Abstract
An efficient synthetic route to highly functionalized naphthalenes and quinolines has been developed using domino reactions between Morita–Baylis–Hillman (MBH) acetates and active methylene compounds (AMCs) promoted by anhydrous K2CO3 in dry N,N-dimethylformamide (DMF) at 23 °C. The substrates incorporate allylic acetates positioned adjacent to a Michael acceptor as well as an aromatic ring activated toward a SNAr ring closure. A control experiment indicated that the initial reaction was an SN2’-type displacement of a side chain acetoxy by the AMC anion to afford the alkene product bearing the added nucleophile trans to the SNAr aromatic ring acceptor. Thus, equilibration of the alkene geometry of the initial product was required prior to cyclization. Products were isolated in good to excellent yields. Numerous cases (24) are reported, and several mechanistic possibilities are discussed.
1. Introduction
One facet of our research program has focused on domino reactions for the rapid assembly of molecules with potential use in drug synthesis. This requires highly functionalized compounds with reactive sites strategically positioned to capture intermediates from an initial reaction in one or more subsequent reactions to produce targets of high value in an efficient, eco-friendly manner. Among the compounds meeting these requirements are the products of the Morita–Baylis–Hillman (MBH) reaction. These highly functionalized adducts have proven quite useful in drug synthesis [1,2]. Additionally, the naphthalenes [3] and quinolines [4,5] targeted in this work could have immense value in medicinal chemistry.
Previous work by others has appeared in this area, but details were lacking, and the diversity of examples reported was limited. One article outlined the formation of a number of naphthalene derivatives [6] from MBH acetates and active methylene compounds (AMCs) (Figure 1A), but the initial adduct was assumed to have the correct geometry for ring closure, and the only molecule eliminated during the final aromatization was benzenesulfinic acid (PhSO2H). A second report described a synthesis of 3-quinolinecarboxylic esters [7] involving the reaction of ethyl 2-((2-chlorophenyl)((tosylsulfonamido)methyl)acrylate acrylate with tosylsulfonamide (Figure 1B). A third study [8] advanced a synthesis of dihydroacridines from MBH acetates derived from 2-chloroquinoline-3-carboxaldehyde and AMCs to fuse a substituted benzene ring to the heterocycle (Figure 1C). The final account detailed the AMC-dependent annulation of substituted benzene rings by reaction of this same MBH precursor to give acridines and phenanthridines [9] (Figure 1D). The current work describes the synthesis of naphthalenes and quinolines with a broader range of functionality, evaluates a selection of different leaving groups, and discusses several mechanistic possibilities.
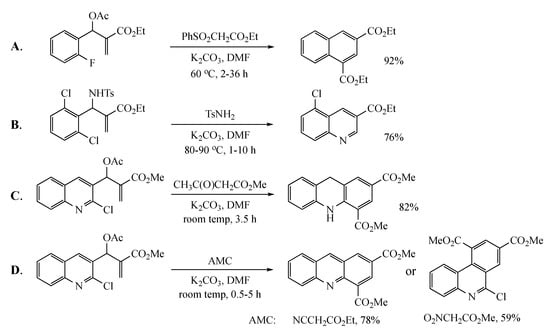
Figure 1.
Previous cyclizations to generate aromatic systems using Morita–Baylis–Hillman (MBH) acetates. (A): Formation of naphthalenes; (B): Formation of quinolines; (C): Formation of dihydroacridines; (D): Formation of acridines and phenanthridines.
2. Results and Discussion
The MBH reaction was first reported in 1968 [10] and has undergone many modifications [11,12,13] to improve its outcome. The generally accepted procedure calls for 1 equiv. of the aldehyde and 1.2 equiv. of the electron-poor alkene in the presence of 1.5 equiv. of 1,4-diazabicyclo [2.2.2]octane (DABCO) in acetonitrile (ACN) at room temperature (23 °C) [14]. For the current project, we have generated these adducts by the reaction of polarized alkenes with aromatic aldehydes incorporating functionality that further activates the aromatic ring toward a SNAr reaction using a 2:1 stoichiometry of alkene:aldehyde with 1.2 equiv. of DABCO (Scheme 1). Thus, 2-fluoro-5-nitrobenzaldehyde (1a), 2-fluoro-5-cyanobenzaldehyde (1b), and 2-fluoronicotinaldehyde (1c) were each treated with ethyl acrylate (2a) and acrylonitrile (2b) in ACN at 23 °C for 2 days to give the MBH alcohols (3a–e) in 86–98% yields. Attempts to acylate the alcohol adduct 3b with acetic anhydride at reflux led to acylation of the alcohol and allylic inversion of the acetate to afford (E)-2-cyano-3-(2-fluoro-5-nitrophenyl)allyl acetate (4). Interestingly, an X-ray structure of this product showed the acetoxymethyl group of the rearranged acetate to be trans to the substrate aromatic ring (see Scheme 1 and the Supplementary Materials). Acylation of the MBH adducts without rearrangement was achieved using trimethylsilyl trifluoromethanesulfonate (TMSOTf) as an acylation catalyst [15] in dichloromethane (DCM). This catalyst permitted the acylation of 3a–e at 0 °C in 30 min and delivered the unrearranged acetates 5–9, respectively, in 95–98% yields.
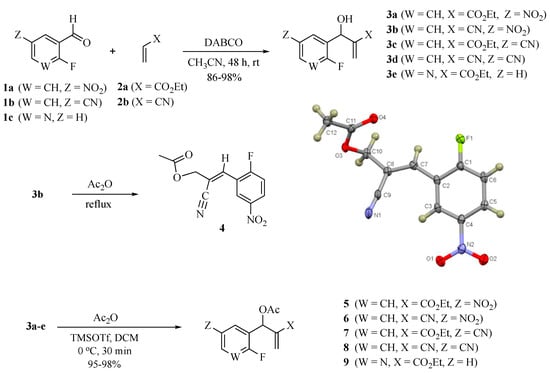
Scheme 1.
Synthesis of MBH acetates for cyclization and rearranged product 4 (CCDC 2035023).
The results of our study are summarized in Table 1 and Table 2. The reaction of substrates 5–8 (1 equiv.) with AMCs 10–14 (1.5 equiv.) yielded 1,3,6-trisubstituted naphthalenes 15–18, while precursor 9 afforded 6,8-disubstituted quinolines 19 with the same panel of nucleophiles. Naphthalene formation was promoted by K2CO3 (1.5 equiv.) in N,N-dimethylformamide (DMF) at 23 °C in 1 h, while quinolines were similarly prepared but required heating at 90 °C for 6 h. Yields were uniformly good to excellent, with the 2-fluoro-5-nitrophenyl SNAr acceptor giving the most efficient ring closure, followed by the 2-fluoro-5-cyano compound and finally 2-fluoropyridine. The overall process appears to involve diastereoselective addition of the nucleophile to the double bond methylene of the MBH substrate with loss of the acetoxy group. Following this addition, deprotonation of the methine hydrogen from the added nucleophile, SNAr cyclization, and elimination fuses a disubstituted benzene ring to the original aromatic nucleus. The exact sequence of events for this process is unclear and is discussed in more detail below. In the current reactions, the final aromatization occurred by the elimination of benzenesulfinic acid (PhSO2H) [6,16], nitrous acid (HNO2) [9,17], or ethoxycarbonyl (CO2Et, presumably by hydrolysis and decarboxylation) [9,18], all of which have precedent in the literature, although the loss of CO2Et in preference to the CN group was not expected. Aromatization by the elimination of CN is known [19,20], but it appears to be quite rare.

Table 1.
Formation of naphthalenes.
Table 2.
Formation of quinolines.
The syntheses are quite efficient, requiring only three steps and one chromatographic purification for the final products 15–19. The products were isolated in good to excellent yields (75–96%) and were fully characterized by spectroscopic methods. The IR spectra indicated the presence of the expected functional groups (conjugated CO2Et or CN, polarized conjugated double bonds, and NO2). The naphthalenes showed three one-proton doublets (J1,3 < 3 Hz or apparent singlets) for the isolated protons at C2, C4, and C5, a doublet of doublets (J1,2 and 1,3 = large and small) for the proton at C7, and a doublet (J1,2 > 8 Hz) for the proton at C8. The chemical shifts were in accordance with those expected for an electron-deficient aromatic system. The quinolines exhibited two doublets (J1,3 < 3 Hz) for the protons at C2 and C4 as well as characteristic chemical shifts and couplings for the protons on C5–C7 of the system. The 13C-NMR were appropriate with respect to the number of carbonyl, aromatic, and aliphatic carbons. The mass spectra showed an ion corresponding to the molecular weight of the compound, and elemental analyses confirmed the formulas and purity.
