1. Introduction
Optical molecular imaging is a powerful tool to gain a better understanding of biological phenomena and the mechanisms of action of therapeutic agents [
1,
2]. However, it has certain limitations for clinical applications due to the limited penetrability of visible light. The highest penetration depth can be achieved when the fluorophore absorbs and emits in a region of the electromagnetic spectrum where biological tissues exhibit less absorption and less autofluorescence (therapeutic window: 650−900 nm) [
3]. In the search for such fluorescent probes, a series of 4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene (abbreviated as BODIPY) dyes could be regarded as perspective candidates. BODIPY dyes are a fluorescent family with unique characteristics such as intense absorption, high quantum yields, tunable emission wavelength, superior stability, and pH insensitivity, which may survive different in vivo environments [
4,
5]. The most prevalent applications of BODIPY dyes are related to different aspects of intracellular imaging of various organelles. Aza-BODIPY is a class of heteroatom-containing BODIPY analogues with near-infrared (NIR) absorption. It allows them to benefit from low autofluorescence of biomolecules, a smaller scattering background, and the applicability of low-cost excitation sources [
6].
Aza-BODIPY derivatives could be considered as promising compounds for the design of functional materials with predictable properties [
7]. The possibility of implementation of a wide range of mechanisms for the fine tuning of the spectral properties of individual compounds, conjugates, and related materials highlights the need for a rational design of new structures [
8,
9].
The introduction of certain substituents into the parent compound allows aimed modification of its physicochemical properties. In this study, we focused on several aza-BODIPY dyes with red-shifted fluorescence and the potential for the environmental response via charge transfer [
10], internal rotation [
11], and aggregation causing mechanisms [
8,
12].
To predict various physicochemical properties of compounds, it is necessary to determine their molecular structure. A large number of works are devoted to the stereochemistry of various classes of compounds. There are experimental data obtained by X-ray diffraction (XRD) analysis [
13], microwave spectroscopy [
14], gas electron diffraction method [
14], etc. Due to the development of computer technology, structural data obtained using quantum chemical (QC) calculations have become dramatically important. As the potential energy surface (PES) might have a large number of local minima, the study of the conformational multiformity of polyatomic molecules is often a difficult task [
15,
16]. At the same time, understanding the features of the molecular structure and the possibilities of low-energy doped changes, caused by light absorption and associative processes, can play a key role in the prediction of the mechanisms of directional changes in the optical properties of compounds for fine tuning and application in molecular sensorics [
17,
18].
There are many works dedicated to modeling new effective materials that have the potential for applied use, for example, as sensitizers for dye-sensitized solar cells (DSSC) [
19]. In an ever-growing field of chemistry, it is important to understand which metrics are appropriate for which properties. To design efficient sensitizers the following information can be used; the type and shape of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), and the energies of LUMO and HOMO relative to the conduction band of TiO
2 and the redox couple [
19]. To achieve the most efficient charge transfer the HOMO should be located on the donor moiety and the LUMO on the acceptor moiety [
19]. Wherein, the HOMO–LUMO gap can be used to study the photooxidative resistance [
20], kinetic stability [
21], and electronic properties [
19]. Still, the most important parameter, determined by the electronic structure of the compounds, is the position of the absorption and emission bands in the electronic spectra. The rational design of new structures for specific practical applications lies in the directional modification of the spectral band positions. At the same time, the number of fluorophores whose working wavelengths lie in the 400–600 nm region is large, in contrast to the compounds with spectral region close to the IR, which are necessary in medicine and molecular sensorics of biological systems. Taking that into account, the main objective of the screening process is to find the compound with the greatest bathochromic shift of absorption/emission compared to the unsubstituted aza-BODIPY.
In this paper, we studied the effect of introducing the following substituents into the different positions; phenyl, 2-thiophenyl, 2-furanyl, 3-pyridinyl, 4-pyridinyl, 2-pyridinyl, and ethyl groups (R
1–R
4,
Figure 1).
The molecule of the original aza-BODIPY possesses C
2v symmetry [
22]. Placing polyatomic substituents in the C
1/C
3/C
5/C
7 positions of aza-BODIPY leads to distortion of the planar structure of the heterocycle. In case of aza-BODIPY derivatives, it is necessary to study the conformational multiformity of a molecule in order to find an appropriate minimum on PES. In addition, the study of rotations of cyclic appendages is relevant as the dihedral angle between dipyrrin and substituent group will have a great influence on the HOMO-LUMO energy gap and absorption spectra [
23]. Therefore, the first aim of the present contribution is to study different conformers of aza-BODIPY derivatives formed as a result of the rotation of substituent groups R
1–R
4 (χ
1, χ
2, χ
3, χ
4).
