1. Introduction
β-lactam antibiotics have been used widely as frontline therapeutics in treating bacteria-related infections and diseases. These molecules specifically target bacterial pathogens by interfering with bacterial cell wall synthesis, which will eventually cause cell lysis [
1]. However, the emergence of antimicrobial resistance (AMR) among bacterial pathogens had raised major concerns in global public health as it can render the commonly used antibiotics and antimicrobial therapy ineffective, prolonging hospital stay and increasing medical expenses. Severe cases of AMR can lead to more complicated medical procedures such as surgery to remove the focal point of infection and even untimely deaths. As such, AMR will exert a huge impact on the world economy in the future if the current situation is not tackled [
2].
AMR among bacteria can be acquired through various means, i.e., through: (1) hydrolysis or inactivation of antibiotics by synthesizing enzymes [
3]; (2) redox process by exploiting the oxidation or reduction of the antibiotics [
4]; (3) modification of antibiotics by chemical substitution [
5]; (4) modification of the inhibition binding site in the target [
6]; (5) mutations on genes that encode the target or efflux pump that affect antibiotics uptake [
7,
8]; (6) horizontal gene transfer where resistant genes are transferred from one pathogen to another via transduction, conjugation or transformation [
9]. Mechanism (1) is one of the most well-studied AMR mechanisms which involves β-lactamase enzymes. β-lactamases deactivates β-lactam antibiotics by hydrolysing the β-lactam ring of the β-lactam antibiotics. Based on the Ambler classification scheme, β-lactamases are classified as Class A, B, C, and D based on their sequence similarities. Class A, C and D β-lactamase are categorised as serine-β-lactamase (SBLs) while Class B β-lactamases are categorised as metallo-β-lactamases (MBLs) [
10]. MBLs can be further divided into four subclasses, B1, B2, B3 and B4, based on their sequence homology [
11]. All MBLs are dependent on Zn
2+ metal ions as co-factor for catalysis. B1, B3 and B4 MBLs require two Zn
2+ ions for their catalytic activity, while B2 MBLs require only one Zn
2+ ion for their activity [
12,
13,
14,
15,
16]. Compared to SBLs, MBLs pose a greater threat to public health due to their broad substrate specificity. Clinically available inhibitors such as clavulanate, sulbactam and tazobactam are effective against Class A SBLs [
17,
18,
19], but not MBLs [
20]. Hence, they have garnered attention in the recent decade due to their inability to be inhibited by commonly used clinical inhibitors or drug combinations [
21].
Previously, a hypothetical protein termed Bleg1_2437 (currently renamed as Bleg1_2478), which has a comparable sequence identity to MBL in the range of 43–65%, was discovered from the pool of hypothetical proteins of
Bacillus lehensis G1 alkaliphile. Its predicted in silico structure revealed similarity to the αββα fold and global topology of MBLs. Analysis of its active site and metal-binding ligands revealed that similarity to B3 MBL. Biochemical characterization of purified recombinant Bleg1_2478 further confirmed that it was indeed capable of degrading β-lactam antibiotics, with ampicillin as the preferred substrate [
22]. However, in terms of evolutionary relationship, it did not exhibit relatedness to other B3 MBLs [
22]. In view of limited inhibitors against B3 MBLs to date and the evolutionary divergent nature of Bleg1_2478, this study aims to design inhibitory peptides against Bleg1_2478 B3 subclass MBL and characterise their inhibitory potential and properties through in vitro and in silico approaches. This is one of few reports on peptide inhibitors targeted against B3 MBLs [
23,
24].