Two basic mechanisms are possible for this domino transformation: (1) initial SNAr attack by the AMC anion on the aromatic ring, followed by conjugate addition to the side chain double bond with an elimination of acetoxy or (2) initial SN2′-type displacement of acetoxy from the side chain, followed by a SNAr cyclization. Option 1 would unquestionably predict the final product of the reaction and would not depend on the selective formation of a single alkene from the SN2′ process. Option 2 would require a diastereoselective addition to give the double bond geometry that positions the active methylene fragment cis to the SNAr acceptor ring. To probe this aspect of the transformation, a control experiment was performed wherein the anion of methyl phenylsulfonylacetate was generated in the presence of a mixture of 2-fluoro-5-nitrotoluene (20) and 2-cyano-1-(2-fluorophenyl)allyl acetate (21) to determine the relative reactivity of the side chain versus the aromatic ring. If the side chain reacted preferentially, we were also interested in assessing any E-Z diastereoselectivity in the allylically rearranged double bond. Under the standard conditions, a reaction occurred exclusively at the side chain of the MBH acetate and also afforded only methyl (Z)-4-cyano-5-(2-fluorophenyl)-2-(phenylsulfonyl)-4-pentenoate (22, 70%) having the added active methylene nucleophile positioned trans to the aromatic SNAr acceptor ring. This was confirmed by an X-ray structure of adduct 22 (see Scheme 2 and the Supplementary Materials). Compound 20 (93%) was recovered unchanged from this experiment. Thus, the overall sequence must proceed via Option 2 above, followed by double bond equilibration prior to SNAr ring closure.
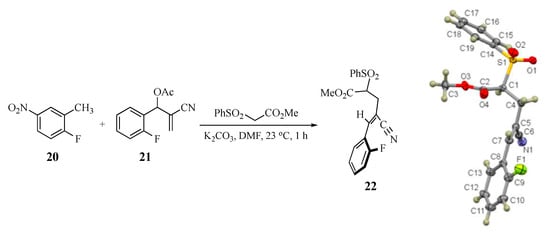
Scheme 2.
Control experiment: Selective reaction of the MBH acetate and formation of the Z alkene 22 (CCDC 2035022).
Analysis of the reactant in the SN2′ reaction (Option 2 above) demands that the starting conformation minimizes the steric interaction between the aryl, the acetoxy, and the electron-withdrawing group (EWG = CO2Et or CN) (Scheme 2). An earlier study describing the reaction of an acrylate-derived MBH alcohol with HBr proposed conformation 23 (Figure 2), which has an additional stabilizing H-bond between the OH and the EWG, to rationalize the products [20]. This model would favor formation of the alkene that places the incoming active methylene fragment cis to the 2-fluoro-5-nitrophenyl moiety in the product. With the current substrates, H-bonding is not possible, and so the large acetoxy group was expected to orient away from the EWG (perhaps more so for CO2Et than CN) to minimize steric crowding (Scheme 3). Then, an initial SN2′ reaction involving a syn orientation of the nucleophile and leaving group invoked in some earlier work [21,22] would give an adduct with the AMC fragment trans to the SNAr acceptor ring as in the aforementioned competitive reaction. Thus, in order for the cyclization to occur, the initial Michael adduct 22 must equilibrate to bring the nucleophilic center cis to the activated aromatic ring to allow cyclization by an intramolecular SNAr reaction. Possible mechanisms for this isomerization are discussed below.
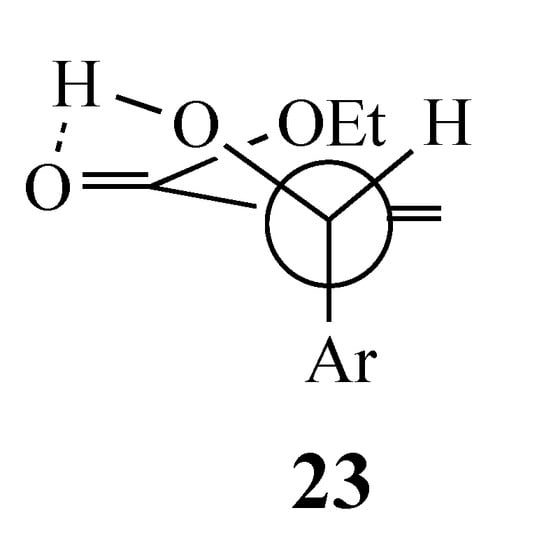
Figure 2.
Conformation of an MBH alcohol with a stabilizing H-bond.
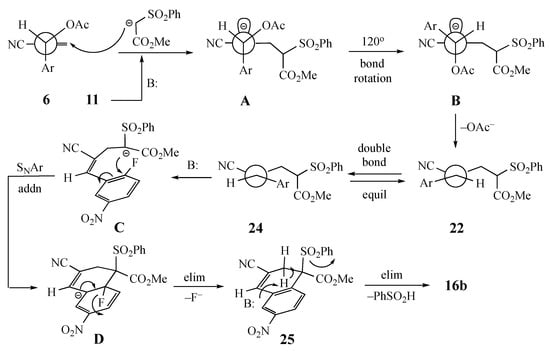
Scheme 3.
SN2′ mechanism leading to the formation of naphthalenes from MBH acetates.
In reactions at the side chain allylic moiety, there are two possible scenarios to explain the addition to the MBH acetate. The first [22] invokes an SN2′ process where a nucleophile attacks C1 (unsubstituted) of the allylic moiety, resulting in an accumulation of electron density on the opposite face of the molecule at C2. Then, this electron density is responsible for assisting in the departure of the acetoxy leaving group at C3. This process has been used to rationalize the reported preference for a syn orientation between the incoming nucleophile and the leaving group often seen in the reaction [23,24,25], although exceptions are known [26] with certain Nu–/L– combinations. The second scenario [27] claims that there is no justification for a syn orientation between the nucleophile and the leaving group. In fact, in highly substituted derivatives, such as 5–9, it has been suggested that the formation of a stabilized carbocation is more likely, in which case the avoidance of steric interactions guides the transformation to give the final product stereochemistry.
The SN2′ initiated mechanism is depicted in Scheme 3 for the reaction of 6 with 11 to give 16b. The initial conformation of 6 would allow minimal steric interaction between the groups on the allylic side chain. In this conformation, the acetoxy substituent should hinder approach to the top face while the planar aromatic ring does not significantly block the bottom face. Introduction of the nucleophile to the bottom face would result in an accumulation of negative charge on the top face of the allylic system, as shown in A. Rotation of the acetoxy group 120° would position it anti to the accumulated negative charge and move the substrate 2-fluoro-5-nitrophenyl (Ar) moiety away from the newly added nucleophile to give B. Then, the elimination of acetoxy would deliver the Z alkene 22. Subsequent equilibration of the double bond geometry to generate 24 and deprotonation of the residual active methine proton to give C would be followed by ipso attack at the fluorine-bearing aromatic carbon to afford Meisenheimer intermediate D. Rearomatization by loss of fluoride should then lead to 25. After ring closure, a conformation aligned for the elimination of benzenesulfinic acid by an E2 process would afford the fused aromatic compound 16b.
The second mechanistic scenario, illustrated in Scheme 4, begins with loss of the acetoxy group to form stabilized 3-arylallylic cations E and F [27]. At this point, it may appear that addition to carbocation F should be easier, since the alkene terminus is less hindered. Furthermore, since the EWG should not be coplanar with the allylic cation, it should have less steric influence on the reaction, and the larger bond angles around the carbocation should permit the formation of both E and F. Addition of the active methylene nucleophile would then lead to both 22 and 24, which could cyclize as above. In both examples cited in this paper, the alkene with the nucleophile trans to the SNAr aromatic system was isolated with no contamination by the cis. This observation could disqualify this mechanism, but if 24 cyclizes very rapidly, it may not be possible to detect or isolate this short-lived intermediate.
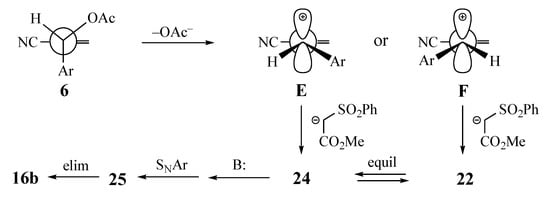
Scheme 4.
Carbocation mechanism leading to the formation of naphthalenes from MBH acetates.
Two pathways can be envisioned for the equilibration of 22 to 24 that must occur before SNAr cyclization: (1) a well-precedented Michael-reverse Michael process [28,29,30,31,32] involving the excess AMC or perhaps (2) an intramolecular addition–elimination reaction (Scheme 5). This alternative process would involve loss of the acidic methine proton from 22 and addition of the resulting anion G to the benzylic double bond to give H. This may be possible, since the electron-deficient aromatic ring should stabilize the benzylic anion. Then, bond rotation to give I and ring opening would deliver the anion needed for cyclization. Once equilibration occurs, ring closure to 25 and elimination to generate 16b should be facile. Even if the equilibrium does not strongly favor the required alkene geometry, product formation should gradually siphon the initial adduct over to the fused aromatic target.
