
SiO2-PVA-Fe(acac)3 Hybrid Based Superparamagnetic Nanocomposites for Nanomedicine: Morpho-textural Evaluation and In Vitro Cytotoxicity Assay

 ,
, 
 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
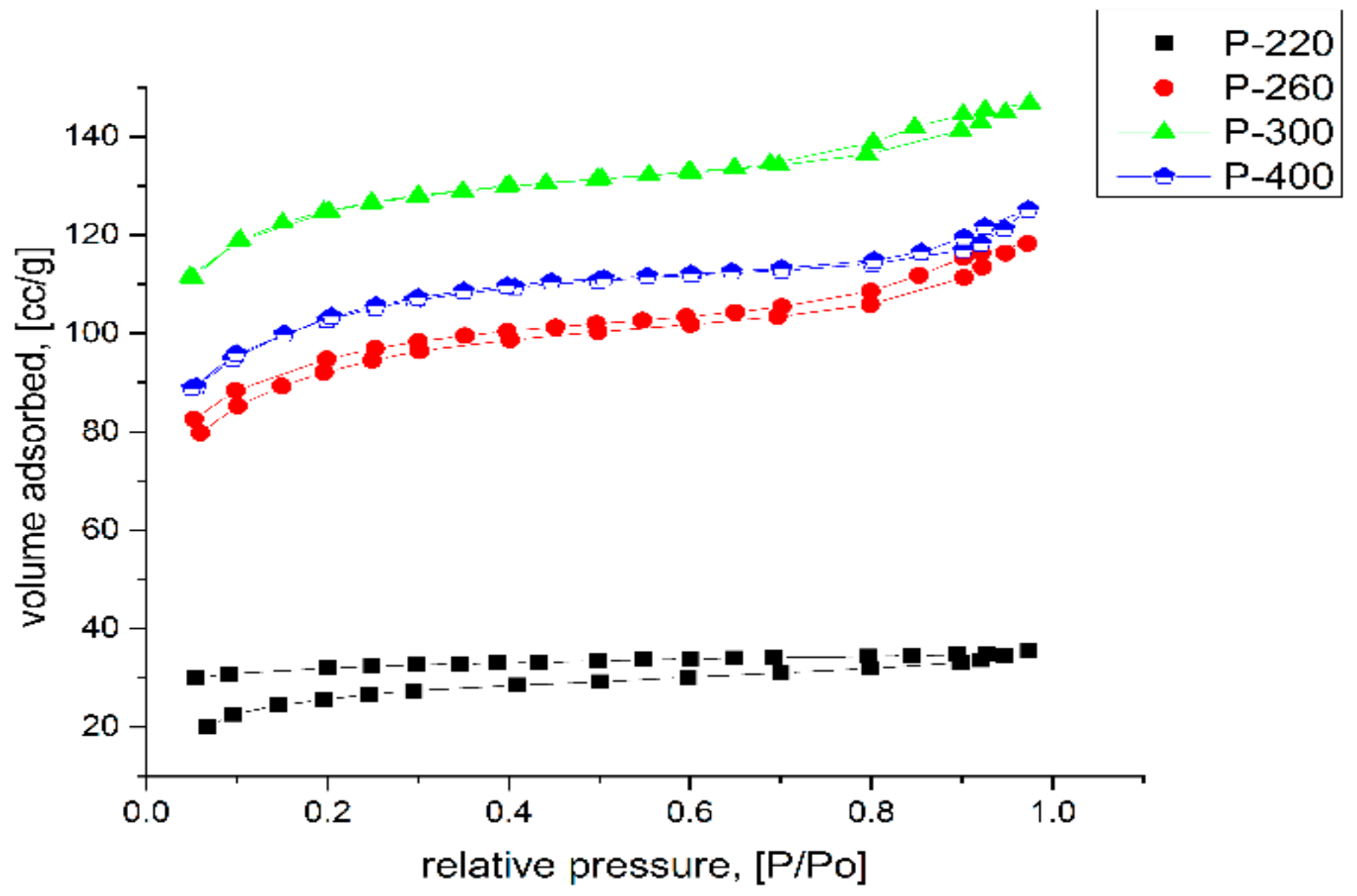
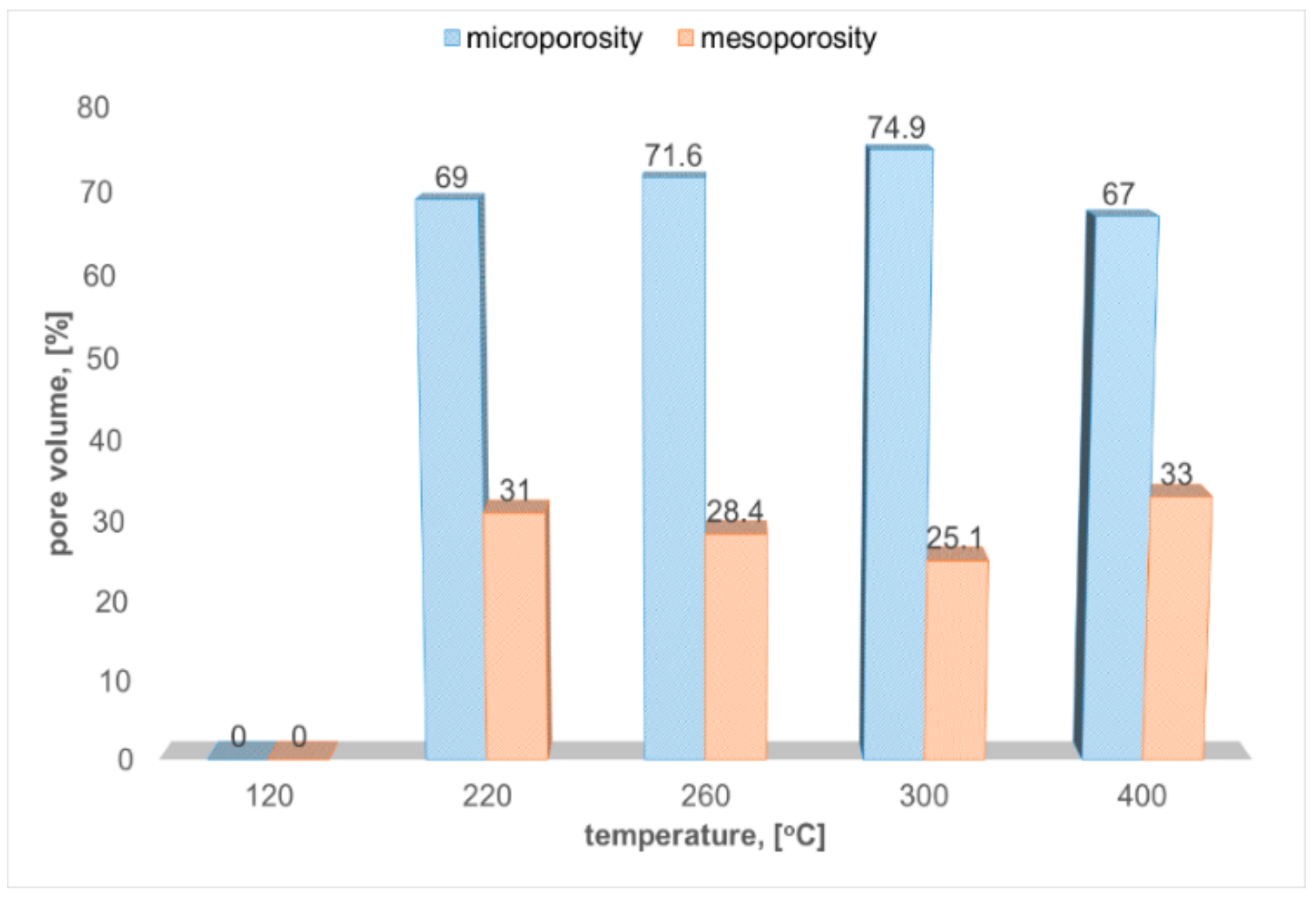
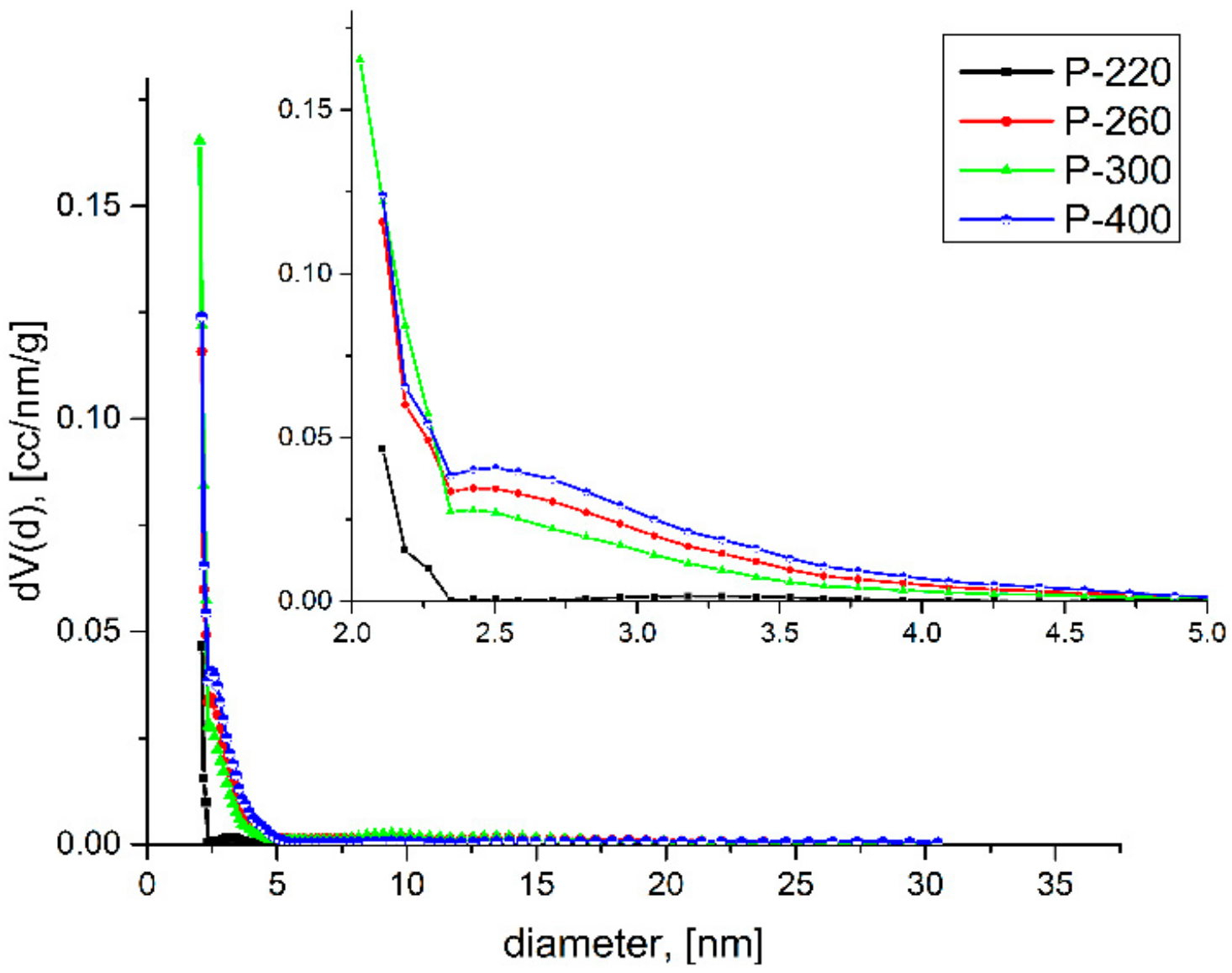
2.1. Nitrogen Porosimetry
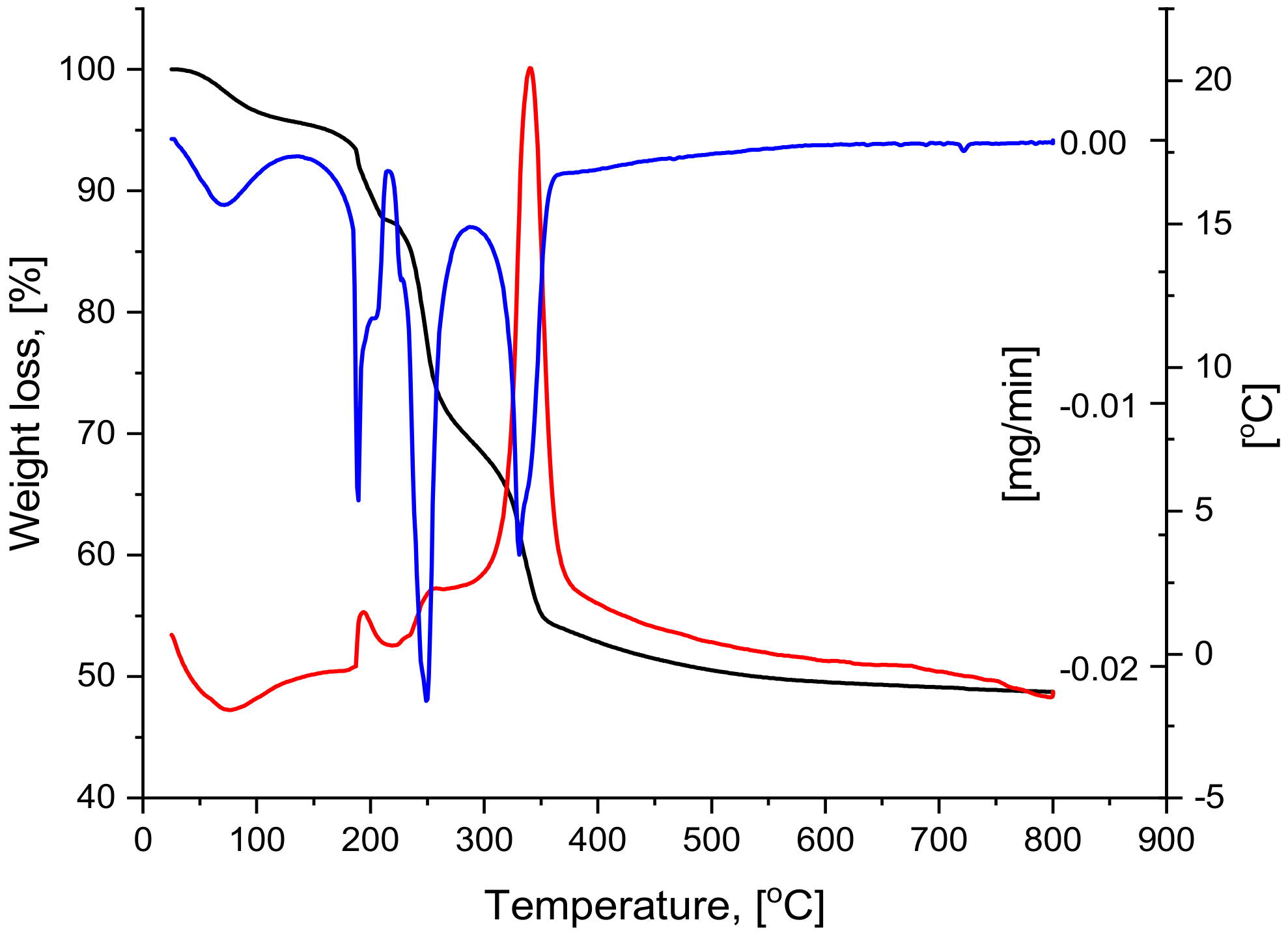
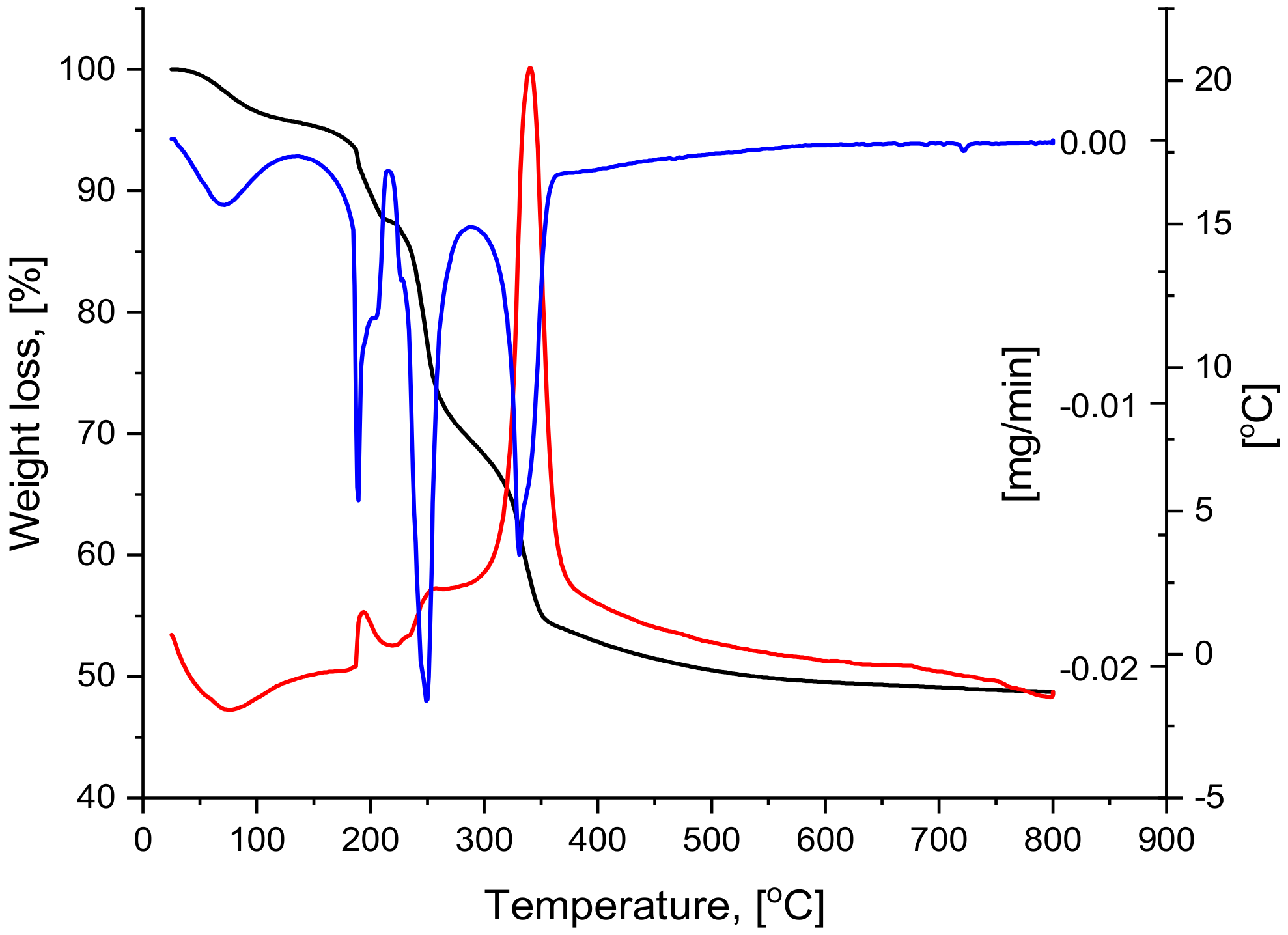
2.2. Thermal Analysis
2.3. Transmission Electron Microscopy
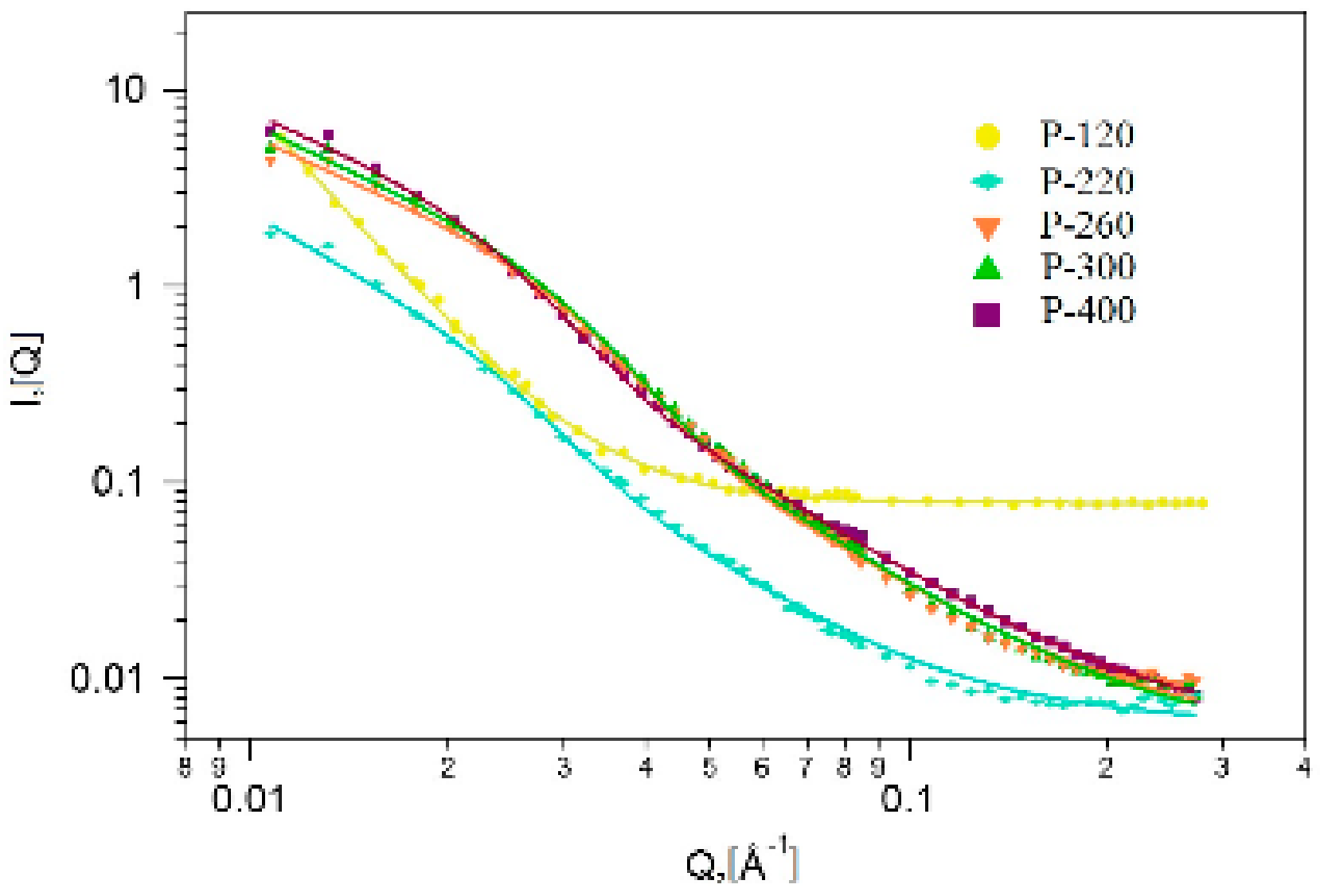
2.4. Small Angle Neutron Scattering (SANS)
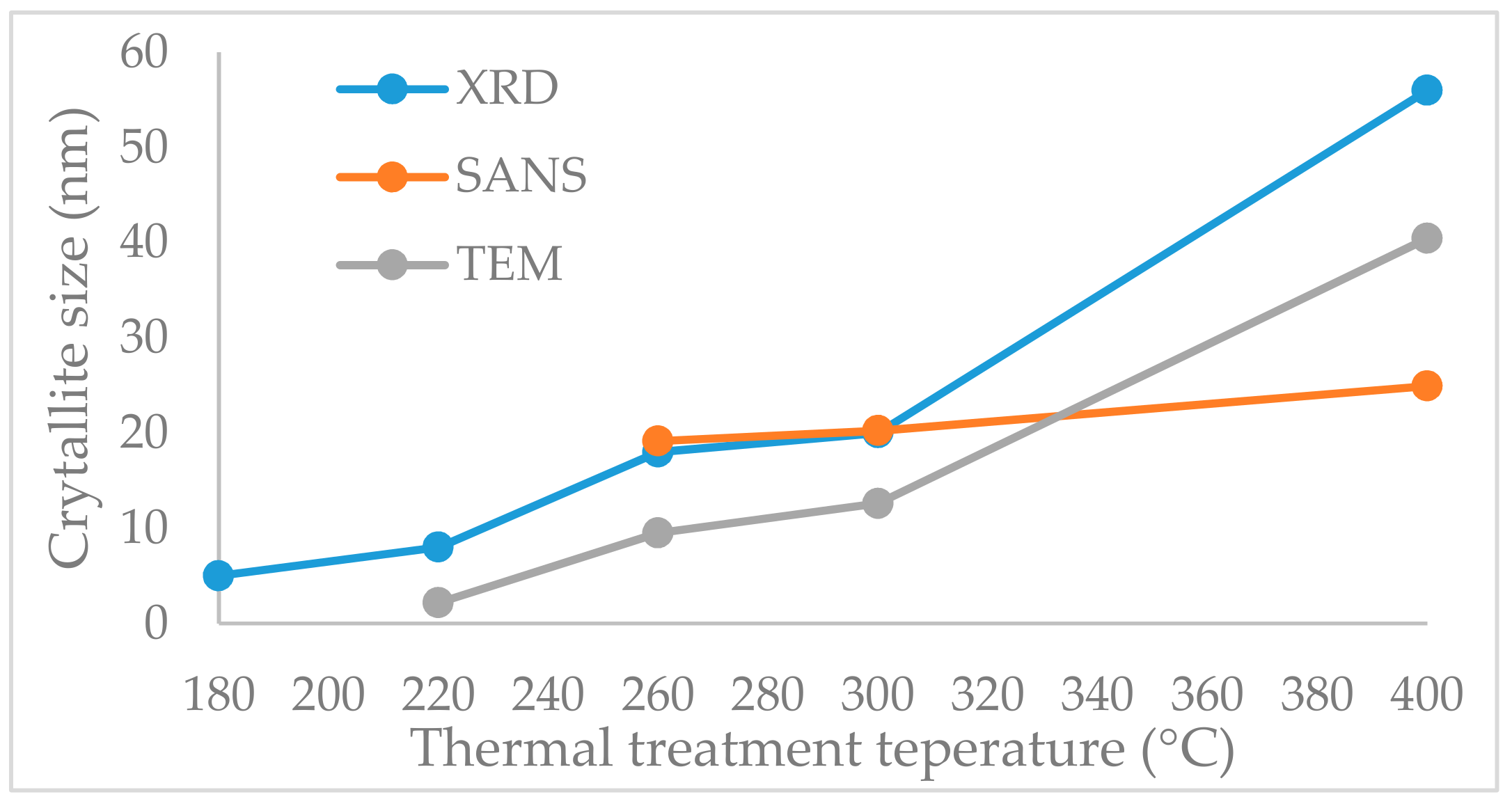
