
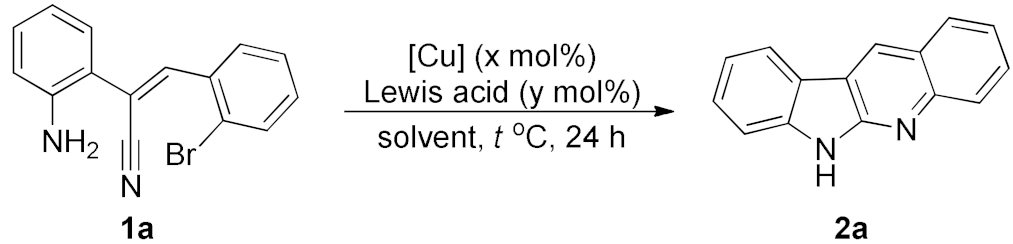
Copper-Catalyzed Dual Cyclization for the Synthesis of Quinindolines


Abstract
:
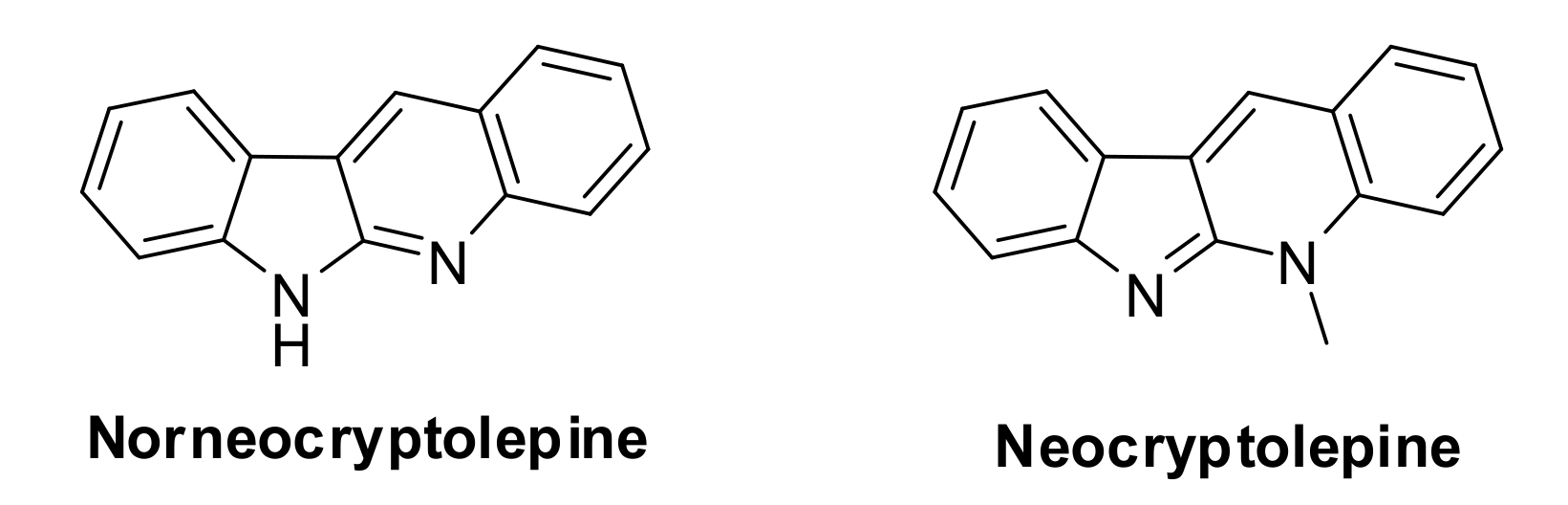
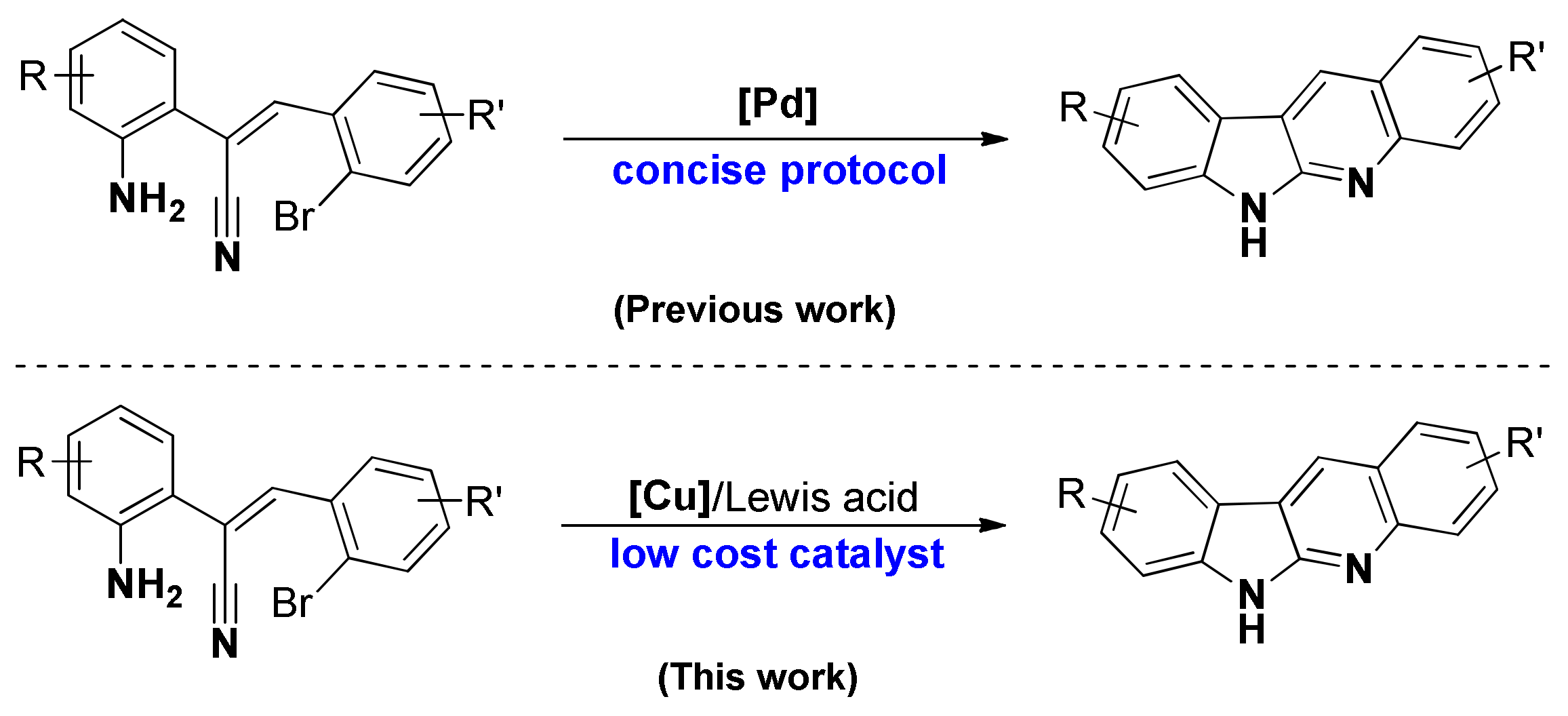
1. Introduction
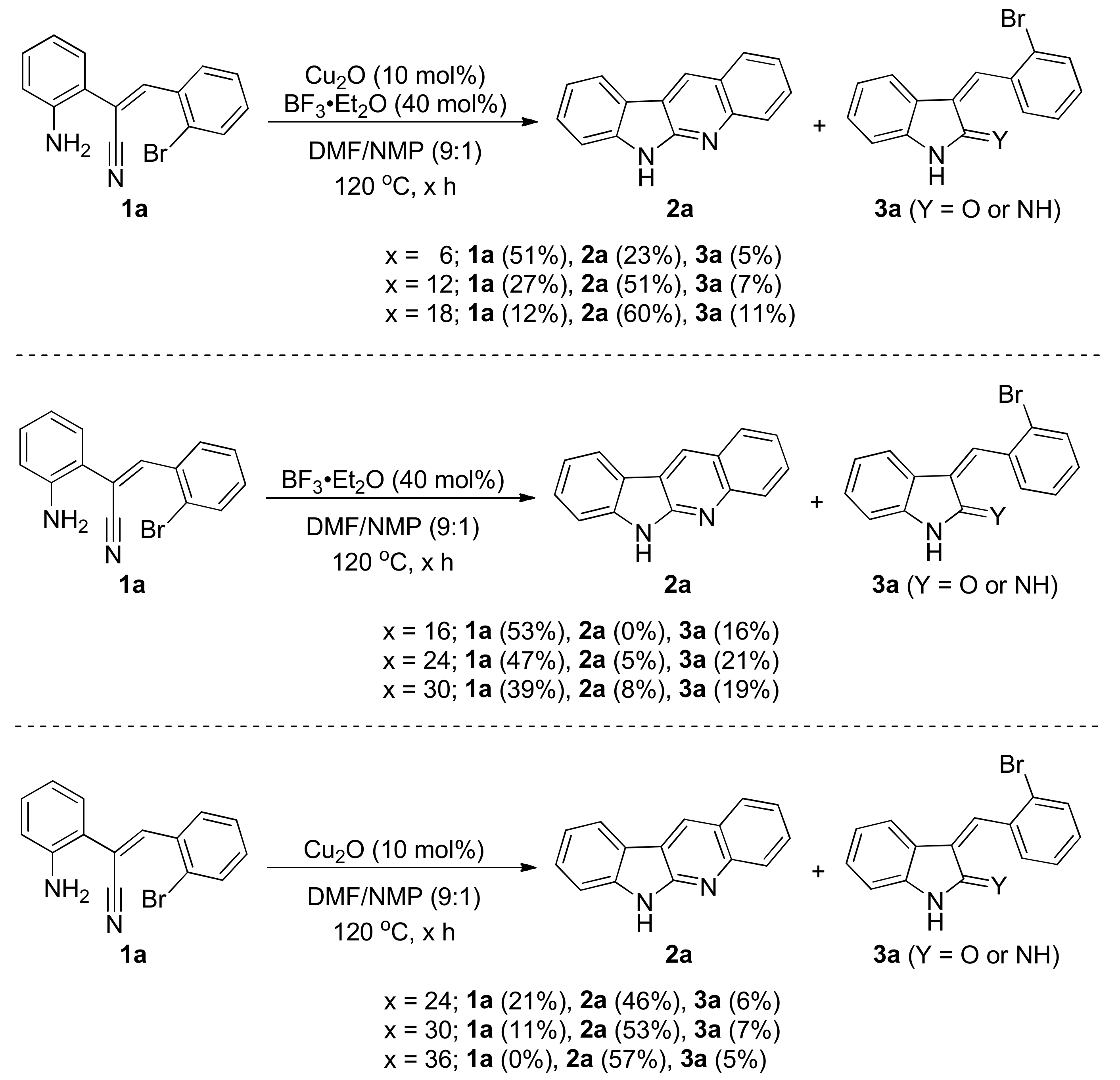
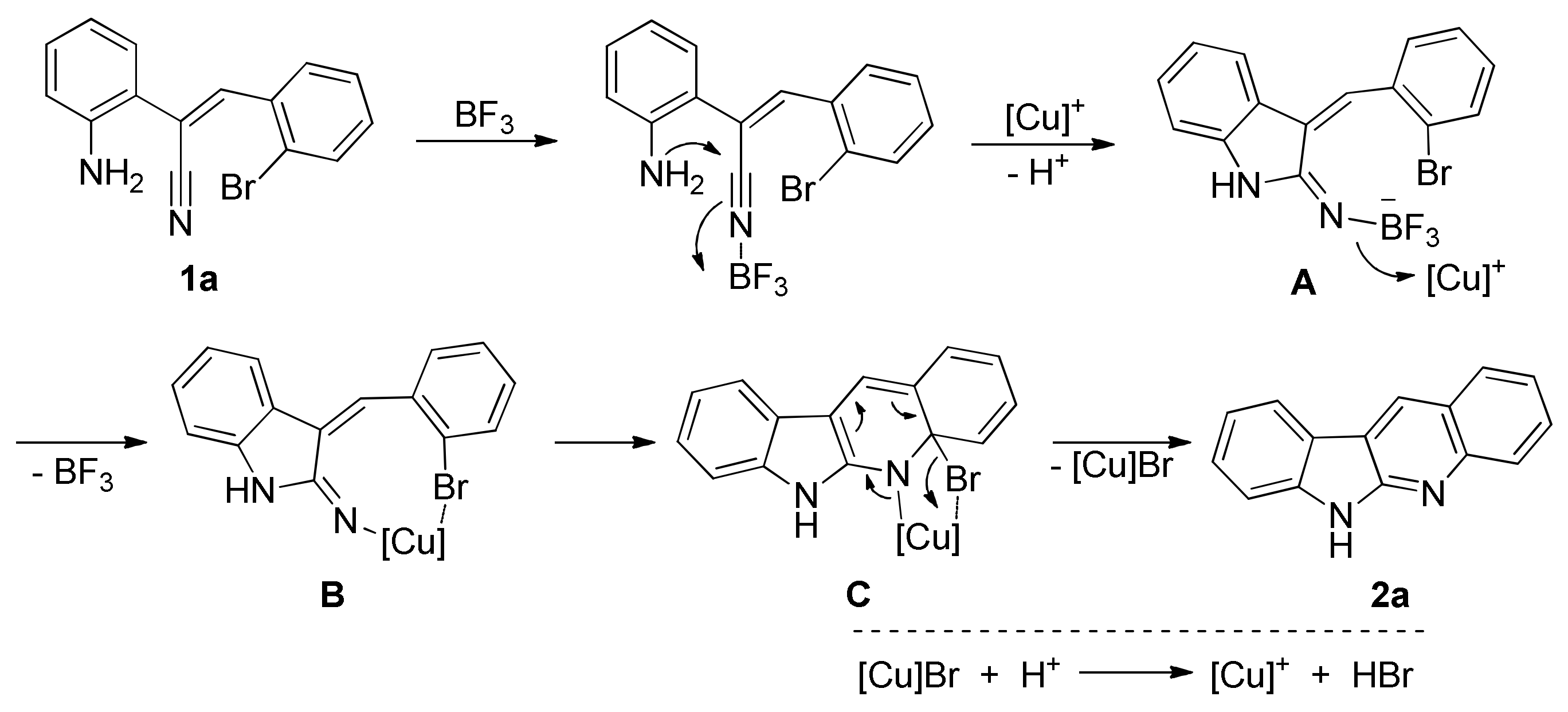
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Subbaraju, G.V.; Kavitha, J.; Rajasekhar, D.; Jimenez, J.I. Jusbetonin, the First Indolo[3,2-b]quinoline Alkaloid Glycoside, from Justicia betonica. J. Nat. Prod. 2004, 67, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, M.H.M.; Schiff, P.L., Jr.; Tackie, A.N.; Phoebe, C.H., Jr.; Martin, G.E. Two new indoloquinoline alkaloids from cryptolepis sanguinolenta: Cryptosanguinolentine and cryptotackieine. J. Heterocycl. Chem. 1996, 33, 239–243. [Google Scholar] [CrossRef]
- Cimanga, K.; De Bruyne, T.; Pieters, L.; Claeys, M.; Vlietinck, A. New alkaloids from Cryptolepis sanguinolenta. Tetrahedron Lett. 1996, 37, 1703–1706. [Google Scholar] [CrossRef]
- Jonckers, T.H.M.; van Miert, S.; Cimanga, K.; Bailly, C.; Colson, P.; De Pauw-Gillet, M.-C.; van der Heuvel, H.; Claeys, M.; Lemière, F.; Esmans, E.L.; et al. Synthesis, Cytotoxicity, and Antiplasmodial and Antitrypanosomal Activity of New Neocryptolepine Derivatives. J. Med. Chem. 2002, 45, 3497–3508. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk, K.; Świtalska, M.; Jaromin, A.; Cmoch, P.; Bujak, I.; Kaczmarska, M.; Wietrzyk, J.; Dominguez, E.G.; Żarnowski, R.; Andes, D.R.; et al. The synthesis of indolo[2,3-b]quinoline derivatives with a guanidine group: Highly selective cytotoxic agents. Eur. J. Med. Chem. 2015, 105, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Godlewska, J.; Luniewski, W.; Zagrodzki, B.; Kaczmarek, Ł.; Bielawska-Pohl, A.; Dus, D.; Wietrzyk, J.; Opolski, A.; Siwko, M.; Jaromin, A.; et al. Biological Evaluation of ω-(Dialkylamino)alkyl Derivatives of 6H-indolo[2,3-b]quinoline-Novel Cytotoxic DNA Topoisomerase II Inhibitors. Anticancer Res. 2005, 25, 2857–2868. [Google Scholar]
- Lavrado, J.; Moreira, R.; Paulo, A. Indoloquinolines as scaffolds for drug discovery. Curr. Med. Chem. 2010, 17, 2348–2370. [Google Scholar] [CrossRef]
- Bracca, A.B.J.; Heredia, D.A.; Larghi, E.L.; Kaufman, T.S. Neocryptolepine (Cryprotackieine), a Unique Bioactive Natural Product: Isolation, Synthesis, and Profile of Its Biological Activity. Eur. J. Org. Chem. 2014, 36, 7979–8003. [Google Scholar] [CrossRef]
- Sundaram, G.S.M.; Venkatesh, C.; Syam Kumar, U.K.; Ila, H.; Junjappa, H. A Concise Formal Synthesis of Alkaloid Cryptotackiene and Substituted 6H-Indolo[2,3-b]quinolines. J. Org. Chem. 2004, 69, 5760–5762. [Google Scholar] [CrossRef]
