
Palladium-Catalyzed Cross-Coupling of Gem-Bromofluoroalkenes with Alkylboronic Acids for the Synthesis of Alkylated Monofluoroalkenes


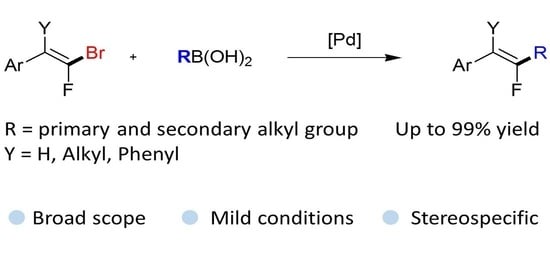
Abstract
:
1. Introduction
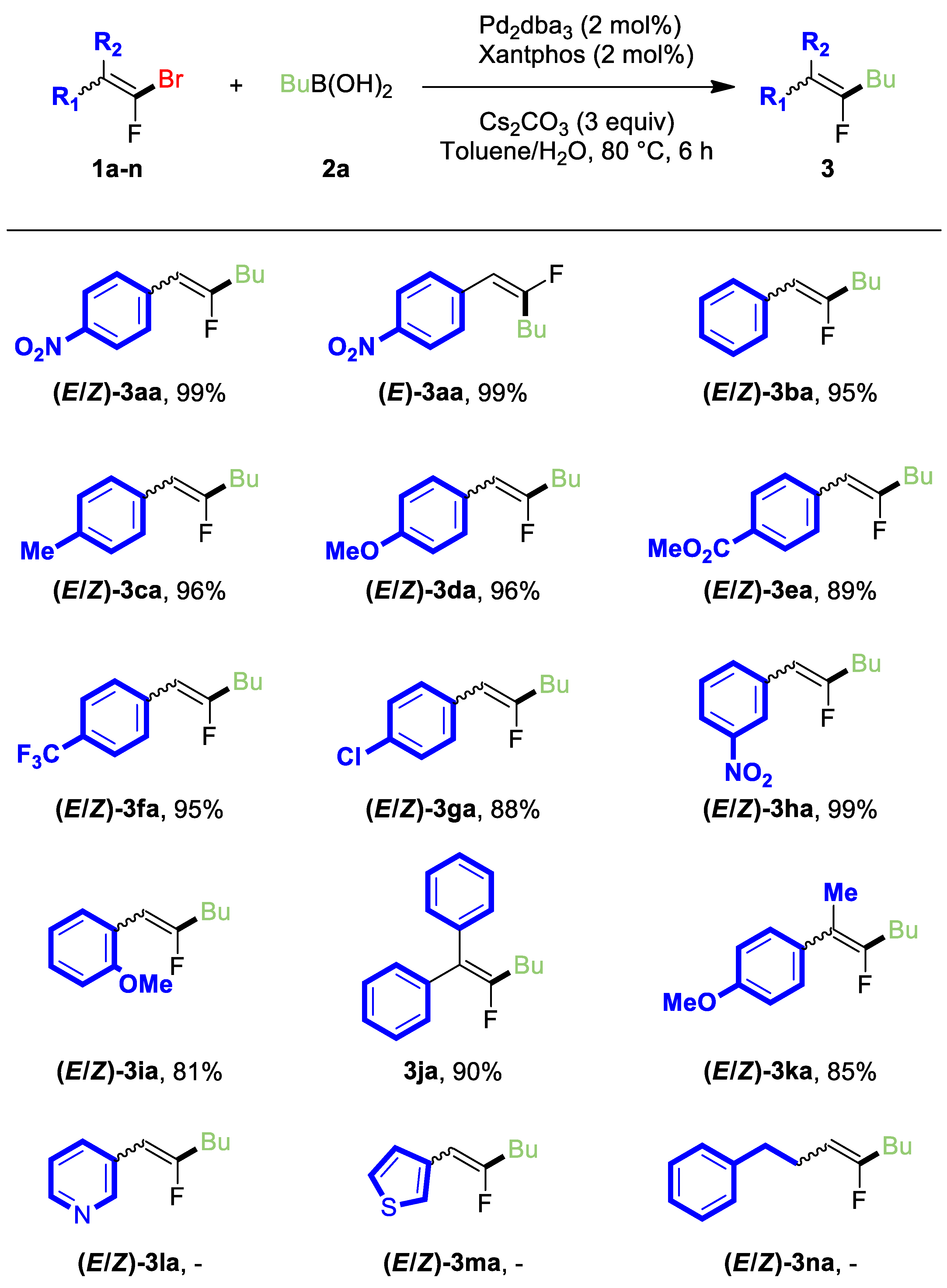
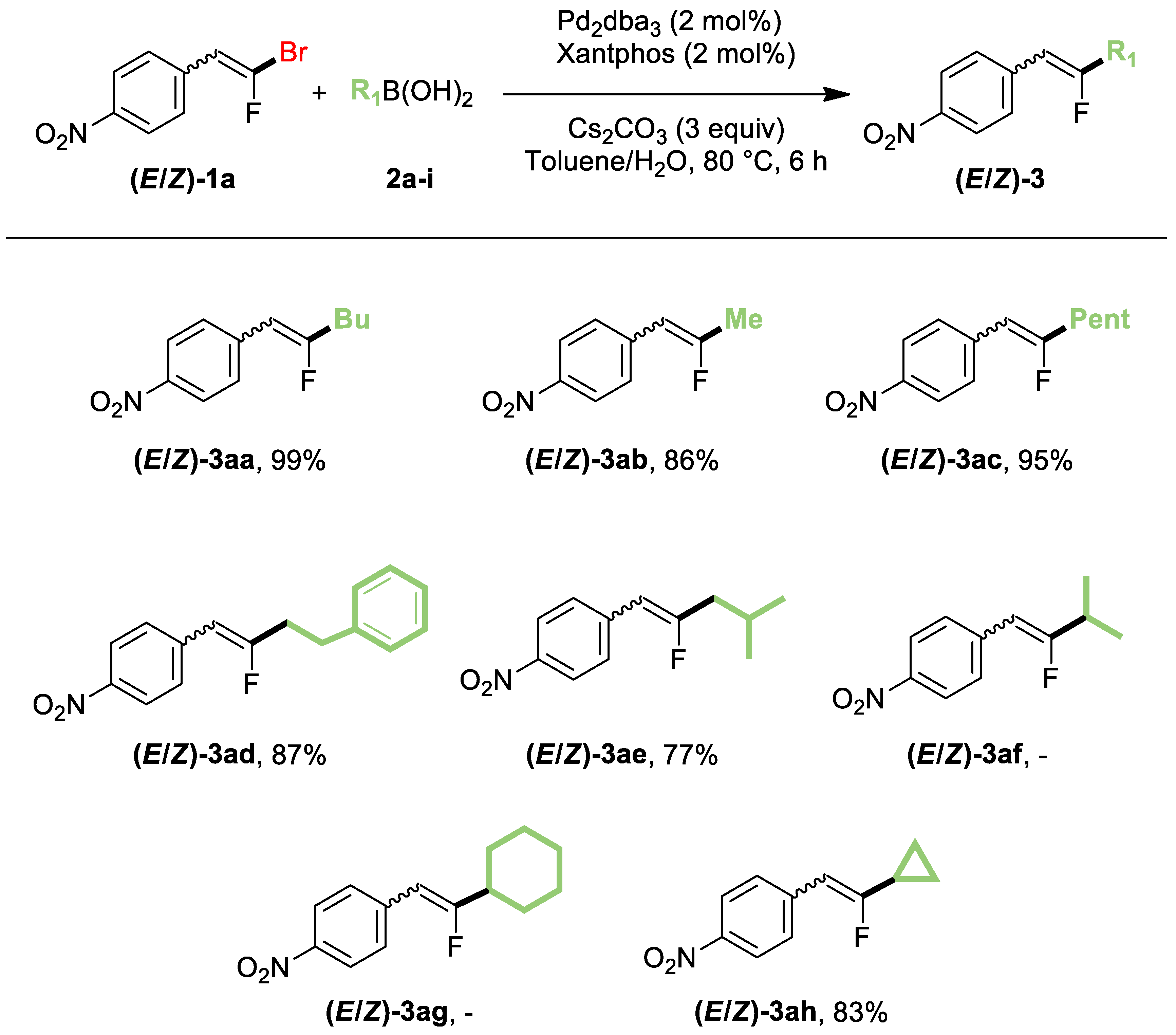
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of Gem-Bromofluoroalkenes 1
3.3. General Procedure for the Synthesis of Monofluoroalkenes 3
3.3.1. (E/Z)-1-(2-fluorohex-1-en-1-yl)-4-nitrobenzene (3aa)
3.3.2. (E)-1-(2-fluorohex-1-en-1-yl)-4-nitrobenzene (E-3aa)
3.3.3. (E/Z)-(2-fluorohex-1-en-1-yl)benzene (3ba)
3.3.4. (E/Z)-1-(2-fluorohex-1-en-1-yl)-4-methylbenzene (3ca)
3.3.5. (E/Z)-1-(2-fluorohex-1-en-1-yl)-4-methoxybenzene (3da)
3.3.6. (E/Z)-methyl 4-(2-fluorohex-1-en-1-yl)benzoate (3ea)
3.3.7. (E/Z)-1-(2-fluorohex-1-en-1-yl)-4-(trifluoromethyl)benzene (3fa)
3.3.8. (E/Z)-1-chloro-4-(2-fluorohex-1-en-1-yl)benzene (3ga)
3.3.9. (E/Z)-1-(2-fluorohex-1-en-1-yl)-3-nitrobenzene (3ha)
3.3.10. (E/Z)-1-(2-fluorohex-1-en-1-yl)-2-methoxybenzene (3ia)
3.3.11. (2-Bromo-2-fluoroethene-1,1-diyl)dibenzene (3ja)
3.3.12. (E/Z)-1-(3-fluorohept-2-en-2-yl)-4-methoxybenzene (3ka)
3.3.13. 1-(2-Fluoroprop-1-en-1-yl)-4-nitrobenzene (3ab)
3.3.14. 1-(2-Fluoropent-1-en-1-yl)-4-nitrobenzene (3ac)
3.3.15. 1-(2-Fluoro-4-phenylbut-1-en-1-yl)-4-nitrobenzene (3ad)
3.3.16. 1-(2-Fluoro-4-methylpent-1-en-1-yl)-4-nitrobenzene (3ae)
3.3.17. 1-(2-Cyclopropyl-2-fluorovinyl)-4-nitrobenzene (3ah)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hagmann, W.K. The Many Roles for Fluorine in Medicinal Chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Wang, J.; Sánchez-Roselló, M.; Aceña, J.L.; Del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluor. Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. Organic fluorine compounds: A great opportunity for enhanced materials properties. Chem. Soc. Rev. 2011, 40, 3496–3508. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Couve-Bonnaire, S.; Cahard, D.; Pannecoucke, X. Chiral dipeptide mimics possessing a fluoroolefin moiety: A relevant tool for conformational and medicinal studies. Org. Biomol. Chem. 2007, 5, 1151–1157. [Google Scholar] [CrossRef]
