
Molecular Docking Study on Several Benzoic Acid Derivatives against SARS-CoV-2



Abstract
:1. Introduction
2. Results and Discussion
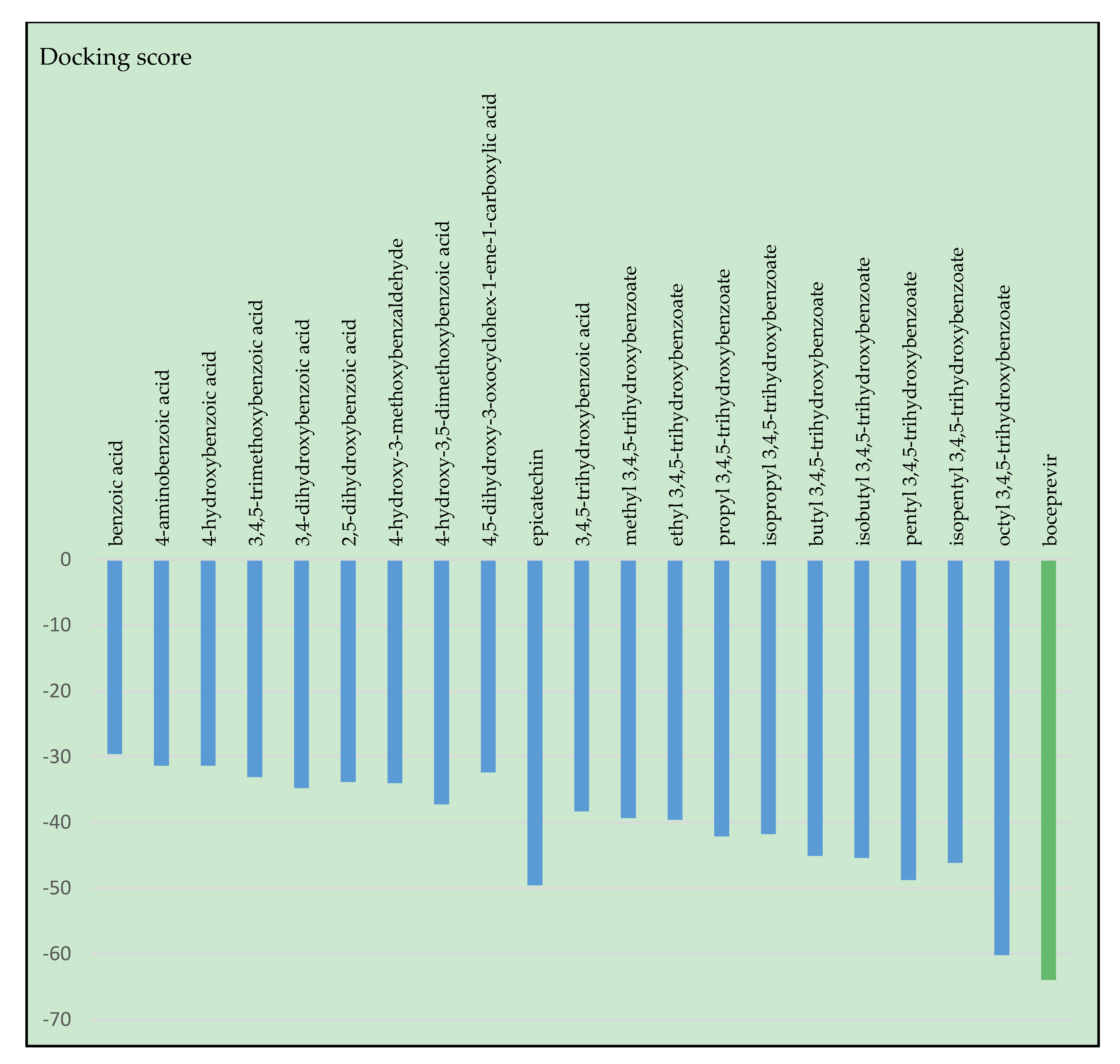
2.1. Results of Molecular Docking Simulations
2.2. Results of Oral Bioavailability Evaluation
2.3. Results of Quantum Reactivty Analysis
2.4. Results of Principal Component Analysis (PCA)
3. Methods
3.1. Methods for Molecular Docking Simulations
3.2. Methods for Energy Minimization and Computation of Molecular Properties
3.3. Methods for Principal Component Analysis (PCA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A | A-electron affinity |
| B3LYP | Becke, 3-parameter, Lee-Yang-Parr |
| DAA | direct-acting antiviral agent |
| DFT | Density Functional Theory |
| EHOMO | energy of the highest occupied molecular orbital |
| ELUMO | energy of the lowest unoccupied molecular orbital |
| FDA | Food and Drug Administration (U.S.) |
| HBA | hydrogen bond acceptor |
| HBD | hydrogen bond donor |
| HCV | hepatitis C virus |
| HOMO | the highest occupied molecular orbital |
| I | ionization potential |
| logP | octanol-water partition coefficient |
| LUMO | the highest occupied molecular orbital |
| LV | Lipinski’s violations |
| MW | Molecular weight |
| PCA | Principal component analysis |
| PSA | polar surface area |
| r | Pearson correlation coefficient |
| rb | rotatable bond |
| RMSD | Root mean square deviation |
| RNA | ribonucleic acid |
| SARS | CoV-2-Severe acute respiratory syndrome coronavirus 2 |
| ΔEgap | energy gap between frontier molecular orbitals |
| µ | chemical potential |
| η | global chemical hardness |
| σ | global softness |
| χ | electronegativity |
| ω | global electrophilicity index |
References
- Vijayakumar, B.G.; Ramesh, D.; Joji, A.; Prakasan, J.J.; Kannan, T. In silico pharmacokinetic and molecular docking studies of natural flavonoids and synthetic indole chalcones against essential proteins of SARS-CoV-2 proteins. Eur. J. Pharmacol. 2020, 886, 173448. [Google Scholar] [CrossRef] [PubMed]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.S.; Ali, D.; Alarifi, S.; Radhakrishnan, S.; Akbar, I. In silico molecular docking: Evaluation of coumarin based derivatives against SARS-CoV-2. J. Infect. Public Health 2020, 13, 1671–1677. [Google Scholar]
- El-hoshoudy, A.N. Investigating the potential antiviral activity drugs against SARS-CoV-2 by molecular docking simulation. J. Mol. Liq. 2020, 318, 113968. [Google Scholar] [CrossRef] [PubMed]
- Narkhede, R.R.; Pise, A.V.; Cheke, R.S.; Shinde, S.D. Recognition of Natural Products as Potential Inhibitors of COVID-19 Main Protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect. 2020, 10, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Kiser, J.J.; Flexner, C. Direct-acting antiviral agents for hepatitis C virus infection. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 427–449. [Google Scholar] [CrossRef] [Green Version]