- Parvatkar, P.T.; Parameswaran, P.S.; Tilve, S.G. An Expeditious I2-Catalyzed Entry into 6H-Indolo[2,3-b]quinoline System of Cryptotackieine. J. Org. Chem. 2009, 74, 8369–8372. [Google Scholar] [CrossRef]
- Yu, S.; Li, Y.; Zhou, X.; Wang, H.; Kong, L.; Li, X. Access to Structurally Diverse Quinoline-Fused Heterocycles via Rhodium(III)-Catalyzed C–C/C–N Coupling of Bifunctional Substrates. Org. Lett. 2016, 18, 2812–2815. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, B. Tandem Rh(III)-Catalyzed C–H Amination/Annulation Reactions: Synthesis of Indoloquinoline Derivatives in Water. Org. Lett. 2016, 18, 2820–2823. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Liu, M.; Ye, Y.; Yin, G. Synthesis of 6-Substituted 6H-Indolo[2,3-b]quinolines from Isoindigos. Org. Lett. 2017, 19, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.J.; Petrow, V. Carbazoles, carbolines, and related compounds. Part III. Quinindoline derivatives. J. Chem. Soc. 1948, 1948, 922–924. [Google Scholar] [CrossRef]
- Kaczmarek, Ł.; Balicki, R.; Nantka-Namirski, P.; Peczyńska-Czoch, W.; Mordarski, M. Cancerostatics, VI. Synthesis and antineoplastic properties of some benzo-iso-alpha-carbolines. Arch. Pharm. 1988, 321, 463–467. [Google Scholar] [CrossRef]
- Timári, G.; Soós, T.; Hajós, G. A convenient synthesis of two new indoloquinoline alkaloids. Synlett 1997, 1067–1068. [Google Scholar] [CrossRef]
