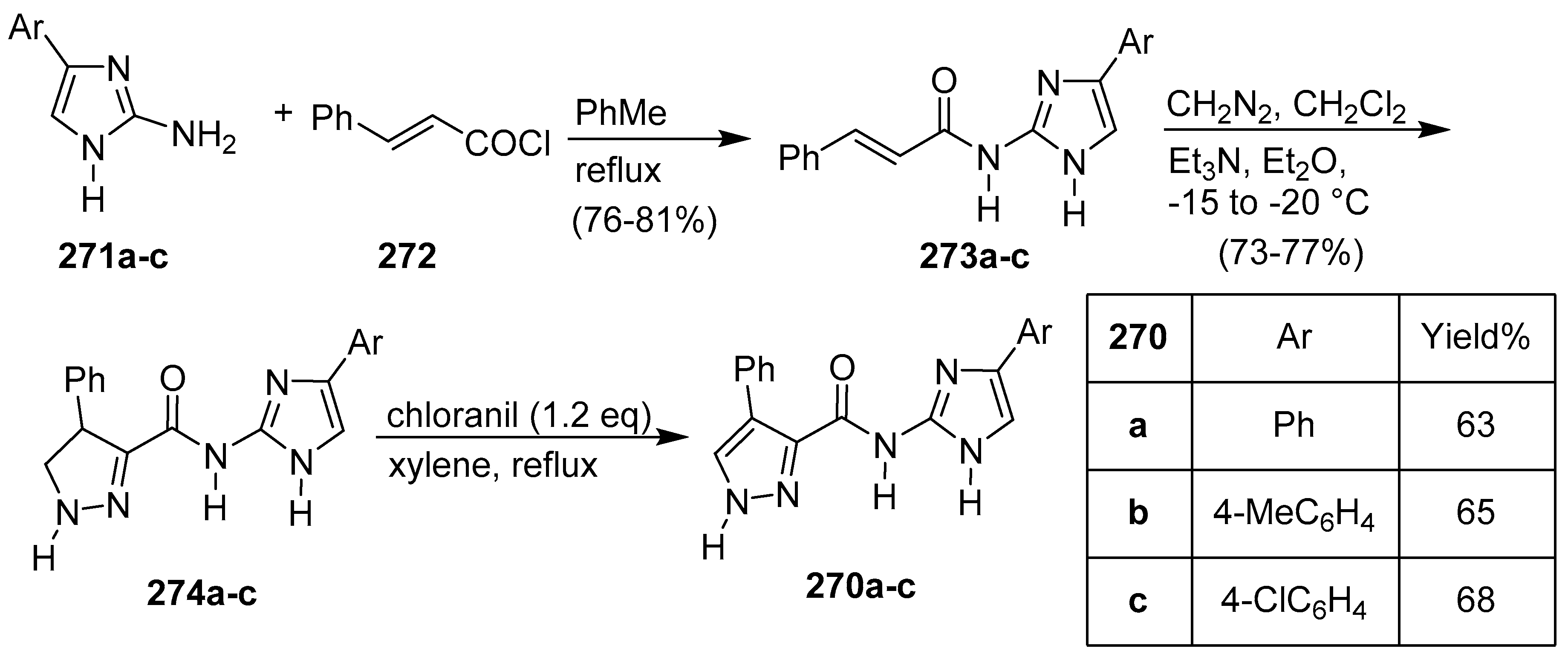
7. Pyrrole/Imidazole and Indole/Imidazole Hybrids
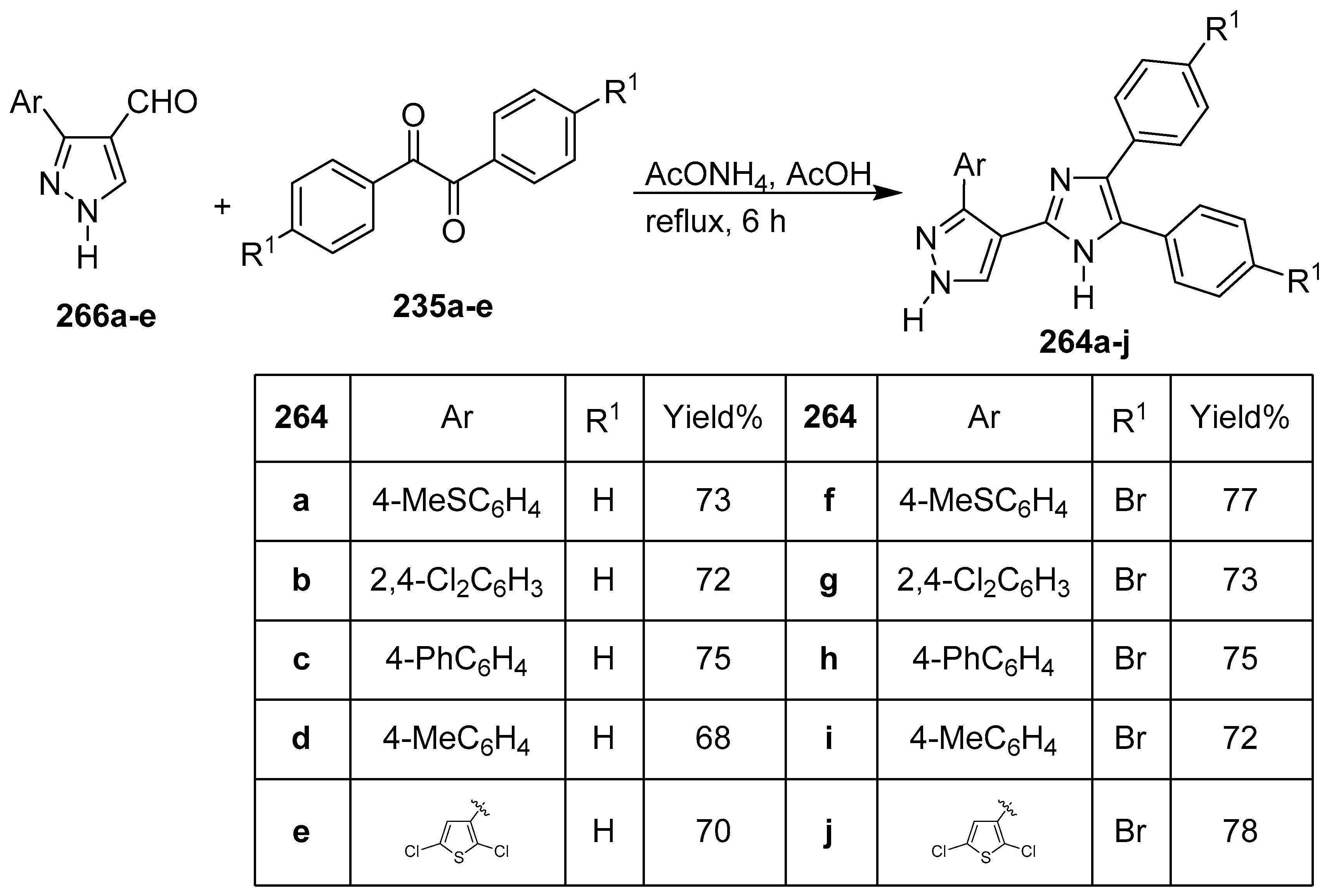
8. Pyrazole/Imidazole Hybrids
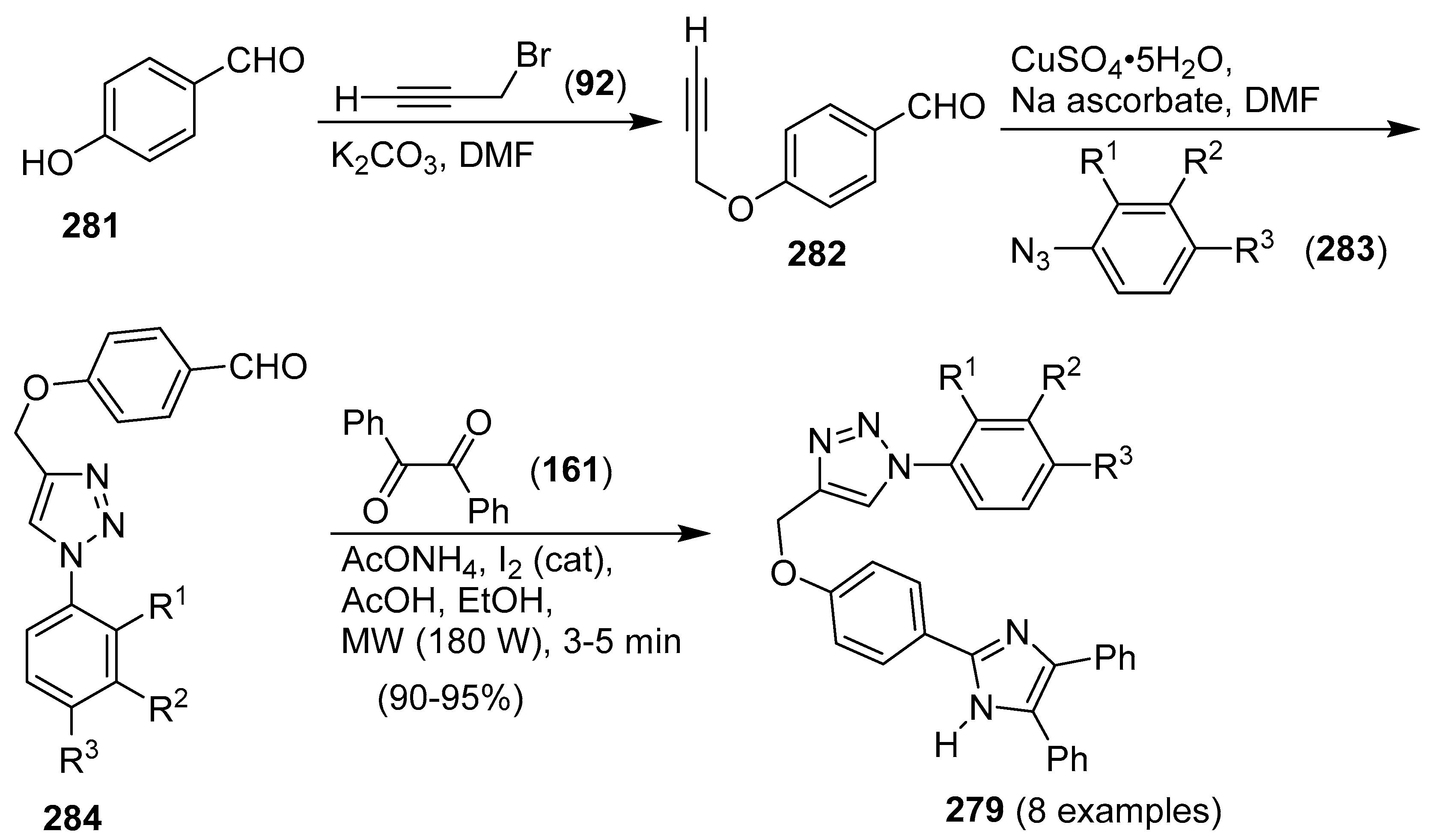
9. 1,2,3- and 1,2,4-Triazole/Imidazole Hybrids and Conjugates
10. Hybrids of Imidazole Derivatives and 5-Membered Heterocycles Containing Oxygen and Nitrogen
11. 1,8-Naphthalimide/Imidazole Hybrids
12. Bis-Imidazoles
13. Pyridine/Imidazole Hybrids
14. Quinoline/Imidazole Hybrids
15. Pyrimidine/Imidazole Hybrids
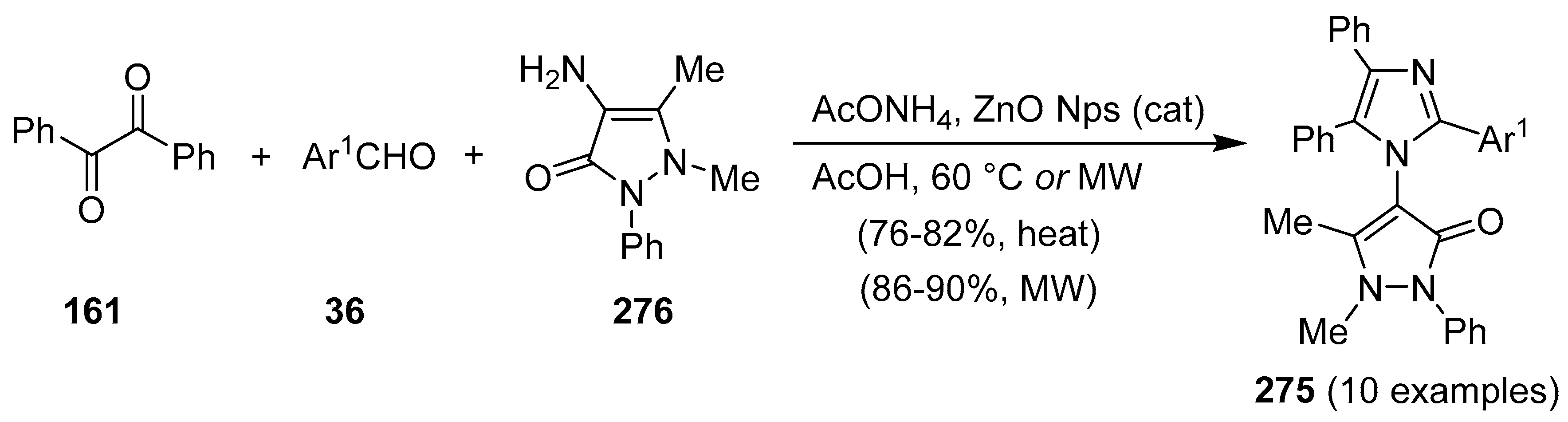
16. Addendum
17. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ac | acetyl |
| Ar | aryl |
| ATCC | American Type Culture Collection |
| Bn | benzyl |
| Boc | tert-butoxycarbonyl |
| BOM | benzyloxymethyl |
| Bu | butyl |
| CAN | cerium(IV) ammonium nitrate |
| Cbz | N-carboxybenzyl |
| CDI | 1,1′-carbonyldiimidazole |
| CFL | compact fluorescent lamp |
| CGMCC | China General Microbiological Culture Collection Center |
| m-CPBA | m-chloroperoxybenzoic acid |
| DBU | 1,5-diazabicyclo[5.4.0]undec-7-ene |
| DEAD | diethyl azodicarboxylate |
| DIBAL-H | diisobutylaluminum hydride |
| DIPEA | N,N-diisopropylethylamine |
| DMSO | dimethyl sulfoxide |
| DPIX | deuterioporphyrin |
| DMF | dimethylformamide |
| ED50 | half effective dose |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| Eh | 2-ethylhexyl |
| eq | (stoichiometric) equivalent |
| Et | ethyl |
| FDA | Food and Drug Administration |
| Fmoc | fluorenylmethyloxycarbonyl |
| HOBt | hydroxybenzotriazole |
| HBTU | O-(benzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate |
| HMTA | hexamethylenetetramine |
| IC50 | half inhibitory concentration (bacterial growth or biochemical) |
| Ir(ppy)3 | tris(2-phenylpyridine)iridium |
| Me | methyl |
| MIC | minimum inhibitory concentration (bacterial growth) |
| MRSA | methicillin-resistant Staphylococcus aureus |
| Ms | methanesulfonyl |
| MTCC | Microbial Type Culture Collection (India) |
| Mtz | metronidazole |
| MW | microwave |
| Nim | 5-nitro-1H-imidazol-1-yl |
| NMM | 4-methylmorpholine |
| Nps | nanoparticles |
| n.r. | not reported |
| Ns | nosyl (p-nitrobenzesulfonyl) |
| PBP | penicillin binding protein |
| PCC | pyridinium chlorochromate |
| Ph | phenyl |
| PNB | p-nitrobenzyl |
| PNZ | p-nitrobenzyloxycarbonyl |
| Pr | propyl |
| Pro | proline |
| PTCC | Persian Type Culture Collection |
| Py | pyridyl |
| rt | room temperature |
| TBAB | tetrabutylammonium bromide |
| TBS | tert-butyldimethylsilyl |
| TBTU | N,N,N′,N′-tetramethyl-O-(benzotriazol-1-yl)uronium tetrafluoroborate |
| Tf | triflyl (trifluoromethanesulfonyl) |
| TFA | trifluoroacetic acid |
| THF | tetrahydrofuran |
| TIPS | trisopropylsilyl |
| Ts | tosyl (p-toluenesulfonyl) |
References and Notes
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breijyeh, Z.; Jubeh, B.; Karaman, R. Resistance of Gram-Negative Bacteria to Current Antibacterial Agents and Approaches to Resolve It. Molecules 2020, 25, 1340. [Google Scholar] [CrossRef] [Green Version]
- Rai, J.; Randhawa, G.K.; Kaur, M. Recent advances in antibacterial drugs. Int. J. Appl. Basic Med. Res. 2013, 3, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, N.; Czaplewski, L.; Piddock, L.J.V. Discovery and development of new antibacterial drugs: Learning from experience? J. Antimicrob. Chemother. 2018, 73, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Datta, P. Next-generation strategy for treating drug resistant bacteria: Antibiotic hybrids. Indian J. Med. Res. 2019, 149, 97–106. [Google Scholar] [CrossRef]
- Decker, M. Design of Hybrid Molecules for Drug Development; Elsevier: Oxford, UK, 2017. [Google Scholar]
- Klahn, P.; Brönstrup, M. Bifunctional antimicrobial conjugates and hybrid antimicrobials. Nat. Prod. Rep. 2017, 34, 832–885. [Google Scholar] [CrossRef]
- Rossi, R.; Ciofalo, M. Current Advances in the Synthesis and Biological Evaluation of Pharmacologically Relevant 1,2,4,5-Tetrasubstituted-1H-Imidazole Derivatives. Curr. Org. Chem. 2019, 23, 2016–2101. [Google Scholar] [CrossRef]
- Rossi, R.; Angelici, G.; Casotti, G.; Manzini, C.; Lessi, M. Catalytic Synthesis of 1,2,4,5-Tetrasubstituted 1H-Imidazole Derivatives: State of the Art. Adv. Synth. Catal. 2019, 361, 2737–2803. [Google Scholar] [CrossRef]
- Rossi, R.; Lessi, M.; Manzini, C.; Bellina, F. Synthesis and Biological Profiles of 4,5-, 1,5-, and 1,2-Diaryl-1H-imidazoles. In Vicinal Diaryl Substituted Heterocycles; Yadav, M.R., Murumkar, P.R., Ghuge, R.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 83–160. [Google Scholar]
- Rossi, R.; Lessi, M.; Manzini, C.; Marianetti, G.; Bellina, F. Direct (Hetero)arylation Reactions of (Hetero)arenes as Tools for the Step- and Atom-Economical Synthesis of Biologically Active Unnatural Compounds Including Pharmaceutical Targets. Synthesis 2016, 48, 3821–3862. [Google Scholar] [CrossRef]
- Bellina, F.; Rossi, R. Regioselective Functionalization of the Imidazole Ring via Transition Metal-Catalyzed C-N and C-C Bond Forming Reactions. Adv. Synth. Catal. 2010, 352, 1223–1276. [Google Scholar] [CrossRef]
- Bonezzi, K.; Taraboletti, G.; Borsotti, P.; Bellina, F.; Rossi, R.; Giavazzi, R. Vascular Disrupting Activity of Tubulin-Binding 1,5-Diaryl-1H-imidazoles. J. Med. Chem. 2009, 52, 7906–7910. [Google Scholar] [CrossRef] [Green Version]
- Bellina, F.; Rossi, R. Recent advances in the synthesis of (hetero)aryl-substituted heteroarenes via transition metal-catalysed direct (hetero)arylation of heteroarene C-H bonds with aryl halides or pseudohalides, diaryliodonium salts, and potassium aryltrifluoroborates. Tetrahedron 2009, 65, 10269–10310. [Google Scholar] [CrossRef]
- Bellina, F.; Cauteruccio, S.; Di Fiore, A.; Rossi, R. Regioselective Synthesis of 4,5-Diaryl-1-methyl-1H-imidazoles Including Highly Cytotoxic Derivatives by Pd-Catalyzed Direct C-5 Arylation of 1-Methyl-1H-imidazole with Aryl Bromides. Eur. J. Org. Chem. 2008, 5436–5445. [Google Scholar] [CrossRef]
- Ang, C.W.; Jarrad, A.M.; Cooper, M.A.; Blaskovich, M.A.T. Nitroimidazoles: Molecular Fireworks That Combat a Broad Spectrum of Infectious Diseases. J. Med. Chem. 2017, 60, 7636–7657. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, A.A.; Johnson, S. Current State of Clostridium difficile Treatment Options. Clin. Infect. Dis. 2012, 55, S71–S76. [Google Scholar] [CrossRef]
- Kazanowski, M.; Smolarek, S.; Kinnarney, F.; Grzebieniak, Z. Clostridium difficile: Epidemiology, diagnostic and therapeutic possibilities-a systematic review. Tech. Coloproctol. 2014, 18, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.J.; Groundwater, P.W.; Todd, A.; Worsley, A.J. Antibacterial Agents. Chemistry, Mode of Action, Mechanisms of Resistance and Clinical Applications; Wiley: Chichester, UK, 2012; pp. 85–101. [Google Scholar]
- Leiros, H.-K.S.; Kozielski-Stuhrmann, S.; Kapp, U.; Terradot, L.; Leonard, G.A.; McSweeney, S.M. Structural basis of 5-nitroimidazole antibiotic resistance: The crystal structure of NimA from Deinococcus radiodurans. J. Biol. Chem. 2004, 279, 55840–55849. [Google Scholar] [CrossRef] [Green Version]
- Kaakoush, N.O.; Asencio, C.; Mégraud, F.; Mendz, G.L. A Redox Basis for Metronidazole Resistance in Helicobacter Pylori. Antimicrob. Agents Chemother. 2009, 53, 1884–1891. [Google Scholar] [CrossRef] [Green Version]
- Peláez, T.; Cercenado, E.; Alcalá, L.; Marín, M.; Martín-López, A.; Martínez-Alarcón, J.; Catalán, P.; Sánchez-Somolinos, M.; Bouza, E. Metronidazole resistance in Clostridium difficile is heterogeneous. J. Clin. Microbiol. 2008, 46, 3028–3032. [Google Scholar] [CrossRef] [Green Version]
- Exner, M.; Bhattacharya, S.; Christiansen, B.; Gebel, J.; Goroncy-Bermes, P.; Hartemann, P.; Heeg, P.; Ilschner, C.; Kramer, A.; Larson, E.; et al. Antibiotic resistance: What is so special about multidrug-resistant Gram-negative bacteria? GMS Hyg. Infect. Control. 2017, 12, 5. [Google Scholar] [CrossRef]
- Demirayak, S.; Karaburun, A.Ç.; Kiraz, N. Synthesis and antibacterial activities of some 1-[2-(substituted pyrrol-1-yl)ethyl]-2-methyl-5-nitroimidazole derivatives. Eur. J. Med. Chem. 1999, 34, 275–278. [Google Scholar] [CrossRef]
- Balakrishna, A.; Aguiar, A.; Sobral, P.J.M.; Wani, M.Y.; Almeida e Silva, J.; Sobral, A.J.F.N. Paal–Knorr synthesis of pyrroles: From conventional to green synthesis. Cat. Rev.-Sci. Eng. 2018, 61, 84–110. [Google Scholar] [CrossRef]
- Hadj-esfandiari, N.; Navidpour, L.; Shadnia, H.; Amini, M.; Samadi, N.; Faramarzi, M.A.; Shafiee, A. Synthesis, antibacterial activity, and quantitative structure–activity relationships of new (Z)-2-(nitroimidazolylmethylene)-3(2H)-benzofuranone derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 6354–6363. [Google Scholar] [CrossRef]
- Pires, J.R.; Saito, C.; Gomes, S.L.; Giesbrecht, A.M.; Amaral, A.T.-d. Investigation of 5-Nitrofuran Derivatives: Synthesis, Antibacterial Activity, and Quantitative Structure-Activity Relationships. J. Med. Chem. 2001, 44, 3673–3681. [Google Scholar] [CrossRef]
- Mirzaei, J.; Siavoshi, F.; Emami, S.; Safari, F.; Khoshayand, M.R.; Shafiee, A.; Foroumadi, A. Synthesis and in vitro anti-Helicobacter pylori activity of N-[5-(5-nitro-2-heteroaryl)-1,3,4-thiadiazol-2-yl]thiomorpholines and related compounds. Eur. J. Med. Chem. 2008, 43, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Foroumadi, A.; Mansouri, S.; Kiani, Z.; Rahmani, A. Synthesis and in vitro antibacterial evaluation of N-[5-(5-nitro-2-thienyl)-1,3,4-thiadiazole-2-yl] piperazinyl quinolones. Eur. J. Med. Chem. 2003, 38, 851–854. [Google Scholar] [CrossRef]
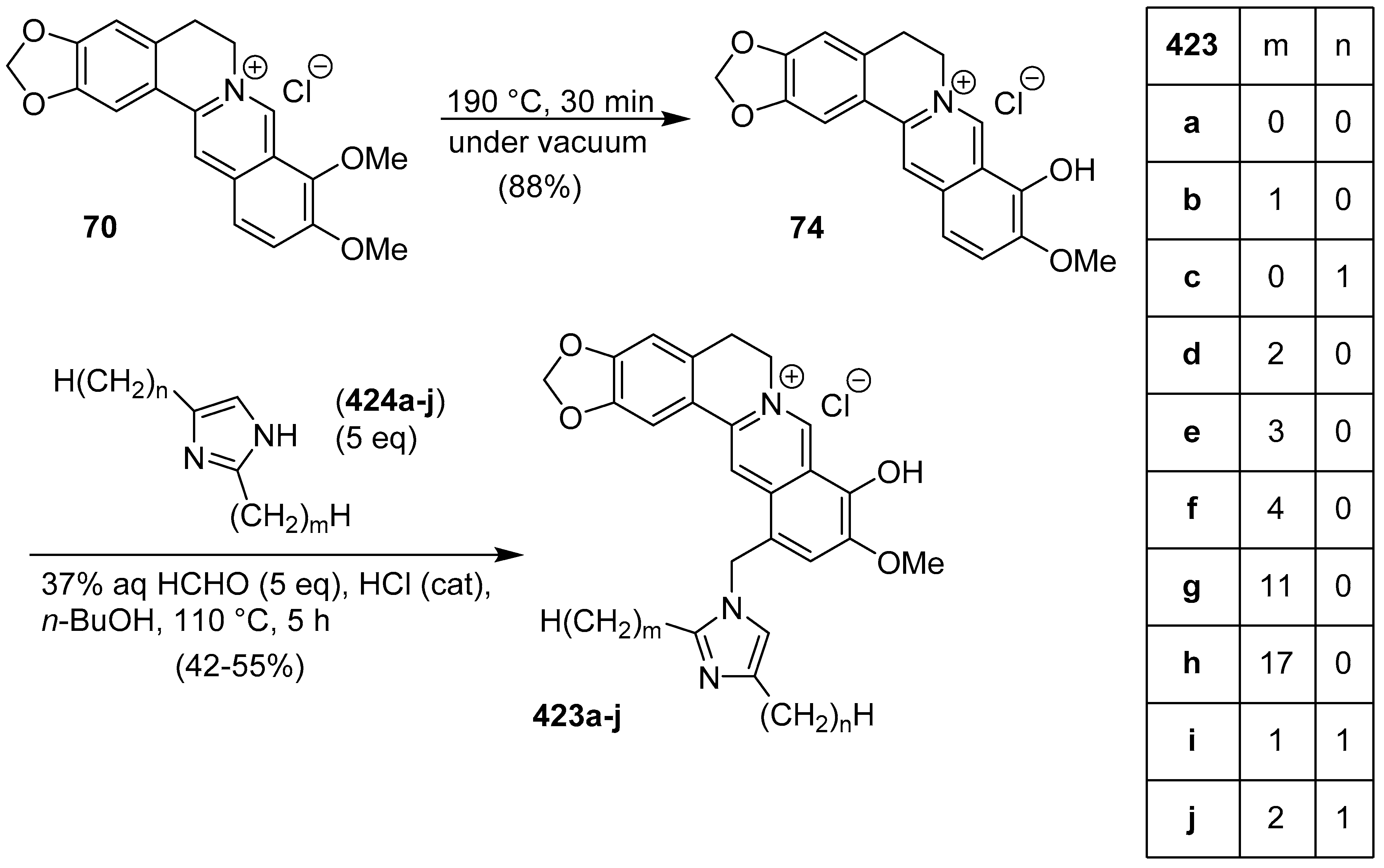
- Pfeil, E. Theorie und Praxis der Sandmeyerschen Reaktion. Angew. Chem. 1953, 65, 155–158. [Google Scholar] [CrossRef]
- Atia, A.J.K. Synthesis and Antibacterial Activities of New Metronidazole and Imidazole Derivatives. Molecules 2009, 14, 2431–2446. [Google Scholar] [CrossRef] [Green Version]
- [Negi], B.; Kumar, N.; Rohilla, R.K.; Roy, N.; Rawat, D.S. Synthesis and antibacterial activity evaluation of metronidazole-triazole conjugates. Bioorg. Med. Chem. Lett. 2009, 19, 1396–1398. [Google Scholar] [CrossRef]
- Meingassner, J.G.; Mieth, H.; Czok, R.; Lindmark, D.G.; Müller, M. Assay Conditions and the Demonstration of Nitroimidazole Resistance in Tritrichomonas foetus. Antimicrob. Agents Chemother. 1978, 13, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Britz, M.L.; Wilkinson, R.G. Isolation and Properties of Metronidazole-Resistant Mutants of Bacteroides fragilis. Antimicrob. Agents Chemother. 1979, 16, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadeh, H.A.; Mosleh, I.M.; Al-Bakri, A.G.; Mubarak, M.S. Synthesis and antimicrobial activity of new 1,2,4-triazole-3-thiol metronidazole derivatives. Monatsh. Chem. 2010, 141, 471–478. [Google Scholar] [CrossRef]
- Clayton, R.; Ramsden, C. N-Vinyl-Nitroimidazole Cycloadditions: Potential Routes to Nucleoside Analogues. Synthesis 2005, 2695–2700. [Google Scholar] [CrossRef]
- Zamani, K.; Faghihi, K.; Tofighi, T.; Shariatzadeh, M.R. Synthesis and Antimicrobial Activity of Some Pyridyl and Naphthyl Substituted 1,2,4-Triazole and 1,3,4-Thiadiazole Derivatives. Turk. J. Chem. 2004, 28, 95–100. [Google Scholar]
- Varshney, V.; Mishra, N.N.; Shukla, P.K.; Sahu, D.P. Synthesis of nitroimidazole derived oxazolidinones as antibacterial agents. Eur. J. Med. Chem. 2010, 45, 661–666. [Google Scholar] [CrossRef]
- Khalaj, A.; Nakhjiri, M.; Negahbani, A.S.; Samadizadeh, M.; Firoozpour, L.; Rajabalian, S.; Samadi, N.; Faramarzi, M.A.; Adibpour, N.; Shafiee, A.; et al. Discovery of a novel nitroimidazolyl-oxazolidinone hybrid with potent anti Gram-positive activity: Synthesis and antibacterial evaluation. Eur. J. Med. Chem. 2011, 46, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Letafat, B.; Mohammadhosseini, N.; Asadipour, A.; Foroumadi, A. Synthesis and In vitro Antibacterial Activity of New 2-(1-Methyl-4-nitro-1H-imidazol-5-ylsulfonyl)-1,3,4-thiadiazoles. J. Chem. 2011, 8, 1120–1123. [Google Scholar] [CrossRef] [Green Version]
- Letafat, B.; Emami, S.; Aliabadi, A.; Mohammadhosseini, N.; Moshafi, M.H.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Synthesis and In-Vitro Antibacterial Activity of 5-Substituted 1-Methyl-4-nitro-1H-imidazoles. Arch. Pharm. 2008, 341, 497–501. [Google Scholar] [CrossRef]
- Li, Y.; Luo, Y.; Hu, Y.; Zhu, D.-D.; Zhang, S.; Liu, Z.-J.; Gong, H.-B.; Zhu, H.-L. Design, synthesis and antimicrobial activities of nitroimidazole derivatives containing 1,3,4-oxadiazole scaffold as FabH inhibitors. Bioorg. Med. Chem. 2012, 20, 4316–4322. [Google Scholar] [CrossRef]
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Duellman, S.; Benink, H.A.; Worzella, T.J.; Minor, L. Cell Viability Assay. In Assay Guidance Manual (Last Updated: June 1, 2020); Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Bejcek, B., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly: Bethesda, MD, USA, 2004; pp. 1–25. [Google Scholar]
- Zhang, L.; Chang, J.-J.; Zhang, S.-L.; Damu, G.L.V.; Geng, R.-X.; Zhou, C.-H. Synthesis and bioactive evaluation of novel hybrids of metronidazole and berberine as new type of antimicrobial agents and their transportation behavior by human serum albumin. Bioorg. Med. Chem. 2013, 21, 4158–4169. [Google Scholar] [CrossRef]
- Gan, R.-Y. Bioactivities of Berberine: An Update. Int. J. Modern Biol. Med. 2012, 1, 48–81. [Google Scholar]
- Zhang, D.; Li, A.; Xie, J.; Ji, C. In vitro antibacterial effect of berberine hydrochloride and enrofloxacin to fish pathogenic bacteria. Aquac. Res. 2010, 41, 1095–1100. [Google Scholar] [CrossRef]
- Yu, X.-T.; Xu, Y.-F.; Huang, Y.-F.; Qu, C.; Xu, L.-Q.; Su, Z.-R.; Zeng, H.-F.; Zheng, L.; Yi, T.-G.; Li, H.-L.; et al. Berberrubine attenuates mucosal lesions and inflammation in dextran sodium sulfate-induced colitis in mice. PLoS ONE 2018, 13, e0194069. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, Y.; Kalisiak, J.; Korthals, K.; Lauwaet, T.; Cheung, D.Y.; Lozano, R.; Cobo, E.R.; Upcroft, P.; Upcroft, J.A.; Berg, D.E.; et al. Expanded therapeutic potential in activity space of next-generation 5-nitroimidazole antimicrobials with broad structural diversity. Proc. Natl. Acad. Sci. USA 2013, 110, 17564–17569. [Google Scholar] [CrossRef] [Green Version]
- Brook, I. Antimicrobial treatment of anaerobic infections. Exp. Opin. Pharmacother. 2011, 12, 1691–1707. [Google Scholar] [CrossRef] [PubMed]
- chloramphenicol is referred to as “chloromycin” in the original article.