In the case of molecules with two substituents (
A7,
B7,
C7,
D1–
D6), the main reasons for the conformational diversity are different mutual orientations of neighboring (R
1 and R
2 or R
3 and R
4) cyclic and ethyl groups (
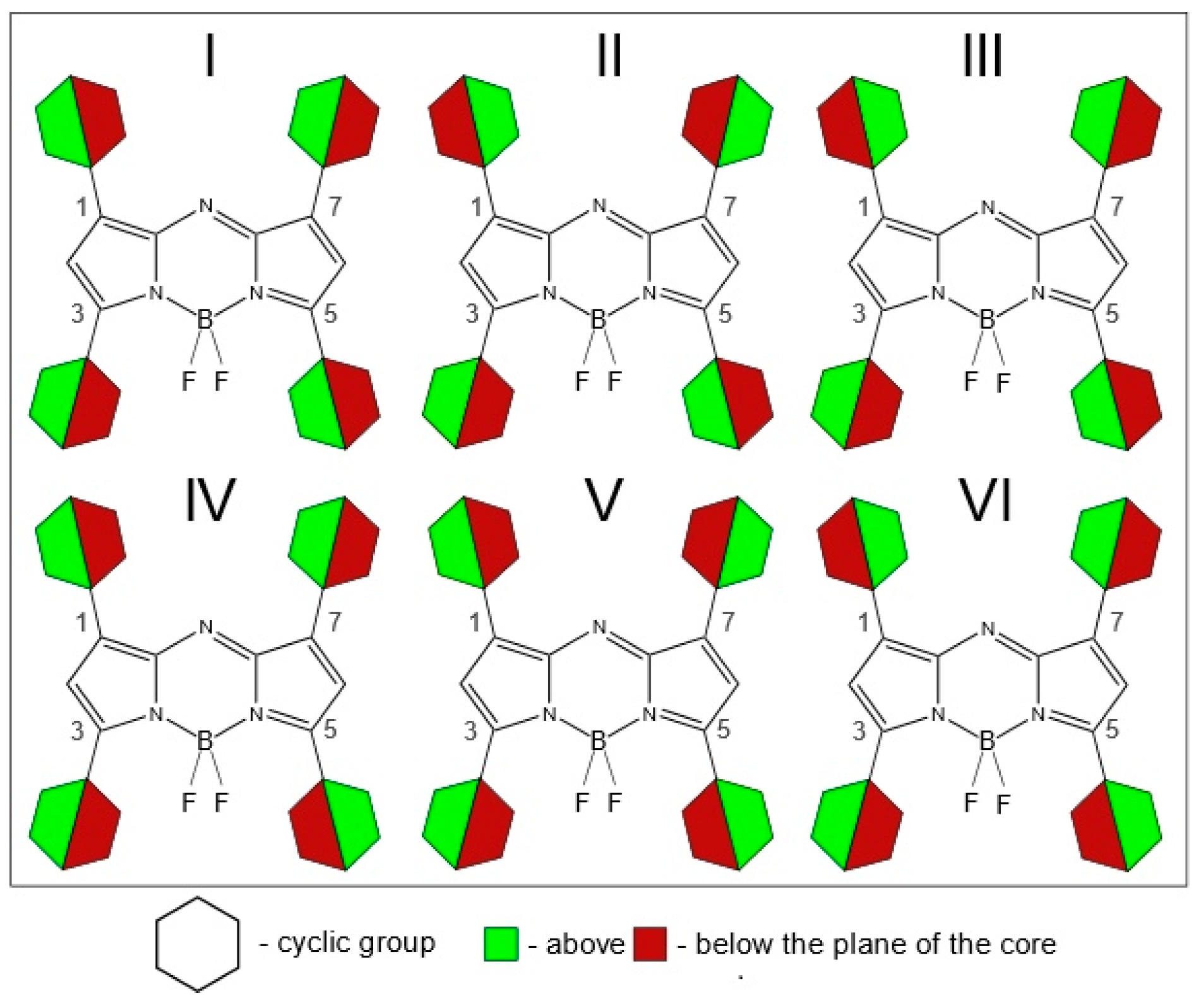
Figure 2). In case of the presence of four cyclic substituents, the number of possible conformers grows.
Figure 3 shows a graphical representation of possible conformer multiformity caused by the mutual arrangement of not only neighboring substituents but also long-distance substituents.
Figure 3 does not take into consideration possible locations of asymmetric cyclic groups with a heteroatom (
Figure 2c–e).
The efficient strategy for conformers searching is to use meta-dynamical simulations. Meta-dynamical simulations work by applying a biasing potential (here it is based on root mean square deviation (RMSD) between the current and reference structure in Cartesian coordinates) that ensures that the collective variables (i.e., all atom coordinates) do not converge to the same local minima often and can, therefore, find other, sometimes unexpected structures. More theoretical explanation is available in [
24]. The semiempirical method used was shown [
25,
26] to be in good agreement to density functional theory (DFT) when it comes to the qualitative results, i.e., which conformer is the lowest in energy. Thereby, meta-dynamical calculations were used as the first step in the search for the most energetically favorable conformers.
In order to learn more about the structural implications of different substituents and to reconfirm our conformational findings at the higher level, we have examined the process of rotation of substituent groups around C-C bonds. To minimize computer time, we decided to analyze conformers of A1–A7, B1–B7, C1–C7, and D1–D7 in several steps:
To study molecules obtained by introducing one substituent (1–6) into the aza-BODIPY (D7) in position C1;
To study molecules D1–D6 obtained by introducing two substituents (1–6) into the aza-BODIPY (D7) in positions C1/C7;
To study molecules obtained by introducing one substituent (A–C) into the aza-BODIPY (D7) in position C3;
To study molecules A7, B7, and C7 obtained by introducing two substituents (A–C) into the aza-BODIPY (D7) in positions C3/C5;
Based on the analysis (steps No. 2 and 4), to select the appropriate conformers of A1–A6, B1–B6, and C1–C6 for further investigation. Generally this step uses the assumption that the substituents in positions C1/C7 only slightly affect the rotations of the groups located in positions C3/C5 (and vice versa).
Due to the need for a large number of calculations in steps 1–4, we selected a combination of PBE functional and 6-31G* basis set. The choice of the PBE functional was made taking into account the small amount of CPU time needed for a single point calculation [
27]. It has been shown [
28] that the 6-31G* basis set is enough to estimate the geometries of the aza-BODIPY dyes correctly.
The second task of this work is to study the simulated electronic absorption and emission spectra. QC calculations of molecules in the solvated state (solvent is CH
2Cl
2) were performed using CAM-B3LYP functional [
29] and 6-31+G(d,p) basis set in the framework of conductor-like polarizable continuum model (CPCM) [
30,
31]. Electronic absorption spectra of aza-BODIPY derivatives were modeled on the time-dependent density-functional theory (TDDFT) calculation results. CAM-B3LYP was chosen for its good performance for both ground and excited electronic state geometries [
32]. The choice of CH
2Cl
2 as a solvent was made for easy comparison to the experimental data due to its prevalence in the aza-BODIPY synthesis procedure.
For calculating vibrationally-resolved absorption and emission spectra we decided to use the 6-31G(d) basis set to minimize the computer time.
2. Results and Discussion
2.1. Searching the Most Energetically Favorable Conformers: Meta-Dynamical Simulations
First, energetically favorable conformers for each molecule are shown in
Figures S1–S4 in the
Supplementary Materials. For
A1–
A7, the algorithm predicts the out-facing position (
Figure 2c) of 2-thiophenyl substituents in positions C
3/C
5 for the lowest conformers. It should be noted the advantage of five-membered rings—because of the difference in ring size, all of them have a larger distance between C
2/C
6 and C
2-sub-H hydrogens in the substituent allowing for a smaller dihedral angle.
For B1–B7, the lowest energy conformation corresponds to the in-plane or “side-facing” position of ethyl groups (R3, R4). The orientation of the R1,R2-substituents is the same as in the “A-series” counterparts. There is a sharp increase in the total amount of unique PES minima for each molecule compared to the aryl substituted dyes. Some of those states are quite low-lying. Those conformers are apparently thermally accessible due to the low barrier of rotation for ethyl groups.