- Schmittel, M.; Steffen, J.-P.; Engels, B.; Lennartz, C.; Hanrath, M. Two Novel Thermal Biradical Cyclizations in Theory and Experiment: New Synthetic Routes to 6H-Indolo[2,3-b]quinolines and 2-Aminoquinolines from Enyne-Carbodiimides. Angew. Chem. Int. Ed. 1998, 37, 2371–2373. [Google Scholar] [CrossRef]
- Schmittel, M.; Rodríguez, D.; Steffen, J.-P. A Highly Efficient Triplet Analogue of a Thermal Biradical Cyclization-The Photochemical C2–C6 Cyclization of Enyne-Heteroallenes. Angew. Chem. Int. Ed. 2000, 39, 2152–2155. [Google Scholar] [CrossRef]
- Zhang, Q.; Shi, C.; Zhang, H.-R.; Wang, K.K. Synthesis of 6H-Indolo[2,3-b][1,6]naphthyridines and Related Compounds as the 5-Aza Analogues of Ellipticine Alkaloids. J. Org. Chem. 2000, 65, 7977–7983. [Google Scholar] [CrossRef]
- Nallapati, S.B.; Prasad, B.; Sreenivas, B.Y.; Sunke, R.; Poornachandra, Y.; Kumar, C.G.; Sridhar, B.; Shivashankar, S.; Mukkanti, K.; Pal, M. Apparent Carbon Monoxide Insertion via Double Isocyanide Incorporation during Palladium-Catalyzed Construction of Indoloquinoline Ring in a Single Pot: Synthesis of New Cytotoxic Agents. Adv. Synth. Catal. 2016, 358, 3387–3393. [Google Scholar] [CrossRef]
- Yan, Z.; Wan, C.; Wan, J.; Wang, Z. An efficient iron-promoted synthesis of 6H-indolo [2,3-b]quinolines and neocryptolepine derivatives. Org. Biomol. Chem. 2016, 14, 4405–4408. [Google Scholar] [CrossRef] [PubMed]