- Makawana, J.A.; Sun, J.; Zhu, H.-L. Schiff’s base derivatives bearing nitroimidazole moiety: New class of antibacterial, anticancer agents and potential EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 6264–6268. [Google Scholar] [CrossRef]
- Gu, W.; Qiao, C.; Wang, S.-F.; Hao, Y.; Miao, T.-T. Synthesis and biological evaluation of novel N-substituted 1H-dibenzo[a,c]carbazole derivatives of dehydroabietic acid as potential antimicrobial agents. Bioorg. Med. Chem. Lett. 2014, 24, 328–331. [Google Scholar] [CrossRef]
- González, M.A.; Pérez-Guaita, D.; Correa-Royero, J.; Zapata, B.; Agudelo, L.; Mesa-Arango, A.; Betancur-Galvis, L. Synthesis and biological evaluation of dehydroabietic acid derivatives. Eur. J. Med. Chem. 2010, 45, 811–816. [Google Scholar] [CrossRef]
- Manner, S.; Vahermo, M.; Skogman, M.E.; Krogerus, S.; Vuorela, P.M.; Yli-Kauhaluoma, J.; Fallarero, A.; Moreira, V.M. New derivatives of dehydroabietic acid target planktonic and biofilm bacteria in Staphylococcus aureus and effectively disrupt bacterial membrane integrity. Eur. J. Med. Chem. 2015, 102, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Wang, S. Synthesis and antimicrobial activities of novel 1H-dibenzo[a,c]carbazoles from dehydroabietic acid. Eur. J. Med. Chem. 2010, 45, 4692–4696. [Google Scholar] [CrossRef]
- Sangani, C.B.; Mak[a]wana, J.A.; Duan, Y.-T.; Tarpada, U.P.; Patel, Y.S.; Patel, K.B.; Dave, V.N.; Zhu, H.-L. Design, synthesis, and antibacterial evaluation of new Schiff’s base derivatives bearing nitroimidazole and pyrazole nuclei as potent E. coli FabH inhibitors. Res. Chem. Intermed. 2015, 41, 10137–10149. [Google Scholar] [CrossRef]
- Sangani, C.B.; Mungra, D.C.; Patel, M.P.; Patel, R.G. Synthesis and in vitro antimicrobial screening of new pyrano[4,3-b]pyrane derivatives of 1H-pyrazole. Chin. Chem. Lett. 2012, 23, 57–60. [Google Scholar] [CrossRef]
- Zhang, L.-X.; Liu, Y.; Cia, L.-H.; Hu, Y.-J.; Yin, J.; Hu, P.-Z. Inhibitory study of some novel Schiff base derivatives on Staphylococcus aureus by microcalorimetry. Thermochim. Acta 2006, 440, 51–56. [Google Scholar] [CrossRef]
- Jarrad, A.M.; Karoli, T.; Debnath, A.; Tay, C.Y.; Huang, J.X.; Kaeslin, G.; Elliott, A.G.; Miyamoto, Y.; Ramu, S.; Kavanagh, A.M.; et al. Metronidazole-triazole conjugates: Activity against Clostridium difficile and parasites. Eur. J. Med. Chem. 2015, 101, 96–102. [Google Scholar] [CrossRef] [Green Version]
- reported as Bacillus proteus ATCC 13315 in the original paper.
- Zhang, L.; Kannekanti, V.K.; Geng, R.-X.; Zhou, C.-H. Design and biological evaluation of novel quinolone-based metronidazole derivatives as potent Cu2+ mediated DNA-targeting antibacterial agents. Bioorg. Med. Chem. Lett. 2015, 25, 3699–3705. [Google Scholar] [CrossRef]
- Cherian, P.T.; Wu, X.; Yang, L.; Scarborough, J.S.; Singh, A.P.; Alam, Z.A.; Lee, R.E.; Hurdle, J.G. Gastrointestinal localization of metronidazole by a lactobacilli-inspired tetramic acid motif improves treatment outcomes in the hamster model of Clostridium difficile infection. J. Antimicrob. Chemother. 2015, 70, 3061–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dingsdag, S.A.; Yap, B.C.M.; Hunter, N.; Crossley, M.J. Amino acid-linked porphyrin-nitroimidazole antibiotics targeting Porphyromonas gingivalis. Org. Biomol. Chem. 2015, 13, 98–109. [Google Scholar] [CrossRef]
- Crossley, M.; Thordarson, P.; Hunter, N.; Yap, B.; Collyer, C.A. Porphyrin Linked Metronidazole Against Gum Disease: Porphyromonas Gingivalis. World patent WO 2006005137A1, 19 January 2006. [Google Scholar]
- Kang, J.; Tangadanchu, V.K.R.; Gopala, L.; Gao, W.-W.; Cheng, Y.; Liu, H.-B.; Geng, R.-X.; Li, S.; Zhou, C.-H. Novel potentially antibacterial naphthalimide-derived metronidazoles: Design, synthesis, biological evaluation and supramolecular interactions with DNA, human serum albumin and topoisomerase II. Chin. Chem. Lett. 2017, 28, 1369–1374. [Google Scholar] [CrossRef]
- Zhou, Y.; Ju, Y.; Yang, Y.; Sang, Z.; Wang, Z.; He, G.; Yang, T.; Luo, Y. Discovery of hybrids of indolin-2-one and nitroimidazole as potent inhibitors against drug-resistant bacteria. J. Antibiot. (Tokyo) 2018, 71, 887–897. [Google Scholar] [CrossRef]
- Gholamzadeh, P.; Mohammadi Ziarani, G.; Badiei, A.; Abolhassani Soorki, A.; Lashgari, N. Efficient green synthesis of isoindigo derivatives using sulfonic-acid-functionalized nanoporous silica (SBA-Pr-SO3H) catalyst and study of their antimicrobial properties. Res. Chem. Intermed. 2013, 39, 3925–3936. [Google Scholar] [CrossRef]
- Hosseinzadeh, N.; Hasani, M.; Foroumadi, A.; Nadri, H.; Emami, S.; Samadi, N.; Faramarzi, M.A.; Saniee, P.; Siavoshi, F.; Abadian, N.; et al. 5-Nitro-heteroarylidene analogs of 2-thiazolylimino-4-thiazolidinones as a novel series of antibacterial agents. Med. Chem. Res. 2013, 22, 2293–2302. [Google Scholar] [CrossRef]
- Zhang, G.-B.; Maddili, S.K.; Tangadanchu, V.K.R.; Gopala, L.; Gao, W.-W.; Cai, G.-X.; Zhou, C.-H. Discovery of natural berberine-derived nitroimidazoles as potentially multi-targeting agents against drug-resistant Escherichia coli. Sci. China Chem. 2018, 61, 557–568. [Google Scholar] [CrossRef]
- Bhanot, S.; Viljoen, A.; Kremer, L.; Kumar, V. Alkylated/aminated nitroimidazoles and nitroimidazole-7-chloroquinoline conjugates: Synthesis and anti-mycobacterial evaluation. Bioorg. Med. Chem. Lett. 2018, 28, 1309–1312. [Google Scholar] [CrossRef]
- Raj, R.; Biot, C.; Carrère-Kremer, S.; Kremer, L.; Guérardel, Y.; Gut, J.; Rosenthal, P.J.; Kumar, V. 4-Aminoquinoline-β-Lactam Conjugates: Synthesis, Antimalarial, and Antitubercular Evaluation. Chem. Biol. Drug Des. 2014, 83, 191–197. [Google Scholar] [CrossRef]
- Singh, A.; Biot, C.; Viljoen, A.; Dupont, C.; Kremer, L.; Kumar, K.; Kumar, V. 1H-1,2,3-triazole-tethered uracil-ferrocene and uracil-ferrocenylchalcone conjugates: Synthesis and antitubercular evaluation. Chem. Biol. Drug Des. 2017, 89, 856–861. [Google Scholar] [CrossRef]
- Bhovi, M.G.; Tanwar, P.; Yadav, G.C. Synthesis and characterization of impurities present in an antimalarial drug piperaquine phosphate. Indian J. Heterocycl. Chem. 2010, 19, 215–220. [Google Scholar]
- Kang, J.; Gopala, L.; Tangadanchu, V.K.R.; Gao, W.-W.; Zhou, C.-H. Novel naphthalimide nitroimidazoles as multitargeting antibacterial agents against resistant Acinetobacter baumannii. Future Med. Chem. 2018, 10, 711–724. [Google Scholar] [CrossRef]
- Morris, F.C.; Dexter, C.; Kostoulias, X.; Uddin, M.I.; Peleg, A.Y. The Mechanisms of Disease Caused by Acinetobacter baumannii. Front. Microbiol. 2019, 10, 1601. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-Y.; Zhou, C.-H. Synthesis and activities of naphthalimide azoles as a new type of antibacterial and antifungal agents. Bioorg. Med. Chem. Lett. 2011, 21, 4349–4352. [Google Scholar] [CrossRef]
- Damu, G.L.V.; Wang, Q.; Zhang, H.; Zhang, Y.; Lv, J.; Zhou, C. A series of naphthalimide azoles: Design, synthesis and bioactive evaluation as potential antimicrobial agents. Sci. China Chem. 2013, 56, 952–969. [Google Scholar] [CrossRef]
