
Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
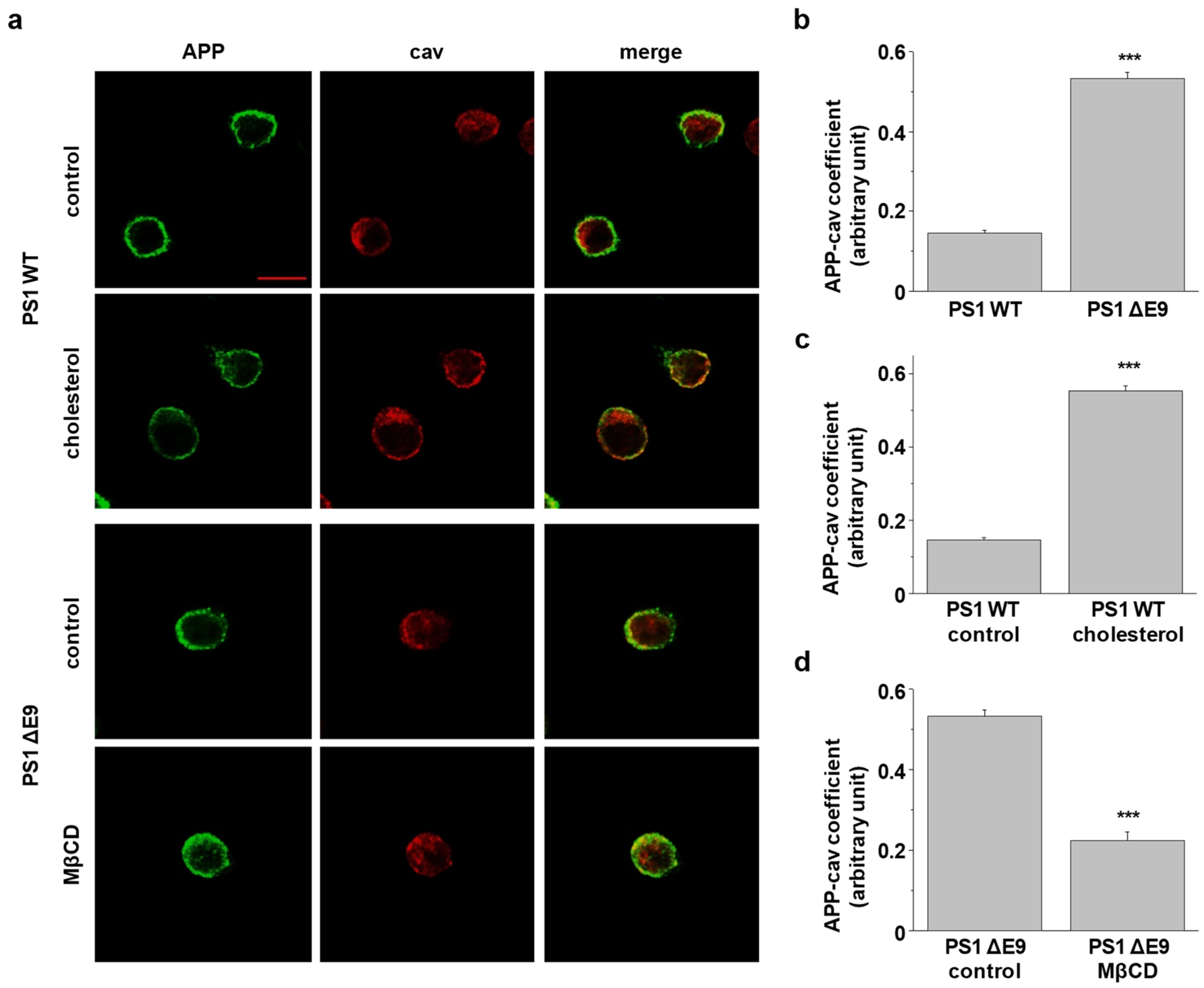
2.1. Cholesterol Levels Regulate the Localization of Cell Surface APP in Lipid Raft Microdomains
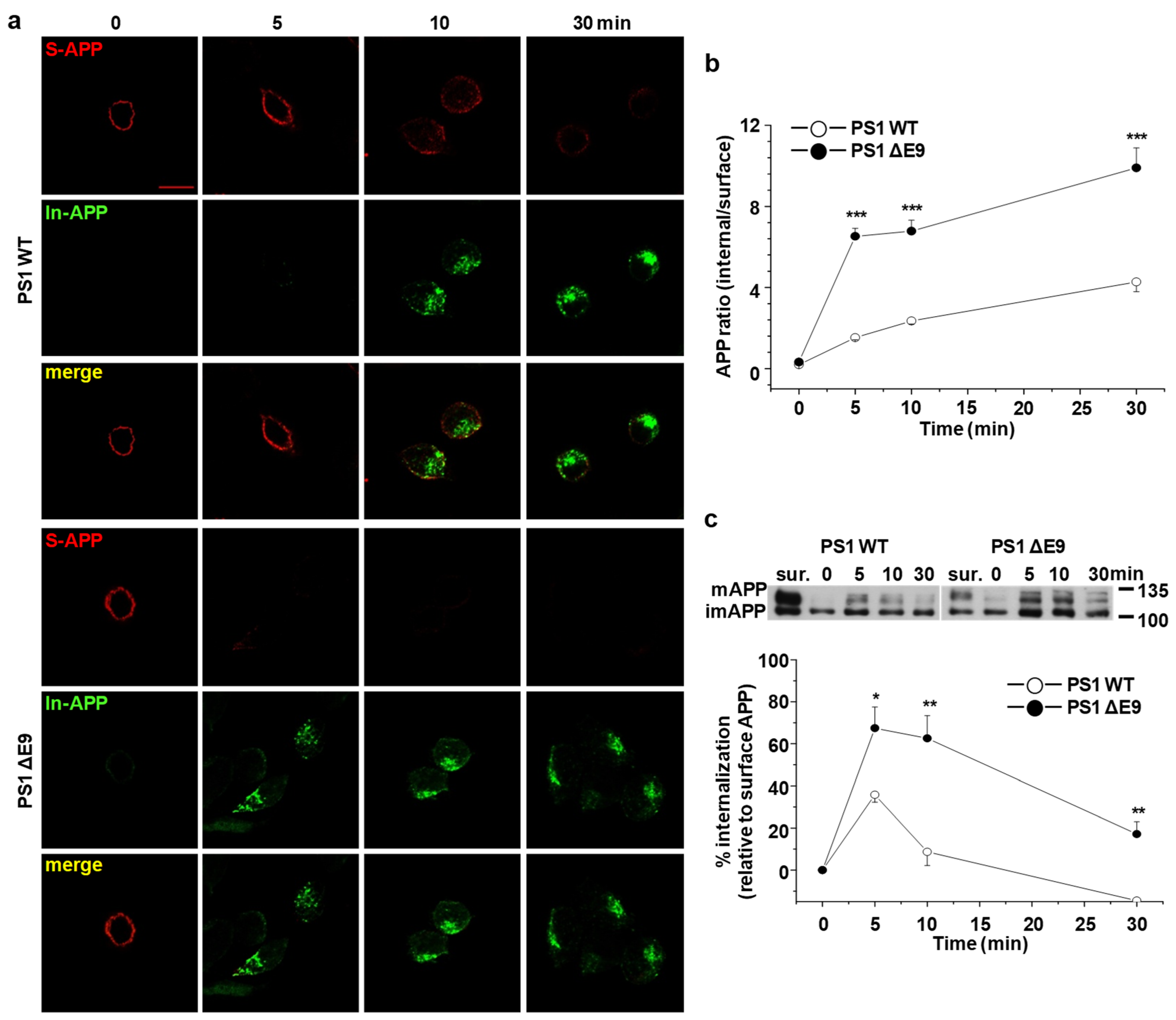
2.2. Endocytosis Rate of APP Is Increased in CHO PS1 ΔE9 Cells