For
C1–
C7, the algorithm predicts unusual mirror-symmetric orientation (
Figure 2b) of phenyl rings (R
3, R
4) in the main conformer.
2.2. Internal Rotation of One Cyclic Substituent around Bonds C-C
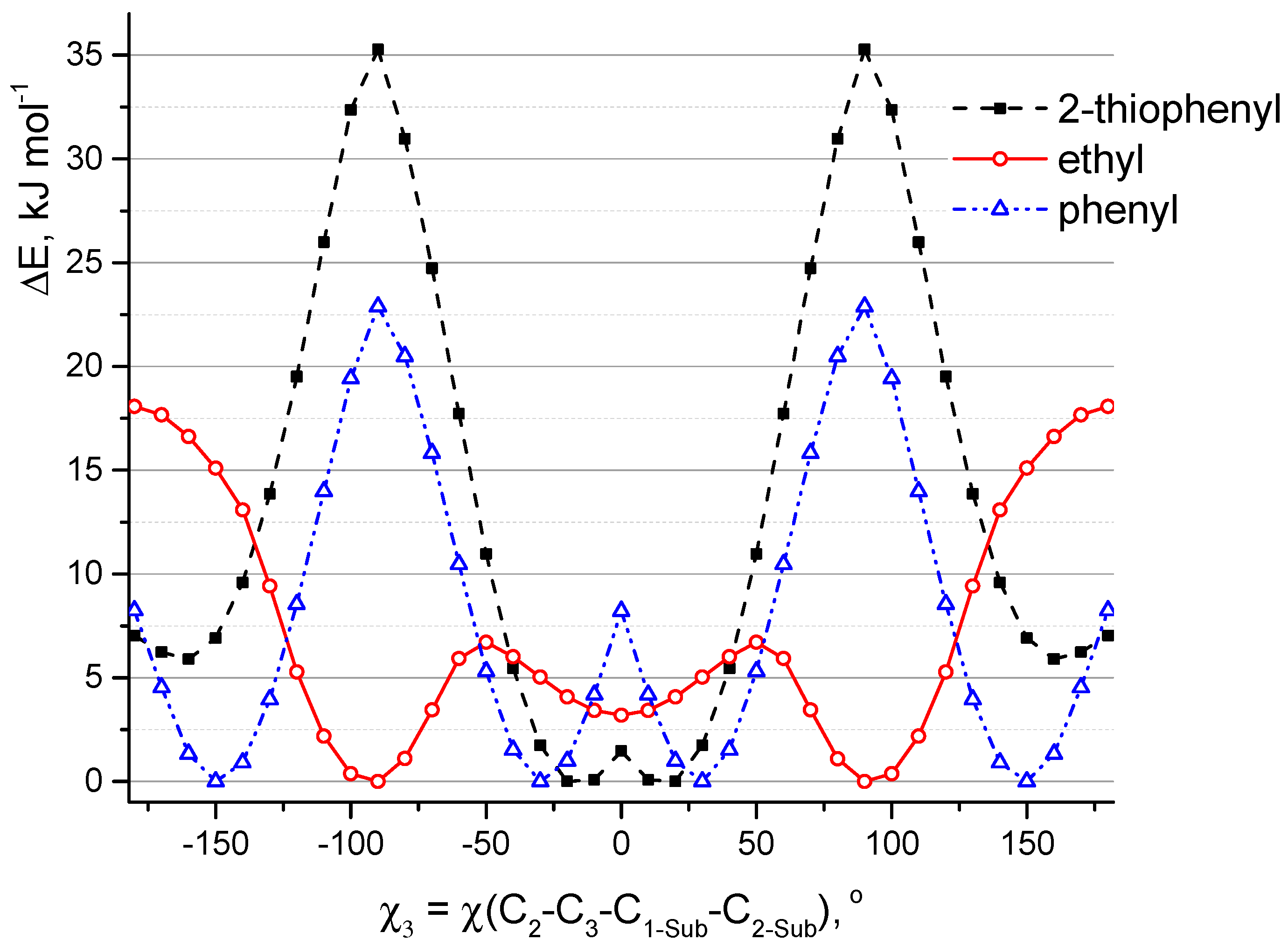
At the first step, QC relaxed PES scan calculations (PBE/6-31G(d)) of aza-BODIPY derivatives with only one substituent R
1 were carried out. Hydrogen atoms in these objects are in positions C
3, C
5, C
7 (
Figure 1). Rotation barriers of these groups increase in the sequence: 4-pyridinyl ≈ 3-pyridinyl ≈ phenyl < 2-thiophenyl ≈ 2-pyridinyl < 2-furanyl (
Figure 4). 2-Furanyl, 2-thiophenyl, and 2-pyridinyl groups are located in the plane of the molecule. Phenyl, 4-pyridinyl, and 3-pyridinyl groups are not located in the plane of the cycle (χ
1 = ∼18°), and therefore lead to a weak distortion of the aza-BODIPY skeleton. This is apparently explained by the steric repulsion associated with a short distance between H
2 and H
2-Sub: leaving the plane of the cycle leads to the distance r
e(H
2···H
2-Sub) increase by ∼0.11 Å for phenyl, 4-pyridinyl, 3-pyridinyl (2.173 → 2.283 Å, 2.217 → 2.315 Å, 2.199 → 2.333 Å, respectively). The location (
Figure 2) of substituents, where a heteroatom is oriented in the “outer” direction (χ
1 ≈ 0°) with respect to the center of the molecule, is energetically favorable for 2-furanyl, 3-pyridinyl, 2-pyridinyl groups.
Similarly, QC calculations of aza-BODIPY derivatives with only one substituent R
3 at position C
3 were carried out (
Figure 5). Positions C
1, C
5, and C
7 in these molecules are occupied by hydrogen atoms. In addition to phenyl, 2-thiophenyl also moves out of the plane of the heterocycle (χ
3 = ∼25°). The location of these groups in the plane of the molecule corresponds to the saddle point on PES. Substituents leave the plane via rotation as a result of the formation of a hydrogen bond between fluorine and hydrogen atoms. For the model in which sulfur atom of 2-thiophenyl is directed towards fluorine atoms, the planar structure is also not preferable. Apparently, this is due to the steric repulsion associated with a short distance r(H
2···H
2-Sub) = 2.192 Å (in planar conformation) and the presence of interaction between sulfur and fluorine atoms: according to QTAIM (Quantum Theory of Atoms in Molecules) analysis of non-flat conformation the value of electron density distribution function ρ(r) in appropriate bond critical point (BCP) is 0.015 and ∇ρ(r) is 0.057.