- Lv, J.-S.; Peng, X.-M.; Kishore, B.; Zhou, C.-H. 1,2,3-Triazole-derived naphthalimides as a novel type of potential antimicrobial agents: Synthesis, antimicrobial activity, interaction with calf thymus DNA and human serum albumin. Bioorg. Med. Chem. Lett. 2014, 24, 308–313. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Gopala, L.; Bheemanaboina, R.R.Y.; Liu, H.-B.; Cheng, Y.; Geng, R.-X.; Zhou, C.-H. Novel Naphthalimide Aminothiazoles as Potential Multitargeting Antimicrobial Agents. ACS Med. Chem. Lett. 2017, 8, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-Z.; Tangadanchu, V.K.R.; Battini, N.; Bheemanaboina, R.R.Y.; Zang, Z.-L.; Zhang, S.-L.; Zhou, C.-H. Indole-nitroimidazole conjugates as efficient manipulators to decrease the genes expression of methicillin-resistant Staphylococcus aureus. Eur. J. Med. Chem. 2019, 179, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Roch, M.; Lelong, E.; Panasenko, O.O.; Sierra, R.; Renzoni, A.; Kelley, W.L. Thermosensitive PBP2a requires extracellular folding factors PrsA and HtrA1 for Staphylococcus aureus MRSA β-lactam resistance. Commun. Biol. 2019, 2, 417. [Google Scholar] [CrossRef]
- Li, Z.-Z.; Gopala, L.; Tangadanchu, V.K.R.; Gao, W.-W.; Zhou, C.-H. Discovery of novel nitroimidazole enols as Pseudomonas aeruginosa DNA cleavage agents. Bioorg. Med. Chem. 2017, 25, 6511–6522. [Google Scholar] [CrossRef]
- Girhepunje, N.S.; Kedar, P.S.; Ittadwar, A.M.; Dumore, N.G. Design, Synthesis and Characterization of Some 5-Nitroimidazole Derivatives. Int. J. Pharm. Pharm. Res. 2016, 6, 456–480. [Google Scholar]
- Kayser, O.; Kolodziej, H. Antibacterial Activity of Simple Coumarins: Structural Requirements for Biological Activity. Z. Naturforsch. C Biosci. 1999, 54, 169–174. [Google Scholar] [CrossRef]
- De Souza, S.M.; Delle Monache, F.; Smânia, A., Jr. Antibacterial Activity of Coumarins. Z. Naturforsch. C Biosci. 2005, 60, 693–700. [Google Scholar] [CrossRef]
- Smyth, T.; Ramachandran, V.N.; Smyth, W.F. A study of the antimicrobial activity of selected naturally occurring and synthetic coumarins. Int. J. Antimicrob. Agents 2009, 33, 421–426. [Google Scholar] [CrossRef]
- Ostrov, D.A.; Hernández Prada, J.A.; Corsino, P.E.; Finton, K.A.; Le, N.; Rowe, T.C. Discovery of Novel DNA Gyrase Inhibitors by High-Throughput Virtual Screening. Antimicrob. Agents Chemother. 2007, 51, 3688–3698. [Google Scholar] [CrossRef] [Green Version]
- Behrami, A.; Krasniqi, I. Antibacterial activity of coumarine derivatives synthesized from 8-amino-4,7-dihydroxy-chromen-2-one and comparison with standard drug. J. Chem. Pharm. Res. 2012, 4, 2495–2500. [Google Scholar]
- RamaGanesh, C.K.; Bodke, Y.D.; Venkatesh, K.B. Synthesis and biological evaluation of some innovative coumarin derivatives containing thiazolidin-4-one ring. Indian J. Chem. 2010, 49B, 1151–1154. [Google Scholar] [CrossRef]
- Tan, N.; Yazıcı-Tütüniş, S.; Bilgin, M.; Tan, E.; Miski, M. Antibacterial Activities of Pyrenylated Coumarins from the Roots of Prangos hulusii. Molecules 2017, 22, 1098. [Google Scholar] [CrossRef]
- De Araújo, R.S.A.; Barbosa-Filho, J.M.; Scotti, M.T.; Scotti, L.; da Cruz, R.M.D.; Falcão-Silva, V.D.S.; de Siqueira-Júnior, J.P.; Mendonça-Junior, F.J.B. Modulation of Drug Resistance in Staphylococcus aureus with Coumarin Derivatives. Scientifica 2016, 2016, 6894758. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.-M.; Kannekanti, V.K.; Damu, G.L.V.; Zhou, C.-H. Coumarin-derived azolyl ethanols: Synthesis, antimicrobial evaluation and preliminary action mechanism. Sci. China Chem. 2016, 59, 878–894. [Google Scholar] [CrossRef]
- Holiyachi, M.; Samundeeswari, S.; Chougala, B.M.; Naik, N.S.; Madar, J.; Shastri, L.A.; Joshi, S.D.; Dixit, S.R.; Dodamani, S.; Jalalpure, S.; et al. Design and synthesis of coumarin–imidazole hybrid and phenyl-imidazoloacrylates as potent antimicrobial and antiinflammatory agents. Monatsh. Chem. 2018, 149, 595–609. [Google Scholar] [CrossRef]
- Holiyachi, M.; Shastri, S.L.; Chougala, B.M.; Shastri, L.A. Effects of Base for the Efficient Synthesis of 4-Formylcoumarins and 4-Formylcarbostyrils. Synth. Commun. 2015, 45, 1002–1008. [Google Scholar] [CrossRef]
- Hu, Y.; Shen, Y.; Wu, X.; Tu, X.; Wang, G.-X. Synthesis and biological evaluation of coumarin derivatives containing imidazole skeleton as potential antibacterial agents. Eur. J. Med. Chem. 2018, 143, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, H.; Ozawa, T.; Takahata, S.; Iida, M.; Saito, J.; Yamada, M. Phenylimidazole Derivatives of 4-Pyridone as Dual Inhibitors of Bacterial Enoyl-Acyl Carrier Protein Reductases FabI and FabK. J. Med. Chem. 2007, 50, 4710–4720. [Google Scholar] [CrossRef]
- Holiyachi, M.; Samundeeswari, S.; Chougala, B.M.; Naik, N.S.; Madar, J.M.; Shaikh, F.; Shastri, L.A.; Joshi, S.D.; Dixit, S.R.; Sunagar, V.A.; et al. Synthesis and molecular docking studies of coumarin-imidazole conjugates as potential antimicrobial agents. Indian J. Chem. 2020, 59B, 110–125. [Google Scholar]
- Araque, P.; Herrera, A.; Montaño, D.; Yepes, A.; García, E.; Sepúlveda, J.; Torijano, S.; Cardona-G, W. Antimicrobial Activity and In Silico Study of Methylimidazolium-Furanchalcone Hybrids and 1-Alkyl-3-methylimidazolium Salts. J. Chil. Chem. Soc. 2019, 64, 4547–4552. [Google Scholar] [CrossRef] [Green Version]
- El-Obeid, H.A.; Elnima, E.I.; Al-Badr, A.A. Synthesis and Antimicrobial Activity of New Furan Derivatives. Pharm. Res. 1985, 2, 42–43. [Google Scholar] [CrossRef]
- Kharb, R.; Birla, S.; Sharma, A.K. Recent Updates on Antimicrobial Potential of Novel Furan Derivatives. Int. J. Pharm. Phytopharmacol. Res. 2014, 3, 451–459. [Google Scholar]
- Vollaro, A.; Catania, M.R.; Iesce, M.R.; Sferruzza, R.; D’Abrosca, B.; Donnarumma, G.; De Filippis, A.; Cermola, F.; DellaGreca, M.; Buommino, E. Antimicrobial and anti-biofilm properties of novel synthetic lignan-like compounds. New Microbiol. 2019, 42, 21–28. [Google Scholar]
- Xu, M.; Wu, P.; Shen, F.; Ji, J.; Rakesh, K.P. Chalcone derivatives and their antibacterial activities: Current development. Bioorg. Chem. 2019, 91, 103133. [Google Scholar] [CrossRef] [PubMed]
- Khodarahmi, G.; Asadi, P.; Hassanzadeh, F.; Khodarahmi, E. Benzofuran as a promising scaffold for the synthesis of antimicrobial and antibreast cancer agents: A review. J. Res. Med. Sci. The Off. J. Isfahan Univ. Med. Sci. 2015, 20, 1094–1104. [Google Scholar] [CrossRef]
- Jafari, E.; Khajouei, M.R.; Hassanzadeh, F.; Hakimelahi, G.H.; Khodarahmi, G.A. Quinazolinone and quinazoline derivatives: Recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci. 2016, 11, 1–14. [Google Scholar]
- Mohamed, M.A.; Ghanem, H.M.; Abd El-Ghaffar, N.F.; Mohamed, S.S. Biological Evaluation and Molecular Docking of Substituted Quinazolinones as Antimicrobial Agents. Aust. J. Basic Appl. Sci. 2013, 7, 263–274. [Google Scholar]
- Asadi, P.; Khodarahmi, G.; Jahanian-Najafabadi, A.; Saghaie, L.; Hassanzadeh, F. Synthesis, characterization, molecular docking studies and biological evaluation of some novel hybrids based on quinazolinone, benzofuran and imidazolium moieties as potential cytotoxic and antimicrobial agents. Iran. J. Basic Med. Sci. 2017, 20, 975–989. [Google Scholar] [CrossRef]
- Pitout, J.D.D.; Sanders, C.C.; Sanders, W.E., Jr. Antimicrobial Resistance with Focus on β-Lactam Resistance in Gram-Negative Bacilli. Am. J. Med. 1997, 103, 51–59. [Google Scholar] [CrossRef]
- Rokade, Y.; Dongre, N.; Engla, G.; Behera, C.; Sayyed, R. Azetidinone (ß-Lactam) Derivatives: An Emerging Antimicrobials. Asian J. Microbiol. Biotechnol. Environ. Sci. 2009, 11, 109–114. [Google Scholar]
- Konaklieva, M.I. Molecular Targets of β-Lactam-Based Antimicrobials: Beyond the Usual Suspects. Antibiotics (Basel) 2014, 3, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Salunkhe, D.S.; Piste, P.B. A Brief Review on Recent Synthesis of 2-Azetidinone Derivatives. Int. J. Pharm. Sci. Res. 2014, 5, 666–689. [Google Scholar] [CrossRef]