2.3. Increased Cholesterol Level Does Not Affect the Endocytosis Rate of Transferrin
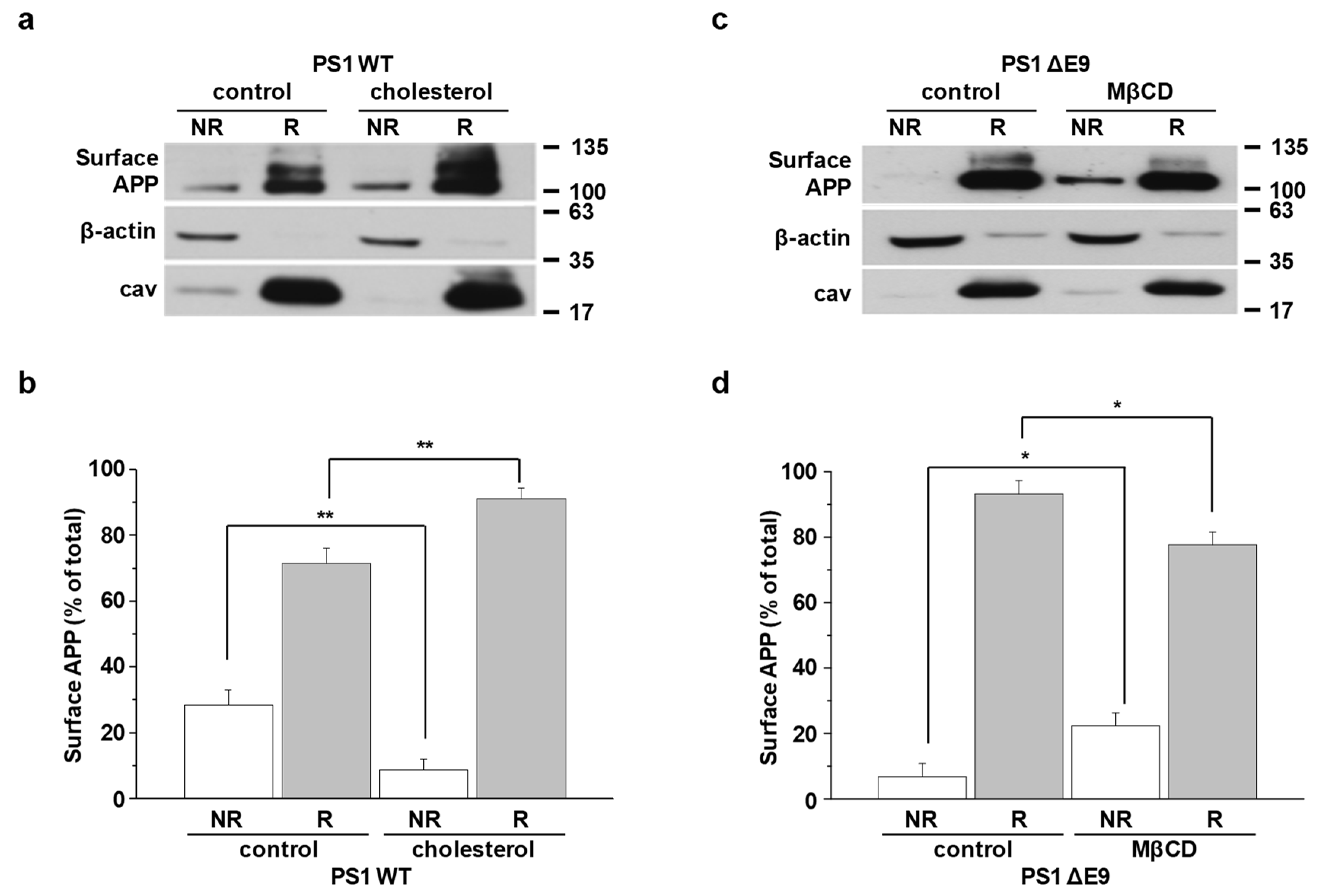
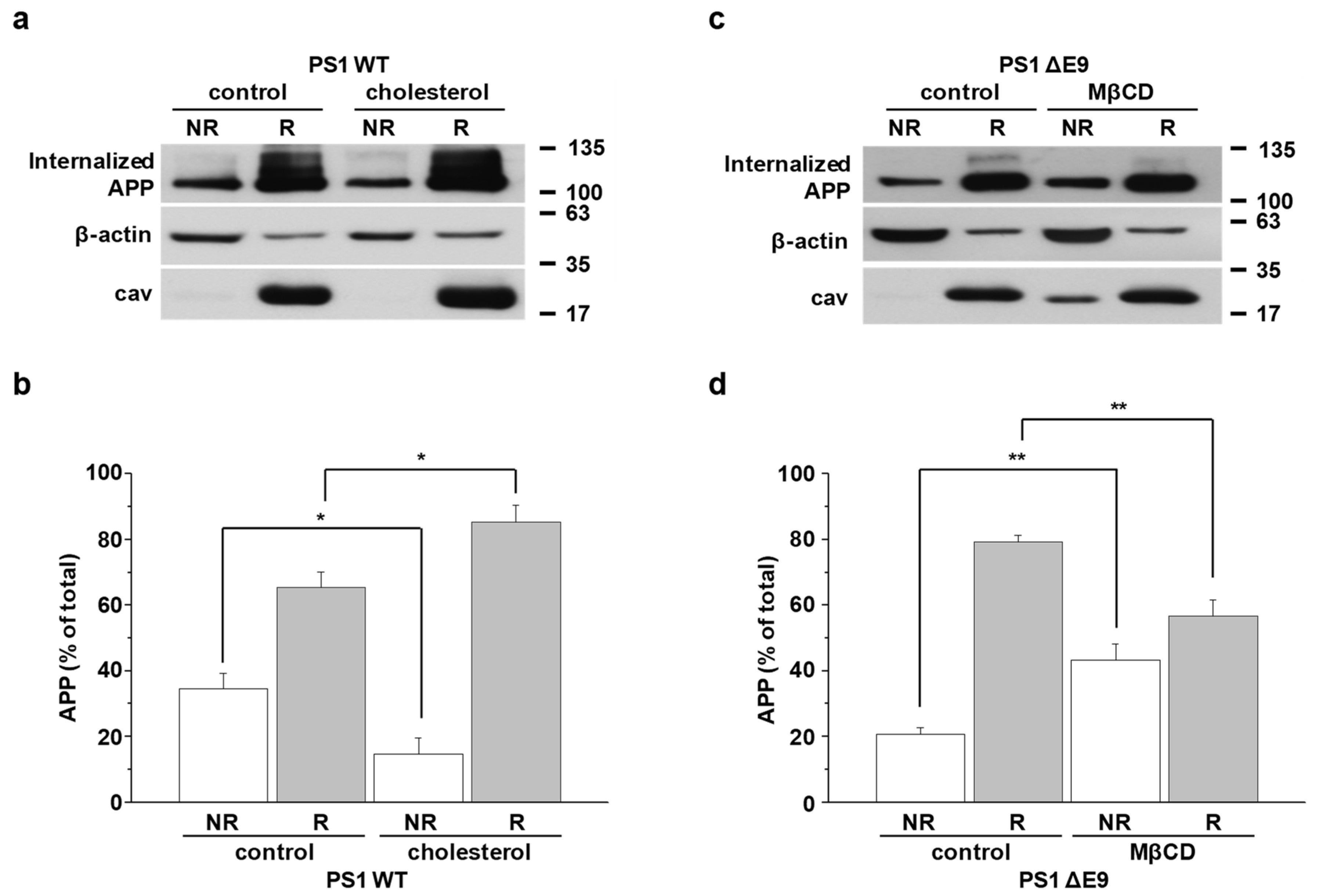
2.4. Cholesterol Levels Regulate Endocytosis Rates of APP
2.5. APP Localization in Lipid Rafts Is Regulated by Cholesterol Levels
2.6. APP Preferentially Internalizes from Lipid Rafts