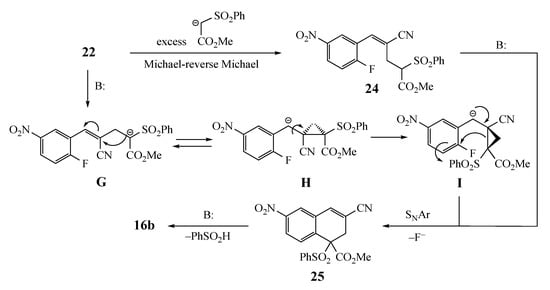
Scheme 5.
Possible mechanisms for double bond equilibration.
Addition to the substrates with formation of the Z alkene (EWG and added AMC trans) was confirmed by both a side reaction observed during the synthesis to form acetate 4 and a control experiment to form 15 (Scheme 2). The formation of a single product has significant precedent in the case of EWG = CO2Et, but it is less probable for the sterically smaller EWG = CN group [33]. Unfortunately, the ester intermediate, while a single isomer, was not a solid and could not be subjected to X-ray structure analysis. Additionally, Nuclear Overhauser enhancement (NOE) measurements were inconclusive. Nevertheless, products incorporating both EWGs formed in high yields. In cases where the initial addition yields the Z alkene, a reversible Michael reaction [28,29,30,31,32] or intramolecular isomerization could establish an E–Z equilibrium that would eventually cyclize and eliminate H–L to afford the aromatic products. A similar rationale can be applied to the pyridine-containing substrates that lead to quinolines. The stereoselective formation of the alkenes with the added nucleophile cis to the aromatic ring has been observed in the past with small nucleophiles [21,34,35,36,37,38,39,40]. This has been confirmed by spectral characterization, and in one instance, X-ray analysis [41]. However, our results, using large stabilized nucleophiles, appear to differ from these earlier findings.
3. Material and Methods
3.1. General Methods
Unless otherwise indicated, all reactions were carried out under dry N2 in oven-dried glassware. All reagents and solvents were used as received. Reactions were monitored by thin layer chromatography on silica gel GF plates (Analtech No 21521, Newark, DE, USA). Preparative separations were performed by flash chromatography on silica gel (Davisil®, grade 62, 60–200 mesh, Sorbent Technologies, Norcross, GA, USA) containing UV-active phosphor (Sorbent Technologies No. UV-05) slurry packed into quartz columns. Band elution for all chromatographic separations was monitored using a hand-held UV lamp (Fisher Scientific, Pittsburgh, PA, USA). Wash solutions used in work-up procedures were all aqueous. Melting points were obtained using a MEL-TEMP apparatus (Cambridge, MA, USA) and are uncorrected. FT-IR spectra were run using a Varian Scimitar FTS 800 spectrophotometer (Randolph, MA, USA) as thin films on NaCl disks. 1H- and 13C-NMR spectra were measured using a Bruker Avance 400 system (Billerica, MA, USA) in the indicated solvents at 400 MHz and 101 MHz, respectively, with (CH3)4Si as the internal standard; coupling constants (J) are given in Hz. Low-resolution mass spectra were obtained using a Hewlett-Packard Model 1800A GCD GC-MS system (Palo Alto, CA, USA). Details of the X-ray structure determinations are given in the Supplementary Materials. Elemental analyses (± 0.4%) were determined by Atlantic Microlabs (Norcross, GA, USA).
3.2. Representative Procedure for the Synthesis of MBH Alcohols (3a–e)
To a stirred solution of the aldehyde 1a–c (1 equiv.) and DABCO (1.2 equiv.) in ACN (8 mL) under N2 was added ethyl acrylate (2a) or acrylonitrile (2b) (2 equiv.) at 23 °C. After 24–48 h, TLC analysis (10% EtOAc/hexane) indicated the reaction was complete. The solution was added to water, and the mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with 0.5 M HCl (2 × 10 mL), saturated NaHCO3, and saturated NaCl and then dried (Na2SO4). Removal of the solvent under vacuum resulted in pure MBH alcohols 3a–e, which were used without further purification.
3.3. Synthesis of (E)-2-cyano-3-(2-fluoro-5-nitrophenyl)allyl acetate (4)
A solution of the MBH alcohol 3b (1.11 g, 5 mmol) was dissolved in acetic anhydride (5.0 mL) and boiled for 6 h. The mixture was cooled, concentrated under vacuum, and the residue was dissolved in DCM (25 mL). The solution was washed with saturated NaHCO3 (3 × 50 mL) and water, dried (Na2SO4), and concentrated under vacuum to afford 4 (1.13 g, 4.3 mmol, 86%) as an off-white solid, m.p. 78–79 °C. IR: 2222, 1749, 1621, 1530, 1352 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.99 (dd, J = 6.3, 2.8 Hz, 1H), 8.35 (ddd, J = 9.0, 4.4, 2.8 Hz, 1H), 7.43 (s, 1H), 7.33 (t, J = 9.0 Hz, 1H), 4.22 (d, J = 1.3 Hz, 2H), 2.19 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 170.1, 163.5 (d, J = 263.3 Hz), 144.4, 135.9 (d, J = 5.4 Hz), 127.8 (d, J = 10.7 Hz), 125.4 (d, J = 3.7 Hz), 122.1 (d, J = 14.3 Hz), 116.1 (d, J = 24.4 Hz), 115.5, 112.4, (d, J = 2.1 Hz), 64.2, 20.7; MS (EI): m/z 264 [M]+·; Anal. Calcd for C12H9FN2O4: C 54.55; H, 3.43; N, 10.60. Found: C, 54.58; H, 3.45; N, 10.53. The X-ray structure of 4 (CCDC 2035023) was obtained and the thermal elipsoid plot is shown in Scheme 1 and the Supplementary Materials.
3.4. Representative procedure for the Synthesis of MBH Acetates (5–9)
A solution of the MBH alcohol 3a–d (1 equiv.) in DCM (2 mL) was treated with acetic anhydride (1.5 equiv.) at 0 °C, followed by a 1 M solution of TMSOTf in DCM (20 μL/mmol substrate) [15]. The MBH alcohol 3e (1 equiv.) required acetic anhydride (3 equiv.) and 1 M TMSOTf in DCM (60 μL/mmol substrate). After 30 min, TLC analysis (10% ether in hexane) showed the reaction was complete. The reaction mixture was added to saturated NaHCO3, and the mixture was extracted with DCM (3 × 5 mL). The organic extracts were washed with water, dried (Na2SO4), and evaporated under vacuum to afford clean MBH acetates 5–9, which did not require further purification. After optimization, this procedure was scaled up by a factor of five with no significant decrease in yield.
3.4.1. Ethyl 2-(acetoxy (2-fluoro-5-nitrophenyl)methyl)acrylate (5)
Yield: 305 mg (0.98 mmol, 98%) as a colorless oil; IR: 1751, 1723, 1639, 1532, 1351 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.26 (dd, J = 6.0, 2.8 Hz, 1H), 8.23 (ddd, J = 8.9, 4.3, 2.9 Hz, 1H), 7.22 (t, J = 8.9 Hz, 2H), 6.91 (s, 1H), 6.54 (s, 1H), 4.18 (qd, J = 7.1, 1.3 Hz, 2H), 2.15 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 169.0, 164.2, 163.7 (d, J = 232.5 Hz ), 144.2 (d, J = 2.9 Hz), 137.4, 127.5, 127.4 (d, J = 3.1 Hz), 126.0 (d, J = 10.5 Hz), 125.1 (d, J = 5.4 Hz), 116.9 (d, J = 24.5 Hz), 66.6 (d, J = 2.8 Hz), 61.3, 20.8 (d, J = 1.6 Hz), 14.0; MS: m/z 311 [M]+·; Anal. Calcd for C14H14FNO6: C, 54.02; H, 4.53; N, 4.50. Found: C, 53.94; H, 4.54; N, 4.39.
3.4.2. 2-Cyano-1-(2-fluoro-5-nitrophenyl) allyl acetate (6)
Yield: 259 mg (0.98 mmol, 98%) as off-white crystals, m.p. 66–67 °C; IR: 2232, 1759, 1632, 1532, 1535, 1352 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.42 (dd, J = 6.1, 2.8 Hz, 1H), 8.31 (ddd, J = 9.0, 4.4, 2.8 Hz, 1H), 7.29 (t, J = 9.0 Hz, 1H), 6.64 (s, 1H), 6.19 (s, 1H), 6.18 (s, 1H), 2.26 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 168.8, 163.1 (d, J = 260.1 Hz), 144.7, 133.7, 126.8 (d, J = 10.3 Hz), 125.3 (d, J = 15.1 Hz), 124.1 (d, J = 4.9 Hz), 120.9, 117.1 (d, J = 23.7 Hz), 115.2, 67.8 (d, J = 3.0 Hz), 20.8; MS: m/z 264 [M]+·; Anal. Calcd for C12H9FN2O4: C, 54.55; H, 3.43; N, 10.60. Found: C, 54.63; H, 3.45; N,10.52.