- Sarkar, S.; Chauhan, R.; Dwivedi, J. Synthesis and Antibacterial Activity of Some Azetidinone Derivatives Containing 2-Amino 6,7 Substituted Benzothiazole. Turk. J. Pharm. Sci. 2015, 12, 39–44. [Google Scholar]
- Askar, F.W.; Ali, R.A.; Al-Mouamin, T.M. Synthesis of New Some Imidazole Derivatives Containing β-Lactam Ring. Baghdad Sci. J. 2016, 13, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Sankar, P.S.; Divya, K.; Padmaja, A.; Padmavathi, V. Synthesis and Antimicrobial Activity of Azetidinone and Thiazolidinone Derivatives from Azolylindolyl Schiff’s Bases. Med. Chem. (Los Angeles) 2017, 7, 340–347. [Google Scholar] [CrossRef]
- Niccolai, D.; Tarsi, L.; Thomas, R.J. The renewed challenge of antibacterial chemotherapy. Chem. Commun. 1997, 2333–2342. [Google Scholar] [CrossRef]
- Poole, K. Resistance to β-lactam antibiotics. Cell. Mol. Life Sci. 2004, 61, 2200–2223. [Google Scholar] [CrossRef]
- Fernandes, R.; Amador, P.; Prudêncio, C. β-Lactams: Chemical structure, mode of action and mechanisms of resistance. Rev. Med. Microbiol. 2013, 24, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Thakuria, B.; Lahon, K. The Beta Lactam Antibiotics as an Empirical Therapy in a Developing Country: An Update on Their Current Status and Recommendations to Counter the Resistance against Them. J. Clin. Diagn. Res. 2013, 7, 1207–1214. [Google Scholar] [CrossRef]
- Shaikh, S.; Fatima, J.; Shakil, S.; Rizvi, S.M.D.; Kamal, M.A. Antibiotic resistance and extended spectrum beta-lactamases: Types, epidemiology and treatment. Saudi J. Biol. Sci. 2015, 22, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, A.M.; Agarwal, A.; Abe, T.; Ushirogochi, H.; Yamamura, I.; Kumagai, T.; Petersen, P.J.; Weiss, W.J.; Lenoy, E.; Yang, Y.; et al. Novel imidazole substituted 6-methylidene-penems as broad-spectrum β-lactamase inhibitors. Bioorg. Med. Chem. 2004, 12, 5807–5817. [Google Scholar] [CrossRef]
- Osborne, N.F.; Atkins, R.J.; Broom, N.J.P.; Coulton, S.; Harbridge, J.B.; Harris, M.A.; Stirling-François, I.; Walker, G. Synthesis of (5R)-(Z)-6-(1-Methyl-1,2,3-triazol-4-ylmethylene)penem-3-carboxylic acid, a Potent Broad Spectrum β-Lactamase Inhibitor, from 6-Aminopenicillanic Acid. J. Chem. Soc., Perkin Trans. 1 1994, 179–188. [Google Scholar] [CrossRef]
- Pagadala, R.; Uppalaiah, K.; Kamatala, C.R.; Meshram, J.S. Synthesis and Antimicrobial Studies of Novel Imidazole Containing Bisazetidinones and Bisthiazolidinone Derivatives. J. Heterocycl. Chem. 2015, 52, 403–410. [Google Scholar] [CrossRef]
- Pagadala, R.; Meshram, J.S.; Chopde, H.N.; Panyala, N.R. A Zeolite Promoted Expeditious One-Pot Synthesis of 1-Arylmethyl-4, 5-dihydro-2-aryl-1H-imidazole. J. Heterocycl. Chem. 2010, 47, 350–353. [Google Scholar] [CrossRef]
- Raju, B.; Ragul, R.; Sivasankar, B.N. A new reagent for selective reduction of nitro group. Indian J. Chem. 2009, 48B, 1315–1318. [Google Scholar] [CrossRef]
- Noori, S.D.; Mousa, M.N.; Al-Jadaan, S.A.N. Synthesis of a New Conjugates of Imidazole with Beta Lactam Moiety and Evaluation of its Expanded Antibacterial Activity. Orient. J. Chem. 2018, 34, 2495–2501. [Google Scholar] [CrossRef] [Green Version]
- Forte, B.; Malgesini, B.; Piutti, C.; Quartieri, F.; Scolaro, A.; Papeo, G. A Submarine Journey: The Pyrrole-Imidazole Alkaloids. Mar. Drugs 2009, 7, 705–753. [Google Scholar] [CrossRef] [Green Version]
- Lindel, T. Chemistry and Biology of the Pyrrole-Imidazole Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: Oxford, UK, 2017; Volume 77, pp. 117–219. [Google Scholar]
- Hoffmann, H.; Lindel, T. Synthesis of the Pyrrole-Imidazole Alkaloids. Synthesis 2003, 1753–1783. [Google Scholar] [CrossRef]
- Weinreb, S.M. Some recent advances in the synthesis of polycyclic imidazole-containing marine natural products. Nat. Prod. Rep. 2007, 24, 931–948. [Google Scholar] [CrossRef]
- Al-Mourabit, A.; Zancanella, M.A.; Tilvi, S.; Romo, D. Biosynthesis, asymmetric synthesis, and pharmacology, including cellular targets, of the pyrrole-2-aminoimidazole marine alkaloids. Nat. Prod. Rep. 2011, 28, 1229–1260. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, Z.; Wang, X.; De, S.; Ma, Y.; Chen, C. Dimeric pyrrole-imidazole alkaloids: Synthetic approaches and biosynthetic hypotheses. Chem. Commun. 2014, 50, 8628–8639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, J.; Bhandari, M.; Lovely, C.J. Isolation, Bioactivity, and Synthesis of Nagelamides. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 50, pp. 341–371. [Google Scholar]
- Gjorgjieva, M.; Masic, L.P.; Kikelj, D. Antibacterial and Antibiofilm Potentials of Marine Pyrrole-2-Aminoimidazole Alkaloids and their Synthetic Analogs. Mini-Rev. Med. Chem. 2018, 18, 1640–1658. [Google Scholar] [CrossRef]
- Araki, A.; Tsuda, M.; Kubota, T.; Mikami, Y.; Fromont, J.; Kobayashi, J.i. Nagelamide J, a Novel Dimeric Bromopyrrole Alkaloid from a Sponge Agelas Species. Org. Lett. 2007, 9, 2369–2371. [Google Scholar] [CrossRef]
- Walker, R.P.; Faulkner, D.J.; Van Engen, D.; Clardy, J. Sceptrin, an Antimicrobial Agent from the Sponge Agelas sceptrum. J. Am. Chem. Soc. 1981, 103, 6772–6775. [Google Scholar] [CrossRef]
- Bernan, V.S.; Roll, D.M.; Ireland, C.M.; Greenstein, M.; Maiese, W.M.; Steinberg, D.A. A study on the mechanism of action of sceptrin, an antimicrobial agent isolated from the South Pacific sponge Agelas mauritiana. J. Antimicrob. Chemother. 1993, 32, 539–550. [Google Scholar] [CrossRef]
- Baran, P.S.; Zografos, A.L.; O’Malley, D.P. Short Total Synthesis of (±)-Sceptrin. J. Am. Chem. Soc. 2004, 126, 3726–3727. [Google Scholar] [CrossRef]
- Birman, V.B.; Jiang, X.-T. Synthesis of Sceptrin Alkaloids. Org. Lett. 2004, 6, 2369–2371. [Google Scholar] [CrossRef] [PubMed]
- Baran, P.S.; Li, K.; O’Malley, D.P.; Mitsos, C. Short, Enantioselective Total Synthesis of Sceptrin and Ageliferin by Programmed Oxaquadricyclane Fragmentation. Angew. Chem. Int. Ed. 2006, 45, 249–252. [Google Scholar] [CrossRef]
- O’Malley, D.P.; Li, K.; Maue, M.; Zografos, A.L.; Baran, P.S. Total Synthesis of Dimeric Pyrrole-Imidazole Alkaloids: Sceptrin, Ageliferin, Nagelamide E, Oxysceptrin, Nakamuric Acid, and the Axinellamine Carbon Skeleton. J. Am. Chem. Soc. 2007, 129, 4762–4775. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, X.; Wang, X.; Rodriguez, R.A.; Moore, C.E.; Gao, S.; Tan, X.; Ma, Y.; Rheingold, A.L.; Baran, P.S.; et al. Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence. Science (New York) 2014, 346, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Yoon, T.P. Visible Light Photocatalysis: The Development of Photocatalytic Radical Ion Cycloadditions. ACS Catal. 2013, 3, 895–902. [Google Scholar] [CrossRef]
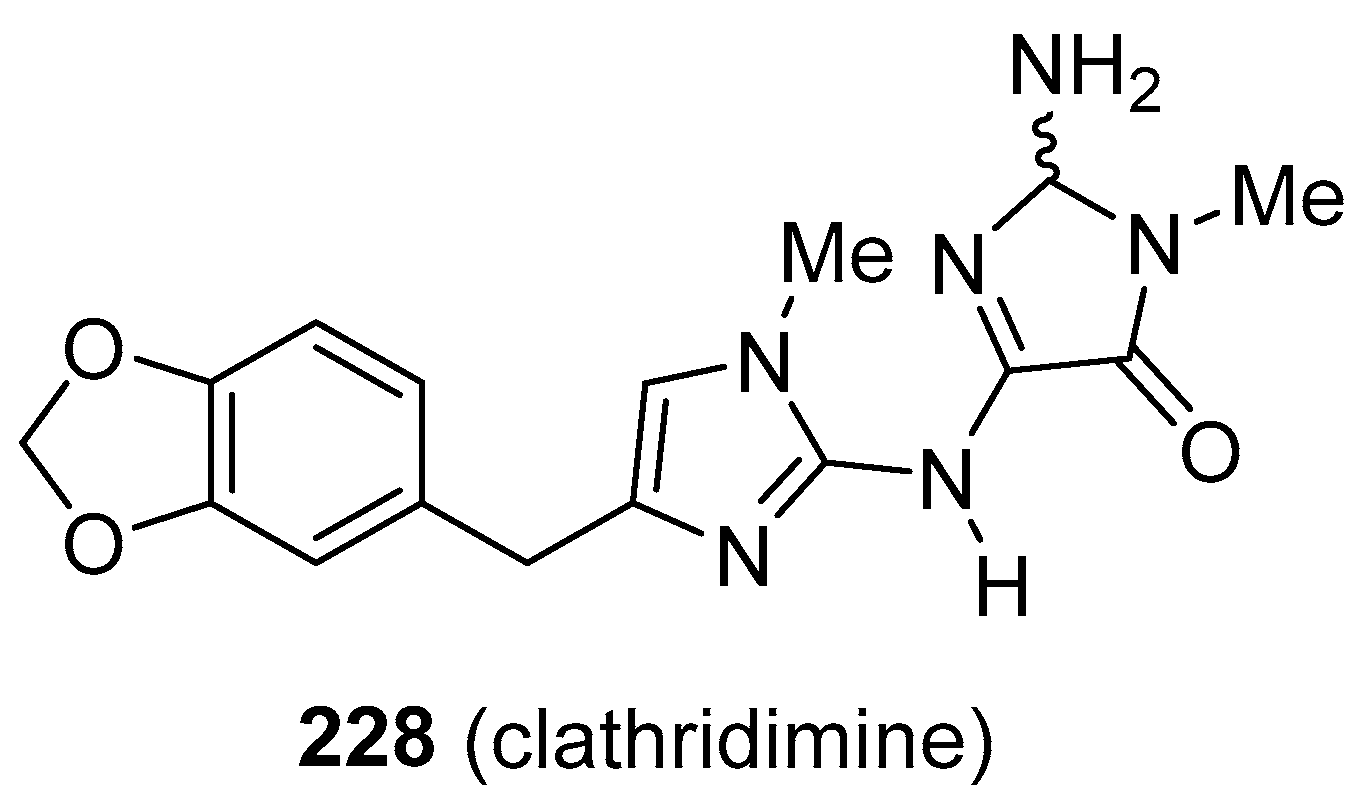
- Roueé, M.l.; Domart-Coulon, I.; Ereskovsky, A.; Djediat, C.; Perez, T.; Bourguet-Kondracki, M.-L. Cellular Localization of Clathridimine, an Antimicrobial 2-Aminoimidazole Alkaloid Produced by the Mediterranean Calcareous Sponge Clathrina clathrus. J. Nat. Prod. 2010, 73, 1277–1282. [Google Scholar] [CrossRef]
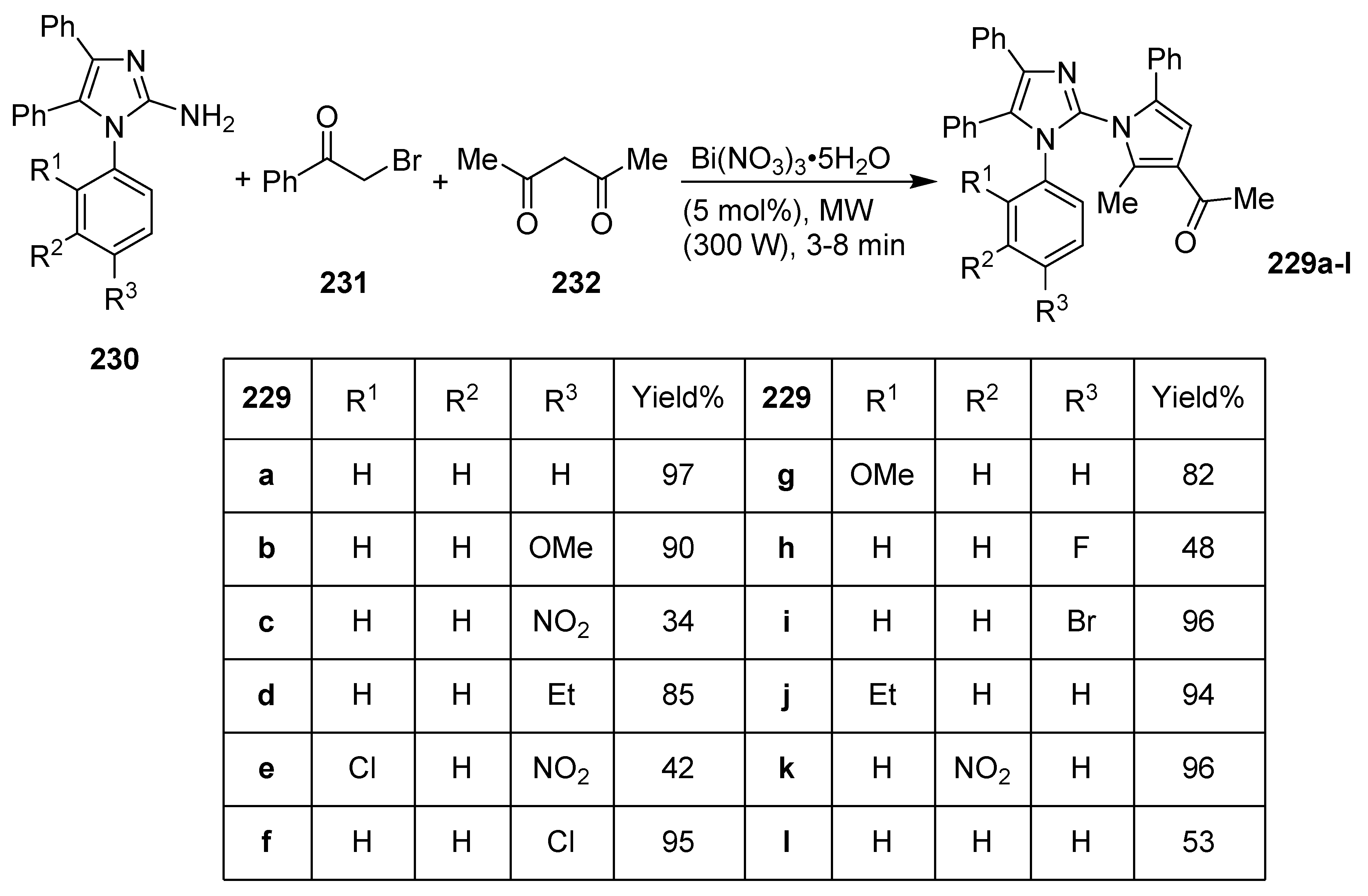
- Chawla, A.; Kapoor, V.K. Microwave Assisted One Pot Synthesis and Antimicrobial Activity of 2-(3′-Acetyl-2′-methyl-5′-phenyl)-pyrrol-1-yl-1,4,5-triphenyl-1H-imidazole Derivatives. Der Pharma Chem. 2018, 10, 27–31. [Google Scholar]
- Chawla, A.; Kapoor, V.K. Microwave Assisted Synthesis of 2-Amino-3,4,5-Trisubstituted Imidazolines Using Radiszewski Method, their Characterisation and Evaluation for Antioxidant Activity. Int. Res. J. Pharm. 2016, 7, 23–30. [Google Scholar] [CrossRef]