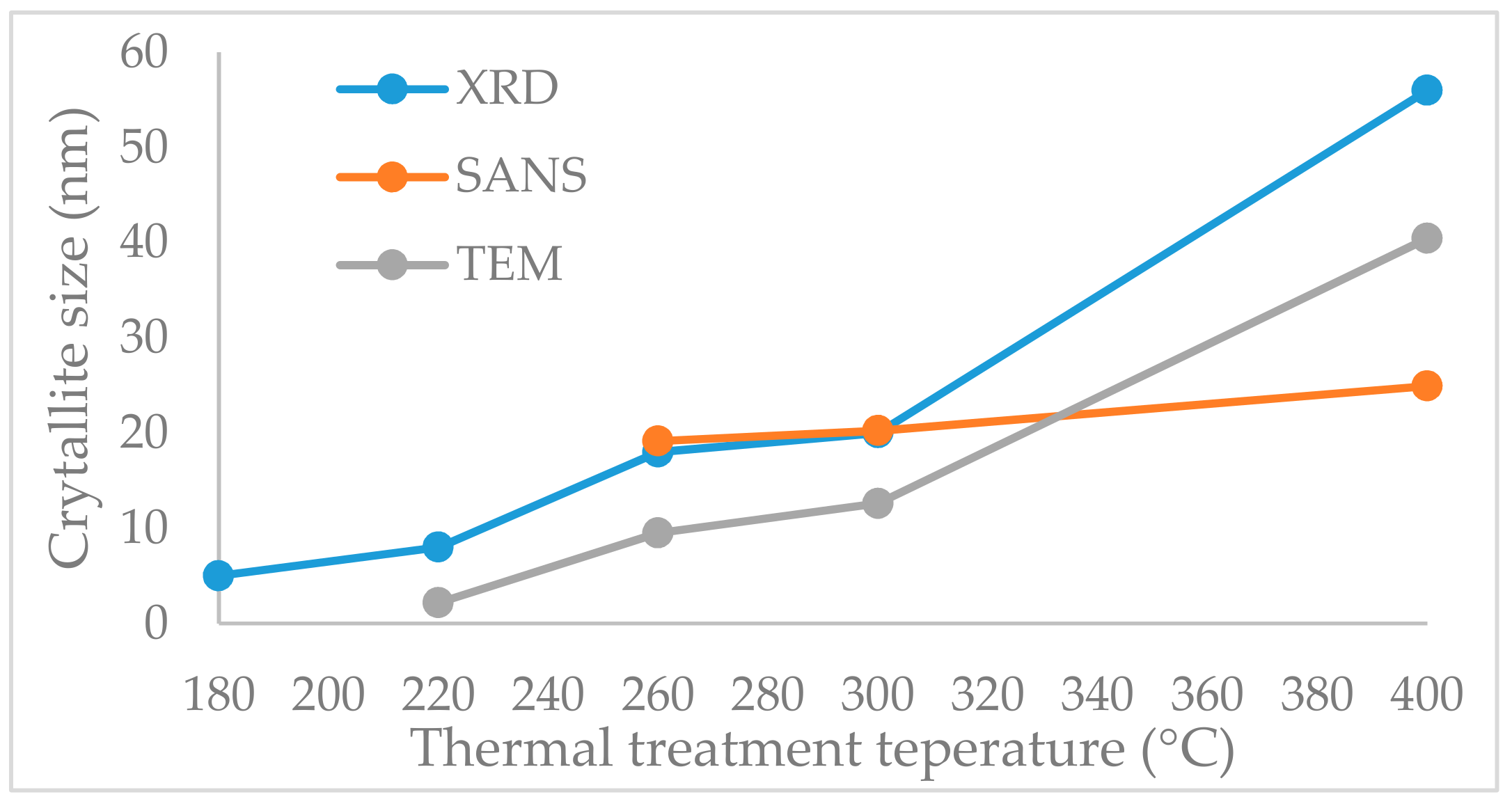
2.5. Crystallite Size
2.6. Zeta Potential Measurements
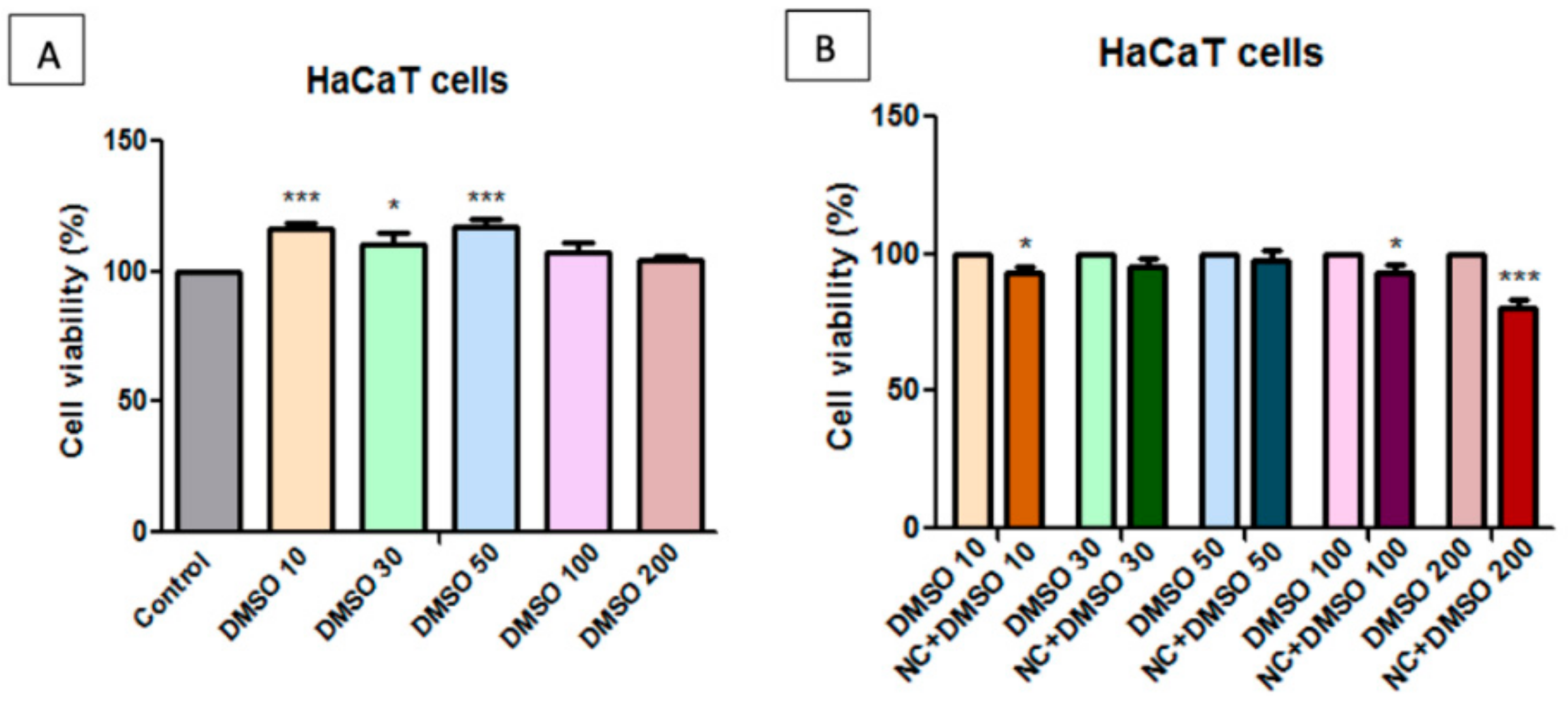
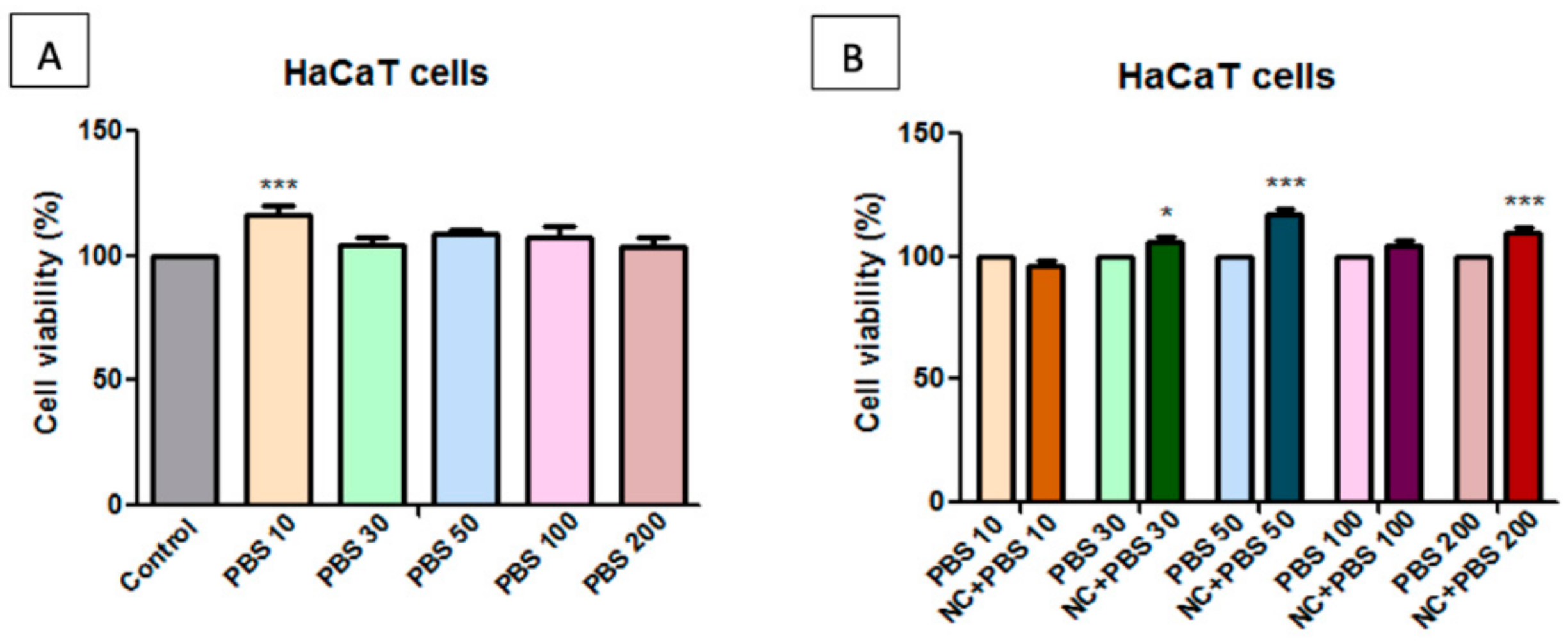
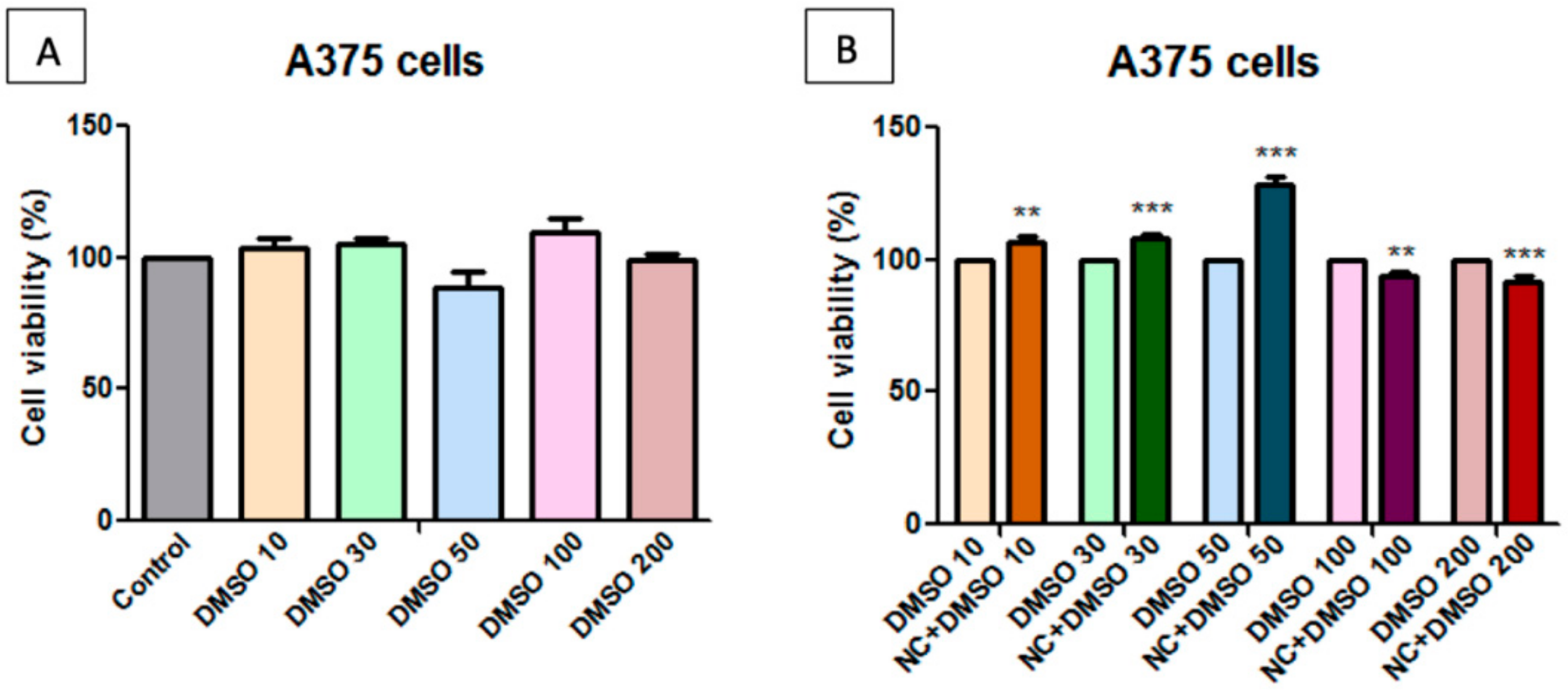
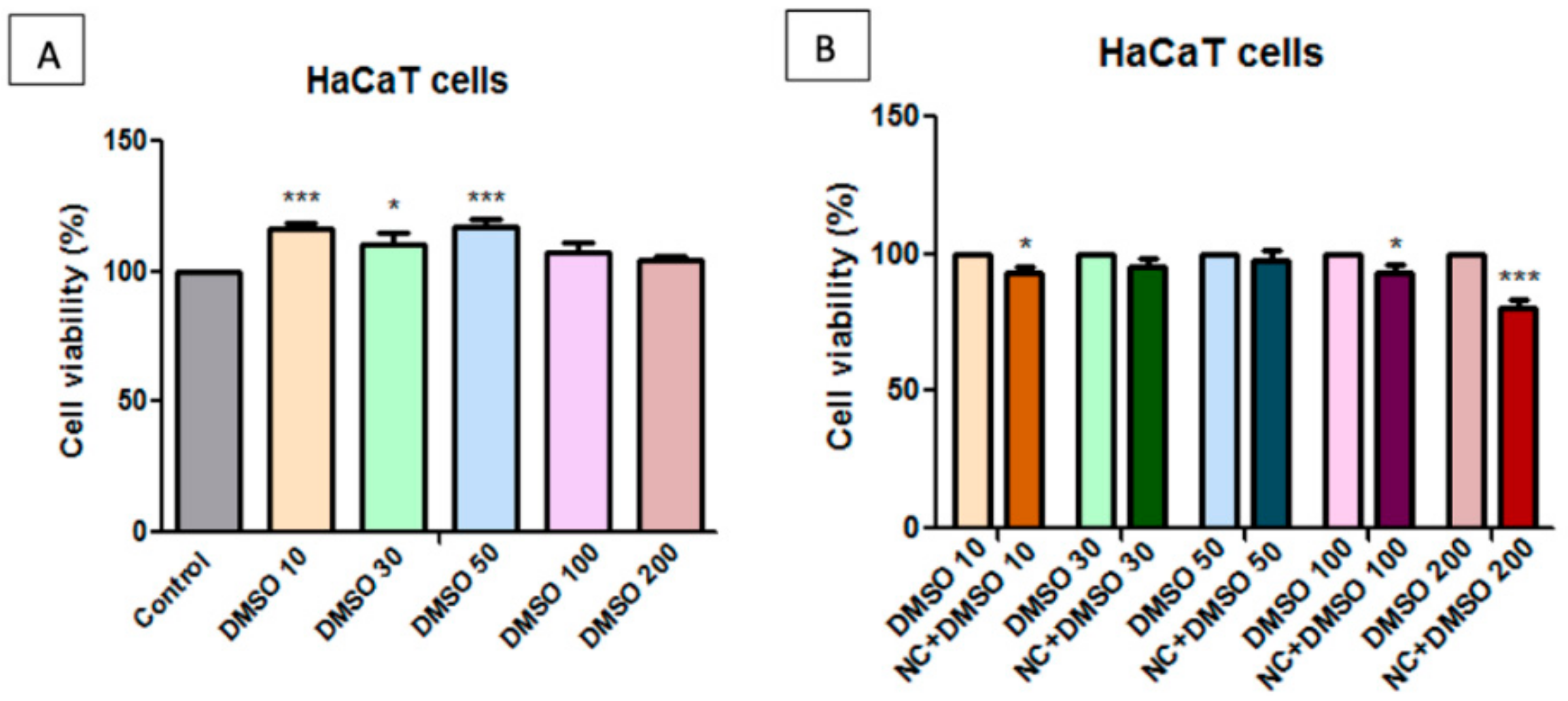
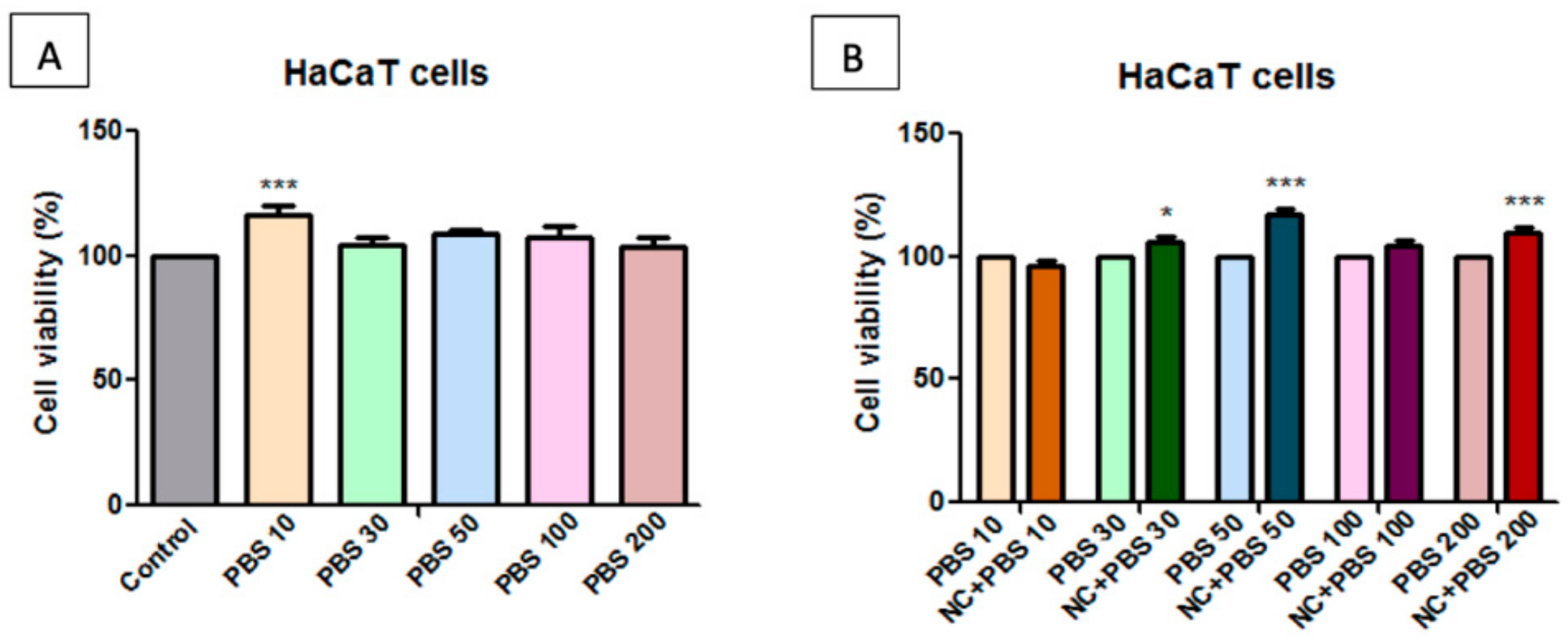
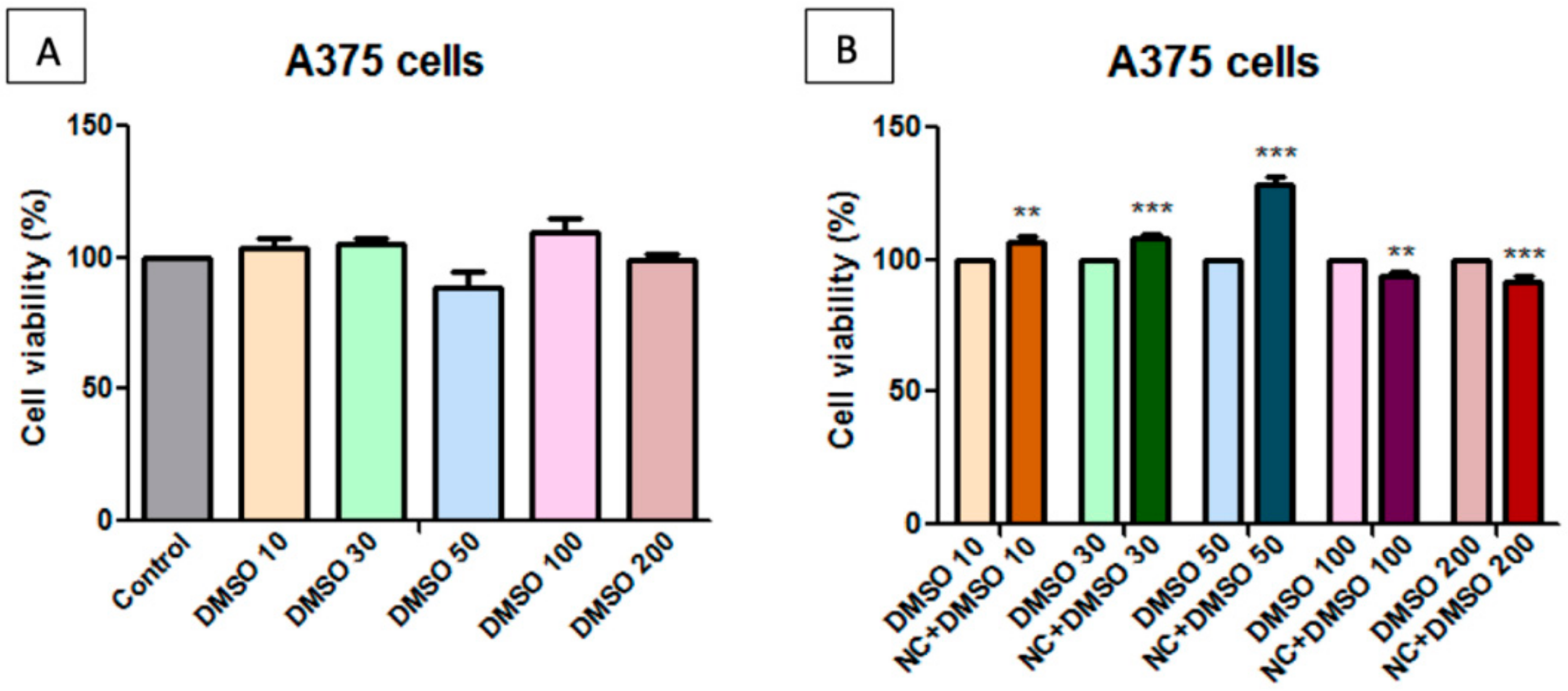
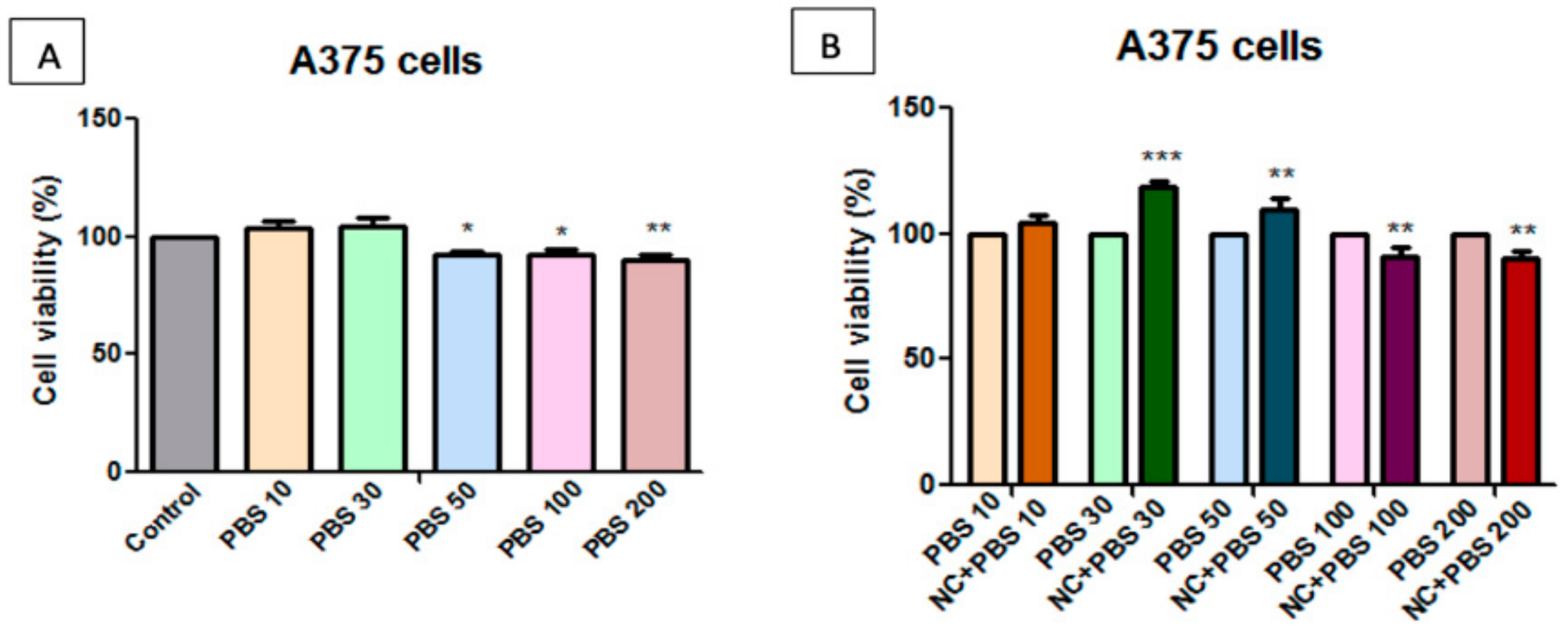
2.7. Toxicological Evaluation of Magnetic Nanocomposite P-260
2.8. Evaluation of P-260 (NC) Nanocomposite by Studying the Effect on Human Keratinocytes Migration and Proliferation Potential
3. Materials and Methods
3.1. Materials and Synthesis Protocol
3.2. Characterization