3.4.3. Ethyl 2-(acetoxy(5-cyano-2-fluorophenyl)methyl)acrylate (7)
Yield: 285 mg (0.98 mmol, 98%) as a colorless oil; IR: 2234, 1752, 1729, 1637 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.69 (dd, J = 6.6, 2.1 Hz, 1H), 7.64 (ddd, J = 8.5, 4.7, 2.1 Hz, 1H), 7.19 (t, J = 9.3 Hz, 1H), 6.88 (s, 1H), 6.51 (s, 1H), 5.92 (s, 1H), 4.19 (q, J = 7.1 Hz, 2H), 2.15 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 169.0, 164.2, 162.6 (d, J = 260.3 Hz), 137.6, 134.4 (d, J = 9.8 Hz), 133.6 (d, J = 4.8 Hz), 127.7 (d, J = 14.9 Hz), 127.3, 117.8, 117.2 (d, J = 23.4 Hz), 108.7 (d, J = 4.0 Hz), 66.6 (d, J = 2.9 Hz), 61.3, 20.8, 14.0; MS: m/z 291 [M]+·; Anal. Calcd for C15H14FNO4: C, 61.85; H, 4.84; N, 4.81. Found: C, 61.77; H, 4.86; N, 4.85.
3.4.4. 2-Cyano-1-(5-cyano-2-fluorophenyl)allyl acetate (8)
Yield: 259 mg (0.98 mmol, 98%) as off-white crystals, m.p. 74–75 °C; IR: 2255, 2235, 1757, 1613 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.86 (dd, J = 6.6, 2.1 Hz, 1H), 7.72 (ddd, J = 8.4, 4.8, 2.1 Hz, 1H), 7.24 (t, J = 9.3 Hz, 1H), 6.60 (s, 1H), 6.16 (s, 1H), 6.15 (s, 1H), 2.24 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 168.8, 161.9 (d, J = 259.0 Hz), 135.2 (d, J = 9.8 Hz), 133.7, 132.3 (d, J = 4.3 Hz), 125.6 (d, J = 14.2 Hz), 120.9, 117.44 (d, J = 22.8 Hz), 117.41, 115.3, 109.6 (d, J = 4.0 Hz), 67.8 (d, J = 3.2 Hz), 20.8; MS: m/z 244 [M]+·; Anal. Calcd for C13H9FN2O2: C, 63.93; H, 3.71; N, 11.47. Found: C, 63.81; H, 3.77; N, 11.35.
3.4.5. Ethyl 2-(acetoxy(2-fluoropyridin-3-yl)methyl)acrylate (9)
Yield: 254 mg (0.95 mmol, 95%) as a colorless oil; IR: 1758, 1727, 1637 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.19 (m, 1H), 7.81 (ddd, J = 9.4, 7.4, 2.0 Hz, 1H), 7.20 (ddd, J = 6.5, 4.8, 1.6 Hz, 1H), 6.81 (s, 1H), 6.50 (s, 1H), 5.92 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 2.14 (s, 3H), 1.24 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 169.1, 164.4, 161.0 (d, J = 241.9 Hz), 147.6 (d, J = 14.9 Hz), 140.1 (d, J = 4.3 Hz), 137.5, 127.2 (d, J = 1.2 Hz), 121.4 (d, J = 4.4 Hz), 120.5 (d, J = 27.9 Hz), 67.8, 61.2, 20.8, 14.0; MS: m/z 267 [M]+·; Anal. Calcd for C13H14FNO4: C, 58.42; H, 5.28; N, 5.24. Found: C, 58.33; H, 5.25; N, 5.16.
3.5. Representative Procedure for the Synthesis of Naphthalene and Quinoline Analogs Using MBH Acetates and Active Methylene Compounds
A 50 mL, round-bottomed flask equipped with a condenser, stir bar, and N2 inlet, was charged with an MBH acetate (1 mmol) in DMF (2 mL). The corresponding active methylene compound (1.5 mmol) in DMF (1 mL) and K2CO3 (207 mg, 1.5 mmol) were added at room temperature with continued stirring. For naphthalenes 15–18, the reaction was complete in 1 h. For quinolines 19, the reaction was stirred at room temperature for 1 h and gradually heated to 90 °C with stirring for 6 h. In each case, TLC analysis (30% EtOAc in hexane) indicated the reaction was complete. The solution was poured into de-ionized water (15 mL), and the mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with saturated NaCl and dried (Na2SO4). Removal of the solvent under vacuum gave the crude product, which was purified by silica gel column chromatography to afford the pure naphthalene/quinoline derivatives.
3.5.1. Ethyl 4-cyano-7-nitro-2-naphthoate (15a) from 5 and ethyl cyanoacetate (10)
Yield: 243 mg (0.90 mmol, 90%) as an off-white solid, m.p. 77–79 °C; IR: 1716, 1536, 1349 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.91 (d, J = 2.3 Hz, 2H), 8.60 (d, J = 1.6 Hz, 1H), 8.48 (dd, J = 9.2, 2.3 HZ, 1H), 8.36 (d, J = 9.2 Hz, 1H), 4.42 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H); 13C-NMR: δ 163.9, 147.1, 136.8, 136.2, 135.2, 131.5, 129.7, 127.4, 126.2, 124.0, 116.1, 111.6, 62.4, 14.3; MS (EI): m/z 270; Anal. Calcd for C14H10N2O4: C, 62.22; H, 3.73; N, 10.37. Found: C, 62.26; H, 3.76; N, 10.28.
3.5.2. 3-Ethyl 1-methyl 6-nitronaphthalene-1,3-dicarboxylate (15b) from 5 and methyl phenylsulfonylacetate (11)
Yield: 276 mg (0.91 mmol, 91%) as an off-white solid, m.p. 203–204 °C; IR: 1707, 1527, 1350 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.20 (d, J = 9.5 Hz, 1H), 8.95–8.90 (complex, 3H), 8.45 (dd, J = 9.5, 2.5 Hz, 1H), 4.51 (q, J = 7.1 Hz, 2H), 4.07 (s, 3H), 1.49 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 166.5, 164.9, 146.0, 137.0, 135.7, 133.0, 132.3, 129.0, 128.2, 127.9, 125.9, 122.9, 62.0, 52.8, 14.4; MS (EI): m/z 303 [M]+·; Anal. Calcd for C15H13NO6: C, 59.41; H, 4.32; N, 4.62. Found: C, 59.48; H, 4.30; N, 4.51.
3.5.3. Diethyl 6-nitronaphthalene-1,3-dicarboxylate (15c) from 5 and ethyl nitroacetate (12)
Yield: 298 mg (0.94 mmol, 94%) as an off-white solid, m.p. 181–182 °C; IR: 1708, 1526, 1350 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.19 (d, J = 9.5 Hz, 1H), 8.95-8.90 (complex, 3H), 8.44 (dd, J = 9.5, 2.4 HZ, 1H), 4.53 (q, J = 7.1 Hz, 2H), 4.51 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H), 1.49 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 166.1, 165.0, 145.9, 136.8, 135.7, 132.8, 132.3, 129.0, 128.4, 128.2, 125.9, 122.8, 62.0, 61.9, 14.4, 14.35; MS (EI): m/z 317 [M]+·; Anal. Calcd for C16H15NO6: C, 60.57; H, 4.77; N, 4.41. Found: C, 60.63; H, 4.73; N, 4.31.
3.5.4. Ethyl 4-benzoyl-7-nitro-2-naphthoate (15d) from 5 and 1-phenyl-2-(phenylsulfonyl)ethan-1-one (13)
Yield: 335 mg (0.96 mmol, 96%) as a white solid, m.p. 160–161 °C; IR: 1724, 1657, 1530, 1350 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.99 (d, J = 2.4 Hz, 1H), 8.93 (s, 1H), 8.36 (m, 2H), 8.27 (d, J = 9.3 Hz, 1H), 7.86 (d, J = 7.4 Hz, 2H), 7.67 (t, J = 7.5 Hz, 1H), 7.51 (t, J = 7.7 Hz, 2H), 4.48 (q, J = 7.1 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 196.1, 165.0, 146.2, 137.2, 137.18, 135.3, 135.0, 134.1, 132.1, 130.5, 130.0, 128.9, 128.8, 127.8, 126.0, 122.5, 62.0, 14.3; MS (EI): m/z 349 [M]+·; Anal. Calcd for C20H15NO5: C, 68.76; H, 4.33; N, 4.01. Found: C, 68.67; H, 4.35; N, 3.95.
3.5.5. Ethyl 4-acetyl-7-nitro-2-naphthoate (15e) from 5 and 1-(phenylsulfonyl)propan-2-one (14)
Yield: 262 mg (0.92 mmol, 92%) as a white flaky solid, m.p. 152–153 °C; IR: 1714, 1690, 1526, 1351 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.01 (br t, J = 8.2 Hz, 1H), 8.90 (m, 2H), 8.73 (br s, 1H), 8.42 (br t, J = 8.4 Hz, 1H), 4.52 (q, J = 7.1 Hz, 2H), 2.84 (s, 3H), 1.49 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 200.3, 165.0, 146.1, 136.6, 135.6, 134.6, 132.5, 131.3, 128.7, 128.5, 125.7, 123.2, 62.1, 29.8, 14.4; MS (EI): m/z 287 [M]+·; Anal. Calcd for C15H13NO5: C, 62.72; H, 4.56; N, 4.88. Found: C, 62.68; H, 4.55; N, 4.79.