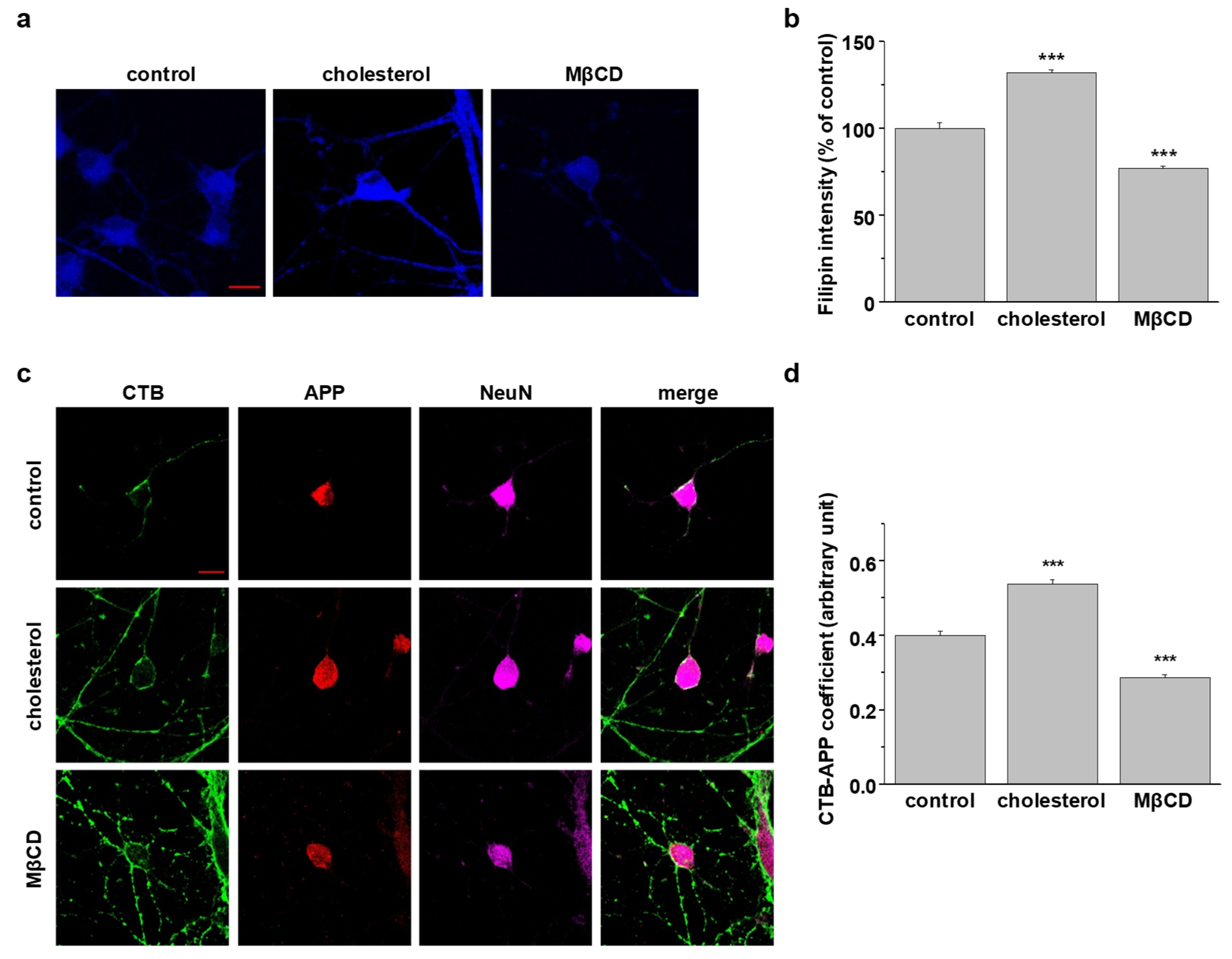
2.7. Localization of Endogenous Neuronal APP in Lipid Rafts at the Plasma Membrane Is Determined by Cellular Cholesterol Levels
2.8. Disruption of Lipid Raft Microdomains Attenuates APP Endocytosis in SH-SY5Y-APP/BACE1 Cells
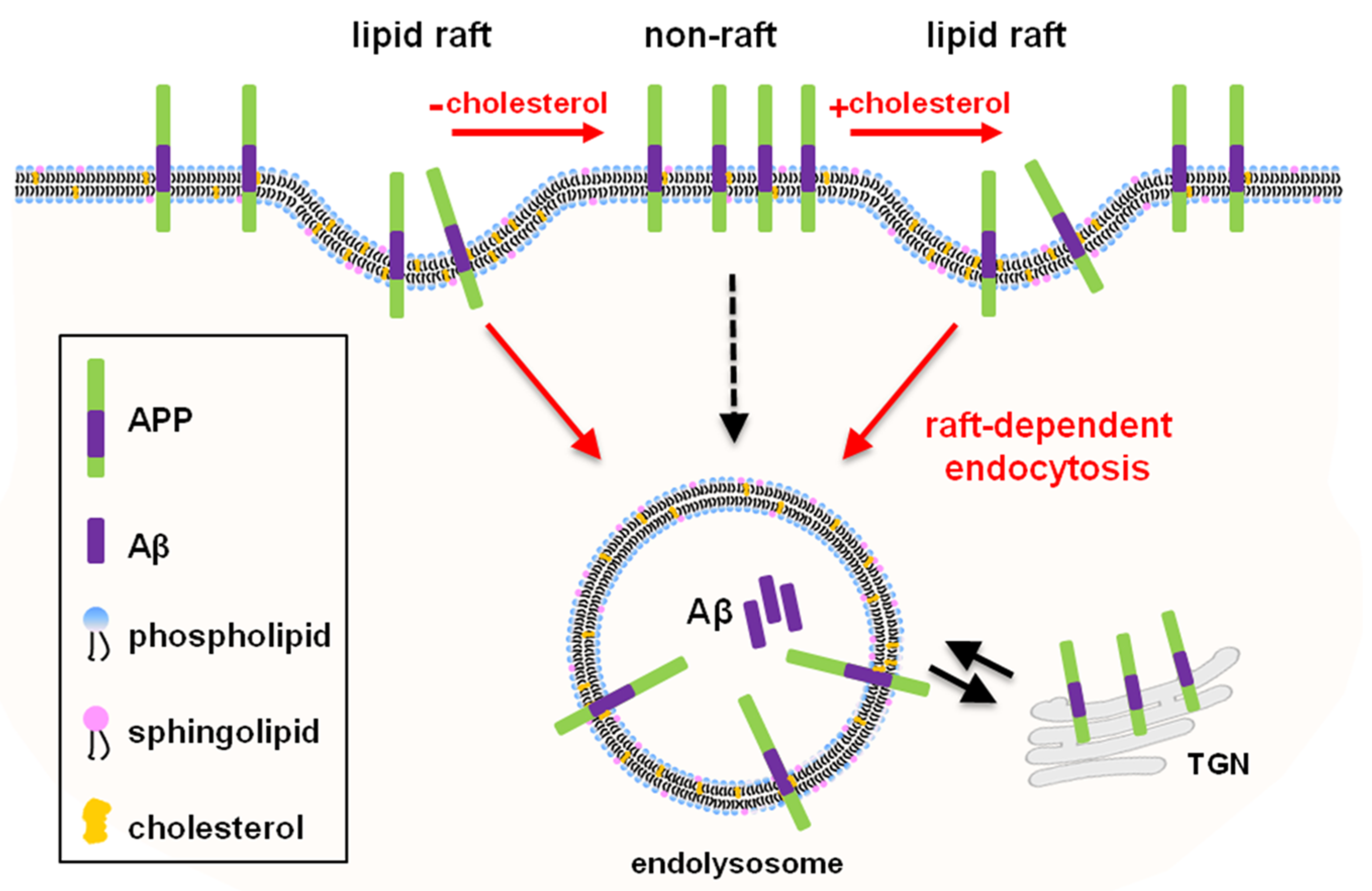
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Experimental Treatments
4.2. Rat Primary Hippocampal Neuron Culture
4.3. Filipin Staining
4.4. Colocalization Experiments
4.5. Primary Antibody Uptake Assay
4.6. Transferrin Uptake
4.7. Reversible Biotinylation Assay
- A: the levels of total surface APP (sulfo-NHS-biotin incubated, nonreduced, and nonquenched).
- B: control (0 min; sulfo-NHS-SS-biotin incubated, kept at 4 °C).
- C: internalized APP at 37 °C (sulfo-NHS-SS-biotin incubated and internalized 5, 10, or 30 min)
- The (C-B)/A ratio represented the rate of internalized APP during each time point.