- Osada, S.; Sano, S.; Ueyama, M.; Chuman, Y.; Kodama, H.; Sakaguchi, K. Fluoroalkene modification of mercaptoacetamide-based histone deacetylase inhibitors. Bioorganic Med. Chem. 2010, 18, 605–611. [Google Scholar] [CrossRef]
- Zajc, B.; Kumar, R. Synthesis of Fluoroolefins via Julia-Kocienski Olefination. Synthesis 2010, 2010, 1822–1836. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Jiang, F.; Hu, J. Spontaneous Resolution of Julia-Kocienski Intermediates Facilitates Phase Separation to Produce Z- and E-Monofluoroalkenes. J. Am. Chem. Soc. 2015, 137, 5199–5203. [Google Scholar] [CrossRef] [PubMed]
- Akana, J.A.; Bhattacharyya, K.X.; Mueller, P.; Sadighi, J.P. Reversible C-F Bond Formation and the Au-Catalyzed Hydrofluorination of Alkynes. J. Am. Chem. Soc. 2007, 129, 7736–7737. [Google Scholar] [CrossRef]
- Gorske, B.C.; Mbofana, C.T.; Miller, S.J. Regio- and Stereoselective Synthesis of Fluoroalkenes by Directed Au(I) Catalysis. Org. Lett. 2009, 11, 4318–4321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.-L.; Wu, J.; Zhao, Y. Divergent reactivities in fluoronation of allylic alcohols: Synthesis of Z-fluoroalkenes via carbon-carbon bond cleavage. Chem. Sci. 2017, 8, 3885–3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, R.; Mamone, M.; Paquin, J. Gold-Catalyzed Hydrofluorination of Internal Alkynes Using Aqueous HF. Org. Lett. 2019, 21, 9024–9027. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Shi, H.; Zhao, X.; Cao, S. Sterically Controlled Cu-Catalyzed or Transition-Metal-Free Cross-Coupling of gem-Difluoroalkenes with Tertiary, Secondary, and Primary Alkyl Grignard Reagents. Org. Lett. 2016, 18, 4284–4287. [Google Scholar] [CrossRef]