3.5.6. 6-Nitronaphthalene-1,3-dicarbonitrile (16a) from 6 and 10
Yield: 178 mg (0.80 mmol, 80%) as a white solid, m.p. 224 °C (dec); IR: 2250, 1541, 1354 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.97 (d, J = 2.2 Hz, 1H), 8.67 (s, 1H), 8.65 (dd, J = 9.1, 2.2 Hz, 1H), 8.51 (d, J = 9.2 Hz, 1H), 8.26 (d, J = 1.6 Hz, 1H); 13C-NMR (101 MHz, CDCl3): δ 147.6, 139.7, 135.7, 135.5, 131.4, 127.9, 125.3, 125.0, 116.0, 114.9, 113.2, 112.0; MS (EI): m/z 223 [M]+·; Anal. Calcd for C12H5N3O2: C, 64.58; H, 2.26; N, 18.83. Found: C, 64.52; H, 2.29; N, 18.74.
3.5.7. Methyl 3-cyano-6-nitro-1-naphthoate (16b) from 6 and 11
Yield: 220 mg (0.86 mmol, 86%) as a white solid, m.p. 219-220 °C; IR: 2254, 1708, 1534, 1350 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.26 (d, J = 9.5 Hz, 1H), 8.89 (d, J = 2.4 Hz, 1H), 8.58 (s, 1H), 8.54 (d, J = 1.7 Hz, 1H), 8.51 (dd, J = 9.5, 2.4 Hz, 1H), 4.08 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 165.3, 146.5, 139.8, 135.0, 133.8, 132.2, 129.1, 128.6, 125.0, 123.8, 117.1, 111.3, 53.1; MS (EI): m/z 256 [M]+·; Anal. Calcd for C13H8N2O4: C, 60.94; H, 3.15; N, 10.93. Found: C, 60.87; H, 3.13; N, 10.87.
3.5.8. Ethyl 3-cyano-6-nitro-1-naphthoate (16c) from 6 and 12
Yield: 238 mg (0.88 mmol, 88%) as a white solid, m.p. 156–157 °C; IR: 2254, 1722, 1629, 1535, 1350 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.26 (d, J = 9.5 Hz, 1H), 8.89 (d, J = 2.4 Hz, 1H), 8.58 (s, 1H), 8.53 (s, 1H), 8.51 (dd, J = 9.5, 2.4 Hz, 1H), 4.54 (q, J = 7.1 Hz, 2H), 1.50 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 164.9, 146.5, 139.6, 135.0, 133.7, 132.1, 129.5, 128.7, 125.0, 123.7, 117.2, 111.3, 62.4, 14.3; MS (EI): m/z 270 [M]+·; Anal. Calcd for C14H10N2O4: C, 62.22; H, 3.73; N, 10.37. Found: C, 62.18; H, 3.72; N, 10.31.
3.5.9. 4-Benzoyl-7-nitro-2-naphthonitrile (16d) from 6 and 13
Yield: 272 mg (0.90 mmol, 90%) as a white solid, m.p. 152–154 °C; IR: 2234, 1663, 1530, 1348 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.94 (d, J = 2.3 Hz, 1H), 8.58 (s, 1H), 8.42 (dd, J = 9.3, 2.3 Hz, 1H), 8.29 (d, J = 9.3 Hz, 1H), 7.90 (d, J = 1.6 Hz, 1H), 7.84 (dd, J = 8.4, 1.7 Hz, 2H), 7.71 (tt, J = 7.5, 1.4 Hz, 1H), 7.54 (t, J = 7.5 Hz, 2H); 13C-NMR (101 MHz, CDCl3): δ 194.7, 146.8, 138.5, 137.8, 136.6, 134.61, 134.55, 132.0, 130.5, 130.4, 129.1, 128.2, 125.0, 123.4, 117.3, 111.1; MS (EI): m/z 302 [M]+·; Anal. Calcd for C18H10N2O3: C, 71.52; H, 3.33; N, 9.27. Found: C, 71.47; H, 3.31; N, 9.37.
3.5.10. 4-Acetyl-7-nitro-2-naphthonitrile (16e) from 6 and 14
Yield: 211 mg (0.88 mmol, 88%) as a white solid, m.p. 202–204 °C; IR: 2228, 1670, 1518, 1344 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.99 (d, J = 9.5 Hz, 1H), 8.88 (d, J = 2.4 Hz, 1H), 8.56 (s, 1H), 8.50 (dd, J = 9.5, 2.4 Hz, 1H), 8.25 (d, J = 1.7 Hz, 1H), 2.82 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 198.9, 146.6, 139.4, 136.8, 133.8, 132.3, 131.7, 128.8, 124.8, 124.0, 117.2, 111.1, 29.8; MS (EI): m/z 240 [M]+·; Anal. Calcd for C13H8N2O3: C, 65.00; H, 3.36; N, 11.66. Found: C, 64.93; H, 3.33; N, 11.59.
3.5.11. Ethyl 4,7-dicyano-2-naphthoate (17a) from 7 and 10
Yield: 200 mg (0.80 mmol, 80%) as a white solid, m.p. 209–210 °C; IR: 2229, 1720 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.87 (s, 1H), 8.66 (d, J = 1.5 Hz, 1H), 8.45 (s, 1H), 8.41 (d, J = 8.7 Hz, 1H), 7.96 (dd, J = 8.7, 1.5 Hz, 1H), 4.51 (q, J = 7.1 Hz, 2H), 1.48 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 164.0, 135.7, 135.6, 135.2, 134.8, 131.5, 131.2, 129.4, 126.8, 117.7, 116.1, 112.5, 111.5, 62.4, 14.3; MS (EI): m/z 250 [M]+·; Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.04; H, 4.06; N, 11.12.
3.5.12. 3-Ethyl 1-methyl 6-cyanonaphthalene-1,3-dicarboxylate (17b) from 7 and 11
Yield: 232 mg (0.82 mmol, 82%) as a white solid, m.p. 181–182 °C; IR: 2224, 1707 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.13 (d, J = 9.0 Hz, 1H), 8.89 (d, J = 1.8 Hz, 1H), 8.77 (s, 1H), 8.38 (d, J = 0.9 Hz, 1H), 7.83 (dd, J = 9.0, 1.6 Hz, 1H), 4.50 (q, J = 7.1 Hz, 2H), 4.05 (s, 3H), 1.48 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 166.5, 165.0, 135.62, 135.55, 134.5, 132.4, 132.2, 130.1, 128.6, 127.8, 127.6, 118.3, 111.0, 61.9, 52.7, 14.4; MS (EI): m/z 283 [M]+·; Anal. Calcd for C16H13NO4: C, 67.84; H, 4.63; N, 4.94. Found: C, 67.76; H, 4.62; N, 4.97.
3.5.13. Diethyl 6-cyanonaphthalene-1,3-dicarboxylate (17c) from 7 and 12
Yield: 247 mg (0.82 mmol, 83%) as a white solid, m.p. 159–160 °C; IR: 2223, 1714 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.12 (d, J = 9.1 Hz, 1H), 8.88 (d, J = 1.9 Hz, 1H), 8.77 (s, 1H), 8.37 (s, 1H), 7.83 (dd, J = 9.0, 1.5 Hz, 1H), 4.52 (q, J = 7.1 Hz, 2H), 4.50 (q, J = 7.1 Hz, 2H), 1.49 (t, J = 7.1 Hz, 3H), 1.48 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 166.1, 165.1, 135.6, 135.4, 134.5, 132.21, 132.18, 130.1, 128.6, 128.3, 127.6, 118.3, 110.9, 61.9, 61.8, 14.36, 14.35; MS (EI): m/z 297 [M]+·; Anal. Calcd for C17H15NO4: C, 68.68; H, 5.09; N, 4.71. Found: C, 68.62; H, 5.09; N, 4.63.
3.5.14. Ethyl 4-benzoyl-7-cyano-2-naphthoate (17d) from 7 and 13
Yield: 280 mg (0.85 mmol, 85%) as a white solid, m.p. 144–145 °C; IR: 2227, 1722, 1656 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.79 (s, 1H), 8.43 (s, 1H), 8.31 (s, 1H), 8.21 (d, J = 8.8 Hz, 1H), 7.85 (d, J = 7.7 Hz, 2H), 7.74 (d, J = 8.8 Hz, 1H), 7.66 (t, J = 7.5 Hz, 1H), 7.51 (t, J = 7.6 Hz, 2H), 4.47 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 196.1, 165.1, 137.24, 137.15, 135.6, 134.2, 134.1, 133.6, 132.1, 130.4, 129.7, 129.4, 128.8, 128.5, 127.3, 118.3, 111.2, 61.9, 14.3; MS (EI): m/z 329 [M]+·; Anal. Calcd for C21H15NO3: C, 76.58; H, 4.59; N, 4.25. Found: C, 76.60; H, 4.63; N, 4.17.