4.8. Western Blotting
4.9. The Localization of Surface APP in Lipid Raft Microdomains
4.10. Rate of Endocytosis of Surface APP in Lipid Raft Microdomains
4.11. Localization of APP in Early Endosomes
4.12. Aβ42 Peptide ELISA Assay
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Probst, A.; Langui, D.; Ulrich, J. Alzheimer’s disease: A description of the structural lesions. Brain Pathol. 1991, 1, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Bergmans, B.A.; de Strooper, B. gamma-secretases: From cell biology to therapeutic strategies. Lancet Neurol. 2010, 9, 215–226. [Google Scholar] [CrossRef]
- Shoji, M.; Golde, T.E.; Ghiso, J.; Cheung, T.T.; Estus, S.; Shaffer, L.M.; Cai, X.D.; McKay, D.M.; Tintner, R.; Frangione, B.; et al. Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 1992, 258, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [Green Version]
- Gandy, S. The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J. Clin. Investig. 2005, 115, 1121–1129. [Google Scholar]
- Tanzi, R.E.; Bertram, L. Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 2005, 120, 545–555. [Google Scholar] [CrossRef]
- Doherty, G.J.; McMahon, H.T. Mechanisms of endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [Green Version]
- El-Sayed, A.; Harashima, H. Endocytosis of gene delivery vectors: From clathrin-dependent to lipid raft-mediated endocytosis. Mol. Ther. 2013, 21, 1118–1130. [Google Scholar] [CrossRef] [Green Version]
- Yue, H.Y.; Xu, J. Cholesterol regulates multiple forms of vesicle endocytosis at a mammalian central synapse. J. Neurochem. 2015, 134, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Burns, M.P.; Rebeck, G.W. Intracellular cholesterol homeostasis and amyloid precursor protein processing. Biochim. Biophys. Acta 2010, 1801, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Chun, Y.S.; Oh, H.G.; Park, M.K.; Kim, T.W.; Chung, S. Increasing Membrane Cholesterol Level Increases the Amyloidogenic Peptide by Enhancing the Expression of Phospholipase C. J. Neurodegener. Dis. 2013, 2013, 407903. [Google Scholar] [PubMed] [Green Version]
- Grimm, M.O.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maulik, M.; Westaway, D.; Jhamandas, J.H.; Kar, S. Role of cholesterol in APP metabolism and its significance in Alzheimer’s disease pathogenesis. Mol. Neurobiol. 2013, 47, 37–63. [Google Scholar] [CrossRef] [PubMed]
- Posse de Chaves, E. Reciprocal regulation of cholesterol and beta amyloid at the subcellular level in Alzheimer’s disease. Can. J. Physiol. Pharmacol. 2012, 90, 753–764. [Google Scholar] [CrossRef]
- Urano, Y.; Ochiai, S.; Noguchi, N. Suppression of amyloid-β production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 2013, 27, 4305–4315. [Google Scholar] [CrossRef]
- Walter, J.; van Echten-Deckert, G. Cross-talk of membrane lipids and Alzheimer-related proteins. Mol. Neurodegener. 2013, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Chun, Y.S.; Chung, S. High-Cholesterol Diet Decreases the Level of Phosphatidylinositol 4,5-Bisphosphate by Enhancing the Expression of Phospholipase C (PLCbeta1) in Rat Brain. Int. J. Mol. Sci. 2020, 21, 1161. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Fabelo, N.; Martin, V.; Marin, R.; Moreno, D.; Ferrer, I.; Diaz, M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef]
- Heverin, M.; Bogdanovic, N.; Lutjohann, D.; Bayer, T.; Pikuleva, I.; Bretillon, L.; Diczfalusy, U.; Winblad, B.; Bjorkhem, I. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer’s disease. J. Lipid Res. 2004, 45, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Schneider, A.; Schulz-Schaeffer, W.; Hartmann, T.; Schulz, J.B.; Simons, M. Cholesterol depletion reduces aggregation of amyloid-beta peptide in hippocampal neurons. Neurobiol. Dis. 2006, 23, 573–577. [Google Scholar] [CrossRef]
- Vanmierlo, T.; Bloks, V.W.; van Vark-van der Zee, L.C.; Rutten, K.; Kerksiek, A.; Friedrichs, S.; Sijbrands, E.; Steinbusch, H.W.; Kuipers, F.; Lutjohann, D.; et al. Alterations in brain cholesterol metabolism in the APPSLxPS1mut mouse, a model for Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 117–127. [Google Scholar] [CrossRef]
- Xiong, H.; Callaghan, D.; Jones, A.; Walker, D.G.; Lue, L.F.; Beach, T.G.; Sue, L.I.; Woulfe, J.; Xu, H.; Stanimirovic, D.B.; et al. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis. 2008, 29, 422–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 2000, 275, 17221–17224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [Green Version]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The mystery of membrane organization: Composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Benjannet, S.; Elagoz, A.; Wickham, L.; Mamarbachi, M.; Munzer, J.S.; Basak, A.; Lazure, C.; Cromlish, J.A.; Sisodia, S.; Checler, F.; et al. Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 2001, 276, 10879–10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, R.; Barren, C.; Kovacs, D.M. Palmitoylation of amyloid precursor protein regulates amyloidogenic processing in lipid rafts. J. Neurosci. 2013, 33, 11169–11183. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.Y.; Welander, H.; Behbahani, H.; Aoki, M.; Franberg, J.; Winblad, B.; Frykman, S.; Tjernberg, L.O. Active gamma-secretase is localized to detergent-resistant membranes in human brain. FEBS J. 2008, 275, 1174–1187. [Google Scholar] [CrossRef]