- Anson, B.; Mesecar, A. 6WNP X-ray Structure of SARS-CoV-2 Main Protease Bound to Boceprevir at 1.45 A. Available online: https://www.rcsb.org/structure/6WNP (accessed on 3 December 2020).
- Fan, K.; Wei, P.; Feng, Q.; Chen, S.; Huang, C.; Ma, L.; Lai, B.; Pei, J.; Liu, Y.; Chen, J.; et al. Biosynthesis, purification, and substrate specificity of severe acute respiratory syndrome coronavirus 3C-like proteinase. J. Biol. Chem. 2004, 279, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Ryu, Y.B.; Park, S.J.; Kim, Y.M.; Lee, J.Y.; Seo, W.D.; Chang, J.S.; Park, K.H.; Rho, M.C.; Lee, W.S. SARS-CoV 3CLpro inhibitory effects of quinone-methide triterpenes from Tripterygium regelii. Bioorg. Med. Chem. Lett. 2010, 20, 1873–1876. [Google Scholar] [CrossRef]
- Kumar, Y.; Singh, H.; Patel, C.N. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation-based drug-repurposing. J. Infect. Public Health 2020. [Google Scholar] [CrossRef]
- Kamboj, A.; Saluja, A.K.; Kumar, M.; Atri, P. Antiviral activity of plant polyphenols. J. Pharm. Res. 2012, 5, 2402–2412. [Google Scholar]
- Park, E.S.; Moon, W.S.; Song, M.J.; Kim, M.N.; Chung, K.H.; Yoon, J.S. Antimicrobial activity of phenol and benzoic acid derivatives. Int. Biodeterior. Biodegrad. 2001, 47, 209–214. [Google Scholar] [CrossRef]
- Kratky, M.; Konecna, K.; Janousek, J.; Brablikova, M.; Jand’ourek, O.; Trejtnar, F.; Stolarikova, J.; Vinsova, J. 4-Aminobenzoic Acid Derivatives: Converting Folate Precursor to Antimicrobial and Cytotoxic Agents. Biomolecules 2019, 10, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manach, C.; Scalbert, A.; Morand, C.; Rémésy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passamonti, S.; Terdoslavich, M.; Franca, R.; Vanzo, A.; Tramer, F.; Braidot, E.; Petrussa, E.; Vianello, A. Bioavailability of flavonoids: A review of their membrane transport and the function of bilitranslocase in animal and plant organisms. Curr. Drug Metab. 2009, 10, 369–394. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.J.; Kremer, M.J.; Luo, M.; Vriend, G.; Arnold, E.; Kamer, G.; Rossman, M.G.; McKinlay, M.A.; Diana, G.D.; Otto, M.J. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science 1986, 233, 1286–1293. [Google Scholar] [CrossRef]
- Kirsten, G.; Joachim, K.; Eva, L.; Wali, H.; Derksena, A.; Detersa, A.; Andreas, H. Proanthocyanidin-enriched extract from Myrothamnus flabellifolia Welw. exerts antiviral activity against herpes simplex virus type-1 by inhibition of viral adsorption and penetration. J. Ethnopharmacol. 2011, 134, 468–474. [Google Scholar]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 252, 117652. [Google Scholar] [CrossRef]
- Cardoso, W.B.; Mendanha, S.A. Molecular dynamics simulation of docking structures of SARS-CoV-2 main protease and HIV protease inhibitors. J. Mol. Struct. 2021, 1225, 129143. [Google Scholar] [CrossRef]