- Yeh, L.-H.; Wang, H.-K.; Pallikonda, G.; Ciou, Y.-L.; Hsieh, J.-C. Palladium-Catalyzed Dual Annulation: A Method for the Synthesis of Norneocryptolepine. Org. Lett. 2019, 21, 1730–1734. [Google Scholar] [CrossRef] [PubMed]
- Thorat, V.H.; Hsieh, J.-C.; Cheng, C.-H. Transition-Metal-Free Tandem Cyclization/N-Arylation Reaction: A Method to Access Biaryl Sultam Derivatives via a Diradical Pathway. Org. Lett. 2020, 22, 6623–6627. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.-C.; Su, H.-L. Synthesis of N-Heterocycles via the Transition-Metal-Catalyzed Tandem Addition/Cyclization of a Nitrile. Synthesis 2020, 52, 819–833. [Google Scholar] [CrossRef]
- Chen, M.-H.; Hsieh, J.-C.; Lee, Y.-H.; Cheng, C.-H. Controlled Synthesis of Enantioselective 1-Aminoindenes via Cobalt-Catalyzed [3+2] Annulation Reaction. ACS Catal. 2018, 8, 9364–9369. [Google Scholar] [CrossRef]
- Jhang, Y.-Y.; Fan-Chiang, T.-T.; Huang, J.-M.; Hsieh, J.-C. Copper-Catalyzed Annulation: A Method for the Systematic Synthesis of Phenanthridinium Bromide. Org. Lett. 2016, 18, 1154–1157. [Google Scholar] [CrossRef]
- Fan-Chiang, T.-T.; Wang, H.-K.; Hsieh, J.-C. Synthesis of Phenanthridine Skeletal Amaryllidaceae Alkaloids. Tetrahedron 2016, 72, 5640–5645. [Google Scholar] [CrossRef]