3.3. In vitro Cytotoxicity Testing
3.4. Cell Culture
3.5. Cell Viability Assessment—MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] Assay
3.6. Migratory and Proliferative Potential – aA Wound Healing Technique
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, L.; Jin, R.; Sun, R.; Du, L.; Liu, L.; Zhang, K.; Ai, H.; Guo, Y. Superparamagnetic iron oxide nanoparticles as magnetic resonance imaging contrast agents and induced autophagy response in endothelial progenitor cells. J. Biomed. Nanotechnol. 2019, 15, 396–404. [Google Scholar] [CrossRef]
- Masdeu, G.; Kralj, S.; Pajk, S.; López-Santín, J.; Makovec, D.; Álvaro, G. Hybrid chloroperoxidase-magnetic nanoparticle clusters: Effect of functionalization on biocatalyst performance. J. Chem. Technol. Biotechnol. 2018, 93, 233–245. [Google Scholar] [CrossRef]
- Gokduman, K.; Bestepe, F.; Li, L.; Yarmush, M.L.; Usta, O.B. Dose-, treatment and time-dependent toxicity of superparamagnetic iron oxide nanoparticles on primary rat hepatocytes. Nanomedicine 2018, 13, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Vallabani, N.V.S.; Singh, S. Recent advances and future prospects of iron oxide nanoparticles in biomedicine and diagnostics. 