- Lu, X.; Wang, Y.; Zhang, B.; Pi, J.-J.; Wang, X.-X.; Gong, T.-J.; Xiao, B.; Fu, Y. Nickel-Catalyzed Defluorinative Reductive Cross-Coupling of gem-Difluoroalkenes with Unactivated Secondary and Tertiary Alkyl Halides. J. Am. Chem. Soc. 2017, 139, 12632–12637. [Google Scholar] [CrossRef]
- Yang, L.; Ji, W.-W.; Lin, E.; Li, J.-L.; Fan, W.-X.; Li, Q.; Zhang, S.-S. Synthesis of Alkylated Monofluoroalkenes via Fe-Catalyzed Defluorinative Cross-Coupling of Donor Alkenes with gem-Difluoroalkenes. Org. Lett. 2018, 20, 1924–1927. [Google Scholar] [CrossRef]
- Yu, L.; Tang, M.-L.; Si, C.-M.; Meng, Z.; Liang, Y.; Han, J.; Sun, X. Zinc-Mediated Decarboxylative Alkylation of gem-Difluoroalkenes. Org. Lett. 2018, 20, 4579–4583. [Google Scholar] [CrossRef]
- Zhou, L.; Zhu, C.; Bi, P.; Feng, C. Ni-catalyzed migratory fluoro-alkenylation of unactivated alkyl bromides with gem-difluoroalkenes. Chem. Sci. 2019, 10, 1144–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Ahmed, E.-A.; Xiao, B.; Lu, Q.-Q.; Wang, Y.-L.; Yu, C.-G.; Fu, Y. ChemInform Abstract: Pd-Catalyzed Regioselective Activation of gem-Difluorinated Cyclopropanes: A Highly Efficient Approach to 2-Fluorinated Allylic Scaffolds. Angew. Chem. 2015, 46, 8231–8235. [Google Scholar] [CrossRef] [PubMed]
- Wenz, J.; Rettenmeier, C.A.; Wadepohl, H.; Gade, L.H. ChemInform Abstract: Catalytic C-F Bond Activation of Geminal Difluorocyclopropanes by Nickel(I) Complexes via a Radical Mechanism. Angew. Chem. 2016, 47, 202–205. [Google Scholar] [CrossRef]
- Ahmed, E.-A.M.A.; Suliman, A.M.Y.; Gong, T.-J.; Fu, Y. Palladium-Catalyzed Stereoselective Defluorination Arylation/Alkenylation/Alkylation of gem-Difluorinated Cyclopropanes. Org. Lett. 2019, 21, 5645–5649. [Google Scholar] [CrossRef]