- Chen, W.-L.; Chen, C.-Y.; Chen, Y.-F.; Hsieh, J.-C. Hydride-Induced Anionic Cyclization: An Efficient Method for the Synthesis of 6-H-Phenanthridines via a Transition-Metal-Free Process. Org. Lett. 2015, 17, 1613–1616. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Hsieh, J.-C. Synthesis of Polysubstituted Phenanthridines via Ligand-Free Copper-Catalyzed Annulation. Org. Lett. 2014, 16, 4642–4645. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Wu, Y.-S.; Jhan, Y.-H.; Hsieh, J.-C. An Efficient Synthesis of (NH)-Phenanthridinones via Ligand-Free Copper-Catalyzed Annulation. Org. Chem. Front. 2014, 1, 253–257. [Google Scholar] [CrossRef]
- Yu, X.; Gao, L.; Jia, L.; Yamamoto, Y.; Bao, M. Synthesis of Quinazolin-4(3H)-ones via the Reaction of 2-Halobenzamides with Nitriles. J. Org. Chem. 2018, 83, 10352–10358. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1a–1t and 2a–2t are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | [Cu] (x) | Lewis Acid (y) | Solvent | Temp (°C) | Yield (%) b |
| 1 | Cu2O (20) | - | DMF | 120 | 36 |
| 2 | Cu2O (20) | - | DMSO | 120 | 29 |
| 3 | Cu2O (20) | - | NMP | 120 | 33 |
| 4 | Cu2O (20) | - | DMSO/NMP (4:1) | 120 | 39 |
| 5 | Cu2O (20) | - | DMF/NMP (4:1) | 120 | 50 |
| 6 | Cu2O (20) | - | DMF/NMP (9:1) | 120 | 65 |
| 7 | Cu2O (20) | - | DMF/NMP (9:1) | 150 | 58 |
| 8 | Cu2O (20) | - | DMF/NMP (9:1) | 100 | 37 |