- Osenkowski, P.; Ye, W.; Wang, R.; Wolfe, M.S.; Selkoe, D.J. Direct and potent regulation of gamma-secretase by its lipid microenvironment. J. Biol. Chem. 2008, 283, 22529–22540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Marquer, C.; Devauges, V.; Cossec, J.C.; Liot, G.; Lecart, S.; Saudou, F.; Duyckaerts, C.; Leveque-Fort, S.; Potier, M.C. Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Cordy, J.M.; Hussain, I.; Dingwall, C.; Hooper, N.M.; Turner, A.J. Exclusively targeting beta-secretase to lipid rafts by GPI-anchor addition up-regulates beta-site processing of the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2003, 100, 11735–11740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquer, C.; Laine, J.; Dauphinot, L.; Hanbouch, L.; Lemercier-Neuillet, C.; Pierrot, N.; Bossers, K.; Le, M.; Corlier, F.; Benstaali, C.; et al. Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol. Neurodegener. 2014, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vetrivel, K.S.; Thinakaran, G. Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim. Biophys. Acta 2010, 1801, 860–867. [Google Scholar] [CrossRef] [Green Version]
- Cossec, J.C.; Simon, A.; Marquer, C.; Moldrich, R.X.; Leterrier, C.; Rossier, J.; Duyckaerts, C.; Lenkei, Z.; Potier, M.C. Clathrin-dependent APP endocytosis and Abeta secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim. Biophys. Acta 2010, 1801, 846–852. [Google Scholar] [CrossRef]
- Cho, Y.Y.; Kwon, O.H.; Park, M.K.; Kim, T.W.; Chung, S. Elevated cellular cholesterol in Familial Alzheimer’s presenilin 1 mutation is associated with lipid raft localization of beta-amyloid precursor protein. PLoS ONE 2019, 14, e0210535. [Google Scholar]
- Chinnapen, D.J.-F.; Chinnapen, H.; Saslowsky, D.; Lencer, W.I. Rafting with cholera toxin: Endocytosis and trafficking from plasma membrane to ER. FEMS Microbiol. Lett. 2007, 266, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Kirkham, M.; Fujita, A.; Chadda, R.; Nixon, S.J.; Kurzchalia, T.V.; Sharma, D.K.; Pagano, R.E.; Hancock, J.F.; Mayor, S.; Parton, R.G. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J. Cell Biol. 2005, 168, 465–476. [Google Scholar] [CrossRef]
- Kirkham, M.; Parton, R.G. Clathrin-independent endocytosis: New insights into caveolae and non-caveolar lipid raft carriers. Biochim. Biophys. Acta 2005, 1746, 349–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, Y.; Ninomiya, H.; Miwa, S.; Masaki, T. Cholesterol oxidation switches the internalization pathway of endothelin receptor type A from caveolae to clathrin-coated pits in Chinese hamster ovary cells. J. Biol. Chem. 2000, 275, 6439–6446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehlers, M.D. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 2000, 28, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Kittler, J.T.; Thomas, P.; Tretter, V.; Bogdanov, Y.D.; Haucke, V.; Smart, T.G.; Moss, S.J. Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking. Proc. Natl. Acad. Sci. USA 2004, 101, 12736–12741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, A.; Konig, G.; Bunke, D.; Fischer, P.; Salbaum, J.M.; Masters, C.L.; Beyreuther, K. Identification, biogenesis, and localization of precursors of Alzheimer’s disease A4 amyloid protein. Cell 1989, 57, 115–126. [Google Scholar] [CrossRef]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef]
- Subtil, A.; Gaidarov, I.; Kobylarz, K.; Lampson, M.A.; Keen, J.H.; McGraw, T.E. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc. Natl. Acad. Sci. USA 1999, 96, 6775–6780. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, S.J.; Aksenova, M.V.; Aksenov, M.Y.; Mactutus, C.F.; Booze, R.M. Endogenous amyloidogenesis in long-term rat hippocampal cell cultures. BMC Neurosci. 2011, 12, 38. [Google Scholar] [CrossRef] [Green Version]
- Ausili, A.; Torrecillas, A.; Aranda, F.J.; Mollinedo, F.; Gajate, C.; Corbalán-García, S.; de Godos, A.; Gómez-Fernández, J.C. Edelfosine is incorporated into rafts and alters their organization. J. Phys. Chem. B 2008, 112, 11643–11654. [Google Scholar] [CrossRef]
- Van der Luit, A.H.; Vink, S.R.; Klarenbeek, J.B.; Perrissoud, D.; Solary, E.; Verheij, M.; van Blitterswijk, W.J. A new class of anticancer alkylphospholipids uses lipid rafts as membrane gateways to induce apoptosis in lymphoma cells. Mol. Cancer Ther. 2007, 6, 2337–2345. [Google Scholar] [CrossRef] [Green Version]