- Xie, J.; Yu, J.; Rudolph, M.; Rominger, F.; Hashmi, A.S.K. Monofluoroalkenylation of Dimethylamino Compounds through Radical-Radical Cross-Coupling. Angew. Chem. Int. Ed. 2016, 55, 9416–9421. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lefebvre, Q.; Yang, H.; Zhao, Y.; Fu, H. Visible light photocatalytic decarboxylative monofluoroalkenylation of α-amino acids with gem-difluoroalkenes. Chem. Commun. 2017, 53, 10299–10302. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Tian, C.; Qiu, D.; Tian, H.; An, G.-H.; Li, G.-M. Organic photoredox catalytic decarboxylative cross-coupling of gem-difluoroalkenes with unactivated carboxylic acids. Org. Chem. Front. 2019, 6, 2365–2370. [Google Scholar] [CrossRef]
- Du, H.-W.; Sun, J.; Gao, Q.-S.; Wang, J.-Y.; Wang, H.; Xu, Z.; Zhou, M. Synthesis of Monofluoroalkenes through Visible-Light-Promoted Defluorinative Alkylation of gem-Difluoroalkenes with 4-Alkyl-1,4-dihydropyridines. Org. Lett. 2020, 22, 1542–1546. [Google Scholar] [CrossRef]
- Chelucci, G. ChemInform Abstract: Synthesis and Metal-Catalyzed Reactions of gem-Dihalovinyl Systems. Angew. Chem. 2012, 43, 1344–1462. [Google Scholar] [CrossRef]
- Landelle, G.; Bergeron, M.; Turcotte-Savard, M.-O.; Paquin, J.-F. Synthetic approaches to monofluoroalkenes. Chem. Soc. Rev. 2011, 40, 2867–2908. [Google Scholar] [CrossRef]
- Lei, X.; Dutheuil, G.; Pannecoucke, X.; Quirion, J.-C. A Facile and Mild Method for the Synthesis of Terminal Bromofluoroolefins via Diethylzinc-Promoted Wittig Reaction. Org. Lett. 2004, 6, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Andrei, D.; Wnuk, S.F. Synthesis of the Multisubstituted Halogenated Olefins via Cross-Coupling of Dihaloalkenes with Alkylzinc Bromides. J. Org. Chem. 2006, 71, 405–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zemmouri, R.; Kajjout, M.; Castanet, Y.; Eddarir, S.; Rolando, C. Palladium-Catalyzed Stereoconvergent Formylation of (E/Z)-β-Bromo-β-fluorostyrenes: Straightforward Access to (Z)-α-Fluorocinnamic Aldehydes and (Z)-β-Fluorocinnamic Alcohols. J. Org. Chem. 2011, 76, 7691–7698. [Google Scholar] [CrossRef] [PubMed]