3.5.15. Ethyl 4-acetyl-7-cyano-2-naphthoate (17e) from 7 and 14
Yield: 224 mg (0.84 mmol, 84%) as a white solid, m.p. 134–135 °C; IR: 2229, 1717, 1678 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.95 (d, J = 9.0 Hz, 1H), 8.76 (s, 1H), 8.68 (t, J = 1.6 Hz, 1H), 8.37 (s, 1H), 7.83 (dt, J = 8.9, 1.8 Hz, 1H), 4.51 (q, J = 7.1 Hz, 2H), 2.82 (s, 3H), 1.49 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 200.4, 165.1, 135.5, 135.4, 135.3, 133.5, 132.4, 130.7, 130.5, 128.4, 127.9, 118.2, 111.2, 62.0, 29.8, 14.4; MS (EI): m/z 267 [M]+·; Anal. Calcd for C16H13NO3: C, 71.90; H, 4.90; N, 5.24. Found: C, 71.84; H, 4.93; N, 5.28.
3.5.16. Naphthalene-1,3,6-tricarbonitrile (18a) from 8 and 10
Yield: 152 mg (0.75 mmol, 75%) as a white solid, m.p. 206–207 °C; IR: 2232 cm−1; 1H-NMR: δ 8.53 (br s, 1H), 8.45 (d, J = 8.7 Hz, 1H), 8.42 (br s, 1H), 8.22 (d, J = 1.6 Hz, 1H), 8.04 (dd, J = 8.7, 1.6 Hz, 1H); 13C-NMR (101 MHz, CDCl3): δ 138.5, 135.2, 134.8, 134.4, 132.2, 131.3, 127.2, 117.1, 116.2, 114.8, 113.8, 113.1, 111.7; MS (EI): m/z 203 [M]+·; Anal. Calcd for C13H5N3: C, 76.84; H, 2.48; N, 20.68. Found: C, 76.89; H, 2.51; N, 20.57.
3.5.17. Methyl 3,6-dicyano-1-naphthoate (18b) from 8 and 11
Yield: 184 mg (0.78 mmol, 78%) as an off-white solid, m.p. 235–236 °C; IR: 2228, 1719 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.18 (d, J = 9.0 Hz, 1H), 8.49 (s, 1H), 8.45 (s, 1H), 8.34 (s, 1H), 7.91 (d, J = 8.9 Hz, 1H), 4.10 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 165.3, 138.5, 134.6, 133.8, 133.3, 132.0, 131.1, 129.0, 127.9, 117.7, 117.2, 112.2, 110.9, 53.1; MS (EI): m/z 236 [M]+·; Anal. Calcd for C14H8N2O2: C, 71.18; H, 3.41; N, 11.86. Found: C, 71.09; H, 3.43; N, 11.79.
3.5.18. Ethyl 3,6-dicyano-1-naphthoate (18c) from 8 and 12
Yield: 200 mg (0.80 mmol, 80%) as a white solid, m.p. 185–186 °C; IR: 2228, 1704 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.18 (d, J = 9.0 Hz, 1H), 8.48 (s, 1H), 8.44 (s, 1H), 8.34 (s, 1H), 7.90 (d, J = 9.0 Hz, 1H), 4.53 (q, J = 7.1 Hz, 2H), 1.49 (t, J = 7.1 Hz, 3H); 13C-NMR: δ 164.9, 138.3, 134.6, 133.9, 133.2, 132.0, 131.0, 129.3, 127.9, 117.8, 117.3, 112.1, 110.9, 62.3, 14.3; MS (EI): m/z 250 [M]+·; Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.05; H, 4.04; N, 11.07.
3.5.19. 4-Benzoylnaphthalene-2,7-dicarbonitrile (18d) from 8 and 13
Yield: 280 mg (0.80 mmol, 80%) as a white solid, m.p. 170–171 °C; IR: 2232, 1664 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.44 (s, 1H), 8.39 (s, 1H), 8.23 (d, J = 8.8 Hz, 1H), 7.88-7.77 (complex, 4H), 7.70 (t, J = 7.5 Hz, 1H), 7.53 (t, J = 7.7 Hz, 2H); 13C-NMR (101 MHz, CDCl3): δ 194.7, 138.4, 136.6, 136.5, 134.59, 134.55, 133.4, 131.9, 130.6, 130.4, 130.0, 129.0, 127.6, 117.7, 117.4, 112.4, 110.7; MS (EI): m/z 282 [M]+·; Anal. Calcd for C19H10N2O: C, 80.84; H, 3.57; N, 9.92. Found: C, 80.83; H, 3.54; N, 9.86.
3.5.20. 4-Acetylnaphthalene-2,7-dicarbonitrile (18e) from 8 and 14
Yield: 224 mg (0.79 mmol, 79%) as a white solid, m.p. 215–216 °C; IR: 2227, 1686 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.91 (d, J = 9.0 Hz, 1H), 8.43 (s, 1H), 8.34 (d, J = 1.7 Hz, 1H), 8.20 (d, J = 1.6 Hz, 1H), 7.89 (dd, J = 9.0, 1.7 Hz, 1H), 2.82 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ 199.0, 138.1, 136.7, 134.4, 132.7, 132.2, 131.3, 131.2, 128.1, 117.7, 117.3, 112.4, 110.7, 29.8; MS (EI): m/z 220 [M]+·; Anal. Calcd for C14H8N2O: C, 76.35; H, 3.66; N, 12.72. Found: C, 76.30; H, 3.63; N, 12.67.
3.5.21. Ethyl 8-cyanoquinoline-6-carboxylate (19a) from 9 and 10
Yield: 170 mg (0.75 mmol, 75%) as a yellow solid, m.p. 142–144 °C; IR: 2233, 1722 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.20 (m, 1H), 8.81 (s, 1H), 8.73 (d, J = 1.7 Hz, 1H), 8.38 (dd, J = 8.3, 1.7 Hz, 1H), 7.65 (dd, J = 8.3, 4.3 Hz, 1H), 4.50 (q, J = 7.1 Hz, 2H), 1.48 (q, J = 7.1 Hz, 3H); 13C-NMR) (101 MHz, CDCl3): δ 164.3, 154.4, 149.0,137.8, 135.3, 135.2, 128.3, 127.5, 123.5, 116.5, 113.8, 62.2, 14.3; MS (EI): m/z 226 [M]+·; Anal. Calcd for C13H10N2O2: C, 69.02; H, 4.46; N, 12.38. Found: C, 69.07; H, 4.49; N, 12.27.
3.5.22. 6-Ethyl 8-methyl quinoline-6,8-dicarboxylate (19b) from 9 and 11
Yield: 212 mg (0.82 mmol, 82%) as an off-white solid, m.p. 75–77 °C; IR: 1722 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.14 (dd, J = 4.2, 1.8 Hz, 1H), 8.70 (d, J = 2.0 Hz, 1H), 8.62 (d, J = 2.0 Hz, 1H), 8.31 (dd, J = 8.4, 1.8 Hz, 1H), 7.54 (dd, J = 8.3, 4.2 Hz, 1H), 4.48 (q, J = 7.1 Hz, 2H), 4.08 (s, 3H), 1.46 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 167.6, 165.3, 153.5, 147.2, 137.6, 133.9, 132.0, 129.9, 127.8, 127.7, 122.3, 61.8, 52.9, 14.4; MS (EI): m/z 259 [M]+·; Anal. Calcd for C14H13NO4: C, 64.86; H, 5.05; N, 5.40. Found: C, 64.80; H, 5.03; N, 5.37.
3.5.23. Ethyl 8-benzoylquinoline-6-carboxylate (19d) from 9 and 13
Yield: 244 mg (0.80 mmol, 80%) as a light yellow solid, m.p. 146–147 °C; IR: 1718, 1671 cm−1; 1H-NMR (400 MHz, CDCl3): δ 8.93 (d, J = 4.2 Hz, 1H), 8.72 (d, J = 1.9 Hz, 1H), 8.36-8.30 (complex, 2H), 7.84 (d, J = 7.3 Hz, 2H), 7.57 (t, J = 7.3 Hz, 1H), 7.50 (dd, J = 8.4, 4.2 Hz, 1H), 7.42 (t, J = 7.7 Hz, 2H), 4.46 (q, J = 7.1 Hz, 2H), 1.44 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 197.1, 165.5, 152.8, 147.9, 139.8, 137.4, 137.3, 133.5, 132.4, 130.2, 128.5, 128.0, 127.62, 127.55, 122.4, 61.7, 14.4; MS (EI): m/z 305 [M]+·; Anal. Calcd for C19H15NO3: C, 74.74; H, 4.95; N, 4.59. Found: C, 74.71; H, 4.95; N, 4.52.