- Peele, K.A.; Durthi, C.P.; Srihansa, T.; Krupanidhi, S.; Sai, A.V.; Babu, D.J.; Indira, M.; Reddy, A.R.; Venkateswarulu, T.C. Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study. Inform. Med. Unlocked. 2020, 19, 100345. [Google Scholar] [CrossRef]
- Wang, J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef]
- Hetal, T.; Bindesh, P.; Sneha, T. A review on techniques for oral bioavailability enhancement of drugs. Int. J. Pharm. Sci. Rev. Res. 2010, 4, 203–223. [Google Scholar]
- Kahkeshani, N.; Farzaei, F.; Fotouhi, M.; Alavi, S.S.; Bahramsoltani, R.; Naseri, R.; Momtaz, S.; Abbasabadi, Z.; Rahimi, R.; Farzaei, M.H.; et al. Pharmacological effects of gallic acid in health and diseases: A mechanistic review. Iran J. Basic Med. Sci. 2019, 22, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Nayeem, N.; Asdaq, S.M.B.; Salem, H.; Alfqy, S.A. Gallic Acid: A Promising Lead Molecule for Drug Development. J. Appl. Pharm. 2016, 8, 1000213. [Google Scholar] [CrossRef] [Green Version]
- Takai, E.; Hirano, A.; Shirak, K. Effects of alkyl chain length of gallate on self-association and membrane binding. J. Biochem. 2011, 150, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Król, E.; de Sousa Borges, A.; da Silva, I.; Polaquini, C.R.; Regasini, L.O.; Ferreira, H.; Scheffers, D.J. Antibacterial activity of alkyl gallates is a combination of direct targeting of FtsZ and permeabilization of bacterial membranes. Front. Microbiol. 2015. [Google Scholar] [CrossRef]
- Fujita, K.; Kubo, I. Plasma membrane injury induced by nonyl gallate in Saccharomyces cerevisiae. J. Appl. Microbiol. 2002, 92, 1035–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A. Lead-and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Koopmans, T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Sastri, V.S.; Perumareddi, J.R. Molecular orbital theoretical studies of some organic corrosion inhibitors. Corrosion 1997, 53, 617–622. [Google Scholar] [CrossRef]
- Yankova, R.; Genieva, S.; Halachev, N.; Dimitrova, G. Molecular structure, vibrational spectra, MEP, HOMO-LUMO and NBO analysis of Hf(SeO3)(SeO4)(H2O)4. J. Mol. Struct. 2016, 1106, 82–88. [Google Scholar] [CrossRef]
- Landeros-Martinez, L.L.; Orrantia-Borunda, E.; Flores-Holguin, N. DFT Chemical Reactivity analysis of biological molecules in the presence of silver ion. Org. Chem. Curr. Res. 2015, 4, 153. [Google Scholar] [CrossRef]
- Pirvu, L.; Neagu, G.; Terchescu, I.; Albu, B.; Stefaniu, A. Comparative studies of two vegetal extracts from Stokesia laevis and Geranium pratense: Polyphenol profile, cytotoxic effect and antiproliferative activity. Open Chem. 2020, 18, 488–502. [Google Scholar] [CrossRef]
- Jolliffe, I.T. Principal Component Analysis; Springer: New York, NY, USA, 2002; ISBN 978-0-387-22440-4. [Google Scholar]
- Jackson, J.E. A Use’s Guide to Principal Components; John Wiley & Sons: New York, NY, USA, 1991; ISBN 9780471725336. [Google Scholar] [CrossRef]
- Saporta, G.; Niang, N. Chapter 1: Principal component analysis: Application to statistical process control. In Data Analysis; Govaert, G., Ed.; John Wiley & Sons: London, UK, 2009; pp. 1–23. [Google Scholar]