- Eddarir, S.; Kajjout, M.; Rolando, C. Stereoselective cyanation of β-bromo-β-fluorostyrenes using potassium cyanide and promoted by (CH3CN)4Cu+ BF4−. Tetrahedron 2012, 68, 603–607. [Google Scholar] [CrossRef]
- Kajjout, M.; Zemmouri, R.; Eddarir, S.; Rolando, C. An efficient access to (Z)-β-fluoroallyl alcohols based on the two carbon homologation of aromatic aldehydes by Horner-Wadsworth-Emmons reaction with 2-(diethoxyphosphinyl)-2-fluoro-ethanethioic acid, S-ethyl ester followed by reduction with sodium borohydride. Tetrahedron 2012, 68, 3225–3230. [Google Scholar] [CrossRef]
- Kajjout, M.; Smietana, M.; Leroy, J.; Rolando, C. A new approach to the synthesis of (Z)-2-fluoro-2-alkenals via Wittig-type carbonyl condensation reactions of 2-(fluoromethyl)-4,4,6-trimethyl-1,3-oxazine phosphonium bromide. Tetrahedron Lett. 2013, 54, 1658–1660. [Google Scholar] [CrossRef]
- Tan, Z.; Negishi, E.-I. Widely Applicable Pd-Catalyzed trans-Selective Monoalkylation of Unactivated 1,1-Dichloro-1-alkenes and Pd-Catalyzed Second Substitution for the Selective Synthesis of (E)- or (Z)- Trisubstituted Alkenes. Angew. Chem. 2006, 37, 762–765. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
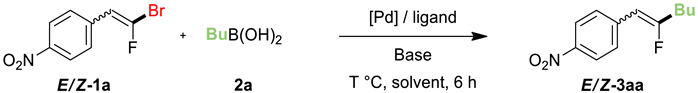 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | E/Z 1a b | [Pd] | Ligand | Base | Solvent | T (°C) | Z/E 3aa b | Yield (%) c |
| 1 | 55:45 | Pd(Ph3)4 | - | Cs2CO3 | toluene/H2O | 80 | - | - |
| 2 | 55:45 | Pd2dba3 CHCl3 | - | Cs2CO3 | toluene/H2O | 80 | - | - |
| 3 | 56:44 | PdCl2dppf | - | Cs2CO3 | toluene/H2O | 80 | 58:42 | 95 |
| 4 | 55:45 | PdCl2dppf | - | K2CO3 | toluene/H2O | 80 | 46:54 | 76 |
| 5 | 55:45 | PdCl2dppf | - | Na2CO3 | toluene/H2O | 80 | 57:43 | 44 |
| 6 | 55:45 | PdCl2dppf | - | K3PO4 | toluene/H2O | 80 | 51:49 | 93 |