| 9 c | Cu2O (20) | BF3·OEt2 (20) | DMF/NMP (9:1) | 120 | 88 (83) d |
| 10 e | Cu2O (10) | BF3·OEt2 (40) | DMF/NMP (9:1) | 120 | 90 (83) d |
| 11 | CuI (20) | BF3·OEt2 (40) | DMF/NMP (9:1) | 120 | 69 |
| 12 | Cu2O (10) | AlCl3 (40) | DMF/NMP (9:1) | 120 | 48 |
| 13 | Cu2O (10) | TiCl4 (40) | DMF/NMP (9:1) | 120 | 37 |
| 14 | Cu2O (10) | FeCl3 (40) | DMF/NMP (9:1) | 120 | trace |
| 15 | - | BF3·OEt2 (40) | DMF/NMP (9:1) | 120 | 11 |
 | ||||
|---|---|---|---|---|
| Entry | 1 | 2 | Condition | Yield (%) b |
| 1 | 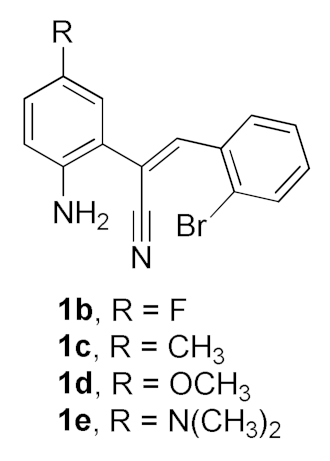 | 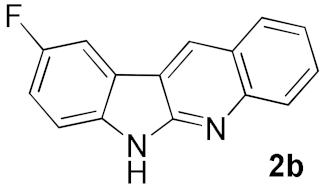 | B | 41 |
| 2 |  | A | 68 | |
| 3 | 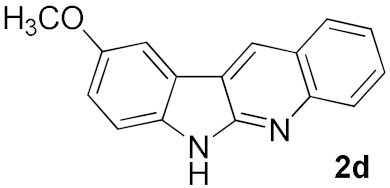 | A | 76 | |
| 4 | 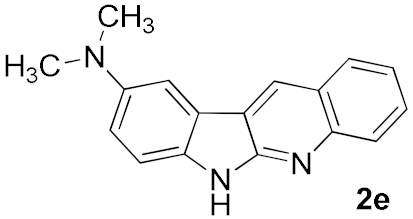 | A | 45 | |
| 5 | 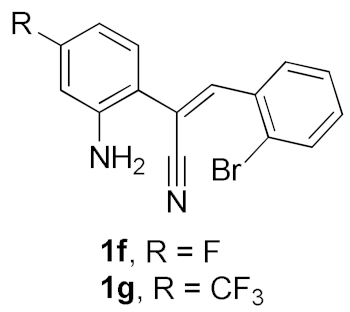 |  | B | 53 |
| 6 |  | B | 47 | |
| 7 | 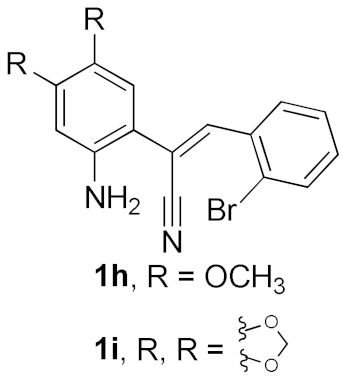 | 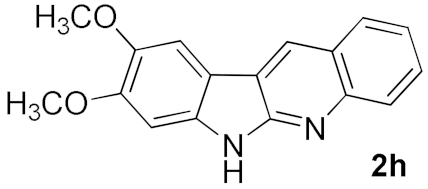 | A | 71 |
| 8 | 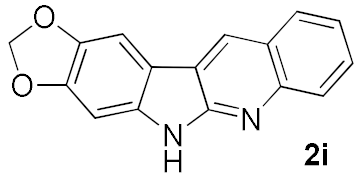 | A | 56 | |
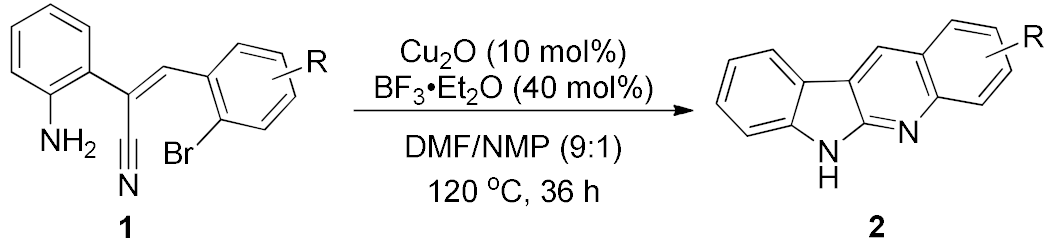 | |||
| Entry | 1 | 2 | Yield (%) b |
| 1 | 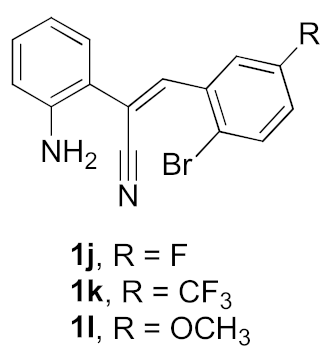 |  | 79 |
| 2 | 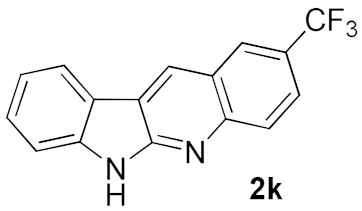 | 57 | |
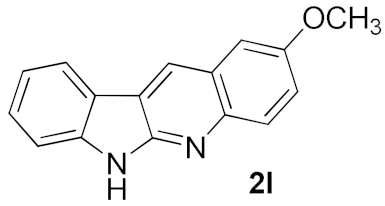 | 45 | ||
| 4 |  | 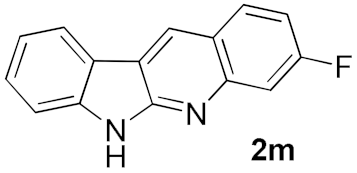 | 75 |
| 5 | 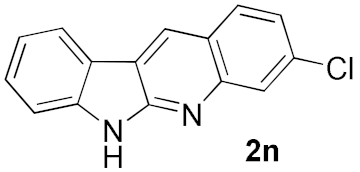 | 73 | |