3 Biotech 2018, 8, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Agnihotri, S.; Roy, I. Preparation and characterization of superparamagnetic iron oxide nanoparticles for magnetically guided drug delivery. Int. J. Nanomed. 2018, 13, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Jin, W.; Chen, H.; Zhang, Q. Superparamagnetic iron oxide nanoparticles for cancer diagnosis and therapy. J. Biomed. Nanotechnol. 2019, 15, 215–235. [Google Scholar] [CrossRef]
- Jung, E.; Kim, S.W.; Cho, A.; Kim, Y.-J.; Jeong, G.-J.; Kim, J.; Bhang, S.H.; Yu, T. Synthesis of Sub 3 nm-Sized Uniform Magnetite Nanoparticles Using Reverse Micelle Method for Biomedical Application. Materials 2019, 12, 3850. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, L.; de la Cueva, L.; Moros, M.; Mazarío, M.; de Bernardo, S.; de la Fuente, J.M.; Morales, M.P.; Salas, G. Aggregation effects on the magnetic properties of iron oxide colloids. Nanotechnology 2019, 30, 11. [Google Scholar] [CrossRef] [Green Version]
- Ling, W.; Wang, M.; Xiong, C.; Xie, D.; Chen, Q.; Chu, X.; Qiu, X.; Li, Y.; Xiao, X. Synthesis, surface modification, and applications of magnetic iron oxide nanoparticles. J. Mater. Res. 2019, 34, 1828–1844. [Google Scholar] [CrossRef]
- Turcheniuk, K.; Tarasevych, A.V.; Kukhar, V.P.; Boukherroub, R.; Szunerits, S. Recent advances