Ethyl group is found to be perpendicular to the plane of the heterocycle. The potential function of C
2H
5-rotation has three maxima corresponding to the values of the torsion angle χ
3 equal to ±50° and 180° (
Figure 5). The planar structure, in which ethyl group is bent toward the fluorine atoms (χ
3 = 180°), corresponds to a saddle point with relative energy ΔE = 18.1 kJ mol
−1. The structure characterized by the location of the ethyl group in the plane of the cycle (χ
3 = 0°) corresponds to a shallow minimum. This goes against meta-dynamical simulations at semi-empirical level (
Section 2.1,
Figure S2). In this regard, additional calculations CAM-B3LYP/6-31+G(d,p) (for isolated molecule and for the solvated state) were carried out. According to the different calculations (PBE/6-31G(d) and CAM-B3LYP/6-31+G(d,p) in the framework of PCM and without PCM) the low barrier height to internal rotation of the ethyl group ranges from 1 to 7 kJ mol
−1 and energy difference between conformers with perpendicular and planar location of ethyl group ranges from 0.3 to 4 kJ mol
−1, respectively. Apparently, calculations with 6-31G(d) overestimate the barrier of ethyl rotation, in addition, according to these calculations, the perpendicular arrangement of ethyl groups is more energetically favorable. Despite this, the subsequent calculations (PCM/CAM-B3LYP/6-31+G(d,p)) of the
B1–
B6 molecules showed that the structure with the arrangement of ethyl groups in the plane of the molecular skeleton is energetically more favorable by ∼1 kJ mol
−1.
2.3. Rotations of Two Cyclic Substituent Groups at Positions 1,7 in D1–D6 Molecules
Adding a second substituent to the aza-BODIPY core predictably increases the non-planarity of the molecule and leads to a higher movement of groups from the heterocycle plane. 2-Thiophenyl, 2-furanyl, and 2-pyridinyl groups in
D2,
D3, and
D6 molecules are located with χ
1 = ∼11°. The rotation value of phenyl, 3-pyridinyl, and 4-pyridinyl groups in
D1,
D4, and
D5 molecules increases by ~5° compared to molecules with one substituent group (
Table S1).
Even when two substituent groups are inserted into the molecule
D7, structural studies are significantly complicated by the need to study the conformational multiformity. Conformational multiformity of derivatives
D1–
D6 is caused for two reasons: (1) different arrangement of heteroatoms with respect to the center of the molecule (
Figure 2c–e), and (2) different mutual orientations of neighboring substituent groups relating to each other (cyclic substituents are located outside the plane of the molecule due to their rotation) (
Figure 2a,b).
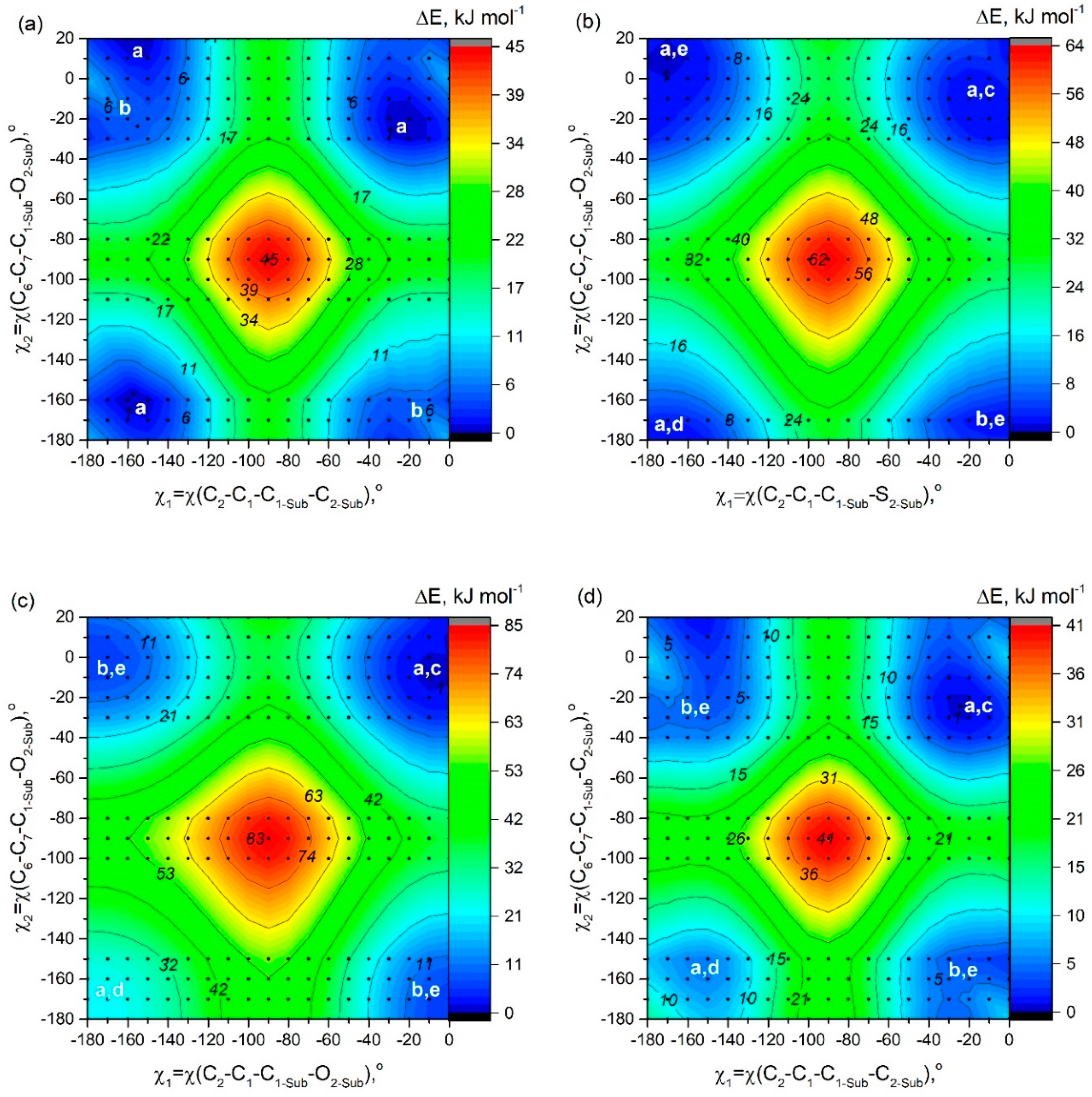
In order to study possible conformers and barriers to synchronous internal rotations of two substituent groups, we performed PES scan using PBE/6-31G(d) calculations. As shown in
Figure 6, in cases of
D1 and
D3–
D6 molecules, points of lowest energy correspond to the area with χ
1 = χ
2 = [−25°; −7°]. It matches the structure of C
2 symmetry according to which heteroatoms in substituents R
1, R
2 are oriented in an “outer” direction and neighboring cyclic groups are quasi-parallel (
Figure 2). The mirror-symmetric structure of C
s symmetry with χ
1 = −χ
2 = ∼−13° is slightly higher by ∼0.8–4.5 kJ mol
−1 (0.8 kJ mol
−1—for
D3; 4.4 kJ mol
−1—for
D4) and corresponds to a saddle point on the PES. In case of
D2 the structure with χ
1 = −169.7° and χ
2 = 11.0° (
Figure 2a,e) possesses the lowest energy, however, the energy of conformer of C
2 symmetry (
Figure 2a,c) is only slightly higher by 0.4 kJ mol
−1.