- Benkli, K.; Demirayak, S.; Gundogdu-Karaburun, N.; Kiraz, N.; Iscan, G.; Ucucu, U. Synthesis and antimicrobial activities of some imidazole substituted indoles. Indian J. Chem. 2004, 43B, 174–179. [Google Scholar] [CrossRef]
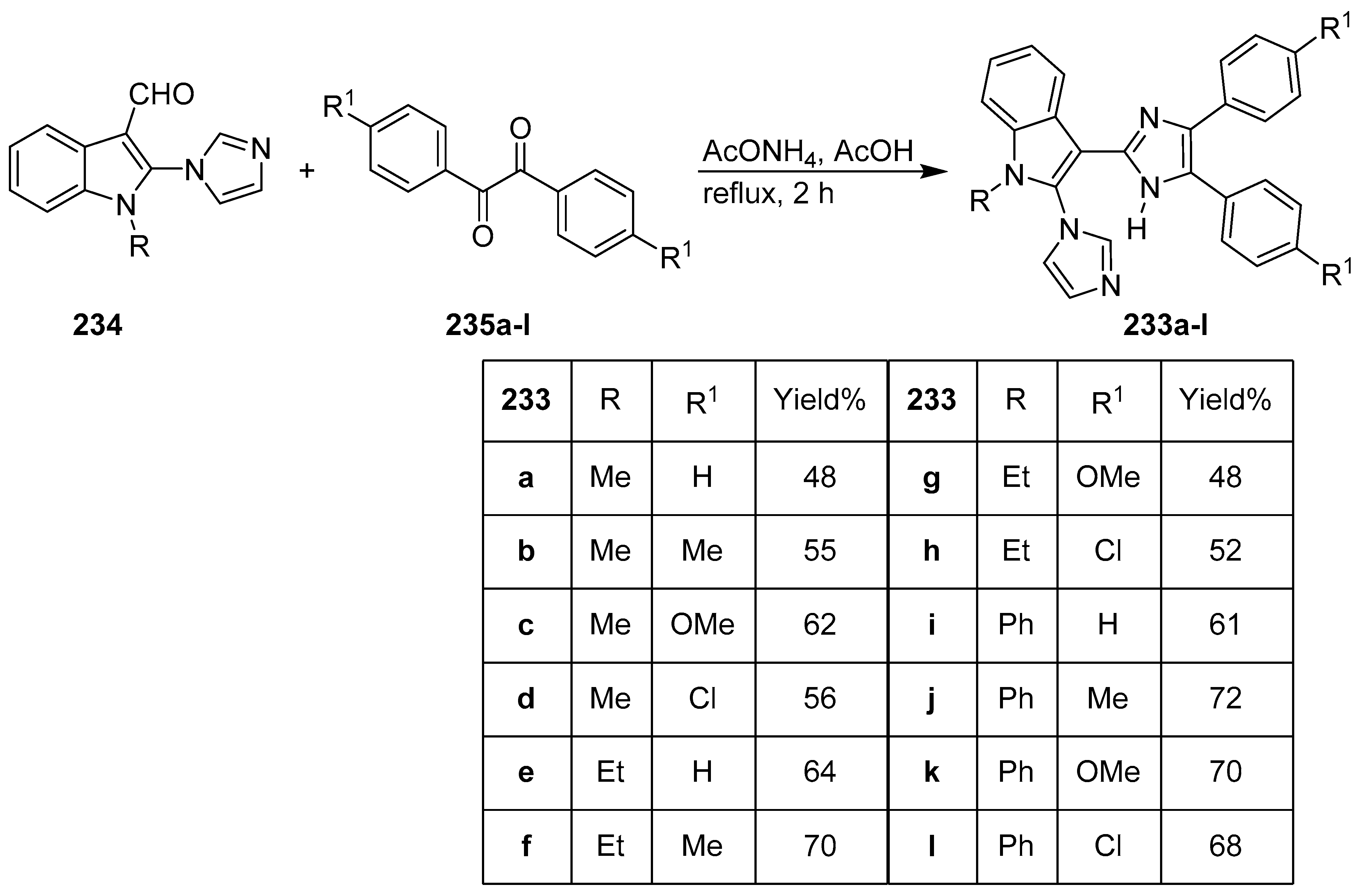
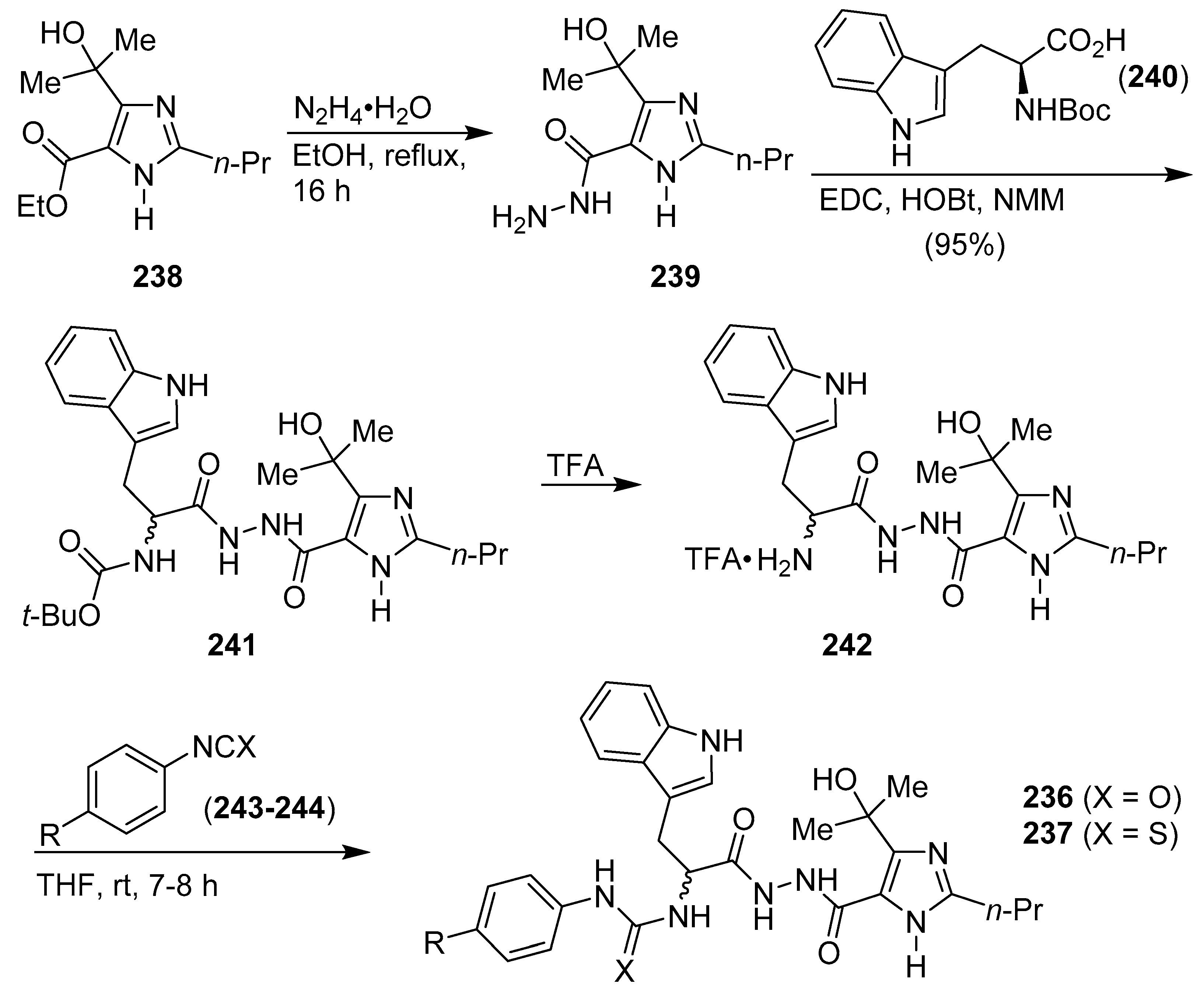
- Kumara, H.K.; Suhas, R.; Suyoga Vardhan, D.M.; Shobha, M.; Channe Gowda, D. Imidazolo and tryptophan-imidazolo hybrid derived ureas/thioureas as potent bioactive agents-SAR and molecular modelling studies. Bioorg. Chem. 2019, 86, 34–38. [Google Scholar] [CrossRef]
- Olofson, A.; Yakushijin, K.; Horne, D.A. Synthesis of Marine Sponge Alkaloids Oroidin, Clathrodin, and Dispacamides. Preparation and Transformation of 2-Amino-4,5-dialkoxy-4,5-dihydroimidazolines from 2-Aminoimidazoles. J. Org. Chem. 1998, 63, 1248–1253. [Google Scholar] [CrossRef]
- Lindel, T.; Hochgürtel, M. Synthesis of the Marine Natural Product Oroidin and Its Z-Isomer. J. Org. Chem. 2000, 65, 2806–2809. [Google Scholar] [CrossRef]
- Berrée, F.; Girard-Le Bleis, P.; Carboni, B. Synthesis of the marine sponge alkaloid oroidin and its analogues via Suzuki cross-coupling reactions. Tetrahedron Lett. 2002, 43, 4935–4938. [Google Scholar] [CrossRef]
- Richards, J.J.; Ballard, T.E.; Melander, C. Inhibition and dispersion of Pseudomonas aeruginosa biofilms with reverse amide 2-aminoimidazole oroidin analogues. Org. Biomol. Chem. 2008, 6, 1356–1363. [Google Scholar] [CrossRef]
- Rasapalli, S.; Kumbam, V.; Dhawane, A.N.; Golen, J.A.; Lovely, C.J.; Rheingold, A.L. Total syntheses of oroidin, hymenidin and clathrodin. Org. Biomol. Chem. 2013, 11, 4133–4137. [Google Scholar] [CrossRef]
- Zidar, N.; Montalvão, S.; Hodnik, Ž.; Nawrot, D.A.; Žula, A.; Ilaš, J.; Kikelj, D.; Tammela, P.; Mašič, L.P. Antimicrobial Activity of the Marine Alkaloids, Clathrodin and Oroidin, and Their Synthetic Analogues. Mar. Drugs 2014, 12, 940–963. [Google Scholar] [CrossRef] [Green Version]
- Forenza, S.; Minale, L.; Riccio, R.; Fattorusso, E. New Bromo-pyrrole Derivatives from the Sponge Agelas oroides. J. Chem. Soc. D 1971, 1129–1130. [Google Scholar] [CrossRef]
- Hao, E.; Fromont, J.; Jardine, D.; Karuso, P. Natural Products From Sponges of the Genus Agelas—On the Trail of a [2+2]-Photoaddition Enzyme. Molecules 2001, 6, 130–141. [Google Scholar] [CrossRef] [Green Version]
- Richelle-Maurer, E.; De Kluijver, M.J.; Feio, S.; Gaudêncio, S.; Gaspar, H.; Gomez, R.; Tavares, R.; Van de Vyver, G.; Van Soest, R.W.M. Localization and ecological significance of oroidin and sceptrin in the Caribbean sponge Agelas conifera. Biochem. Syst. Ecol. 2003, 31, 1073–1091. [Google Scholar] [CrossRef]
- Regalado, E.L.; Laguna, A.; Mendiola, J.; Thomas, O.P.; Nogueiras, C. Bromopyrrole Alkaloids from the Caribbean Sponge Agelas cerebrum. Quim. Nova 2011, 34, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Satheesha Rai, N.; Kalluraya, B. A Novel synthesis of nitrofuran containing 1,3,4,5-tetrasubstituted pyrazoles via 1,3-dipolar addition reaction. Indian J. Chem. 2007, 46B, 375–378. [Google Scholar]
- Bekhit, A.A.; Fahmy, H.T.Y.; Rostom, S.A.F.; Bekhit, A.E.-D.A. Synthesis and biological evaluation of some thiazolylpyrazole derivatives as dual anti-inflammatory antimicrobial agents. Eur. J. Med. Chem. 2010, 45, 6027–6038. [Google Scholar] [CrossRef]
- Mor, S.; Mohil, R.; Kumar, D.; Ahuja, M. Synthesis and antimicrobial activities of some isoxazolyl thiazolyl pyrazoles. Med. Chem. Res. 2012, 21, 3541–3548. [Google Scholar] [CrossRef]
- Pervaram, S.; Ashok, D.; Rao, B.A.; Sarasija, M.; Reddy, C.V.R. Design and Synthesis of New 1,2,3-Triazole-pyrazole Hybrids as Antimicrobial Agents. Russ. J. Gen. Chem. 2017, 87, 2454–2461. [Google Scholar] [CrossRef]
- Menozzi, G.; Merello, L.; Fossa, P.; Schenone, S.; Ranise, A.; Mosti, L.; Bondavalli, F.; Loddo, R.; Murgioni, C.; Mascia, V.; et al. Synthesis, antimicrobial activity and molecular modeling studies of halogenated 4-[1H-imidazol-1-yl(phenyl)methyl]-1,5-diphenyl-1H-pyrazoles. Bioorg. Med. Chem. 2004, 12, 5465–5483. [Google Scholar] [CrossRef]
- Zampieri, D.; Mamolo, M.G.; Laurini, E.; Scialino, G.; Banfi, E.; Vio, L. Antifungal and antimycobacterial activity of 1-(3,5-diaryl-4,5-dihydro-1H-pyrazol-4-yl)-1H-imidazole derivatives. Bioorg. Med. Chem. 2008, 16, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Palomino, J.-C.; Martin, A.; Camacho, M.; Guerra, H.; Swings, J.; Portaels, F. Resazurin Microtiter Assay Plate: Simple and Inexpensive Method for Detection of Drug Resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002, 46, 2720–2722. [Google Scholar] [CrossRef] [Green Version]
- Vijesh, A.M.; Isloor, A.M.; Telkar, S.; Peethambar, S.K.; Rai, S.; Isloor, N. Synthesis, characterization and antimicrobial studies of some new pyrazole incorporated imidazole derivatives. Eur. J. Med. Chem. 2011, 46, 3531–3536. [Google Scholar] [CrossRef]
- Padmavathi, V.; Prema kumari, C.; Venkatesh, B.C.; Padmaja, A. Synthesis and antimicrobial activity of amido linked pyrrolyl and pyrazolyl-oxazoles, thiazoles and imidazoles. Eur. J. Med. Chem. 2011, 46, 5317–5326. [Google Scholar] [CrossRef]
- Jayashree, A.; Narayana, B.; Uppine, G.B.; Ghate, V.M.; Lewis, S.A.; Prakash, B.; Kunhanna, S.B.; Kumar, M.S. ZnO Nanocatalyst Mediated Convergent Synthesis of Highly Substituted Imidazole and Imidazole-derived Bi-heterocyclic Scaffolds as Potential Antibacterial Agents. J. Heterocycl. Chem. 2019, 56, 2398–2410. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Kaplancıklı, Z.A.; Yıldız, M.T.; Chevallet, P.; Kaya, D. Synthesis and antimicrobial activity of 4-phenyl/cyclohexyl-5-(1-phenoxyethyl)-3-[N-(2-thiazolyl)acetamido]thio-4H-1,2,4-triazole derivatives. Eur. J. Med. Chem. 2005, 40, 607–613. [Google Scholar] [CrossRef]
- Sumangala, V.; Poojary, B.; Chidananda, N.; Fernandes, J.; Kumari, N.S. Synthesis and Antimicrobial Activity of 1,2,3-Triazoles Containing Quinoline Moiety. Arch. Pharm. Res. 2010, 33, 1911–1918. [Google Scholar] [CrossRef]
- Kumar, D.; [Negi], B.; Khare, G.; Kidwai, S.; Tyagi, A.K.; Singh, R.; Rawat, D.S. Synthesis of novel 1,2,3-triazole derivatives of isoniazid and their in vitro and in vivo antimycobacterial activity evaluation. Eur. J. Med. Chem. 2014, 81, 301–313. [Google Scholar] [CrossRef]
- Kant, R.; Singh, V.; Nath, G.; Awasthi, S.K.; Agarwal, A. Design, synthesis and biological evaluation of ciprofloxacin tethered bis-1,2,3-triazole conjugates as potent antibacterial agents. Eur. J. Med. Chem. 2016, 124, 218–228. [Google Scholar] [CrossRef]
- Aouad, M.R.; Mayaba, M.M.; Naqvi, A.; Bardaweel, S.K.; Al-blewi, F.F.; Messali, M.; Rezki, N. Design, synthesis, in silico and in vitro antimicrobial screenings of novel 1,2,4-triazoles carrying 1,2,3-triazole scaffold with lipophilic side chain tether. Chem. Cent. J. 2017, 11, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddili, S.K.; Katla, R.; Kannekanti, V.K.; Bejjanki, N.K.; Tuniki, B.; Zhou, C.-H.; Gandham, H. Molecular interaction of novel benzothiazolyl triazolium analogues with calf thymus DNA and HSA-their biological investigation as potent antimicrobial agents. Eur. J. Med. Chem. 2018, 150, 228–247. [Google Scholar] [CrossRef]
- Bi, F.; Ji, S.; Venter, H.; Liu, J.; Semple, S.J.; Ma, S. Substitution of terminal amide with 1H-1,2,3-triazole: Identification of unexpected class of potent antibacterial agents. Bioorg. Med. Chem. Lett. 2018, 28, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Xiao, J.; Huang, G. Antibacterial activity study of 1,2,4-triazole derivatives. Eur. J. Med. Chem. 2019, 173, 274–281. [Google Scholar] [CrossRef]