3.5.24. Ethyl 8-acetylquinoline-6-carboxylate (19e) from 9 and 14
Yield: 185 mg (0.76 mmol, 76%) as a white crystals, m.p. 103–104 °C; IR: 1704, 1689 cm−1; 1H-NMR (400 MHz, CDCl3): δ 9.06 (dd, J = 4.2, 1.8 Hz, 1H), 8.69 (d, J = 1.9 Hz, 1H), 8.50 (d, J = 1.9 Hz, 1H), 8.32 (dd, J = 8.4, 1.8 Hz, 1H), 7.54 (dd, J = 8.3, 4.2 Hz, 1H), 4.47 (q, J = 7.1 Hz, 2H), 2.94 (s, 3H), 1.46 (t, J = 7.1 Hz, 3H); 13C-NMR (101 MHz, CDCl3): δ 203.2, 165.5, 152.4, 147.2, 140.1, 137.6, 133.7, 128.6, 128.1, 127.7, 122.2, 61.7, 32.6, 14.4; MS (EI): m/z 243 [M]+·; Anal. Calcd for C14H13NO3: C, 69.12; H, 5.39; N, 5.76. Found: C, 69.06; H, 5.37; N, 5.69.
3.6. Competitive Reaction Control Experiment. Formation of Methyl (Z)-4-cyano-5-(2-fluorophenyl)-2-(phenylsulfonyl)-4-pentenoate (22)
A 50-mL, round-bottomed flask equipped with a condenser, stir bar, and N2 inlet was charged with a 2-fluoro-5-nitrotoluene (20, 155 mg, 1 mmol) and 2-cyano-1-(2-fluorophenyl)allyl acetate (21, 219 mg, 1 mmol) in DMF (2 mL) under N2. Methyl phenylsulfonylacetate (321 mg, 1.5 mmol) and K2CO3 (207 mg, 1.5 mmol) were added at room temperature with stirring. TLC analysis (20% EtOAc in hexane) indicated that the reaction was complete in 1 h. The solution was poured into de-ionized water (15 mL), and the mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with saturated NaCl and dried (Na2SO4). Removal of the solvent under vacuum gave the crude product, which was purified by silica gel column chromatography to afford 20 (144 mg, 93%) and 22 (261 mg, 0.7 mmol, 70%) as a white solid, m.p. 95–97 °C; IR: 2215, 1744, 1637, 1149 cm−1; 1H-NMR (400 MHz, CDCl3): δ 7.98 (td, J = 7.8, 1.7 Hz, 1H), 7.92 (d, J = 7.8 Hz, 2H), 7.74 (tt, J = 7.5, 1.9 Hz, 1H), 7.62 (t, J = 8.2 Hz, 2H), 7.44–7.36 (complex, 1H), 7.31 (s, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.10 (t, J = 9.5 Hz, 1H), 4.34 (dd, J = 10.7, 4.4 Hz, 1H), 3.67 (s, 3H), 3.20 (dd, J = 14.3, 4.4 Hz, 1H), 3.14 (dd, J = 14.3, 10.7 Hz, 1H); 13C-NMR (101 MHz, CDCl3): δ 164.9, 160.4 (d, J = 253.0 Hz), 139.3 (d, J = 6.5 Hz), 136.8, 134.8, 132.6 (d, J = 8.7 Hz), 129.4, 129.2, 128.3 (d, J = 1.6 Hz), 124.6 (d, J = 3.7 Hz), 121.1 (d, J = 11.7 Hz), 117.0, 115.8 (d, J = 21.7 Hz), 107.2 (d, J = 2.0 Hz), 68.7, 53.3, 33.0; MS (EI): m/z 373 [M]+·; Anal. Calcd for C19H16FNO4S: C, 61.12; H, 4.32; N, 3.75. Found: C, 61.22; H, 4.25; N, 3.68. The X-ray structure for compound 22 (CCDC 2035022) and the thermal elipsoid plot is shown in Scheme 2 and the Supplementary Materials.
4. Conclusions
We have investigated the synthesis of naphthalenes and quinolines from Morita–Baylis–Hillman acetates and active methylene compounds promoted by K2CO3 in DMF. The formation of naphthalenes occurs at 23 °C, while quinolines required heating to 90 °C. Substrates for the naphthalenes were MBH acetates bearing 2-fluoroaromatic rings activated toward SNAr ring closure by C5 NO2 or CN groups. Quinoline precursors were activated only by the electron-withdrawing nitrogen in a 2-fluoropyridine ring. The transformation most likely involves a domino SN2′-SNAr process. A control experiment indicated that the initial reaction occurs by an SN2′-type substitution of the side chain acetate to yield the alkene having the aromatic ring trans to the aromatic SNAr acceptor ring. Thus, under the reaction conditions, a reversible Michael addition or an intramolecular addition–elimination must occur to equilibrate the geometry of the double bond to yield the alkene isomer needed for ring closure. The isolation of intermediates from the reaction disputes an earlier report that the reaction directly delivers the alkene needed for cyclization. Once equilibrated, subsequent deprotonation of the active methine proton, SNAr ring closure, and the elimination of SO2Ph, NO2, or CO2Et then aromatizes the product. Much of the selectivity appears to be guided by steric considerations. The loss of SO2Ph or NO2 in the aromatization process has good precedent in the literature, but the loss of CO2Et in preference to CN was unexpected. Good to excellent yields were isolated for all examples.
Supplementary Materials
The following are available online, Copies of 1H-NMR and 13C-NMR spectra for all compounds and tables of crystal data for compounds 4 and 22.
Author Contributions
Formal analysis, R.A.B.; Investigation, J.K.A.-G., E.A. and K.M.; Methodology, J.K.A.-G., E.A. and K.M.; Project administration, R.A.B.; Writing—original draft, R.A.B.; Writing—review & editing, R.A.B., J.K.A.-G., E.A. and K.M. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support for this work was obtained from the Oklahoma State University Foundation and the College of Arts and Sciences at Oklahoma State University. The University of Oklahoma Chemical Crystallography Laboratory was upgraded with support from the NSF (CHE-1726630). The Oklahoma State University State-wide NMR Facility was established with funds from the NSF (BIR-9512269) the OSU Regents for Higher Education the W. M. Keck Foundation and Conoco, Inc.
Acknowledgments
J.K.A.-G. and K.M. wish to thank K. Darrell Berlin and the OSU Foundation for Summer Scholarships. The authors also wish to thank Douglas Powell at the University of Oklahoma Chemical Crystallography Laboratory for obtaining the X-ray crystal structures. This facility was established with support from the NSF (CHE-1726630). Finally, the authors are indebted to the OSU College of Arts and Sciences for funds to purchase several departmental instruments including an FT-IR and a 400 MHz NMR unit for the State-wide NMR facility. The NMR facility was initially established with support from the NSF (BIR-9512269), the Oklahoma State Regents for Higher Education, the W. M. Keck Foundation, and Conoco, Inc.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Narendar Reddy, T.; Jayathirtha Rao, V. Importance of Baylis-Hillman adducts in modern drug discovery. Tetrahedron Lett. 2018, 59, 2859–2875. [Google Scholar] [CrossRef]
- Lima-Junior, C.G.; Vasconcellos, M.A.A. Morita Baylis-Hillman adducts: Biological activities and potentialities to the discovery of new cheaper drugs. Bioog. Med. Chem. 2012, 20, 3954–3971. [Google Scholar] [CrossRef] [PubMed]
- Makar, S.; Saha, T.; Singh, S.K. Naphthalene, a versatile platform in medicinal chemistry: Sky high perspective. Eur. J. Med. Chem. 2019, 161, 252–276. [Google Scholar] [CrossRef] [PubMed]
- Ofzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef]
- Beena; Rawat, D.S. Antituberculosis drug research: A critical review. Med. Res. Rev. 2013, 33, 693–764. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Chung, Y.M.; Gong, J.H.; Kim, J.N. Synthesis of naphthalenes from the reaction of Baylis-Hillman acetates and sulfonyl group-containing active methylene compounds. Bull. Korean Chem. Soc. 2002, 23, 787–788. [Google Scholar] [CrossRef][Green Version]
- Kim, J.N.; Lee, H.J.; Lee, K.Y.; Kim, H.S. Synthesis of 3-quinolinecarboxylic acid esters from the Baylis-Hillman adducts of 2-halobenzaldehyde N-tosylimines. Tetrahedron Lett. 2001, 42, 3737–3740. [Google Scholar] [CrossRef]