Barrier of internal rotation increases in the following order;
D5 (∼19 kJ mol
−1) <
D4 (∼20 kJ mol
−1) <
D1 (∼21 kJ mol
−1) <
D6 (∼28 kJ mol
−1) <
D2 (∼29 kJ mol
−1) <
D3 (∼39 kJ mol
−1). This order agrees with the information presented in
Figure 2 and is the reverse of the dihedral angle order—
D3 has lowest χ
1/χ
2 and
D5 has the highest. This seems to be caused by the additional conjugation between R
1, R
2, and aza-BODIPY core needs to be broken during rotation, which takes a lot of energy. One of the more relevant areas of fluorophore research is viscosity measurement and molecular rotors. 8-Substituted BODIPY dyes are already a staple of such research, and maybe our aza-BODIPY dyes could also become useful tools.
2.4. Rotation of Two Substituent Groups at Positions 3,5 in Molecules A7, B7, and C7
The PES shown in
Figure 7 has been constructed to study conformational multiformity of
A7,
B7, and
C7 coupled to rotations of 2-thiophenyl, ethyl, and phenyl groups. For
A7, the minimum of potential energy corresponds to the structure characterized by quasi-parallel location of thiophene groups (
Figure 2a) and an “outer” direction of sulfur atoms in substituents R
3 and R
4 (
Figure 2c). The structure of C
s symmetry (
Figure 2b,c) with another location of thiophene group is characterized by relative energy of 1.5 kJ mol
−1 and a very small barrier of no more than 3 kJ mol
−1—a fairly low-lying thermally accessible conformer.
In contradiction to A7, for C7 QC PES scan calculations match the mirror-symmetric equilibrium structure of Cs symmetry predicted by meta-dynamical simulations: one fluorine atom forms hydrogen bonds with two phenyl rings -C6H5···F···H5C6-. The structure (C2 symmetry) with quasi-parallel location of phenyl groups is higher in energy only by 0.7 kJ mol−1. Quasi-parallel arrangement of substituent groups leads to the formation of two separate contacts F···H5C6- with the participation of two fluorine atoms and two phenyl groups. The barrier of internal rotation for phenyl groups (C7) is about 8 kJ mol−1.
In case of
B7, the position of the minima corresponds to almost perpendicular orientation of the ethyl groups to the plane of the aza-BODIPY cycle. However, as described in
Section 2.2, the use CAM-B3LYP/6-31+G(d,p) calculations led to another conclusion—a conformer with ethyl groups in the plane of the molecule is energetically more favorable.
2.5. Molecules of Tetra-Substituted Aza-BODIPYs: Conformational Multiformity and Molecular Structure
It is important to mention that a typical RMSD for the relative energies of conformers for many DFT functionals is well above 1 kJ mol
−1: RMSD is equal to 0.61 kcal·mol
−1 for PBE calculations of relative energies of alkanes [
33]. This can lead to difficulties in changing from an initial calculation methodology to a final one. The results of calculations PBE/6-31G* and CAM-B3LYP/6-31+G(d,p) have several differences in the determination of the most favorable conformers (see for example,
Table S6), as well as in the determination of the type of critical points on the PES. The mirror-symmetric arrangements of phenyl rings (
Figure 2b) in
A1,
B1,
C1, and
D1 correspond to saddle points on PES by PBE/6-31G(d), whereas such structures correspond to minimum on PES by CAM-B3LYP/6-31+G(d,p). Besides, according to CAM-B3LYP calculations of
A1 structures conformer V of C
s symmetry is energetically equal to conformer I. These differences are principal, but at the same time it should be considered insignificant for subsequent spectrum modeling. For instance, the wavelengths, corresponding to transitions to the first 25 lowest excited states, calculated using structures of conformer I and V of
C1 molecule, differ by no more than 4 nm (the average difference is 1 nm).
For further investigations at the level of the theory of CAM-B3LYP/6-31+G(d,p), conformers were selected and tested in accordance with the mentioned above molecular structure analysis of
A7,
B7,
C7, and
D1–
D6. The conformer I (
Figure 3) was chosen for a further study of the molecules
A1–
A6: all four cyclic substituents are quasi-parallel to each other. According to CAM-B3LYP/6-31+G(d,p) calculations the energy differences between conformers I–II are 5–10 kJ mol
−1 for
A1–
A6 (
Table S6). It should be noted that for
A1–
A6,
B1–
B6, and
C1–
C6 oxygen and nitrogen atoms in 2-furanyl, 2-pyridinyl, 3-pyridinyl groups are oriented in an “outer” direction with respect to center of the molecules (
Figure 2c). Sulfur atoms are oriented in the same way in 2-thiophenyl group placed in 3, 5 positions; substitutes R
1 and R
2, and 2-thiophenyl groups are arranged as shown in a
Figure 2e. For further calculations of molecular structure and spectra for
C1–
C6 the conformer IV (
Figure 3) was used: one fluorine atom forms two hydrogen-bonding interactions with two H
2-Sub atoms of C
6H
5-groups. For
B1–
B6 and
C1–
C6 molecules, substituents R
1 and R
2 are located quasi-parallel to each other (
Figure 2a).