- Abdi, H.; Williams, L.J. Principal component analysis. WIREs Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef]
- Hehre, W.J. A Guide to Molecular Mechanics and Quantum Chemical Calculations; Wavefunction, Inc.: Irvine, CA, USA, 2003. [Google Scholar]
- Halgren, A.T. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | MW | PSA | HBD | HBA | LogP | rb | LV |
|---|---|---|---|---|---|---|---|
| Benzoic acid | 122.123 | 33.690 | 1 | 2 | 0.79 | 1 | 0 |
| 4-Aminobenzoic acid | 137.138 | 58.471 | 3 | 3 | −0.93 | 1 | 0 |
| 4-Hydroxybenzoic acid | 138.122 | 53.444 | 2 | 3 | −0.29 | 1 | 0 |
| 3,4,5-Trimethoxybenzoic acid | 212.201 | 53.223 | 1 | 5 | −2.14 | 4 | 0 |
| 3,4-Dihydroxybenzoic acid | 154.121 | 71.217 | 3 | 4 | −1.37 | 1 | 0 |
| 2,5-Dihydroxybenzoic acid | 154.121 | 71.262 | 3 | 4 | 0.81 | 1 | 0 |
| 4-Hydroxy-3-methoxybenzaldehyde | 152.149 | 41.012 | 1 | 3 | −1.53 | 2 | 0 |
| 4-Hydroxy-3,5-dimethoxybenzoic acid | 198.174 | 64.706 | 2 | 5 | −2.24 | 3 | 0 |
| 4,5-Dihydroxy-3-oxocyclohex-1-ene-1-carboxylic acid | 172.136 | 83.671 | 3 | 5 | −0.92 | 1 | 0 |
| Epicatechin | 290.271 | 101.294 | 5 | 6 | −3.72 | 1 | 0 |
| 3,4,5-Trihydroxybenzoic acid | 170.12 | 89.408 | 4 | 5 | −2.46 | 1 | 0 |
| Methyl 3,4,5-trihydroxybenzoate | 184.147 | 75.752 | 3 | 5 | −2.19 | 2 | 0 |
| Ethyl 3,4,5-trihydroxybenzoate | 198.174 | 75.425 | 3 | 5 | −1.86 | 3 | 0 |
| Propyl 3,4,5-trihydroxybenzoate | 212.201 | 75.433 | 3 | 5 | −1.37 | 4 | 0 |
| i-Propyl 3,4,5-trihydroxybenzoate | 212.201 | 75.068 | 3 | 5 | −1.54 | 3 | 0 |
| Butyl 3,4,5-trihydroxybenzoate | 226.228 | 75.433 | 3 | 5 | −0.95 | 5 | 0 |
| i-Butyl 3,4,5-trihydroxybenzoate | 226.228 | 75.149 | 3 | 5 | −0.97 | 4 | 0 |
| Pentyl 3,4,5-trihydroxybenzoate | 240.255 | 75.426 | 3 | 5 | −0.54 | 6 | 0 |
| i-Pentyl 3,4,5-trihydroxybenzoate | 240.255 | 75.415 | 3 | 5 | −0.62 | 5 | 0 |
| Octyl 3,4,5-trihydroxybenzoate | 282.336 | 75.390 | 3 | 5 | 0.72 | 9 | 0 |
| F1 | F2 | F3 | F4 | F5 | F6 | |
|---|---|---|---|---|---|---|
| Eigenvalue | 6.529 | 1.440 | 1.087 | 0.480 | 0.291 | 0.084 |
| Variability, % | 65.29% | 14.40% | 10.87% | 4.80% | 2.91% | 0.84% |
| Cumulative, % | 65.29% | 79.69% | 90.56% | 95.36% | 98.27% | 99.11% |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stefaniu, A.; Pirvu, L.; Albu, B.; Pintilie, L. Molecular Docking Study on Several Benzoic Acid Derivatives against SARS-CoV-2. Molecules 2020, 25, 5828. https://doi.org/10.3390/molecules25245828
Stefaniu A, Pirvu L, Albu B, Pintilie L. Molecular Docking Study on Several Benzoic Acid Derivatives against SARS-CoV-2. Molecules. 2020; 25(24):5828. https://doi.org/10.3390/molecules25245828
Chicago/Turabian StyleStefaniu, Amalia, Lucia Pirvu, Bujor Albu, and Lucia Pintilie. 2020. "Molecular Docking Study on Several Benzoic Acid Derivatives against SARS-CoV-2" Molecules 25, no. 24: 5828. https://doi.org/10.3390/molecules25245828