3. Discussion
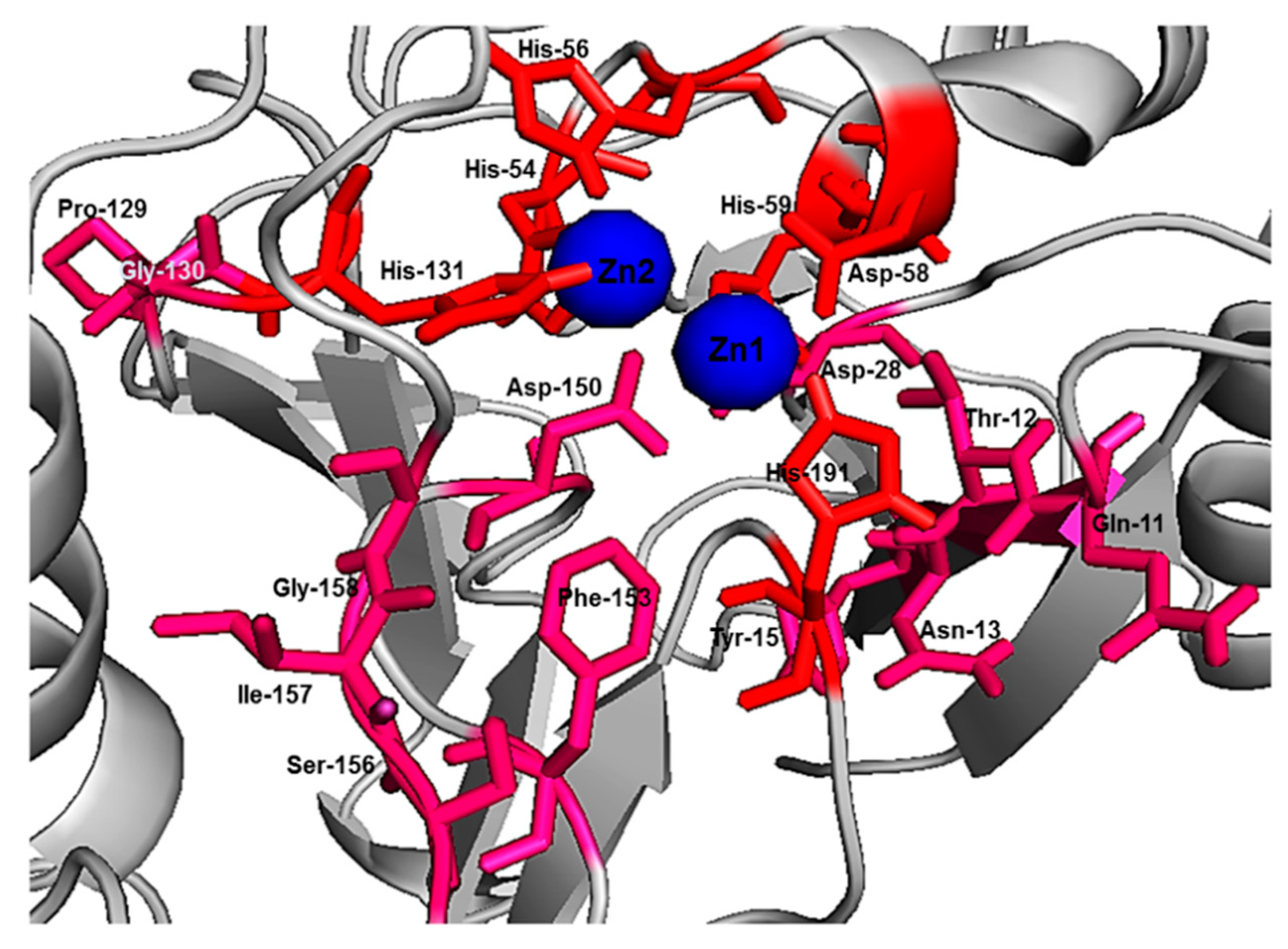
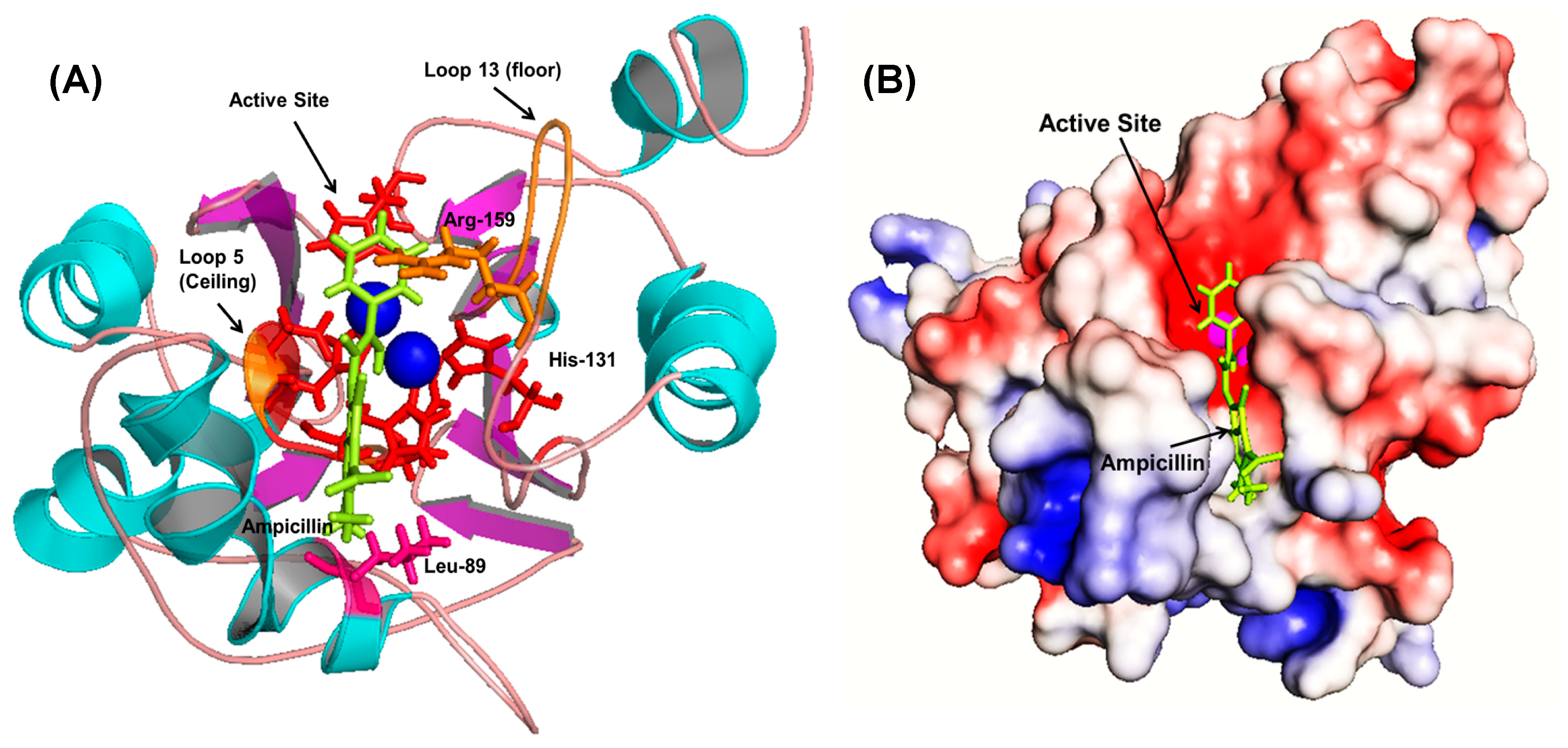
Initial fixed and random docking analyses of ampicillin with Bleg1_2478 were undertaken to determine the binding properties of the β-lactam antibiotic to the protein. Within a distance of 5.0 Å from the binding site, results from both docking analysis showed whilst Zn
2+ ions interacted with His54, 56, 59, 131, 191 and Asp58 of Bleg1_2478 active site, ampicillin interacted with other residues in the active site namely Leu89 (on α4), Arg159 (on loop 13 which acted as floor, shown as an orange stick) and Arg163 (on loop 15) (
Figure A1). The hydrophobic environment surrounding the active site of Bleg1_2478 and ampicillin (
Figure A2) helps to retain the substrate in the active site by interacting with the hydrophobic β side chain of ampicillin, similar to observations related to B3 MBLs interactions with β-lactam antibiotics [
21,
22,
34]. The hydrophobic residues were Pro9 and Ile10 (on loop 2, postulated to be part of a doorkeeper structure), Phe57 (on loop 5, the ceiling structure), Phe153 and Ser156 (on loop 13, the floor structure), similar to previous observations made by Tan et al., 2017 [
22]. Other than this, π–π, cation–π and hydrophobic interactions formed by His131, Arg159 and Leu89 with the benzene and β-lactam rings of ampicillin further facilitate the binding of the molecule in Bleg1_2478 active site. Other than the metal-binding ligands, none of the other residues mentioned above are well conserved in B3 MBLs.