- Nikalje, A.P.G.; Ghodke, M.S.; Kalam Khan, F.A.; Sangshetti, J.N. CAN catalyzed one-pot synthesis and docking study of some novel substituted imidazole coupled 1,2,4-triazole-5-carboxylic acids as antifungal agents. Chin. Chem. Lett. 2015, 26, 108–112. [Google Scholar] [CrossRef]
- Subhashini, N.J.P.; Praveen Kumar, E.; Gurrapu, N.; Yerragunta, V. Design and synthesis of imidazolo-1, 2,3-triazoles hybrid compounds by microwave-assisted method: Evaluation as an antioxidant and antimicrobial agents and molecular docking studies. J. Mol. Struct. 2019, 1180, 618–628. [Google Scholar] [CrossRef]
- Chauhan, S.; Verma, V.; Kumar, D.; Kumar, A. Facile Synthesis, Antimicrobial Activity and Molecular Docking of Novel 2,4,5-Trisubstituted-1H-Imidazole-Triazole Hybrid Compounds. J. Heterocycl. Chem. 2019, 56, 2571–2579. [Google Scholar] [CrossRef]
- Ravi Kumar, K.R.; Mallesha, H.; Rangappa, K.S. Synthesis of Novel Isoxazolidine Derivatives and Their Antifungal and Antibacterial Properties. Arch. Pharm. 2003, 336, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Ravi Kumar, K.R.; Mallesha, H.; Rangappa, K.S. Synthesis and Characterization of 5-Substituted Novel Isoxazolidines Derived from 1,3-Dipolar Cycloaddition of Nitrones with Olefins: Studies of Antibacterial and Antifungal Activities. Synth. Commun. 2003, 33, 1545–1555. [Google Scholar] [CrossRef]
- Damodiran, M.; Sivakumar, P.M.; SenthilKumar, R.; Muralidharan, D.; Phani Kumar, B.V.N.; Doble, M.; Perumal, P.T. Antibacterial Activity, Quantitative Structure-Activity Relationship and Diastereoselective Synthesis of Isoxazolidine Derivatives Via 1,3-Dipolar Cycloaddition of D-Glucose Derived Nitrone with Olefin. Chem. Biol. Drug Des. 2009, 74, 494–506. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Giofrè, S.V.; Romeo, R.; Romeo, G.; Chiacchio, U. Isoxazolidines as Biologically Active Compounds. Curr. Org. Synth. 2016, 13, 726–749. [Google Scholar] [CrossRef] [Green Version]
- Sadashiva, M.P.; Mallesha, H.; Hitesh, N.A.; Rangappa, K.S. Synthesis and microbial inhibition study of novel 5-imidazolyl substituted isoxazolidines. Bioorg. Med. Chem. 2004, 12, 6389–6395. [Google Scholar] [CrossRef]
- Watson, S.P. A Convenient Synthesis of 2-Butyl-4(5)-chloro-1H-imidazole-5(4)-carboxaldehyde. Synth. Commun. 1992, 22, 2971–2977. [Google Scholar] [CrossRef]
- Griffiths, G.J.; Hauck, M.B.; Imwinkelried, R.; Kohr, J.; Roten, C.A.; Stucky, G.C.; Gosteli, J. Novel Syntheses of 2-Butyl-5-chloro-3H-imidazole-4-carbaldehyde: A Key Intermediate for the Synthesis of the Angiotensin II Antagonist Losartan. J. Org. Chem. 1999, 64, 8084–8089. [Google Scholar] [CrossRef] [PubMed]
- Mallesha, H.; Ravi Kumar, K.R.; Vishu Kumar, B.K.; Mantelingu, K.; Rangappa, K.S. Histidine as a catalyst in organic synthesis: A facile in situ synthesis of α,N-diarylnitrones. J. Chem. Sci. 2001, 113, 291–296. [Google Scholar] [CrossRef]
- Frank, P.V.; Kalluraya, B. Synthesis of 1,3,4-oxadiazoles carrying imidazole moiety. Indian J. Chem. 2005, 44B, 1456–1459. [Google Scholar] [CrossRef]
- Frank, P.V.; Girish, K.S.; Kalluraya, B. Solvent-free microwave-assisted synthesis of oxadiazoles containing imidazole moiety. J. Chem. Sci. 2007, 119, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.-L.; Baathulaa, K.; Kannekanti, V.K.; Zhou, C.-H.; Cai, G.-X. Novel benzimidazole derived naphthalimide triazoles: Synthesis, antimicrobial activity and interactions with calf thymus DNA. Sci. China Chem. 2015, 58, 483–494. [Google Scholar] [CrossRef]
- Shaki, H.; Khosravi, A.; Gharanjig, K.; Mahboubi, A. Investigation of synthesis, characterization, photophysical and biological properties of novel antimicrobial fluorescent naphthalimide derivatives. Mater. Technol. 2016, 31, 322–331. [Google Scholar] [CrossRef]
- Marinov, M.; Kostova, I.; Naydenova, E.; Stoyanov, N. Synthesis and Antimicrobial Activity of 1,8-Naphthalimide Derivatives of Nalidixic Acid. J. Chem. Technol. Metall. 2019, 54, 1146–1156. [Google Scholar]
- Al-Azzawi, A.M.; Hamd, A.S. Synthesis and Antimicrobial Screening of New Naphthalimides Linked to Oxadiazole, Thiadiazole and Triazole Cycles. Int. J. Res. Pharm. Chem. 2014, 4, 283–290. [Google Scholar]
- Al-Azzawi, A.M.; Sulaiman, A. Synthesis, Characterization and Evaluation of Antibacterial Activity of Several New 1,8-Naphthalimides Containing Benzothiazole Moiety. Karbala J. Pharm. Sci. 2011, 111–123. [Google Scholar]
- reported as Bacillus typhi in the original paper.
- Gong, H.-H.; Baathulaa, K.; Lv, J.-S.; Cai, G.-X.; Zhou, C.-H. Synthesis and biological evaluation of Schiff base-linked imidazolyl naphthalimides as novel potential anti-MRSA agents. MedChemComm 2016, 7, 924–931. [Google Scholar] [CrossRef]
- Elmorsy, A.; Hebishy, A.M.S.; Elwahy, A.; Abdelfattah, M.S. Synthesis and Chemistry of Bis-imidazole Derivatives: A Review. GJSFR B Chem. 2018, 18, 25–31. [Google Scholar]
- Zhao, G.; Yin, F.-J.; Ge, H.-Y.; Li, S.-A. Synthesis, Structure and Antibacterial Properties of Bis-Imidazole-bis(naphthalene-1-yl-acetato)copper(II). Asian J. Chem. 2014, 26, 2550–2552. [Google Scholar] [CrossRef]
- Zampieri, D.; Mamolo, M.G.; Vio, L.; Banfi, E.; Scialino, G.; Fermeglia, M.; Ferrone, M.; Pricl, S. Synthesis, antifungal and antimycobacterial activities of new bis-imidazole derivatives, and prediction of their binding to P45014DM by molecular docking and MM/PBSA method. Bioorg. Med. Chem. 2007, 15, 7444–7458. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Zhou, C.-H.; Rao, X.-C. Synthesis and biological activities of novel amine-derived bis-azoles as potential antibacterial and antifungal agents. Eur. J. Med. Chem. 2010, 45, 4388–4398. [Google Scholar] [CrossRef]
- Al-Mohammed, N.N.; Alias, Y.; Abdullah, Z.; Shakir, R.M.; Taha, E.M.; Hamid, A.A. Synthesis and Antibacterial Evaluation of Some Novel Imidazole and Benzimidazole Sulfonamides. Molecules 2013, 18, 11978–11995. [Google Scholar] [CrossRef]
- Al-Mohammed, N.N.; Alias, Y.; Abdullah, Z. Bis-imidazolium and benzimidazolium based gemini-type ionic liquids structure: Synthesis and antibacterial evaluation. RSC Adv. 2015, 5, 92602–92617. [Google Scholar] [CrossRef]
- Antoci, V.; Cucu, D.; Zbancioc, G.; Moldoveanu, C.; Mangalagiu, V.; Amariucai-Mantu, D.; Aricu, A.; Mangalagiu, I.I. Bis-(imidazole/benzimidazole)-pyridine derivatives: Synthesis, structure and antimycobacterial activity. Future Med. Chem. 2020, 12, 207–222. [Google Scholar] [CrossRef] [Green Version]
- Nagarajan, N.; Vanitha, G.; Ananth, D.A.; Rameshkumar, A.; Sivasudha, T.; Renganathan, R. Bioimaging, antibacterial and antifungal properties of imidazole-pyridine fluorophores: Synthesis, characterization and solvatochromism. J. Photochem. Photobiol. B Biol. 2013, 127, 212–222. [Google Scholar] [CrossRef]
- Abbas, I.; Gomha, S.; Elaasser, M.; Bauomi, M. Synthesis and biological evaluation of new pyridines containing imidazole moiety as antimicrobial and anticancer agents. Turk. J. Chem. 2015, 39, 334–346. [Google Scholar] [CrossRef]
- Dhawas, A.K.; Thakare, S.S.; Thakare, N.R. Synthesis and characterization of some new 1,4,5-trisubstituted imidazole-2-thiols derivatives. J. Chem. Pharm. Res. 2012, 4, 866–871. [Google Scholar]
- Fu, H.-G.; Li, Z.-W.; Hu, X.-X.; Si, S.-Y.; You, X.-F.; Tang, S.; Wang, Y.-X.; Song, D.-Q. Synthesis and Biological Evaluation of Quinoline Derivatives as a Novel Class of Broad-Spectrum Antibacterial Agents. Molecules 2019, 24, 548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Von Rosenstiel, N.; Adam, D. Quinolone Antibacterials. An Update of their Pharmacology and Therapeutic Use. Drugs 1994, 47, 872–901. [Google Scholar] [CrossRef]