- Gupta, T.; Bharadwaj, K.C.; Singh, R.M. Cascade SN2’-SNAr, Elimination and 1,5-hydride shift reactions by acetylacetone/acetoacetic esters: Synthesis of 9,10-dihydroacridines. Eur. J. Org. Chem. 2016, 4981–4984. [Google Scholar] [CrossRef]
- Gupta, T.; Singh, J.B.; Mishra, K.; Singh, R.M. Active methylene compounds (AMCs)controlled facile synthesis of acridine and phenanthridine from morita Baylis-Hillman acetate. RSC Adv. 2017, 7, 54581–54585. [Google Scholar] [CrossRef]
- Morita, K.; Suzuki, Z.; Hirose, H. A tertiary phosphine-catalyzed reaction of acrylic compounds with aldehydes. Bull. Soc. Chem. Jpn. 1968, 41, 2815. [Google Scholar] [CrossRef]
- Baylis, A.B.; Hillman, M.E.D. Acrylic Compounds. German Patent 2155113, 1972. [Google Scholar]
- Hillman, M.E.D.; Baylis, A.B. The reaction of acrylic type compounds with aldehydes and certain ketones. U.S. Patent 3,743,669, 3 July 1973. [Google Scholar]
- Kürti, L.; Czakó, B. Strategic Applications of Named Reaction in Organic Chemistry; Elsevier: New York, NY, USA, 2005; pp. 48–49. [Google Scholar]
- Singh, B.; Chandra, A.; Singh, R.H. Base-free amination of BH acetates of 2-chloroquinolinyl-3- carboxaldehydes: A facile route to the synthesis of N-substituted-1,2-dihydrobenzo[b][1.8]naphthyridines. Tetrahedron 2011, 67, 2441–2446. [Google Scholar] [CrossRef]
- Procopiou, P.A.; Baugh, S.P.D.; Flack, S.S.; Inglis, G.G.A. An extremely powerful acylation reaction of alcohols with acid anhydrides catalyzed by trimethylsilyl trifluoromethanesulfonate. J. Org. Chem. 1998, 63, 2342–2347. [Google Scholar] [CrossRef]
- Love, B.E.; Raje, P.S. Preparation of 1-aryl-carbolines. J. Org. Chem. 1994, 59, 3219–3222. [Google Scholar] [CrossRef]
- Paredes, E.; Biolatta, B.; Kneetemen, M.; Mancini, P. One-step synthesis of 2,9-disubstituted phenanthrenes via Diels-Alder reactions using 1,4-disubstituted naphthalenes as dienophiles. Tetrahedron Lett. 2002, 43, 4601–4603. [Google Scholar] [CrossRef]
- Schenck, L.W.; Kuna, S.K.; Frank, W.; Alber, A.; Kucklaender, U. Dialkyl quinone-2,3-dicarboxylates in the Nenitzescu reaction. Tetrahedron 2005, 61, 9129–9139. [Google Scholar] [CrossRef]
- Kucukdisli, M.; Ferenc, D.; Heinz, M.; Wiebe, C.; Opatz, T. Simple two-step synthesis of 2,4-disubstituted pyrroles and 3,5-disubstituted pyrrole-2-carbonitriles from enones. BeilsteinJ. Org. Chem. 2014, 10, 466–470. [Google Scholar] [CrossRef]
- Wang, Y.-W.; Fang, J.-M.; Wang, Y.-K.; Wang, M.-H.; Ko, T.-Y.; Cherng, Y.-J. Addition reactions of 2-amino-4-methoxypenta-2,4-dienenitrile with electrophiles containing electron-deficient multiple bonds. J. Chem. Soc., Perkin Trans. 1992, 1, 1209–1213. [Google Scholar] [CrossRef]
- Buchholz, R.; Hoffmann, H.M.R. α-Methylidene and α-Alkylidene-β-lactams from nonproteinogenic amino acids. Helv. Chim. Acta 1991, 74, 1213–1220. [Google Scholar] [CrossRef]
- Stohrer, W.-D. On the stereochemistry of the SN2’ reaction. Angew. Chem. Int. Ed. 1983, 22, 613–614. [Google Scholar] [CrossRef]
- Magid, R.M.; Fruchey, O.S. Stereochemistry of the SN2’ reaction of an acyclic allylic chloride with a secondary amine. J. Am. Chem. Soc. 1977, 99, 8368–8369. [Google Scholar] [CrossRef]
- Oritani, T.; Overton, K.H. Stereochemistry of the SN2’ reaction with acyclic allylic esters. J. Chem. Soc. Chem. Commun. 1978, 454–455. [Google Scholar] [CrossRef]
- Magid, R.M. Nucleophilic and organometallic displacement reactions of allylic compounds: Stereo- and regiochemistry. Tetrahedron 1980, 36, 1901–1930. [Google Scholar] [CrossRef]
- Stork, G.; Kreft III, A.F. Concerning the stereochemistry of the SN2’ reaction. “Concerted” allylic displacement in an acyclic system: Anti displacement with thiolate anion. J. Am. Chem. Soc. 1977, 99, 3851–3852. [Google Scholar] [CrossRef]
- Bach, R.D.; Wolber, G.J. Stereochemistry of the concerted SN2’ reaction of 3-chloropropene: A theoretical study. J. Am. Chem. Soc. 1985, 107, 1352–1357. [Google Scholar] [CrossRef]
- Bergman, E.D.; Ginsburg, D.; Pappo, R. The Michael Reaction. Org. React. 1959, 10, 179–555. [Google Scholar]
- Werner, R.M.; Williams, L.M.; Davis, J.T. The C-glycosyl analog of an N-linked glycoamino acid. Tetrahedron Lett. 1998, 39, 9135–9138. [Google Scholar] [CrossRef]
- Shao, H.; Ekthawatchai, S.; Chen, C.-S.; Wu, S.-H.; Zou, W. 1,2-Migration of 2’-oxoalkyl group and concomitant synthesis of 2-C-branched O-, S-glycosides and glycosyl azides via 1,2-cyclopropanated sugars. J. Org. Chem. 2005, 70, 4726–4734. [Google Scholar] [CrossRef]
- Henderson, A.P.; Bleasdale, C.; Delaney, K.; Lindstrom, A.B.; Rappaport, S.M.; Waidyanatha, S.; Watson, W.P.; Golding, B.T. Evidence for the formation of Michael adducts from reactions of [E,E]-muconaldehyde with glutathione and other thiols. Bioorg. Chem. 2005, 33, 363–373. [Google Scholar] [CrossRef]
- Hintermann, L.; Dittmer, C. Asymmetric ion-pairing catalysis of the reversible cyclization of 2’-hydroxychalcone to flavanone: Asymmetric catalysis of an equilibrating reaction. Eur. J. Org. Chem. 2012, 5573–5584. [Google Scholar] [CrossRef]
- McKenna, E.G.; Walker, B.J. Wittig reactions of ylide anions derived from stabilized ylides. J. Chem. Soc. Chem. Commun. 1989, 568–569. [Google Scholar] [CrossRef]
- Hoffmann, H.M.R.; Rabe, J. DABCO-catalyzed coupling of aldehydes with activated double bonds. 4. Stereoselective synthesis of trisubstituted olefins and terpenoid building blocks via 2-(hydroxyalkyl)-2-propenoic esters. J. Org. Chem. 1985, 50, 3849–3859. [Google Scholar] [CrossRef]
- Ameer, F.; Drewes, S.E.; Houston-McMillan, M.S.; Kaye, P.T. Necic acid synthons. Part 4. Regioselectivity in the reactions of chloro and iodo derivatives of selected 3-hydroxy-2-methylene-alkanoate esters with ethyl 3-methyl-3-oxobutanoate. J. Chem. Soc. Perkin Trans. 1985, 1, 1143–1145. [Google Scholar] [CrossRef]
- Basavaiah, D.; Pandiaraju, S.; Padmaja, K. The Friedel-Crafts chemistry: Acetates of the Baylis-Hillman adducts as novel stereodefined-electrophiles. Synlett 1996, 393–395. [Google Scholar] [CrossRef]
- Chavan, S.P.; Ethiraj, K.S.; Kamat, S.K. Facile synthesis of 2Z-2-chloromethyl aryl-2-enoates. Tetrahedron Lett. 1997, 38, 7415–7416. [Google Scholar] [CrossRef]
- Yadav, J.S.; Subba Reddy, B.V.; Madan, C. Montmorillonite clay-catalyzed stereoselective synthesis of aryl-substituted (E)- and (Z)-allyl iodides and bromides. New J. Chem. 2001, 25, 1114–1117. [Google Scholar] [CrossRef]
- Krishna, P.R.; Kannan, V.; Sharma, G.V.M. FeCl3 and Yb(OTf)3 mediated conversion of acetates of the Baylis-Hillman adducts into (Z) and (E) trisubstituted alkenes. Synth. Commun. 2004, 34, 55–64. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Zhang, Y. Direct and highly efficient synthesis of (Z)-allyl iodides from Baylis-Hillman adducts promoted by TMSCl/NaI system. Synlett 2005, 1039–1041. [Google Scholar] [CrossRef]
- Gohain, M.; Lin, S.; Bezuidenhoudt, B.C.B. Al(OTf)3-catalyzed SN2’ substitution of the-hydroxy group in Morita-Baylis-Hillman adducts with indoles. Tetrahedron Lett. 2015, 56, 2579–2582. [Google Scholar] [CrossRef]














Sample Availability: Samples of the compounds are not available from the authors. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).