The different arrangement of the ethyl groups (
Figure 2f,g) does not introduce large energy changes. According to the results of CAM-B3LYP/6-31+G(d,p) calculations, the energy differences between C
s and C
2 conformers (
Figure 2f,g) of
B7 do not exceed 0.03 kJ mol
−1. However, both of these conformers lie on the PES higher by ∼0.3 kJ mol
−1 than the conformer h (
Figure 2h), in which the ethyl groups are located in a plane of the molecule and are oriented by “outer” direction with respect to center of molecule.
The optimized structures from QC calculations at CAM-B3LYP/6-31+G(d,p) level are given in
Supplementary Materials. In
Tables S1–S4, the calculated structural parameters of aza-BODIPY derivatives of the considered molecules are compared to each other.
2.6. Substitution Effect in Tetra-Substituted Aza-BODIPYs
The introduction of substituents R
1 and R
2 into the parent compound
D7 in positions C
1/C
7 leads to elongation of the bond length of the C
1–C
8a by ∼0.013 Å (
Table S4), while the substitution of hydrogen atoms by groups R
3 and R
4 in positions 3 and 5 leads to some shortening of these bonds (
Tables S1–S3). Bond lengths of the C
2–C
3 are elongated with the introduction of substituents R
3, R
4 and shortened with the introduction of the substituents R
1, R
2. The most significant changes in the internuclear distance with the introduction of substituents are observed for the internuclear distances N
4a-C
3 and C
1–C
8a, on average 0.018 Å and 0.011 Å for
A1–
A6,
B1–
B6, and
C1–
C6. Geometry of the central 6-membered ring is only slightly affected by the introduction of substituents: the maximum change in the distances is 0.006 Å compared to analogous values for
D7.
Compounds, which are equipped with aromatic rings in different positions of the dipyrrin backbone have extended π-delocalization. Apparently, the greatest delocalization is characteristic to the molecules containing 2-thiophenyl and 2-furanyl substituents. This is confirmed by slightly increased values of Wiberg bond indexes (WI) obtained by natural atomic orbital analysis: for molecules
A1–
A7,
A2–
D2, and
A3–
D3—WI(C
3-C
thiophenyl) ≈ WI(C
3-C
furanyl) ≈ 1.12, whereas for 2-methylthiophene and 2-methylfuran—WI(C(CH
3)-C(ring)) = 1.04. Moreover, the dihedral angle for the 2-thiophenyl and 2-furanyl groups is smaller than for other groups; it can also lead to π-delocalization. It is known that the aza-BODIPY is seen to exhibit less aromatic character than the BODIPY [
34].
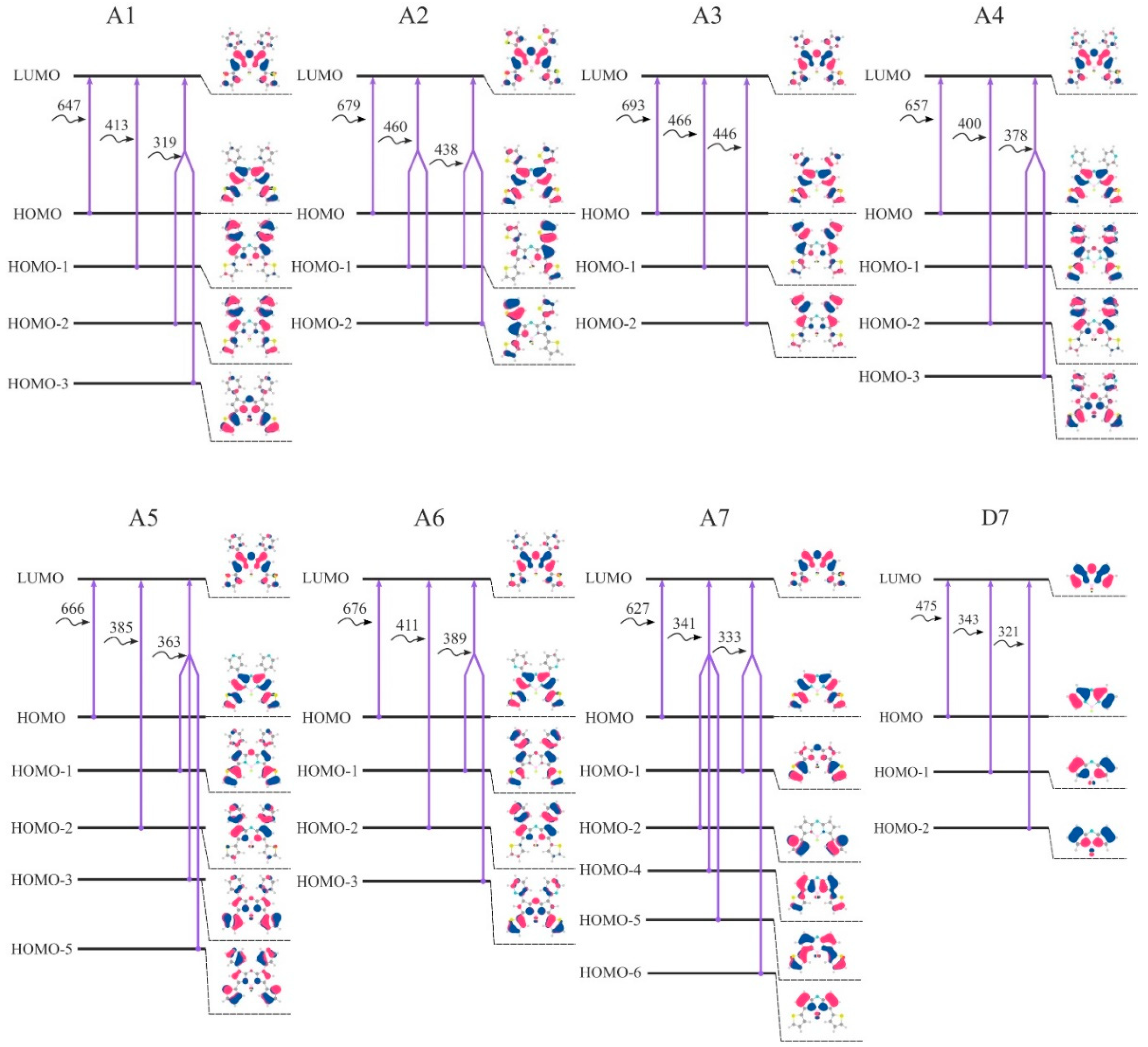
A HOMO–LUMO gap is basically the energy that must be fed to the molecule to kick it from the ground state to an excited state [
19]. A smaller energy gap leads to a redshift in the absorption spectrum. Thereby, studying a HOMO–LUMO gap is of great importance during the design of effective dyes. Introduction of the considered aryl substituents increases the energy of the HOMO orbitals, lowers the energy of the LUMO and reduces the HOMO-LUMO gap. The distribution patterns of frontier canonical MOs of the molecules, studied in this work, are shown in
Figure 8 and
Figures S5–S7. The HOMO of unsubstituted
D7 is localized on the carbon atoms. The HOMOs of aza-BODIPY derivatives display a pronounced resemblance with a strong contribution of the core (the contributions to the HOMOs exceeds 50%) and additionally on aryl moieties (
Figure 8 and
Table S15). Especially for ethyl-substituted, the predicted HOMOs and LUMOs are predominantly localized at the aza-BODIPY’s core. For
B1–
B7, HOMO-LUMO-gaps decrease slightly compared to
A1–
A7 and
C1–
C7. The smallest HOMO–LUMO energy gaps are found for
A3 and
A2 molecules (
Figure 9). It is noteworthy that the molecules with 2-thiophenyl and 2-furanyl groups are characterized by the biggest contribution of substituent groups to the frontier MOs: for
A3—43% and 31% to HOMO and LUMO, respectively.