| 6 |  | 69 | |
| 7 | 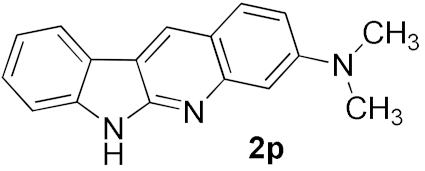 | 51 | |
| 8 | 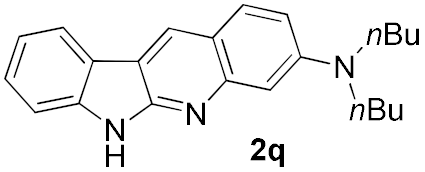 | 32 | |
| 9 | 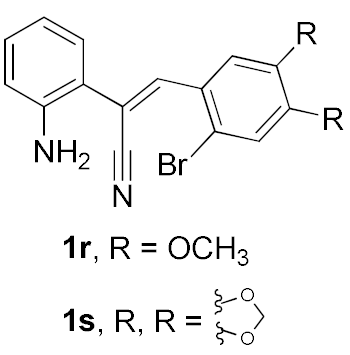 | 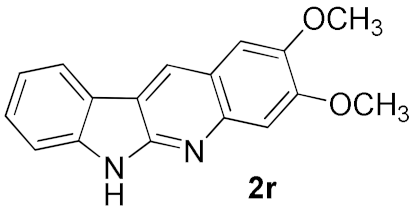 | 62 |
| 10 | 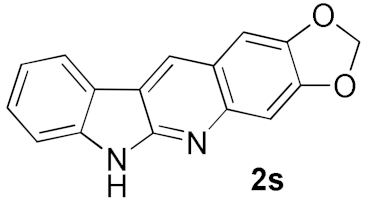 | 57 | |
| 11 | 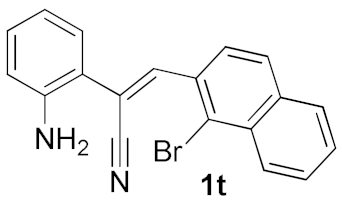 | 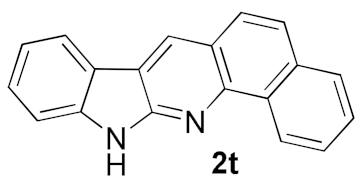 | 49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.-K.; Chio, Y.-L.; Pallikonda, G.; Wu, H.-L.; Su, H.-L.; Hsieh, J.-C. Copper-Catalyzed Dual Cyclization for the Synthesis of Quinindolines. Molecules 2020, 25, 5303. https://doi.org/10.3390/molecules25225303
Wang H-K, Chio Y-L, Pallikonda G, Wu H-L, Su H-L, Hsieh J-C. Copper-Catalyzed Dual Cyclization for the Synthesis of Quinindolines. Molecules. 2020; 25(22):5303. https://doi.org/10.3390/molecules25225303
Chicago/Turabian StyleWang, Hung-Kai, Yu-Lun Chio, Gangaram Pallikonda, Hsyueh-Liang Wu, Haw-Lih Su, and Jen-Chieh Hsieh. 2020. "Copper-Catalyzed Dual Cyclization for the Synthesis of Quinindolines" Molecules 25, no. 22: 5303. https://doi.org/10.3390/molecules25225303
APA StyleWang, H.-K., Chio, Y.-L., Pallikonda, G., Wu, H.-L., Su, H.-L., & Hsieh, J.-C. (2020). Copper-Catalyzed Dual Cyclization for the Synthesis of Quinindolines. Molecules, 25(22), 5303. https://doi.org/10.3390/molecules25225303