- Van Blitterswijk, W.J.; Verheij, M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim. Biophys. Acta 2013, 1831, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Busto, J.V.; Del Canto-Jañez, E.; Goñi, F.M.; Mollinedo, F.; Alonso, A. Combination of the anti-tumour cell ether lipid edelfosine with sterols abolishes haemolytic side effects of the drug. J. Chem. Biol. 2008, 1, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hac-Wydro, K.; Dynarowicz-Latka, P. Searching for the role of membrane sphingolipids in selectivity of antitumor ether lipid-edelfosine. Colloids Surfaces B Biointerfaces 2010, 81, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Heczkova, B.; Slotte, J.P. Effect of anti-tumor ether lipids on ordered domains in model membranes. FEBS Lett. 2006, 580, 2471–2476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiménez-López, J.M.; Ríos-Marco, P.; Marco, C.; Segovia, J.L.; Carrasco, M.P. Alterations in the homeostasis of phospholipids and cholesterol by antitumor alkylphospholipids. Lipids Health Dis. 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Del Canto-Jañez, E.; Acuña, A.U.; Amat-Guerri, F.; Geijo, E.; Santos-Beneit, A.M.; Veldman, R.J.; Mollinedo, F. Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis. J. Exp. Med. 2004, 200, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2007, 109, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; Campanero, M.A.; Blanco-Prieto, M.J. Lipid raft-targeted therapy in multiple myeloma. Oncogene 2010, 29, 3748–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allinquant, B.; Clamagirand, C.; Potier, M.C. Role of cholesterol metabolism in the pathogenesis of Alzheimer’s disease. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 319–323. [Google Scholar] [CrossRef]
- Park, J.; Kim, H.; Kim, J.; Cheon, M. A practical application of generative adversarial networks for RNA-seq analysis to predict the molecular progress of Alzheimer’s disease. PLoS Comput. Biol. 2020, 16, e1008099. [Google Scholar] [CrossRef]
- Hicks, D.A.; Nalivaeva, N.N.; Turner, A.J. Lipid rafts and Alzheimer’s disease: Protein-lipid interactions and perturbation of signaling. Front. Physiol. 2012, 3, 189. [Google Scholar] [CrossRef] [Green Version]
- Kojro, E.; Gimpl, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef] [Green Version]
- Beel, A.J.; Mobley, C.K.; Kim, H.J.; Tian, F.; Hadziselimovic, A.; Jap, B.; Prestegard, J.H.; Sanders, C.R. Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): Does APP function as a cholesterol sensor? Biochemistry 2008, 47, 9428–9446. [Google Scholar] [CrossRef] [Green Version]
- Beel, A.J.; Sakakura, M.; Barrett, P.J.; Sanders, C.R. Direct binding of cholesterol to the amyloid precursor protein: An important interaction in lipid-Alzheimer’s disease relationships? Biochim. Biophys. Acta 2010, 1801, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Hustedt, E.J.; Brandon, S.; Sanders, C.R. Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 2013, 52, 5051–5064. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Kenworthy, A.K.; Sanders, C.R. Cholesterol as a co-solvent and a ligand for membrane proteins. Protein Sci. 2014, 23, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.Y.; Liu, P.H.; Ruan, C.T.; Chiu, L.; Kung, F.L. The intracellular domain of amyloid precursor protein interacts with flotillin-1, a lipid raft protein. Biochem. Biophys. Res. Commun. 2006, 342, 266–272. [Google Scholar] [CrossRef]
- Schneider, A.; Rajendran, L.; Honsho, M.; Gralle, M.; Donnert, G.; Wouters, F.; Hell, S.W.; Simons, M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 2008, 28, 2874–2882. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Baek, S.H.; Chun, Y.S.; Moore, A.Z.; Landman, N.; Berman, D.; Yang, H.O.; Morishima-Kawashima, M.; Osawa, S.; Funamoto, S.; et al. Modulation of lipid kinase PI4KIIalpha activity and lipid raft association of presenilin 1 underlies gamma-secretase inhibition by ginsenoside (20S)-Rg3. J. Biol. Chem. 2013, 288, 20868–20882. [Google Scholar] [CrossRef] [Green Version]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Sample Availability: Samples of the compounds are not available. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, Y.Y.; Kwon, O.-H.; Chung, S. Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains. Molecules 2020, 25, 5490. https://doi.org/10.3390/molecules25235490
Cho YY, Kwon O-H, Chung S. Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains. Molecules. 2020; 25(23):5490. https://doi.org/10.3390/molecules25235490
Chicago/Turabian StyleCho, Yoon Young, Oh-Hoon Kwon, and Sungkwon Chung. 2020. "Preferred Endocytosis of Amyloid Precursor Protein from Cholesterol-Enriched Lipid Raft Microdomains" Molecules 25, no. 23: 5490. https://doi.org/10.3390/molecules25235490