Table S15 shows the calculated characteristics of contributions of different parts of molecules to HOMO and LUMO.
The HOMO–LUMO gap changes if the position of aryl groups varies from 3 and 5 positions to 1 and 7 positions. In case of 2-thiophenyl groups it is found to be 3.90 and 4.20 eV for
A7 and
D2, respectively; in case of phenyl groups—4.27 and 4.54 eV for
C7 and
D1, respectively. It is important to note that the pyridyl group, depending on the location of the nitrogen atom, has a different effect on the energetic properties: the HOMO-LUMO gap for 2-pyridinyl substituted aza-BODIPYs (series
A6–
D6) is smaller than the corresponding molecules from the series
A4–
D4 and
A5–
D5 (
Figure 9).
2.7. Spectral Properties: TDDFT Electronic Absorption Spectra
Calculated (CAM-B3LYP/6-31+G(d,p)) TDDFT electronic absorption spectra of the studied substances are presented in
Figure 10. The calculated oscillator strengths for the lowest allowed excited states along with their composition are given in
Table S16. The comparison of the calculated spectra of aza-BODIPY dyes demonstrates a strong influence of the substituents. A strong bathochromic shift occurs with a change of hydrogen atoms by aryl groups (
Figure 10). The absorption bands in the theoretical spectra are underestimated in comparison with the experimental data for
A1–
A2,
C1–
C2 [
35,
36,
37,
38,
39] by ∼64 nm. The observed discrepancy is due to the high multi-reference character of the dipyrrin complexes, as was previously shown by Brown and Momeni [
40]. They have found that any TDDFT functional will systematically underestimate exicitation energies by ~0.2–0.5 eV.
The first electronic transition is the most interesting one, as for aza-BODIPY dyes it tends to be the strongest one. If one takes a look at
Figure 8, where the first three transitions for each considered dye are summed up in terms of canonical orbitals contributing to each spectral line, it becomes apparent that the first absorption peak is always due mainly to the HOMO-LUMO transition. Nevertheless, the energy of the first and all other transitions is varying quite significantly.
Having examined the orbitals of B1–B6, it becomes apparent that 3,5-ethyl substituents have a minimal effect on the electronic transitions beyond the first one. Even for the first one, B1–B6 consistently show the lowest red shift of absorption in each respective group. It is clear that the aryl substituents have the ability to conjugate their π-electron system, if one is present, with the π-electron system of the dipyrrin. As such, to get the maximum bathochromic shift it is necessary to use aromatic substituents.
Another clear trend emerges between the
A1–
A7 and respective
C1–
C7 compounds.
A1–
A7 dyes consistently have a bigger red-shift of the first peak of absorption compared to the
C1–
C7 dyes. This is in line with the previously reported experimental data on the subset of compounds considered here [
41]. This is also confirmed by the higher contribution of substituent-localized orbitals to the HOMO/LUMO of
A1–
A7 compounds compared to
C1–
C7 compounds.
It is interesting to note that when it comes to the C1/C7 substituents, phenyl ring turns out to perform the worst compared to all considered heterocycles outperforming only the hydrogen-substituted A7, B7, or C7 dyes. This is very promising for the aza-BODIPY chemistry and opens up a lot of opportunities for different heterocycle-substituted dyes and their more functionalized derivatives.
The biggest red shift of the first electronic transition is achieved by A3—2-furanyl substituted dye. In fact, 2-furanyl-substituted dyes are the best in their respective series. Their orbital compositions are very close to the closest analogues (such as A2), but the lower dihedral angle between the dipyrrin and furanyl fragments allows for a stronger π-electron conjugation. This correlation holds for all considered substituents.
In this regard, the case of A4–A6, B4–B6, and C4–C6 dyes is of great interest. Here, a clear progression of red shift of first absorption peak is present, namely, A6 > A5 > A4, B6 > B5 > B4, and C6 > C5 > C4. HOMO and LUMO compositions between all of them are quite similar. However, the dihedral angle between 2-pyridinyl and dipyrrin is much less than between 3- and 4-pyridinyl and dipyrrins. The reason why A5–C5 dyes possess slightly higher bathochromic shift than A4–C4 dyes, however, cannot be explained so that the difference in the relevant angle is minimal. The reason for the shift is due to the fact that in 3-pyridinyl compound HOMO is localized on a nitrogen atom instead of a carbon one. This opens up a lot of opportunities for multiple-heteroatom heterocycles, such as pyrimidine, to become potentially valuable aza-BODIPY substituents.
When it comes to the second transition and beyond, the picture becomes less clear. In the CI singles determinant expansion, each determinant, and, by extension, each virtual orbital will have an associated weight; most of the time, it will be small but not zero. Here, we chose to only show contributions from determinants that make up at least 5% of all contributions. More often than not only one determinant has a significant contribution with additional ones giving 7–16%. However, this is not always the case.