- Gómez, C.M.M.; Kouznetsov, V.V. Recent Developments on Antimicrobial Quinoline Chemistry. In Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education; Méndez-Vilas, A., Ed.; Formatex Research Center: Badajoz, Spain, 2013; pp. 666–677. [Google Scholar]
- Kharb, R.; Kaur, H. Therapeutic Significance of Quinoline Derivatives as Antimicrobial Agents. Int. Res. J. Pharm. 2013, 4, 63–69. [Google Scholar] [CrossRef]
- Teng, P.; Li, C.; Peng, Z.; Anne Marie, V.; Nimmagadda, A.; Su, M.; Li, Y.; Sun, X.; Cai, J. Facilely accessible quinoline derivatives as potent antibacterial agents. Bioorg. Med. Chem. 2018, 26, 3573–3579. [Google Scholar] [CrossRef]
- Parab, R.H.; Dixit, B.C. Synthesis, Characterization and Antimicrobial Activity of Imidazole Derivatives Based on 2-chloro-7-methyl-3-formylquinoline. E-J. Chem. 2012, 9, 1188–1195. [Google Scholar] [CrossRef]
- Pawar, R.A.; Kohak, A.L.; Gogte, V.G. Synthesis of Benzoxazolo-1,8- & 1,6-naphthapyridines, Benzoxazolylbenzo[h]quinoline & 1,9-Bisbenzoxazolophenanthroline. Indian J. Chem. 1976, 14B, 375–376. [Google Scholar]
- Rajput, A.P. Studies on Vilsmeier-Haack Reaction: Preparation and Synthetic Applications of Synthones 4-Chloro-2-arylaminothiazole-5-carboxaldehydes. Asian J. Chem. 2004, 16, 1374–1380. [Google Scholar]
- Desai, N.C.; Maheta, A.S.; Rajpara, K.M.; Joshi, V.V.; Vaghani, H.V.; Satodiya, H.M. Green synthesis of novel quinoline based imidazole derivatives and evaluation of their antimicrobial activity. J. Saudi Chem. Soc. 2014, 18, 963–971. [Google Scholar] [CrossRef] [Green Version]
- Cui, S.-F.; Peng, L.-P.; Zhang, H.-Z.; Rasheed, S.; Kannekanti, V.K.; Zhou, C.-H. Novel hybrids of metronidazole and quinolones: Synthesis, bioactive evaluation, cytotoxicity, preliminary antimicrobial mechanism and effect of metal ions on their transportation by human serum albumin. Eur. J. Med. Chem. 2014, 86, 318–334. [Google Scholar] [CrossRef]
- reported as Eberthella typhosa in the original paper.
- Zhang, L.; Kannekanti, V.K.; Rasheed, S.; Geng, R.-X.; Zhou, C.-H. Design, Synthesis, and Antimicrobial Evaluation of Novel Quinolone Imidazoles and Interactions with MRSA DNA. Chem. Biol. Drug Des. 2015, 86, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Al-Qawasmeh, R.A.; Huthail, B.B.; Sinnokrot, M.O.; Semreen, M.H.; Odeh, R.A.; Abu-Zarga, M.H.; Tarazi, H.; Yousef, I.A.; Al-Tel, T.H. Design, Synthesis and Qualitative Structure Activity Relationship Evaluations of Quinoline-Based Bisarylimidazoles as Antibacterial Motifs. Med. Chem. (Shariqah) 2016, 12, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Shobhashana, P.G.; Prasad, P.; Kalola, A.G.; Patel, M.P. Synthesis of Imidazole Derivatives Bearing Quinoline Nucleus Catalysed by CAN and their Antimicrobial, Antitubercular and Molecular Docking Studies. Res. J. Life Sci. Bioinform. Pharm. Chem. Sci. 2018, 4, 175–186. [Google Scholar] [CrossRef]
- Insuasty, D.; Vidal, O.; Bernal, A.; Marquez, E.; Guzman, J.; Insuasty, B.; Quiroga, J.; Svetaz, L.; Zacchino, S.; Puerto, G.; et al. Antimicrobial Activity of Quinoline-Based Hydroxyimidazolium Hybrids. Antibiotics 2019, 8, 239. [Google Scholar] [CrossRef] [Green Version]
- Laali, K.K.; Insuasty, D.; Abonia, R.; Insuasty, B.; Bunge, S.D. Novel quinoline–imidazolium adducts via the reaction of 2-oxoquinoline-3-carbaldehyde and quinoline-3-carbaldehydes with 1-butyl-3-methylimidazolium chloride [BMIM][Cl]. Tetrahedron Lett. 2014, 55, 4395–4399. [Google Scholar] [CrossRef]
- Meth-Cohn, O. The Synthesis of Pyridines, Quinolines and Other Related Systems by the Vilsmeier and the Reverse Vilsmeier Method. Heterocycles (Sendai) 1993, 35, 539–557. [Google Scholar] [CrossRef]
- Cieplik, J.; Stolarczyk, M.; Pluta, J.; Gubrynowicz, O.; Bryndal, I.; Lis, T.; Mikulewicz, M. Synthesis and Antibacterial Properties of Pyrimidine Derivatives. Acta Pol. Pharm. 2015, 72, 53–64. [Google Scholar] [PubMed]
- Sharma, V.; Chitranshi, N.; Agarwal, A.K. Significance and Biological Importance of Pyrimidine in the Microbial World. Int. J. Med. Chem. 2014, 2014, 202784. [Google Scholar] [CrossRef] [Green Version]
- Dansena, H.; Dhongade, H.J.; Chandrakar, K. Pharmacological Potentials of Pyrimidine Derivative: A Review. Asian J. Pharm. Clin. Res. 2015, 8, 171–177. [Google Scholar]
- Ibraheem, F.; Saddique, F.A.; Aslam, S.; Mansha, A.; Farooq, T.; Ahmad, M. Recent synthetic methodologies for pyrimidine and its derivatives. Turk. J. Chem. 2018, 42, 1421–1458. [Google Scholar] [CrossRef]
- Botta, M.; Artico, M.; Massa, S.; Gambacorta, A.; Marongiu, M.E.; Pani, A.; La Colla, P. Synthesis, antimicrobial and antiviral activities of isotrimethoprim and some related derivatives. Eur. J. Med. Chem. 1992, 27, 251–257. [Google Scholar] [CrossRef]
- Agarwal, N.; Srivastava, P.; Raghuwanshi, S.K.; Upadhyay, D.N.; Sinha, S.; Shukla, P.K.; Ram, V.J. Chloropyrimidines as a New Class of Antimicrobial Agents. Bioorg. Med. Chem. 2002, 10, 869–874. [Google Scholar] [CrossRef]
- Prakash, O.; Bhardwaj, V.; Kumar, R.; Tyagi, P.; Aneja, K.R. Organoiodine (III) mediated synthesis of 3-aryl/hetryl-5,7-dimethyl-1,2,4-triazolo[4,3-a]pyrimidines as antibacterial agents. Eur. J. Med. Chem. 2004, 39, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Desai, N.C.; Kotadiya, G.M.; Trivedi, A.R. Studies on molecular properties prediction, antitubercular and antimicrobial activities of novel quinoline based pyrimidine motifs. Bioorg. Med. Chem. Lett. 2014, 24, 3126–3130. [Google Scholar] [CrossRef]
- Rathod, A.K. Antifungal and Antibacterial activities of Imidazolylpyrimidines derivatives and their QSAR Studies under Conventional and Microwave-assisted. Int. J. PharmTech Res. 2011, 3, 1942–1951. [Google Scholar]
- Desai, N.C.; Vaghani, H.V.; Rajpara, K.M.; Joshi, V.V.; Satodiya, H.M. Novel approach for synthesis of potent antimicrobial hybrid molecules containing pyrimidine-based imidazole scaffolds. Med. Chem. Res. 2014, 23, 4395–4403. [Google Scholar] [CrossRef]
- Pathan, N.B.; Rahatgaonkar, A.M. Solid supported microwave induced synthesis of imidazole–pyrimidine hybrids: Antimicrobial evaluation and docking study as 14DM-CPY51 inhibitors. Arab. J. Chem. 2016, 9, S100–S108. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, B.F.; Awad, G.E.A.; Badria, F.A. Synthesis, antimicrobial, antioxidant, anti-hemolytic and cytotoxic evaluation of new imidazole-based heterocycles. Eur. J. Med. Chem. 2011, 46, 1505–1511. [Google Scholar] [CrossRef]
- Veronese, A.C.; Cavicchioni, G.; Servadio, G.; Vecchiati, G. Syntheses of 2-Arylimidazole Derivatives through Annelations Employing Benzylamines. J. Heterocycl. Chem. 1980, 17, 1723–1725. [Google Scholar] [CrossRef]
- Addla, D.; [Bhukya], B.; Sridhar, B.; Devi, A.; Kantevari, S. Design, synthesis and antimicrobial evaluation of novel 1-benzyl 2-butyl-4-chloroimidazole embodied 4-azafluorenones via molecular hybridization approach. Bioorg. Med. Chem. Lett. 2012, 22, 7475–7480. [Google Scholar] [CrossRef]
- Prachayasittikul, S.; Manam, P.; Chinworrungsee, M.; Isarankura-Na-Ayudhya, C.; Ruchirawat, S.; Prachayasittikul, V. Bioactive Azafluorenone Alkaloids from Polyalthia debilis (Pierre) Finet & Gagnep. Molecules 2009, 14, 4414–4424. [Google Scholar] [CrossRef]
- Koyama, J.; Morita, I.; Kobayashi, N.; Osakai, T.; Usuki, Y.; Taniguchi, M. Structure–activity relations of azafluorenone and azaanthraquinone as antimicrobial compounds. Bioorg. Med. Chem. Lett. 2005, 15, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