Non-covalent interactions such as hydrogen bonds, π–π aromatic stacking, cation–π interactions, hydrophobic interactions, halogen bonds, and salt bridges are vital in drug design, particularly to improve the molecular recognition and binding affinity between the protein–ligand interfaces [
35]. Taking this into account as well as the hydrophobic nature of the Bleg1_2478 active site, several peptides with hydrophobic residues were screened, designed and further derivatized in silico. In vitro assay showed that two peptides, namely RSWPWH and SSWWDR, inhibited Bleg1_2478 by approximately 50% (IC
50) at only 0.90 and 0.50 μM, respectively (
Figure 4 and
Figure 5,
Table 5). Almost complete inhibition of Bleg1_2478 was achieved when 10 µM RSWPWH and 20 µM SSWWDR were used. The IC
50 concentrations of the peptides are lower compared to other inhibitors such as pyrrozole derivative compound [
36] and hydroxamic acid derivatives: 2,5-substituted benzophenone hydroxamic acid [
37] and cysteine-containing peptides [
23]; and are comparable to other reported inhibitors against B3 MBL such as penicillin (β-lactam) derived inhibitors [
38], dicarboxylic acid derivatives, i.e., N-heterocyclic dicarboxylic acid [
39], and thioester-based inhibitors, i.e., amino acid thioester derivatives [
40]. To date, homo-cysteinyl peptide inhibitor recorded the lowest inhibitory concentration of 2 nM [
24].
Prediction of possible binding sites of the inhibitory peptides on Bleg1_2478 via random docking showed that RSWPWH might bind to a site slightly away from Bleg1_2478 active site (
Figure 7A,B) while SSWWDR might bind to a site that was far away from the active site (
Figure 9A,B). It may be unlikely that such binding sites could cause inhibition of Bleg1_2478 due to their distance away from key structural and functional residues important for Bleg1_2478. Fixed docking analyses showed that RSWPWH bound at the vicinity of Bleg1_2478 active site (
Figure 8A,B) while SSWWDR bound at the active site itself (
Figure 10A,B). Such positionings of the peptides would reduce or block the accessibility of the active site from ampicillin. Analyses of the interactions between the inhibitory peptides and Bleg1_2478 from fixed docking simulations revealed that both peptides bound to several key residues postulated to be important for substrate binding and catalytic activity of Bleg1_2478. They were Phe153, Ser156, His167, His191 and Phe206. Phe153 is a hydrophobic residue in Bleg1_2478 substrate binding site. Hydrophobic residues in the binding cavity of MBLs were predicted to enable the interaction between the enzymes and β-lactams to allow the hydrophobic β side chain of β-lactams into the binding pocket [
22,
34]. Ser156, on the other hand, was predicted to provide a second shell effect in lodging β-lactam substrates in the binding pocket by forming an extended network of hydrogen bonds with the backbone of nitrogen of Asp58 and His191 of Bleg1_2478 [
22]. Hydrogen bonds and hydrophobic interactions can support each other mutually. When a hydrogen bond is present next to the side chain of the ligand, it elevates the strength of the hydrophobic interaction by holding the side chain closer and firmly against the hydrophobic pocket. The improved strength and stabilized geometry of the hydrophobic side chain help to increase the strength of the hydrogen bond [
41]. His-191 is a putative Zn
2+-binding ligand important for catalysis [
22]. As these functionally important residues in the active site of Bleg1_2478 are predicted to form interactions with the inhibitory peptides, this, in turn, hindered the binding and hydrolysis of ampicillin by the enzyme, as observed from the results of the inhibition assay. Based on these results, it may be more probable that the inhibitory peptides bind near or at the active site of Bleg1_2478 compared to the secondary sites observed from random docking.
The basic or cationic nature of RSWPWH inhibitory peptide, as well as its predicted binding site at an accessible area of Bleg1_2478 (
Figure 8A,B), may have contributed to its ease of interaction with the enzyme, hence, giving forth more favourable and spontaneous binding (
Figure 6). However, it may be more exposed to pH changes caused by the cellular environment, making it more susceptible to dissociate from the protein (
Table 8). As for SSWWDR, its neutral net charge and its predicted binding site at a less accessible narrow groove of Bleg1_2478 active site (
Figure 10A,B) may have resulted in less spontaneous binding (
Figure 6). As the binding site is less exposed to the cellular environment, particularly to pH changes, SSWWDR is less susceptible to dissociate from the protein; hence giving forth a K
d value that is significantly higher than RSWPWH (
Table 6).
Future studies including in vitro stability of the inhibitory peptides, mutational analyses of Bleg1_2478 secondary binding sites and X-ray crystallography of the Bleg1_2478-peptide complexes will be undertaken to gain more detailed insights of the peptides, their binding sites and key interactions involved. This in turn will enable the mechanism of action of the inhibitory peptides to be deduced.

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}