Notable exceptions include the fourth and the fifth transitions of C1–C7 dyes, as well as the fourth transitions of almost all of B1–B7 dyes and the fifth transitions of A1–A7 dyes. One interesting derivative is B7, with its third, fourth and fifth transitions having notable multi-determinant character. A direct correlation between the value of red-shift and the “complexity” of the relevant transition does not appear to exist. Similarly, there is no clear correlation between structural parameters, such as dihedral angles, and transitions multi-determinant character.
2.8. Vibrationally Resolved Absorption and Emission Spectra
To better understand the relationship between spectral properties and structures of examined compounds, we have computed the vibrationally resolved absorption and emission spectra. It should be noted that in order to minimize CPU time, the calculations were carried out with a smaller basis set (6-31G(d)) compared to TDDFT calculations described in the
Section 2.7.
Obtained data for molecules
A1–
A7 is illustrated in
Figure 11 and
Figures S8–S10 for the other ones. Herein, a notable clarification is due. Because of the non-planarity of the lowest conformers for
B2,
D2 compounds in the ground state and their planarity in the first excited state, the FC (and even FCHT) approximation breaks down. This shortcoming of “constant dipole” or “first order change only” approximations has been previously noted [
42]. New approaches, such as Nuclear Electronic Orbital TDDFT (NEO-TDDFT) [
43] are being developed, but as of right now we have chosen to instead focus on other conformations for affected compounds. For those molecules, instead of a preferred position “e” for 2-thiophenyl groups (
Figure 2), position “c” was taken instead.
For the four previously obtained compounds (
A1,
A2, and
C2) calculated spectra are in reasonable agreement with experiment (
Table S17) [
41]. Although the characteristic shoulder is present for all compounds, its intensity is slightly overestimated. For substituted derivatives (all except
D7) the separation between the main band and the shoulder is overestimated. Those issues are inevitable due to the approximate nature of exchange-correlation functionals which results in an underestimation of vibrational frequencies by ~5–10% for ground state (S0) and even more for excited state (S1).
Optimization of excited state geometries allows including some spurious orbital relaxation. It usually means that for excitation energies ΔE
adia < ΔE
vert. However, all of the trends observed for vertical excitations (
Figure 11) still hold for the adiabatic ones, i.e., for aryl substituted compounds, such as
A1–
A6 and
C1–
C6, there is still a notable red shift of both absorption and fluorescence compared to others. It is clear then that the additional conjugation of the dipyrrin π-electronic system with aromatic substituents is necessary to achieve near-IR absorption and emission, with positions C
3/C
5 being more important than C
1/C
7. The largest bathochromic shift is observed for compound
A3 with the absorption maximum being around 788 nm and emission maximum around 800 nm. A3 is followed closely by
A2, which has maxima of 781 and 795 for absorption and emission respectively. It is interesting that
A6 displays higher red shift than
A5 and
A4. This correlates nicely with the dihedral angle between the substituents and the dipyrrin. Indeed, the lower angle can allow for the increased conjugation and higher red-shift.
Another nod to the importance of C3/C5 substitution of Aza-BODIPY dyes is the D1–D6 series. Many of those compounds have vibronic absorption peaks at wavelengths lower than 600 nm even though they have aromatic substituents. In contrast, A7 and C7, which also have only two substituents but in positions C3 and C5, show absorption maxima at 703 nm and 622 nm, respectively. Introduction of ethyl groups into the C3/C5 (B1–B7) does not change the position of the peaks significantly.
A useful feature of the vibronic spectra is the ability to analyze which normal modes are coupled to the electronic excitations. A rational examination can help to explain the observed line shapes and design a new set of molecules that better suit a given application. To that end, we have examined the primary molecular vibrations, which affect the spectra of Aza-BODIPY dyes. This task is difficult, as many of the vibrational levels give small contributions.
Figures S11–S14 show the scaled displacement vectors for some of the main molecular vibrations affecting the line shapes of absorption/emission in given series of Aza-BODIPY derivatives. The main absorption and emission bands involve 1–2 high intensity transitions—rotations of C
3/C
5 and C
1/C
7 substituents around the substituent-dipyrrin bond. For
D7, which has no substituents, the vibronic structure is completely different—main contribution comes from out-of-plane dipyrrin distortions. Quite interesting is the fact that for pyridine-substituted molecules, there is a higher number of average-to-low intensity vibronic transitions making it complicated to analyze, so much so for
A4–
A6 that there is essentially no dominant contributions. For shoulder in spectra, the most intense transition involves in-plane stretching of R3, R4-substituent C-H bonds (C
2-sub-H, C
3-sub-H, etc.). In case of
D1–
D7, which do not have any C
3/C
5 substituents, the shoulder corresponds to the in-plane stretching of dipyrrin C
2-C
3, C
3-C
4 and C
2-H, C
3-H bonds.
Based on the calculated vibronic spectra, we have compared Stokes shifts of studied compounds. Stokes shift is the difference between the wavelengths of absorption and emission peaks. Here, to simulate our line shapes, we have used Gaussian broadening with HWHM of 250 cm−1. If we chose to use different half widths at half maximums (HWHM), the Stokes shift values would change too. However, by using the same HWHM for all compounds we can obtain qualitative information about the Stokes shift trends.
Introduction of ethyl substituents into the C
3/C
5 positions generally affects Stokes shift very little as
B7 has very close line shape to
D7. Introduction of aromatic substituents into those positions increases the calculated shift—a little bit for the 2-thiophenyls (
A7) and more so for the phenyls (
C7). When it comes to the C
1/C
7 substituents, all molecules display similar amount of Stokes shift given the same C
3/C
5 group. 2-pyridine-substituted molecules (
A6,
B6,
C6, and
D6) have the smallest, if only barely, increase in their respective rows (
Figure 11 and
Figures S8–S10). There exists a subtle correlation between the dihedral angle between the substituent and the dipyrrin and the amount of calculated Stokes shift—the more planar the molecule overall the less shift there is. This, however, is not due to the change in the vibrational levels of compounds. If we compare 0
0 → 1
5 (here electronic
vibrational notation is used) transition contributions between
A4,
A5, and
A6 we obtain values of 3.5, 3.5 and 3.8 a.u. Rather than this, a significantly increased 0
0 → 1
0, usually referred to as the 0-0, transition contribution (6.5, 6.4, and 8.1 a.u., respectively) results in a lower shift. The higher end of the calculated shifts (~20 nm) is not enough for some specific uses in medicine and biochemistry that require cell imaging with high signal-to-noise ratio.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}