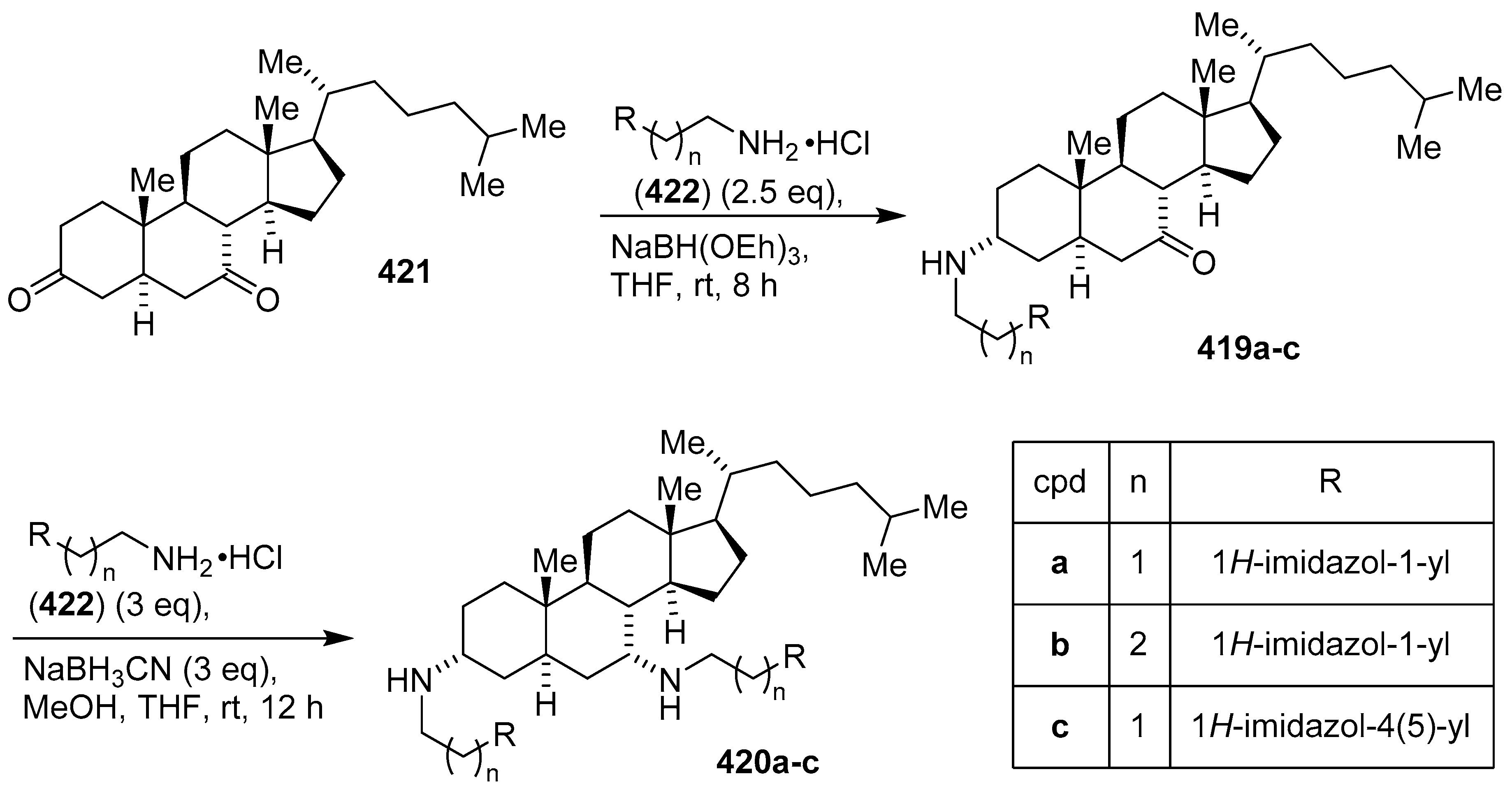
- Kim, H.-S.; Jadhav, J.R.; Jung, S.-J.; Kwak, J.-H. Synthesis and antimicrobial activity of imidazole and pyridine appended cholestane-based conjugates. Bioorg. Med. Chem. Lett. 2013, 23, 4315–4318. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.-Q.; Jeyakkumar, P.; Srinivasa Rao, A.; Zhang, L.; Zhou, C.-H. Discovery of novel berberine imidazoles as safe antimicrobial agents by down regulating ROS generation. Bioorg. Med. Chem. Lett. 2016, 26, 2768–2773. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, C.; Ying, Y.; Luo, L.; Huang, D.; Luo, Z. Metformin and berberine, two versatile drugs in treatment of common metabolic diseases. Oncotarget 2018, 9, 10135–10146. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.-B.; Yan, Y.; Liang, Y.-Z.; Bao, Z. Evaluation of the antimicrobial mode of berberine by LC/ESI-MS combined with principal component analysis. J. Pharm. Biomed. Anal. 2007, 44, 301–304. [Google Scholar] [CrossRef]
- Yan, D.; Jin, C.; Xiao, X.-H.; Dong, X.-P. Antimicrobial properties of berberines alkaloids in Coptis chinensis Franch by microcalorimetry. J. Biochem. Bioph. Methods 2008, 70, 845–849. [Google Scholar] [CrossRef]
- Peng, L.; Kang, S.; Yin, Z.; Jia, R.; Song, X.; Li, L.; Li, Z.; Zou, Y.; Liang, X.; Li, L.; et al. Antibacterial activity and mechanism of berberine against Streptococcus agalactiae. Int. J. Clin. Exp. Pathol. 2015, 8, 5217–5223. [Google Scholar] [PubMed]
- Iwasa, K.; Lee, D.-U.; Kang, S.-I.; Wiegrebe, W. Antimicrobial Activity of 8-Alkyl- and 8-Phenyl-Substituted Berberines and Their 12-Bromo Derivatives. J. Nat. Prod. 1998, 61, 1150–1153. [Google Scholar] [CrossRef]
- Li, R.; Wu, J.; He, Y.; Hai, L.; Wu, Y. Synthesis and in vitro evaluation of 12-(substituted aminomethyl) berberrubine derivatives as anti-diabetics. Bioorg. Med. Chem. Lett. 2014, 24, 1762–1765. [Google Scholar] [CrossRef]
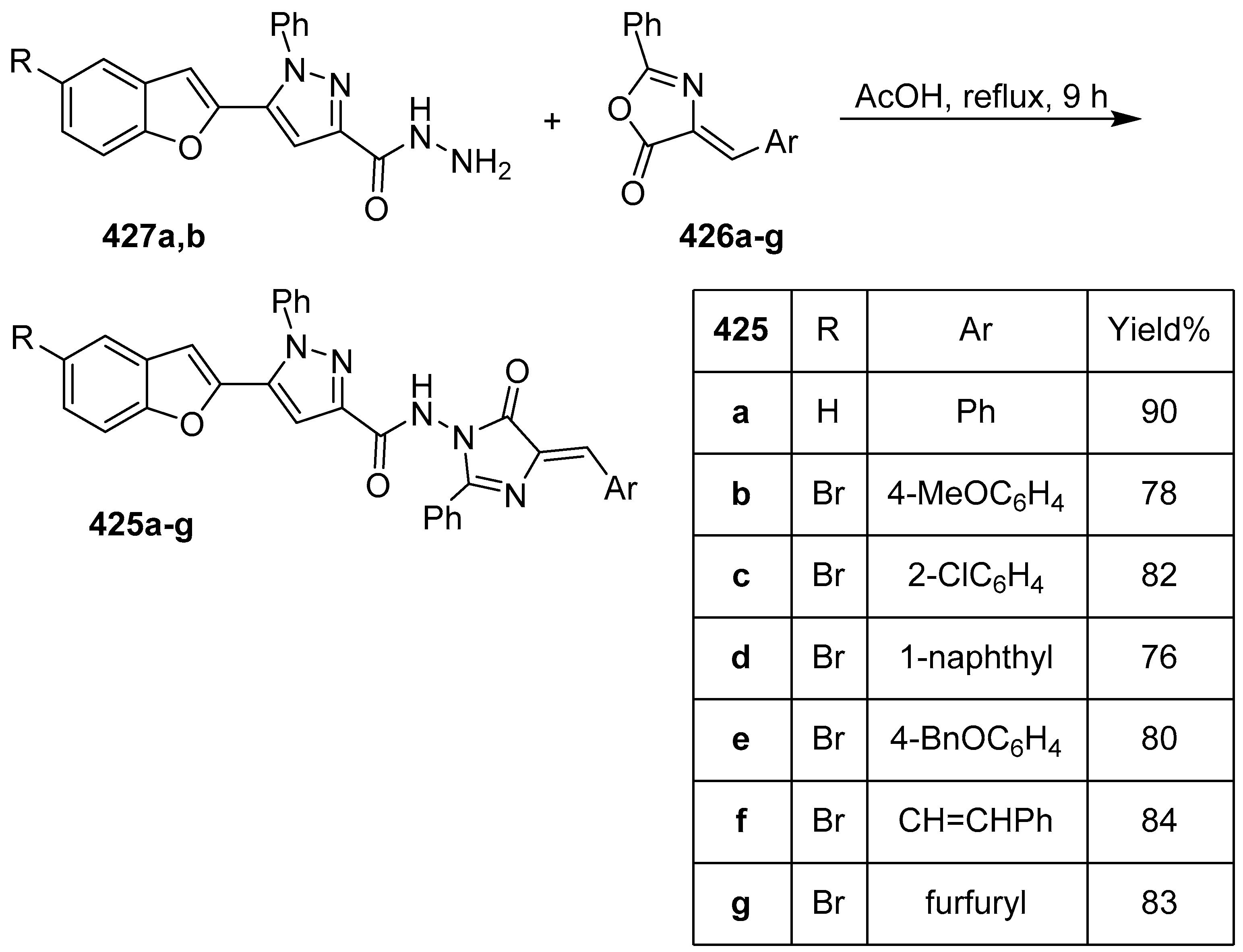
- Idrees, M.; Nasare, R.D.; Siddiqui, N.J. Synthesis and Antibacterial Screening of 4-Arylidene-5-oxo-imidazoles having Carboxamide Linkage with 5-(benzofuran-2-yl)-1-phenylpyrazole Moiety. Chem. Sci. Trans. 2016, 5, 1090–1095. [Google Scholar] [CrossRef]
- Sharma, S.; Sharma, V.; Singh, G.; Kaur, H.; Srivastava, S.; Ishar, M.P.S. 2-(chromon-3-yl)imidazole derivatives as potential antimicrobial agents: Synthesis, biological evaluation and molecular docking studies. J. Chem. Biol. 2017, 10, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biggs-Houck, J.E.; Younai, A.; Shaw, J.T. Recent advances in multicomponent reactions for diversity-oriented synthesis. Curr. Opin. Chem. Biol. 2010, 14, 371–382. [Google Scholar] [CrossRef]
- Dömling, A.; Wang, W.; Wang, K. Chemistry and Biology of Multicomponent Reactions. Chem. Rev. 2012, 112, 3083–3135. [Google Scholar] [CrossRef] [Green Version]
- Malinakova, H.C. Recent advances in the discovery and design of multicomponent reactions for the generation of small-molecule libraries. Rep. Org. Chem. 2015, 75–90. [Google Scholar] [CrossRef] [Green Version]
- Graebin, C.S.; Ribeiro, F.V.; Rogério, K.R.; Kümmerle, A.E. Multicomponent Reactions for the Synthesis of Bioactive Compounds: A Review. Curr. Org. Synth. 2019, 16, 855–899. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled Microwave Heating in Modern Organic Synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Bowman, M.D.; Holcomb, J.L.; Kormos, C.M.; Leadbeater, N.E.; Williams, V.A. Approaches for Scale-Up of Microwave-Promoted Reactions. Org. Process. Res. Dev. 2008, 12, 41–57. [Google Scholar] [CrossRef]
- Loftin, A.; Armstrong, D. Review: Microwave-Promoted Organic Synthesis. Am. J. Undergrad. Res. 2009, 8, 5–7. [Google Scholar] [CrossRef]
- Hügel, H.M. Microwave Multicomponent Synthesis. Molecules 2009, 14, 4936–4972. [Google Scholar] [CrossRef] [Green Version]
- Kokel, A.; Schäfer, C.; Török, B. Microwave-Assisted Reactions in Green Chemistry. In Green Chemistry and Chemical Engineering. A Volume in the Encyclopedia of Sustainability Science and Technology, 2nd ed.; Han, B., Wu, T., Eds.; Springer: New York, NY, USA, 2019; pp. 573–612. [Google Scholar]
- Liang, L.; Astruc, D. The copper(I)-catalyzed alkyne-azide cycloaddition (CuAAC) “click” reaction and its applications. An overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Zheng, X.; Ma, Z.; Zhang, D. Synthesis of Imidazole-Based Medicinal Molecules Utilizing the van Leusen Imidazole Synthesis. Pharmaceuticals 2020, 13, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed. Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [Green Version]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef]
- Butler, M.S.; Paterson, D.L. Antibiotics in the clinical pipeline in October 2019. J. Antibiot. (Tokyo) 2020, 73, 329–364. [Google Scholar] [CrossRef]
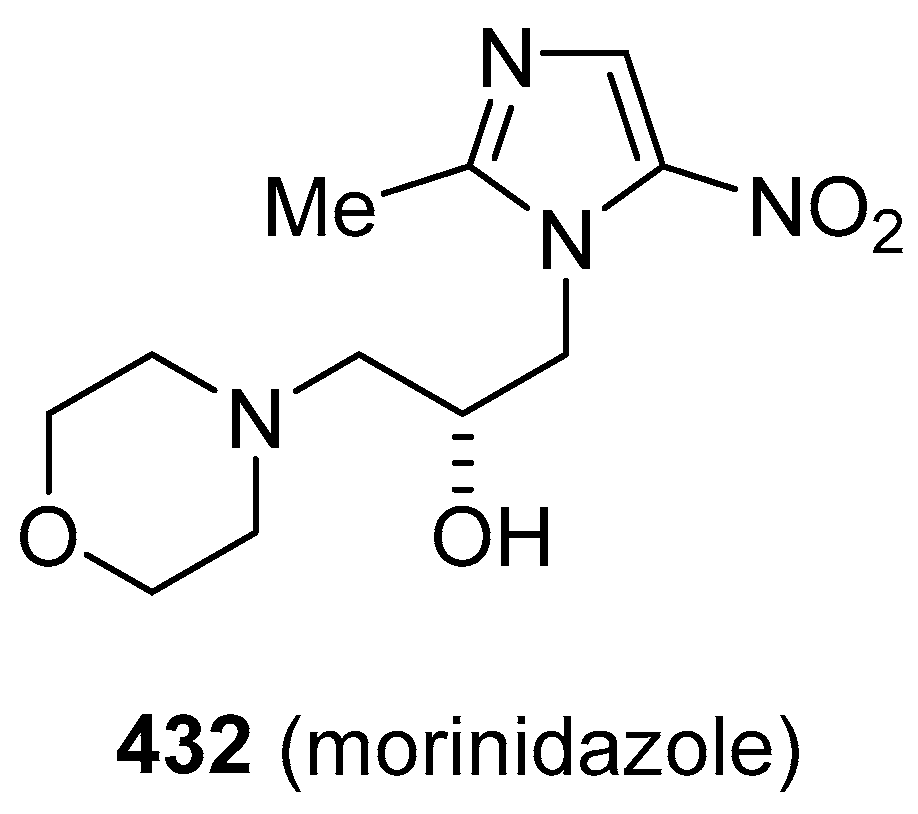
- Cao, C.; Luo, A.; Wu, P.; Weng, D.; Zheng, H.; Wang, S. Efficacy and safety of morinidazole in pelvic inflammatory disease: Results of a multicenter, double-blind, randomized trial. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1225–1230. [Google Scholar] [CrossRef]
- Ocakoglu, K.; Tasli, H.; Hosgor-Limoncu, M.; Lambrecht, F.Y. Synthesis and antimicrobial activity of imidazolium salts. Trends Cancer Res. Chemother. 2018, 1. [Google Scholar] [CrossRef] [Green Version]

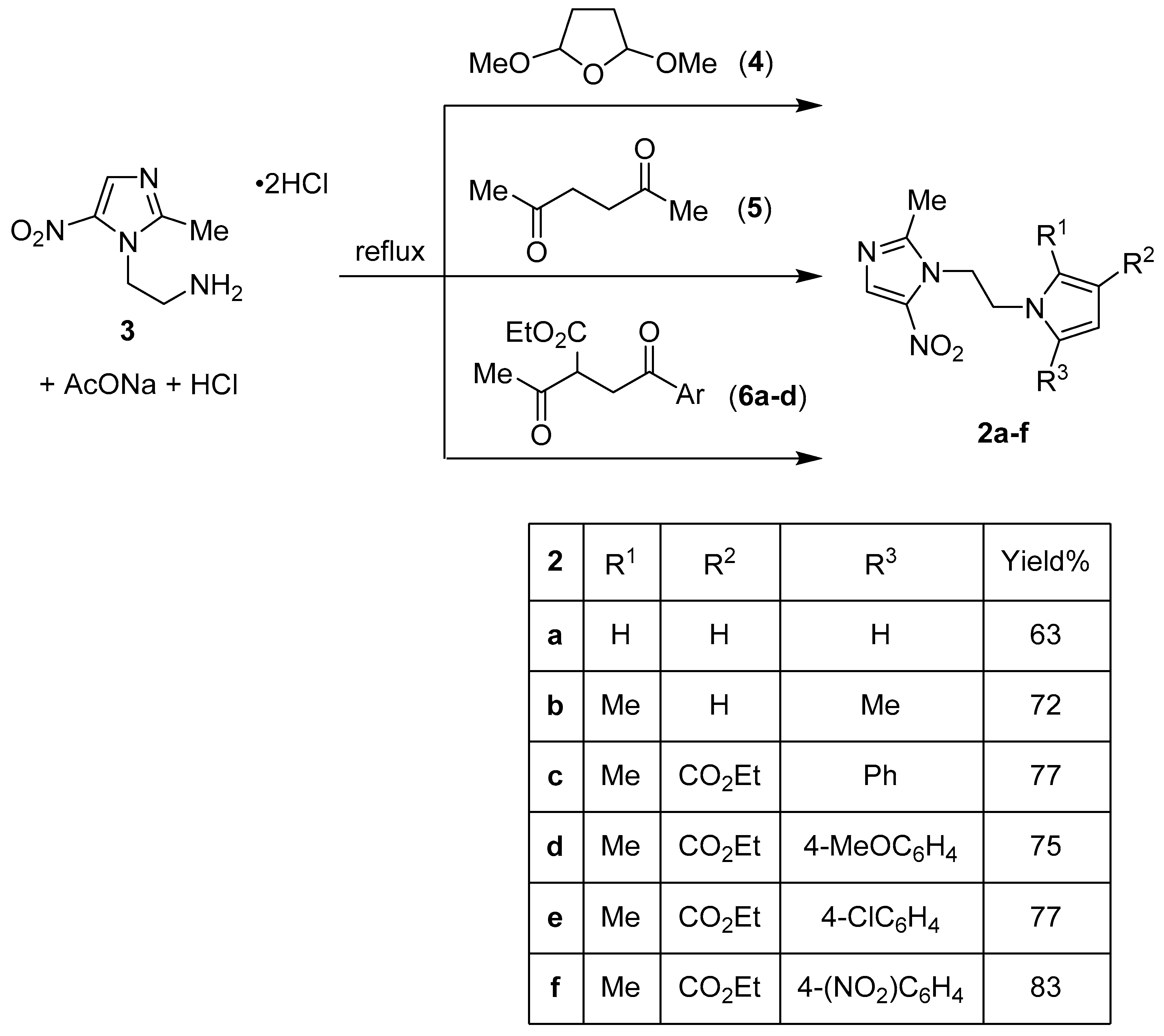




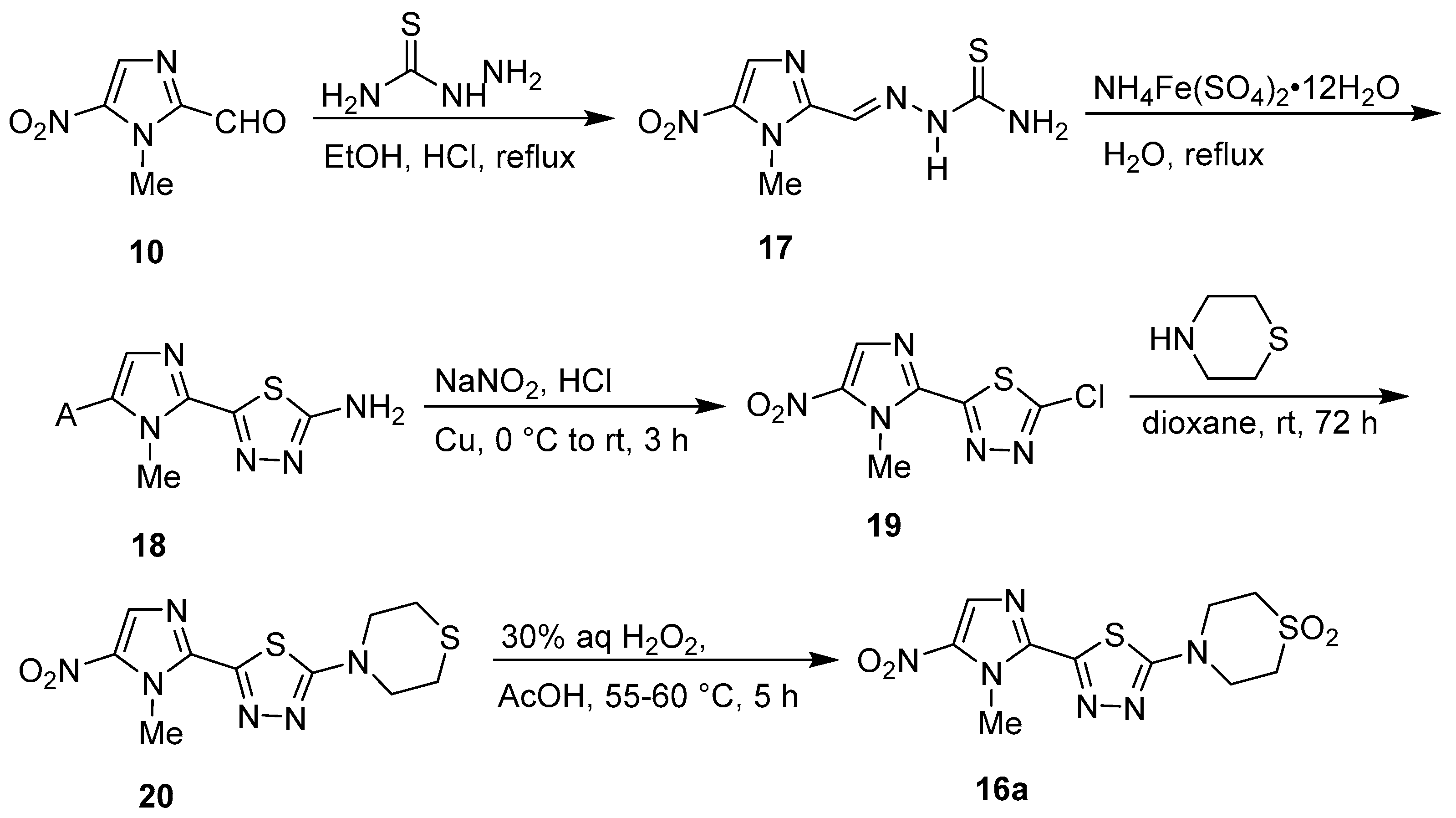
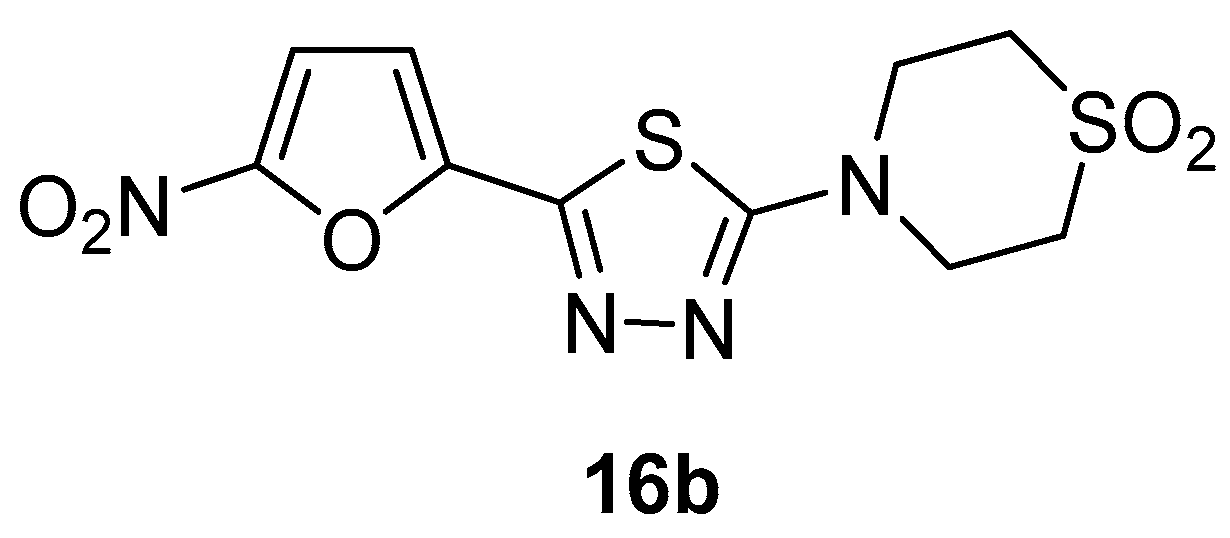

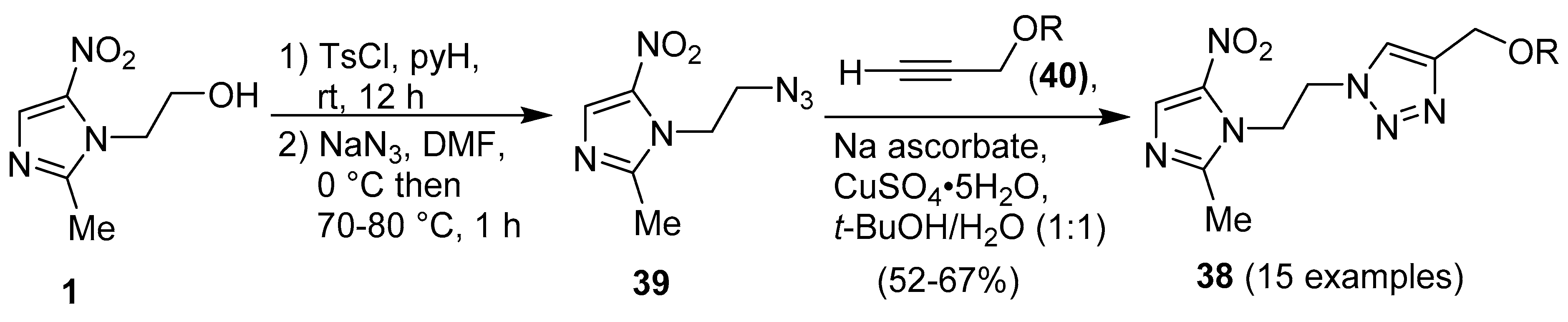
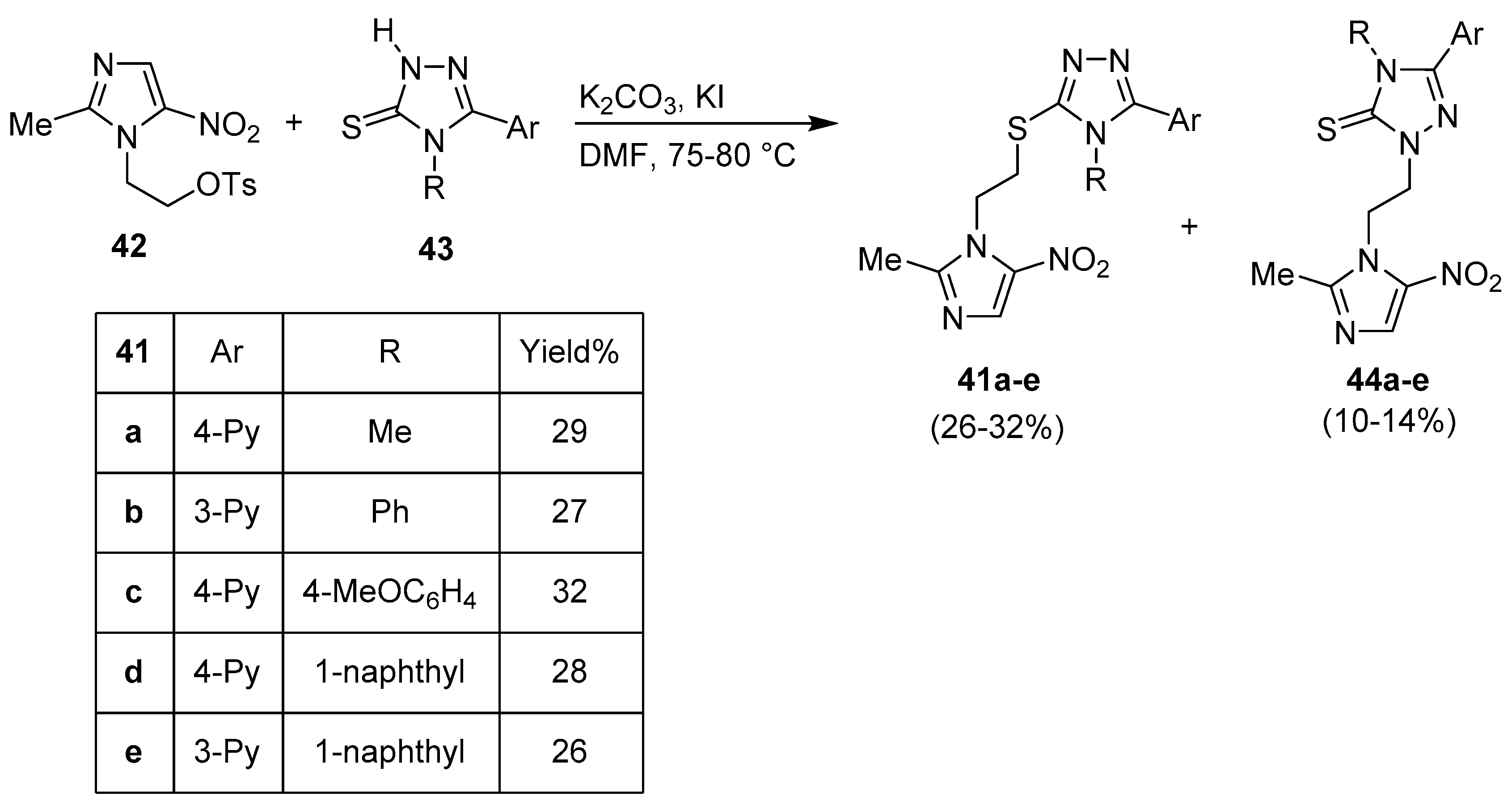
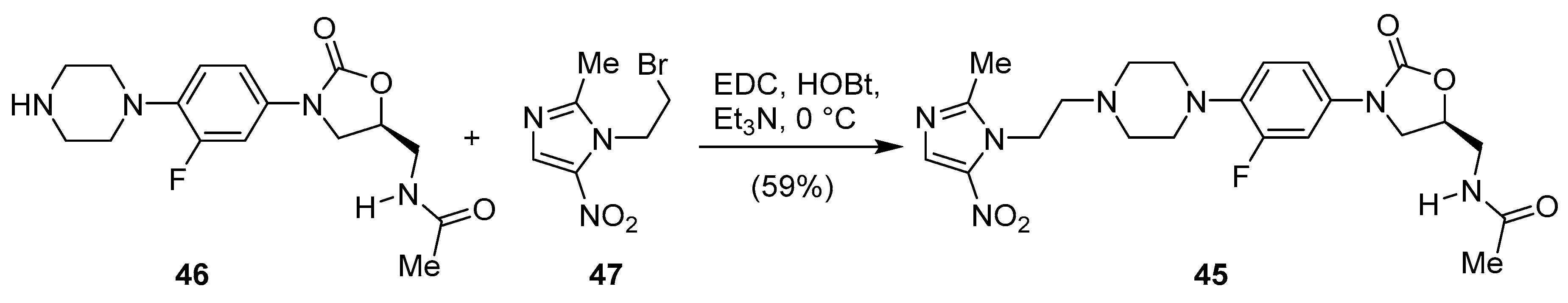
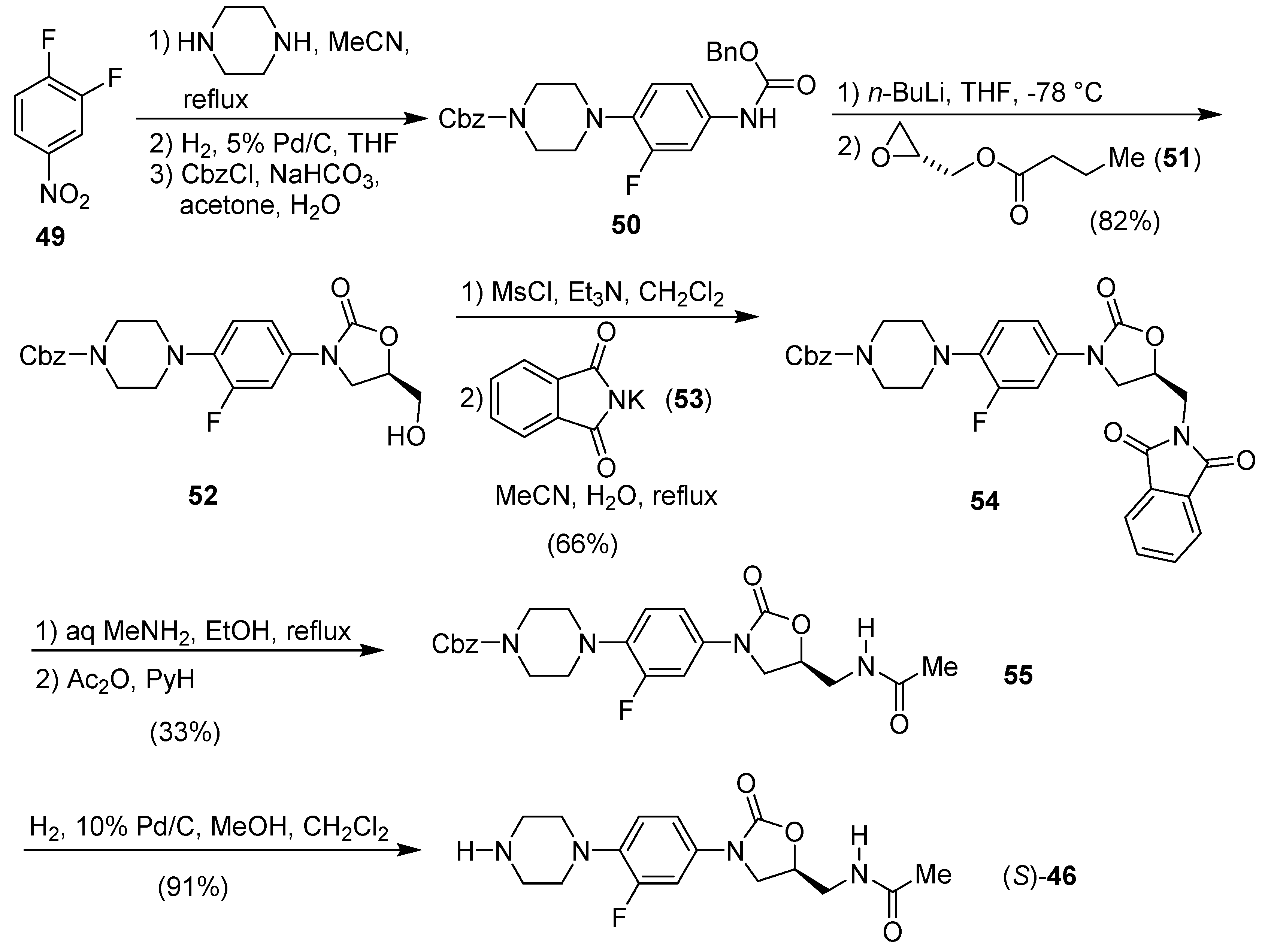



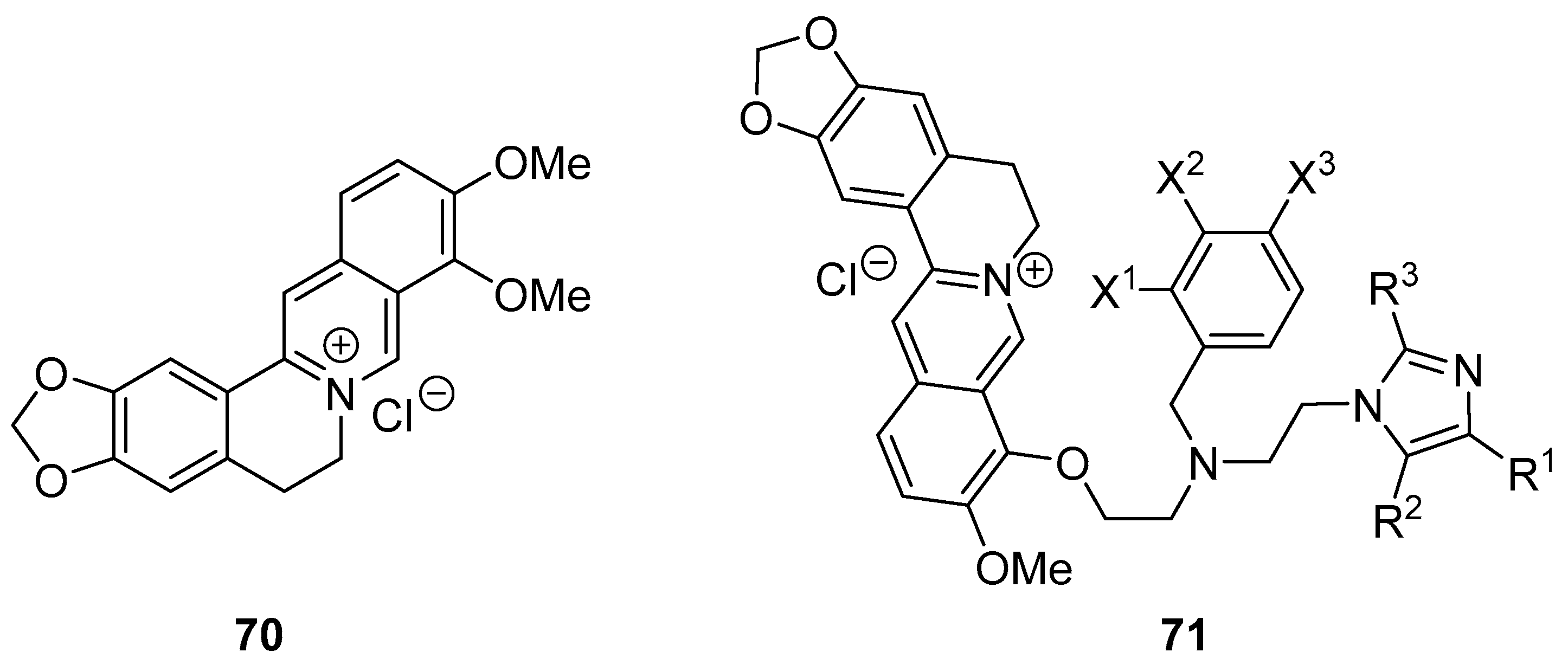
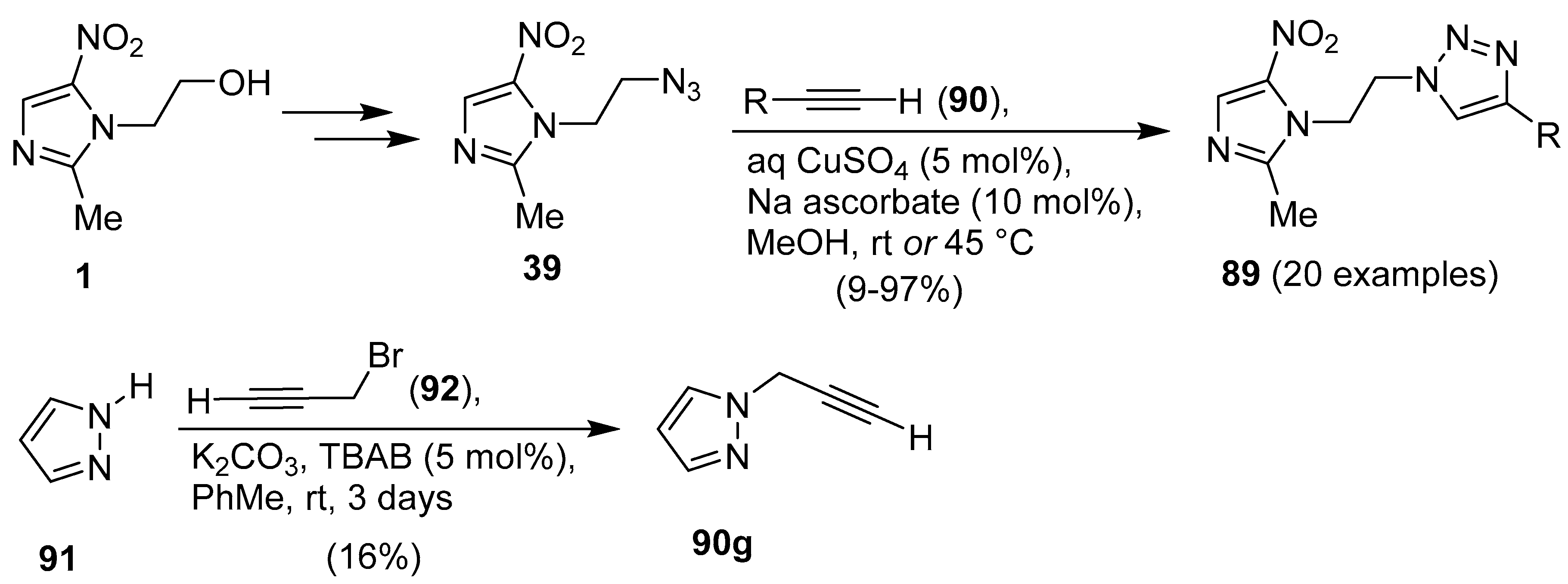
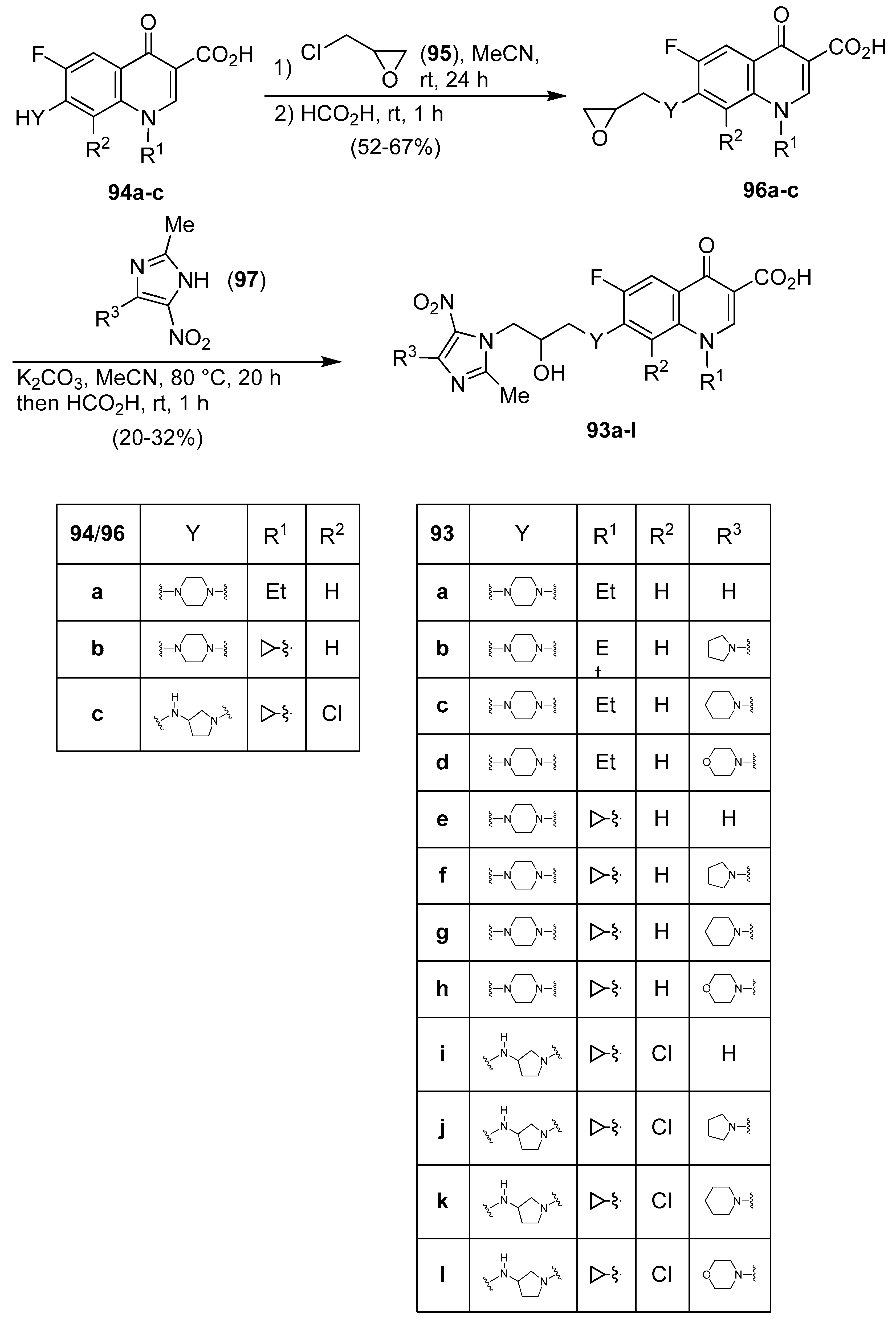

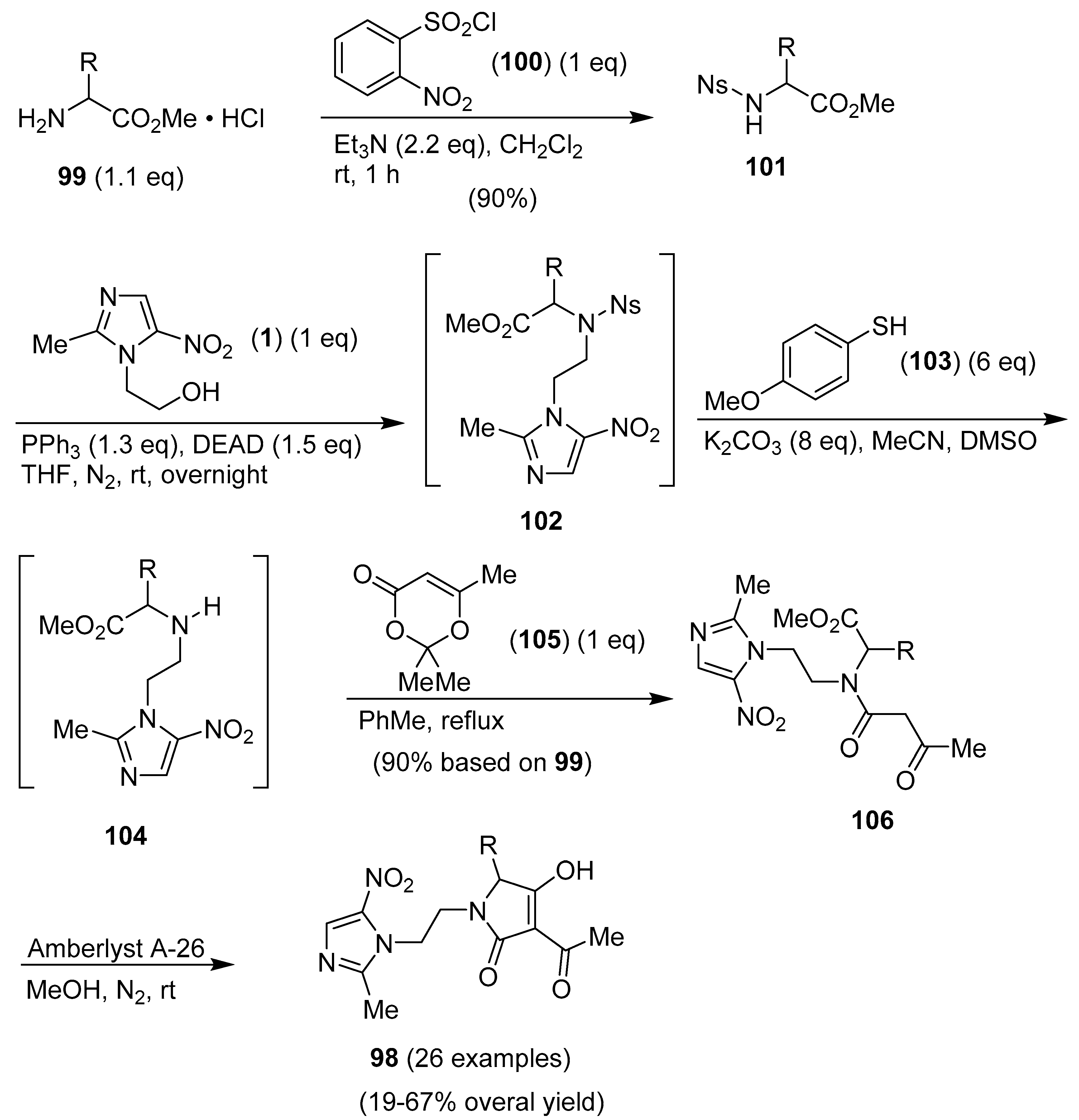

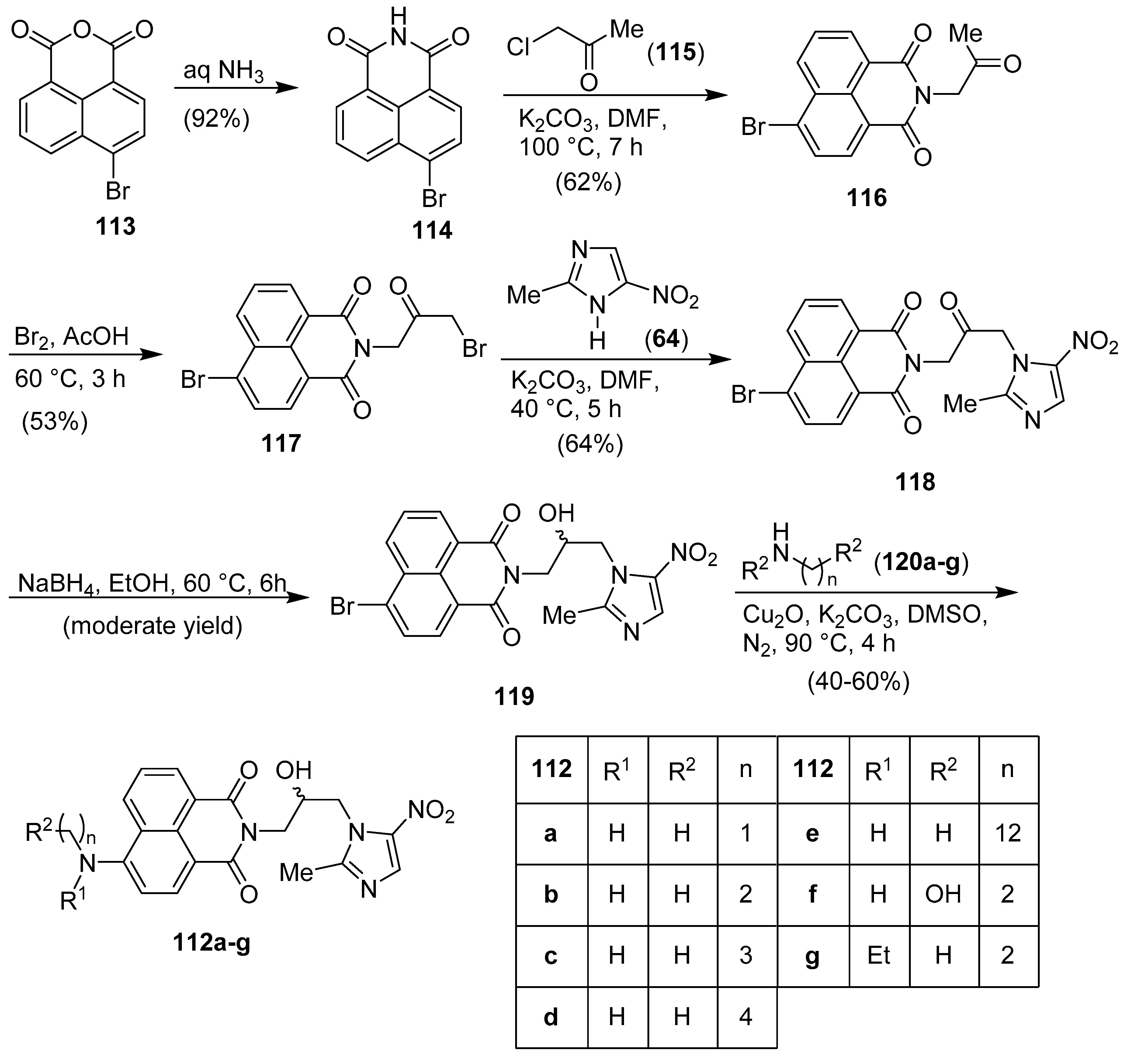







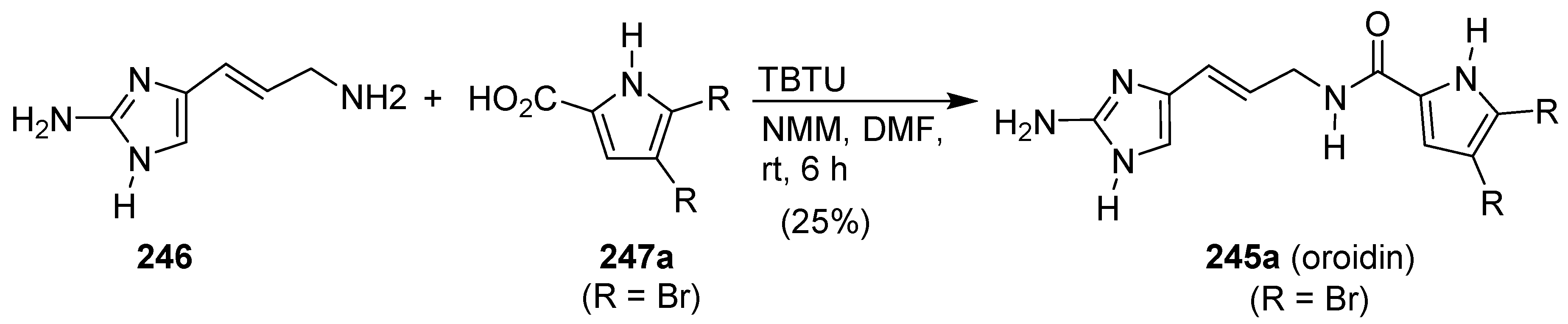
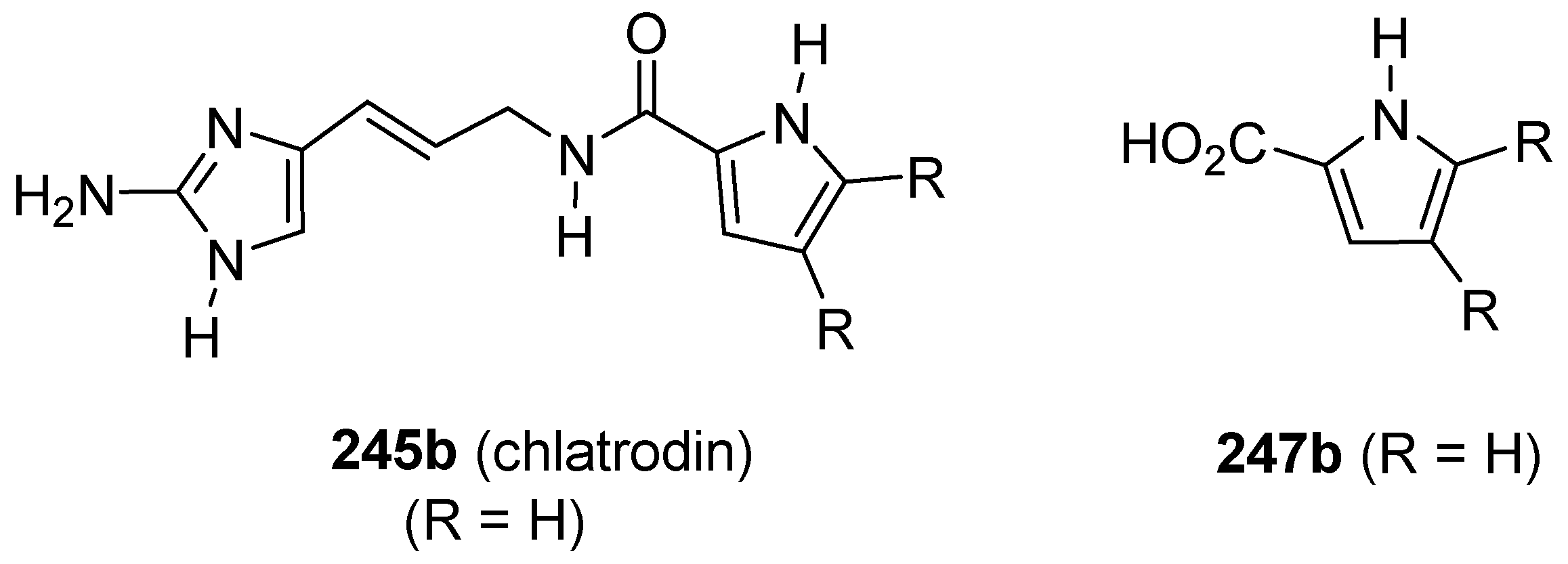
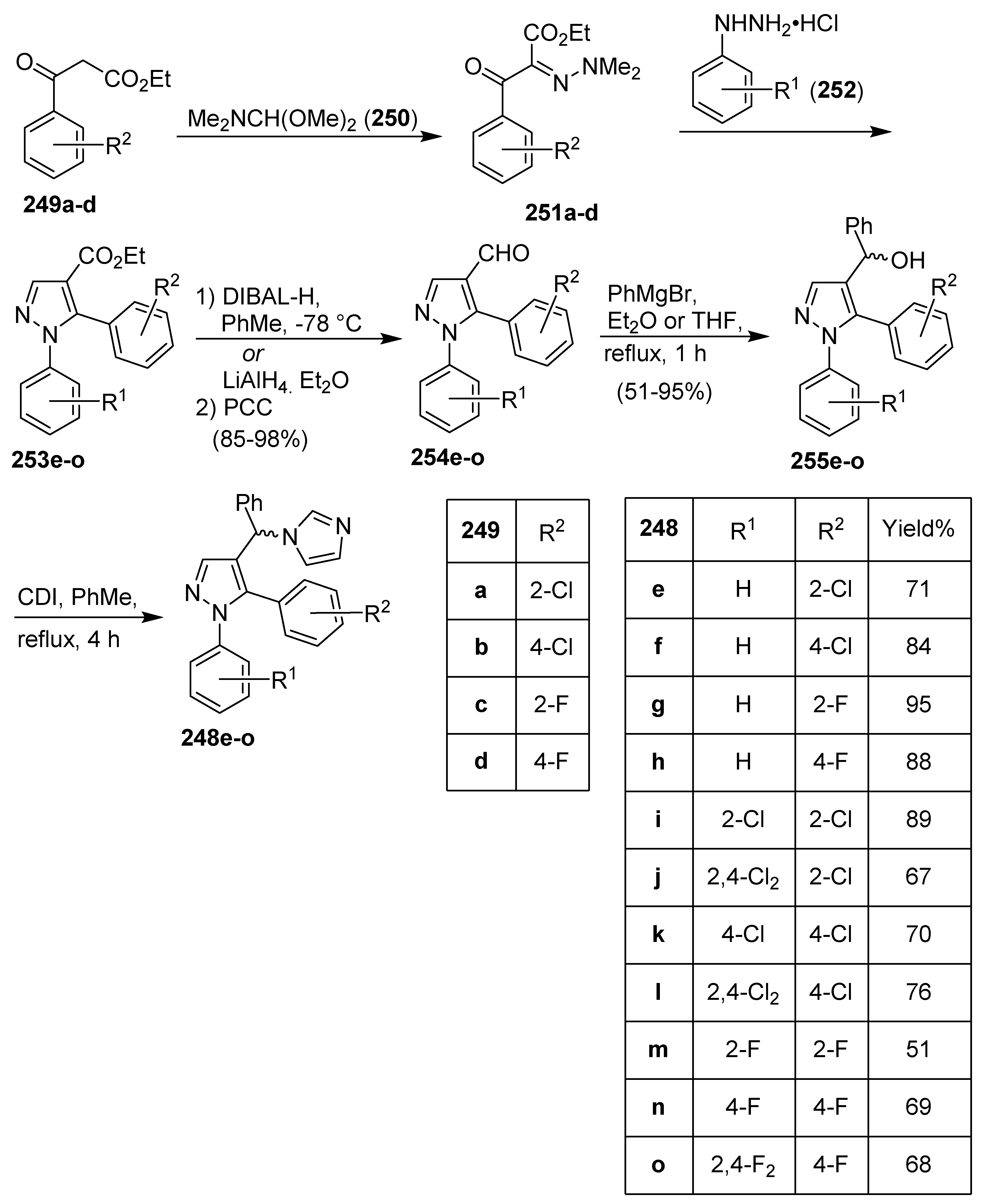
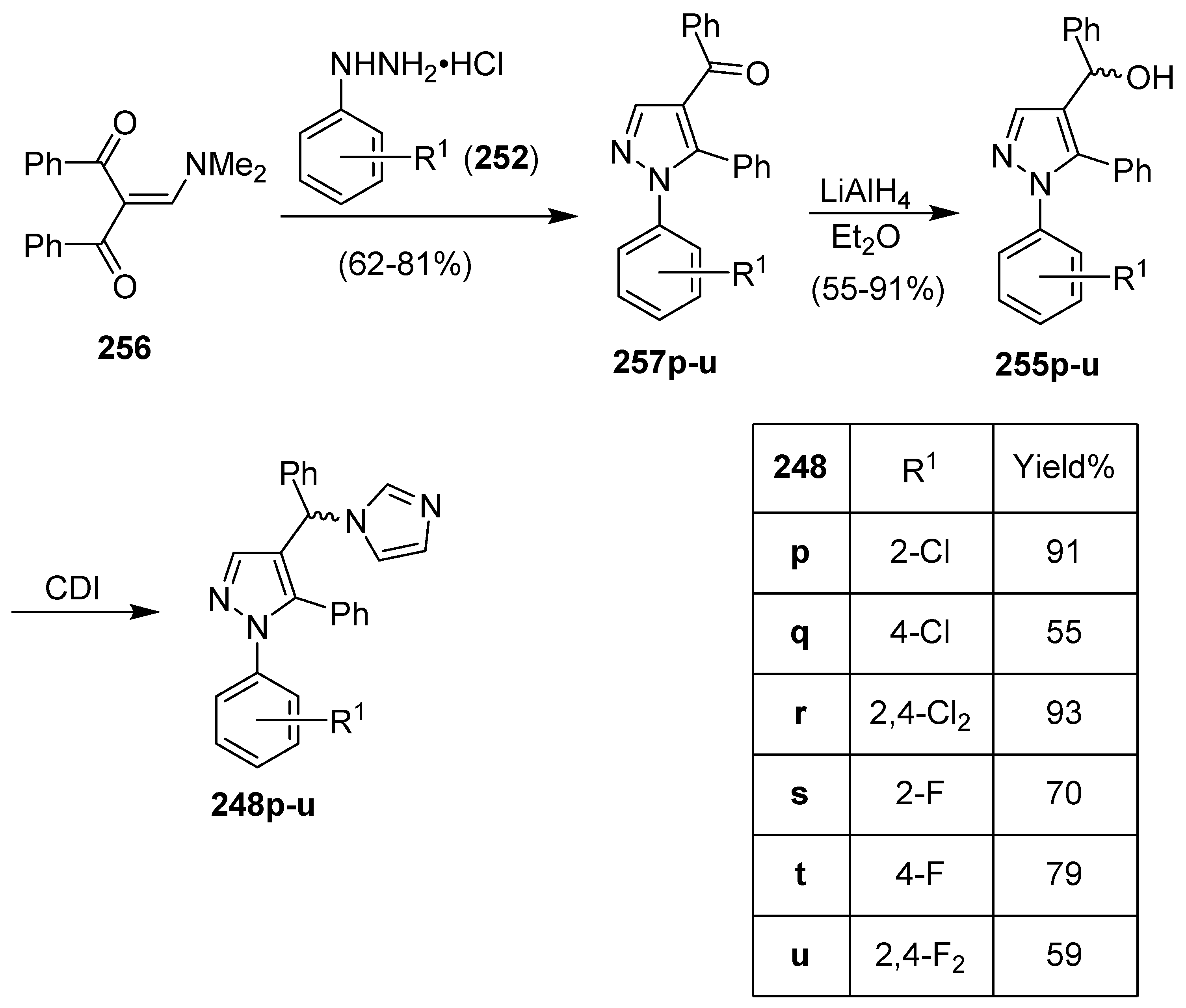







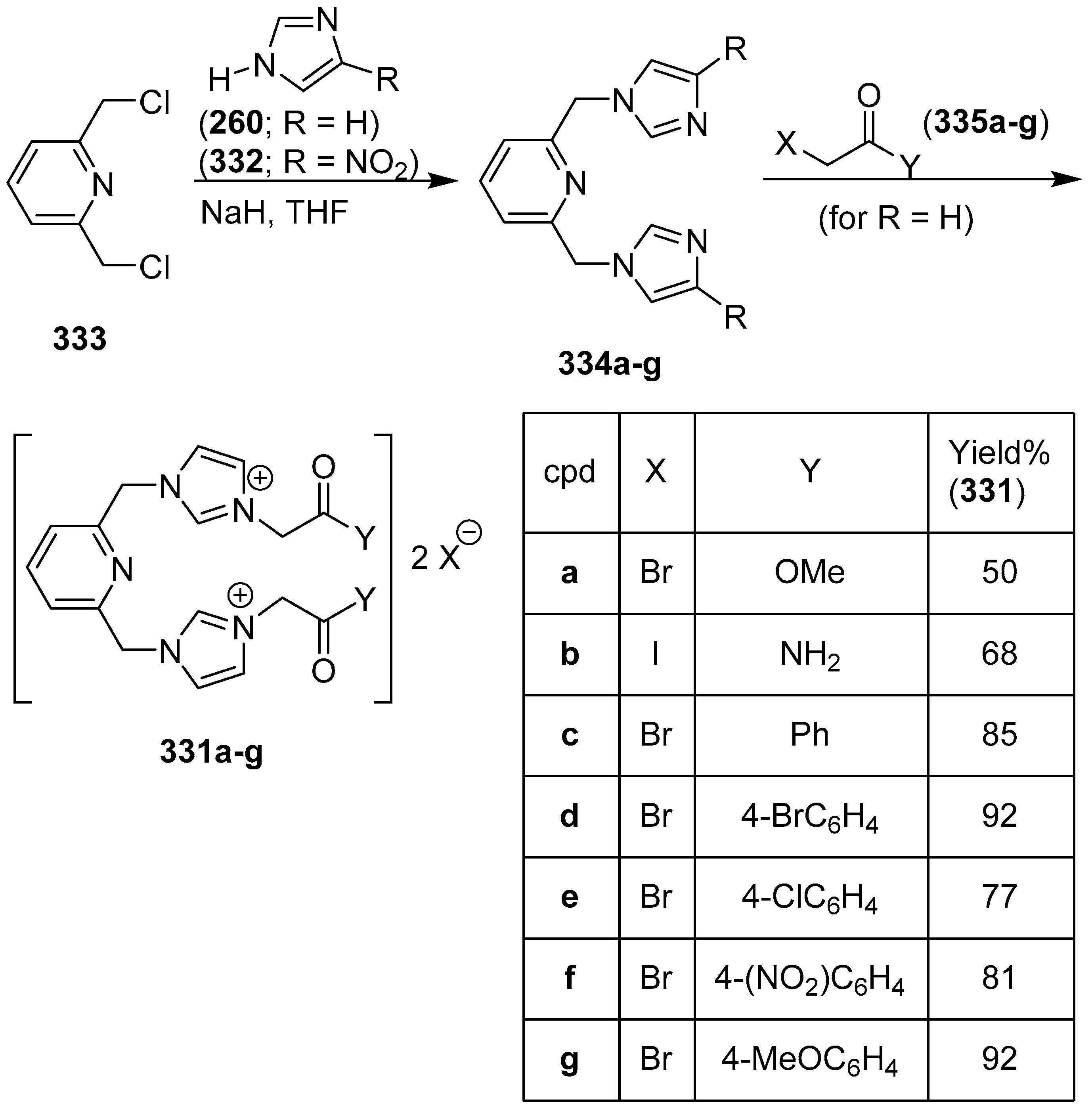

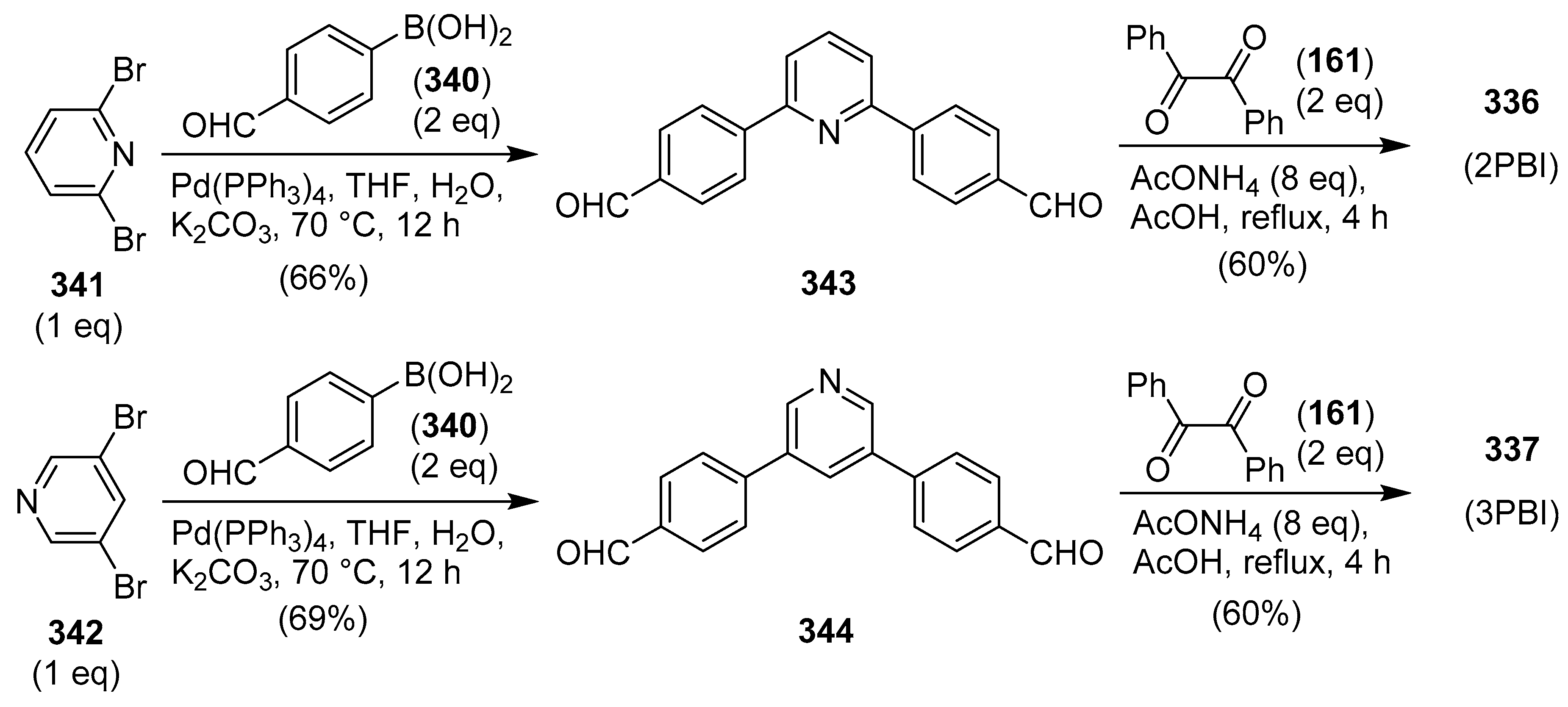
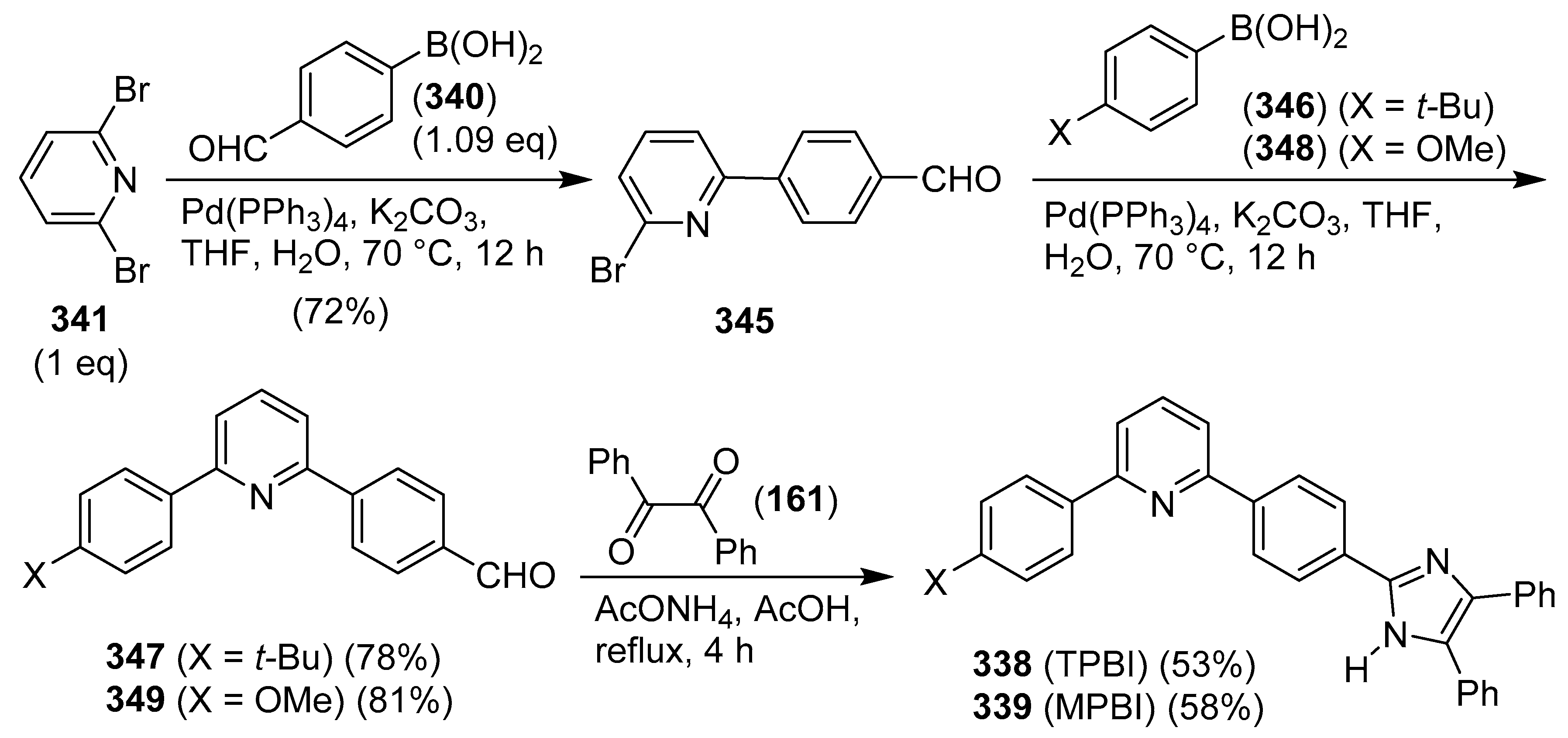
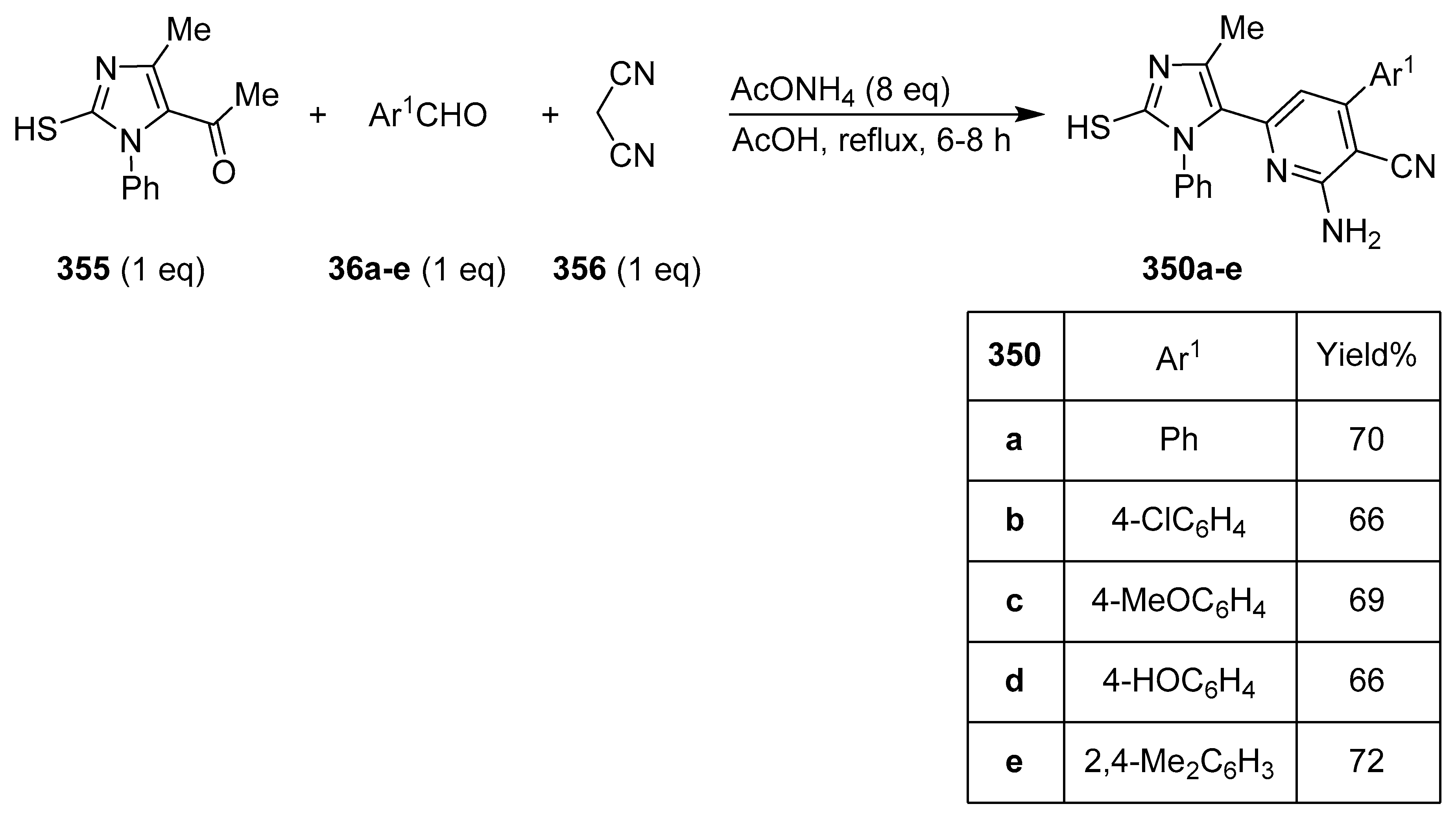
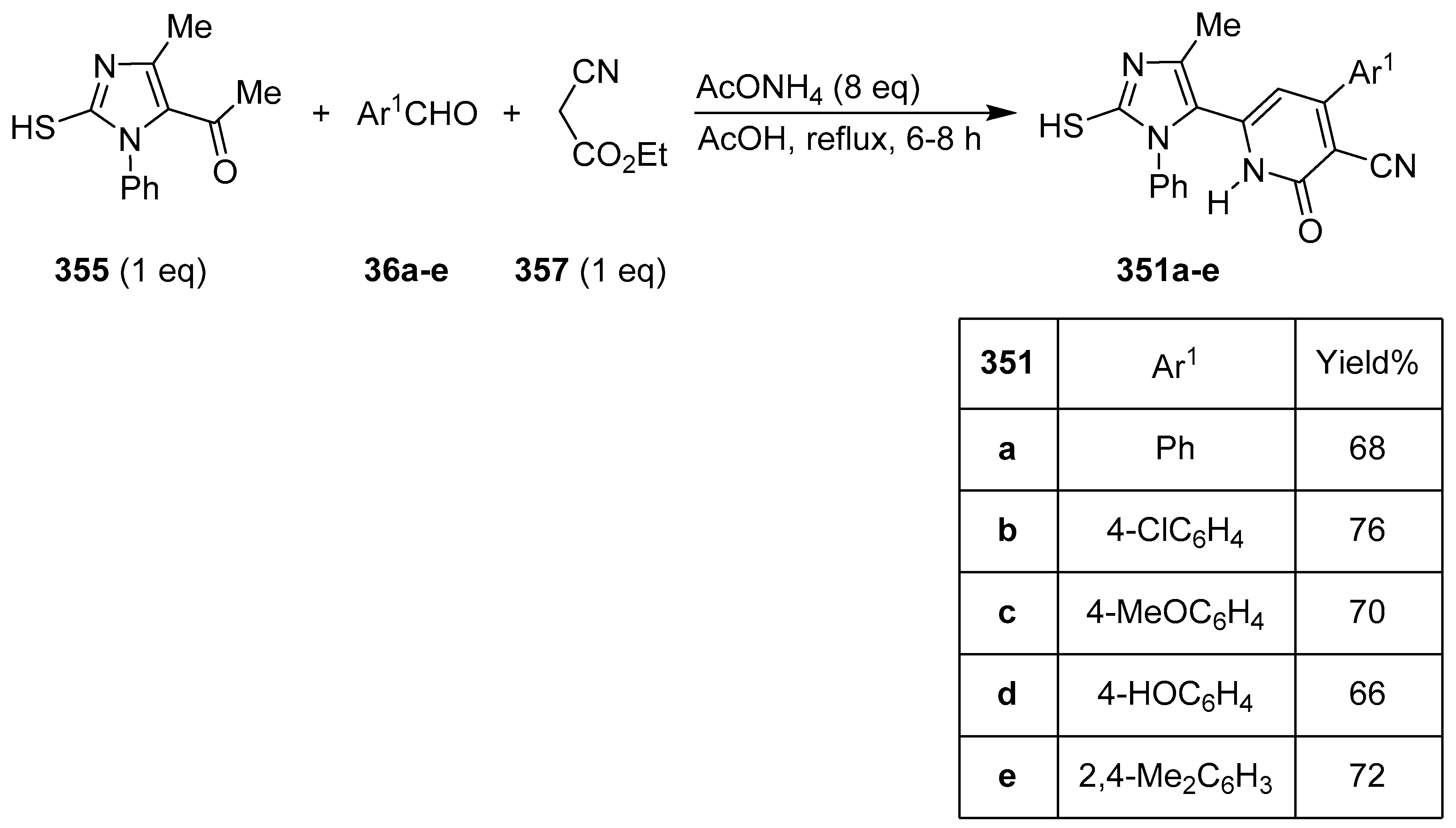

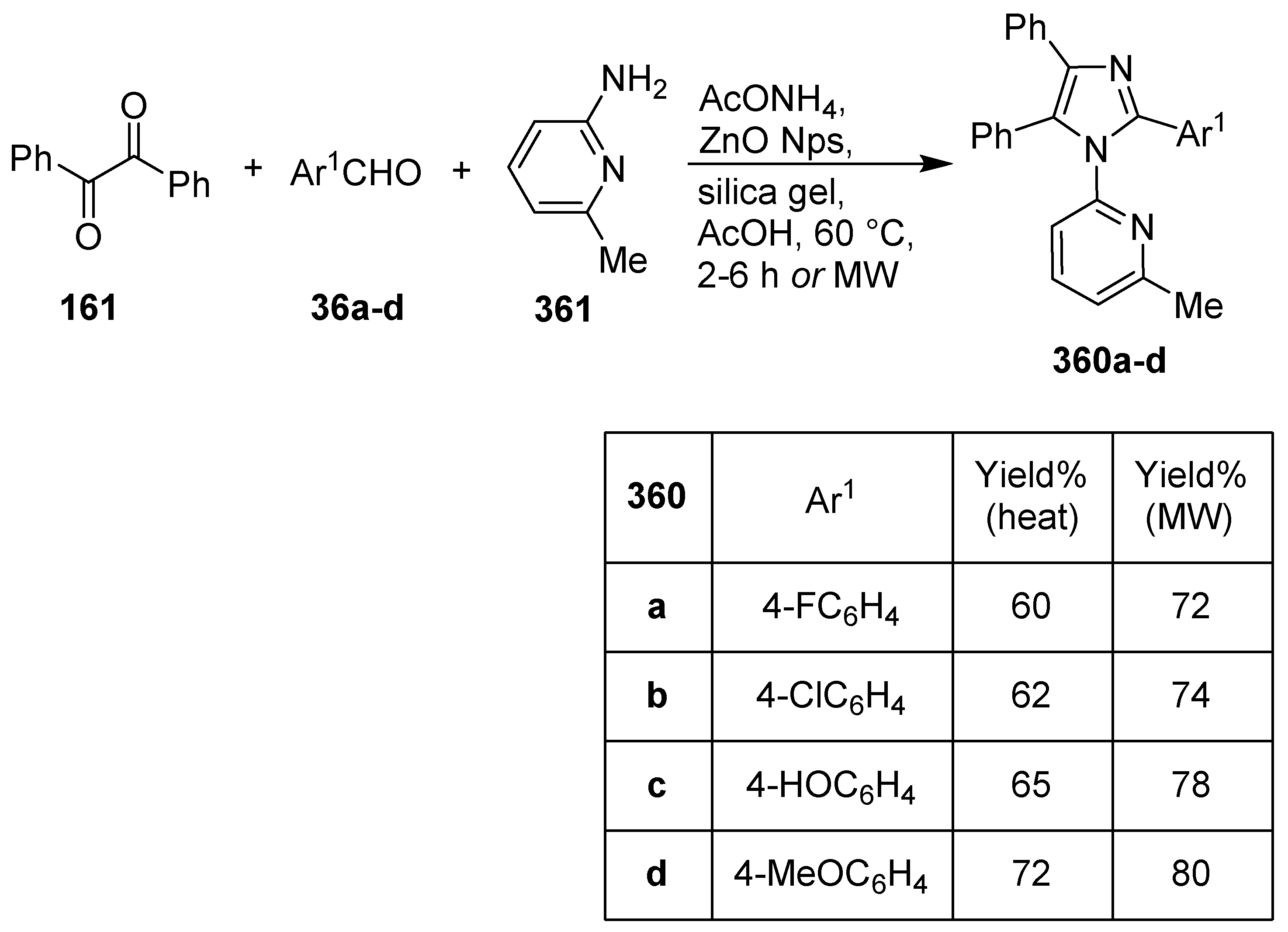
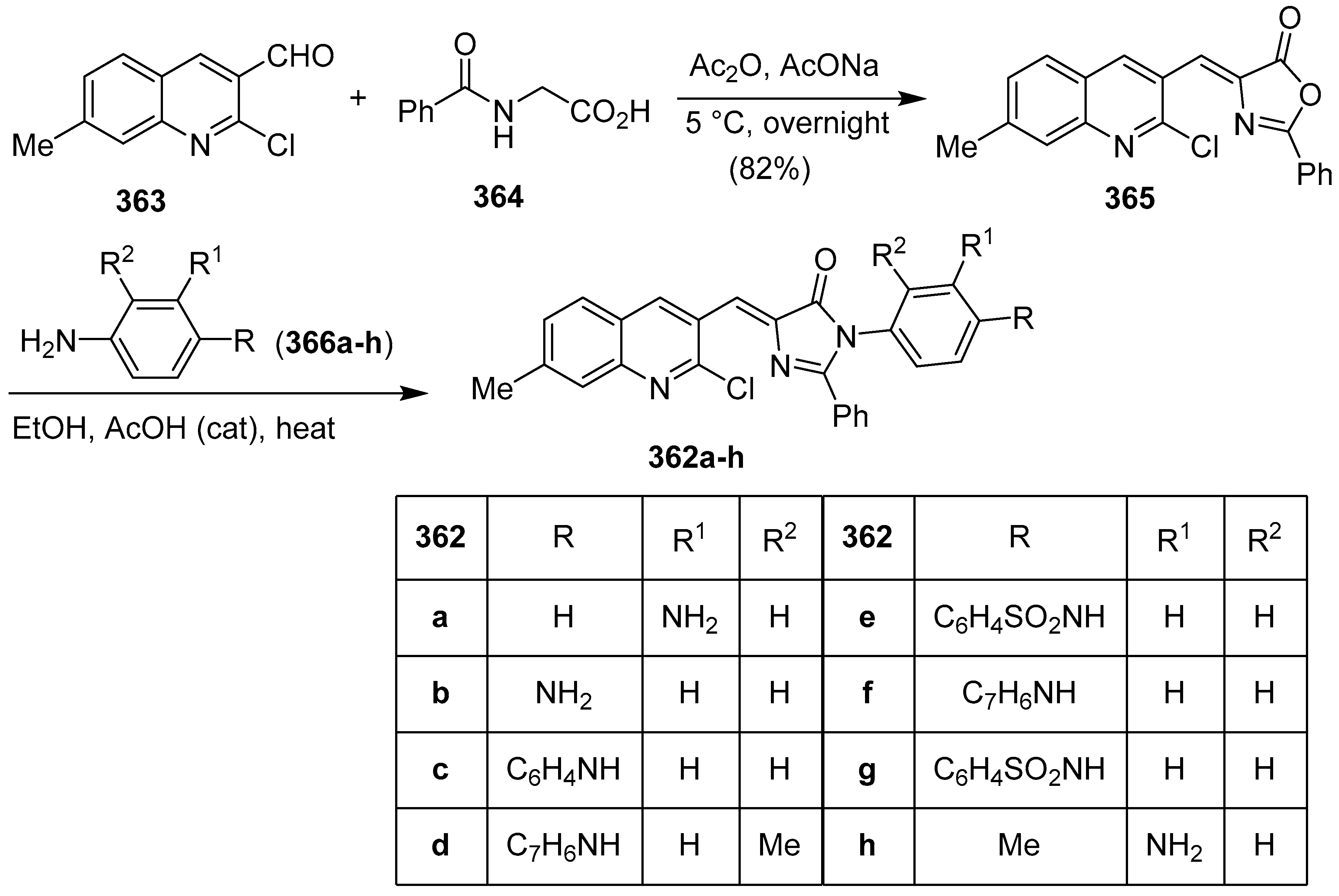
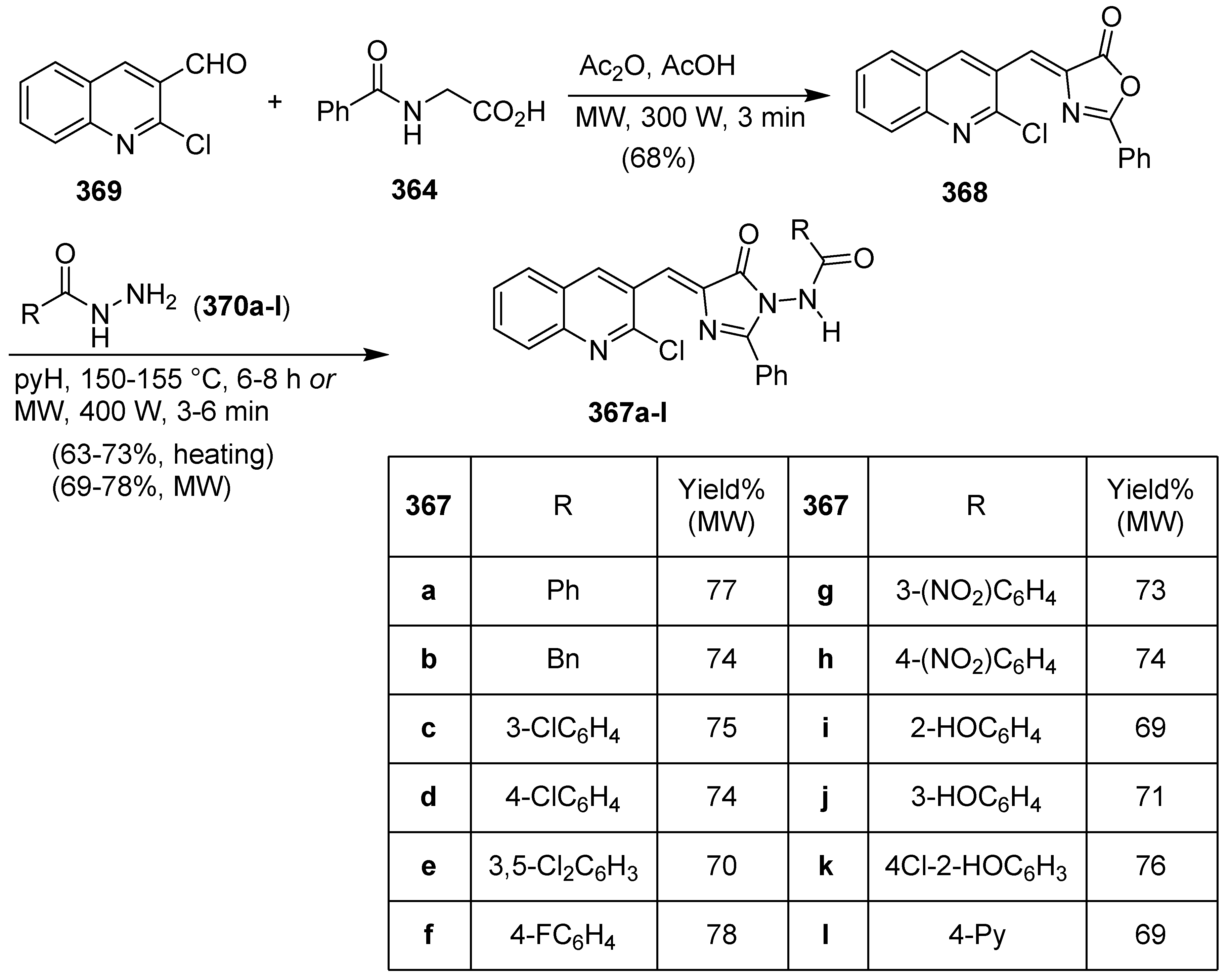

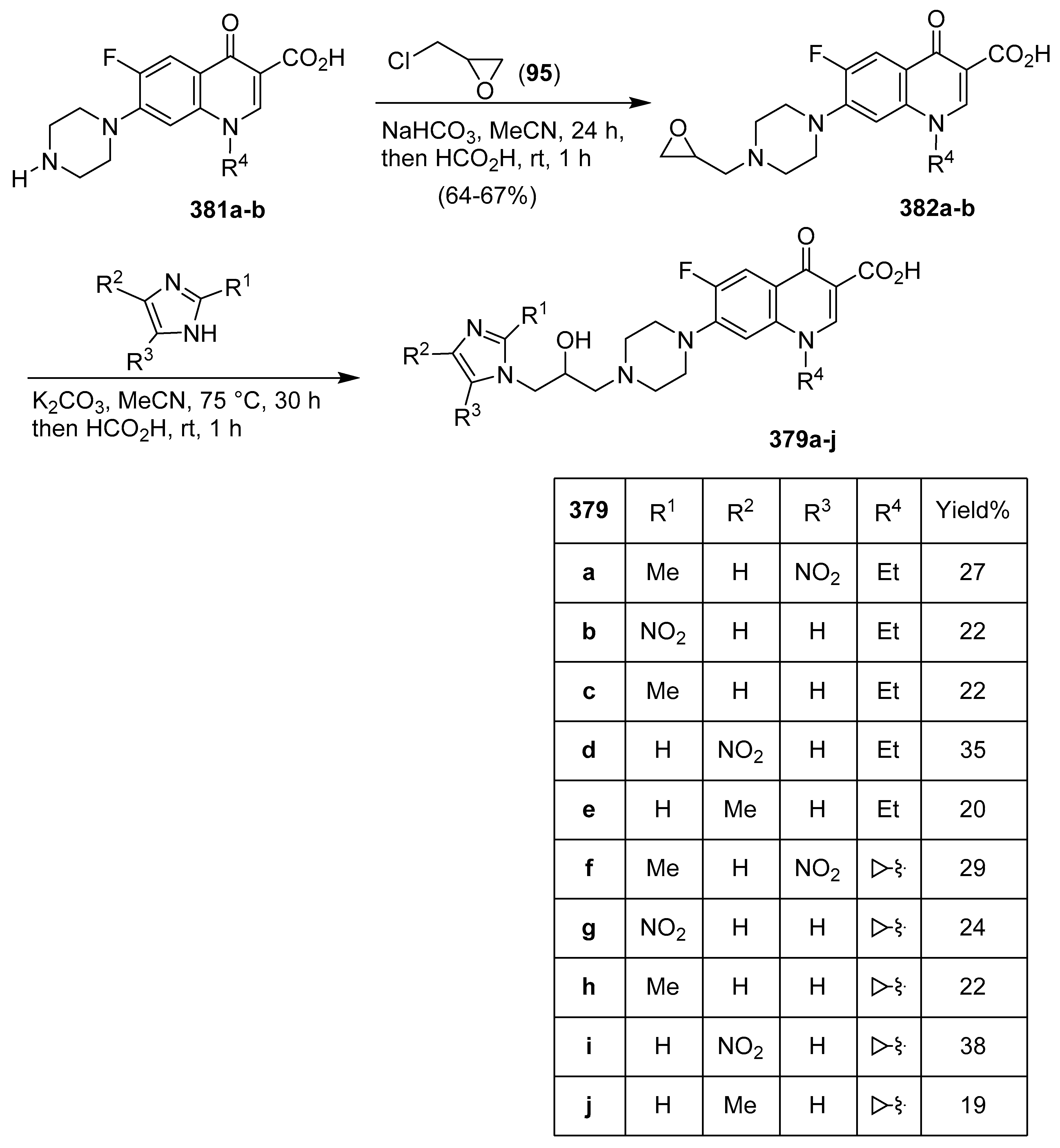

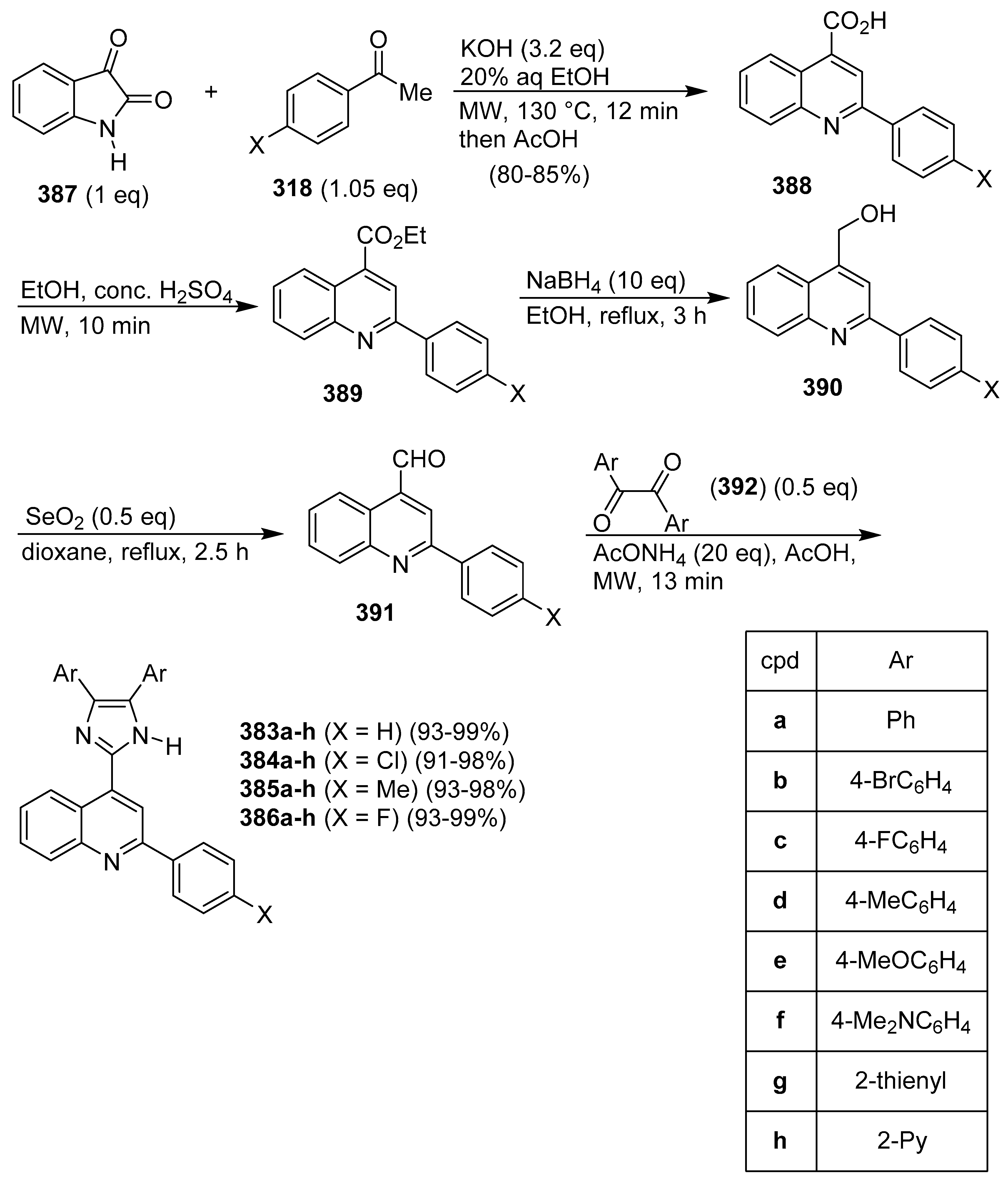




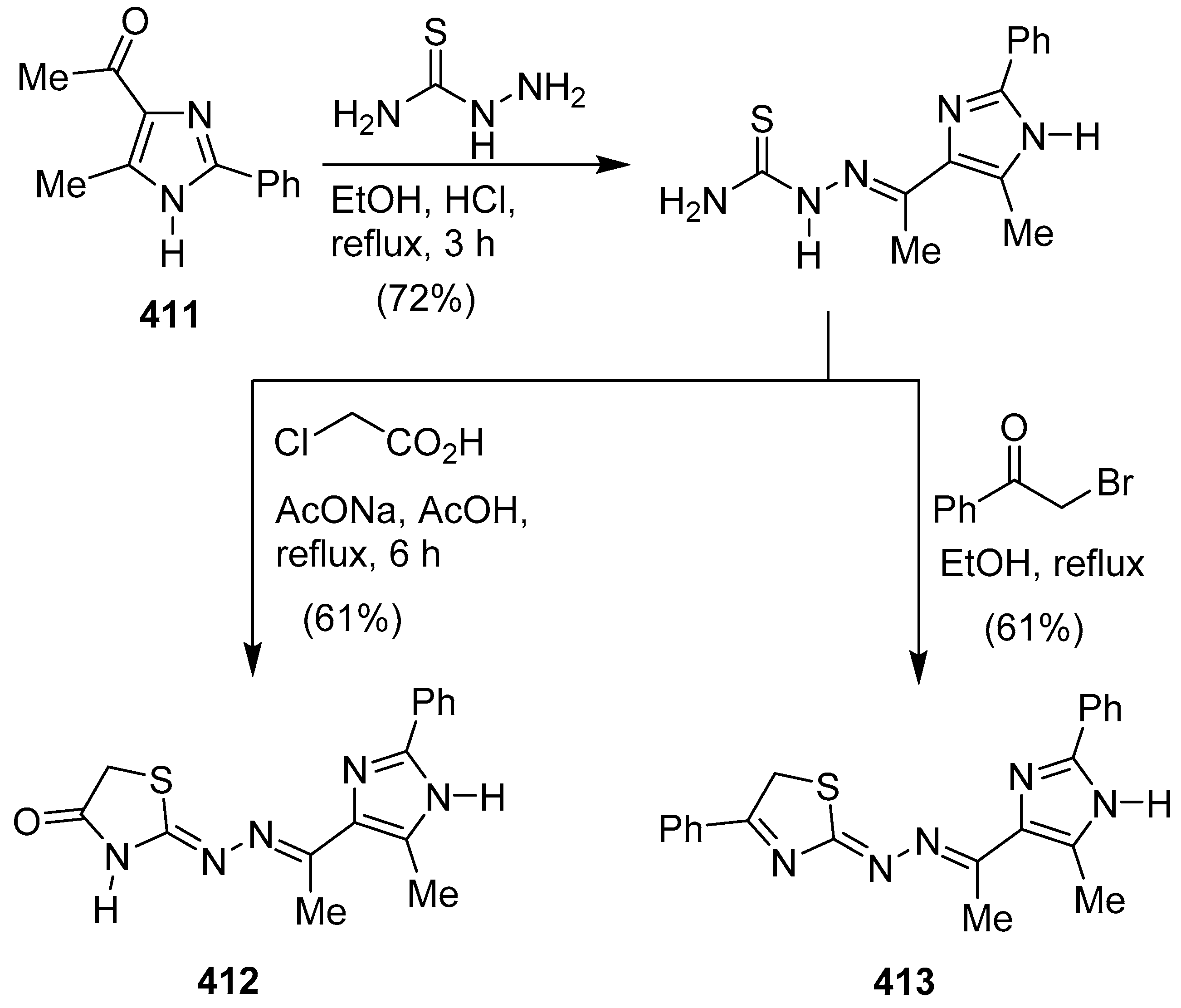
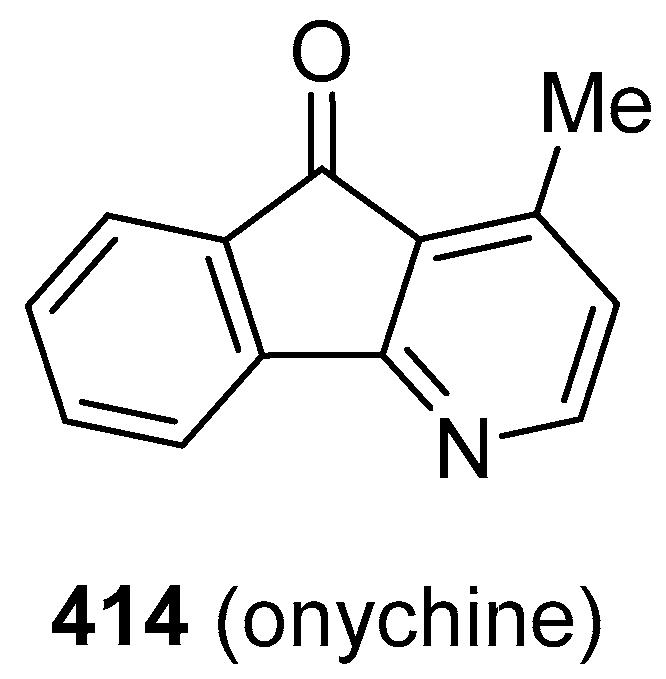


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossi, R.; Ciofalo, M. An Updated Review on the Synthesis and Antibacterial Activity of Molecular Hybrids and Conjugates Bearing Imidazole Moiety. Molecules 2020, 25, 5133. https://doi.org/10.3390/molecules25215133
Rossi R, Ciofalo M. An Updated Review on the Synthesis and Antibacterial Activity of Molecular Hybrids and Conjugates Bearing Imidazole Moiety. Molecules. 2020; 25(21):5133. https://doi.org/10.3390/molecules25215133
Chicago/Turabian StyleRossi, Renzo, and Maurizio Ciofalo. 2020. "An Updated Review on the Synthesis and Antibacterial Activity of Molecular Hybrids and Conjugates Bearing Imidazole Moiety" Molecules 25, no. 21: 5133. https://doi.org/10.3390/molecules25215133
APA StyleRossi, R., & Ciofalo, M. (2020). An Updated Review on the Synthesis and Antibacterial Activity of Molecular Hybrids and Conjugates Bearing Imidazole Moiety. Molecules, 25(21), 5133. https://doi.org/10.3390/molecules25215133