| 7 | 55:46 | PdCl2dppf | - | Ba(OH)2 | toluene/H2O | 80 | - | - |
| 8 | 39:61 | PdCl2dppf | - | Cs2CO3 | toluene | 80 | 38:62 | 89 |
| 9 | 39:61 | PdCl2dppf | - | Cs2CO3 | Dioxane | 80 | - | - |
| 10 | 39:61 | PdCl2dppf | - | Cs2CO3 | THF | 80 | - | - |
| 11 | 55:45 | PdCl2dppf | - | Cs2CO3 | toluene/H2O | RT | - | - |
| 12 | 55:45 | PdCl2dppf | - | Cs2CO3 | toluene/H2O | 60 | 45:55 | 51 |
| 13 | 55:45 | Pd2dba3.CHCl3 | Dppf | Cs2CO3 | toluene/H2O | 80 | 51:49 | 91 |
| 14 | 48:52 | Pd2dba3 CHCl3 | Dppe | Cs2CO3 | toluene/H2O | 80 | 48:52 | 41 |
| 15 | 48:52 | Pd2dba3 CHCl3 | Xantphos | Cs2CO3 | toluene/H2O | 80 | 49:51 | 99 |
| 16 | 48:52 | Pd2dba3 CHCl3 | Xphos | Cs2CO3 | toluene/H2O | 80 | 60:40 | 35 |
| 17 | 48:52 | Pd2dba3 CHCl3 | TFP | Cs2CO3 | toluene/H2O | 80 | 42:58 | 78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chausset-Boissarie, L.; Cheval, N.; Rolando, C. Palladium-Catalyzed Cross-Coupling of Gem-Bromofluoroalkenes with Alkylboronic Acids for the Synthesis of Alkylated Monofluoroalkenes. Molecules 2020, 25, 5532. https://doi.org/10.3390/molecules25235532
Chausset-Boissarie L, Cheval N, Rolando C. Palladium-Catalyzed Cross-Coupling of Gem-Bromofluoroalkenes with Alkylboronic Acids for the Synthesis of Alkylated Monofluoroalkenes. Molecules. 2020; 25(23):5532. https://doi.org/10.3390/molecules25235532
Chicago/Turabian StyleChausset-Boissarie, Laëtitia, Nicolas Cheval, and Christian Rolando. 2020. "Palladium-Catalyzed Cross-Coupling of Gem-Bromofluoroalkenes with Alkylboronic Acids for the Synthesis of Alkylated Monofluoroalkenes" Molecules 25, no. 23: 5532. https://doi.org/10.3390/molecules25235532
APA StyleChausset-Boissarie, L., Cheval, N., & Rolando, C. (2020). Palladium-Catalyzed Cross-Coupling of Gem-Bromofluoroalkenes with Alkylboronic Acids for the Synthesis of Alkylated Monofluoroalkenes. Molecules, 25(23), 5532. https://doi.org/10.3390/molecules25235532