in surface chemistry strategies for the fabrication of functional iron oxide based magnetic nanoparticles. Nanoscale 2013, 5, 10729–10752. [Google Scholar] [CrossRef]
- Sodipo, K.B.; Aziz, A.A. Recent advances in synthesis and surface modification of superparamagnetic iron oxide nanoparticles with silica. J. Magn. Magn. Mater. 2016, 416, 275–291. [Google Scholar] [CrossRef]
- Dudás, Z.; Almásy, L. Effect of the Organic Groups on the Performance of Hybrid Silica Based Materials used as Supports for Biomolecules. Curr. Org. Chem. 2017, 21, 2760–2767. [Google Scholar] [CrossRef]
- Gonçalves, M.C. Sol-Gel Silica Nanoparticles in Medicine: A Natural Choice. Design, Synthesis and Products. Molecules 2018, 23, 2021. [Google Scholar]
- Chen, F.; Hableel, G.; Zhao, E.R.; Jokerst, J.V. Multifunctional nanomedicine with silica: Role of silica in nanoparticles for theranostic, imaging, and drug monitoring. J. Colloid. Interface Sci. 2018, 521, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.F.; Ciciliatti, M.A.; Hechenleitner, A.A.W.; Penalva, R.; Agueros, M.; Irache, J.M.; Oliveira, A.J.A.; Farooqi, A.A.; Pineda, E.A.G. Superparamagnetic maghemite loaded poly (ε- caprolactone) nanocapsules: Characterization and synthesis optimization. Materials 2014, 19, 40–52. [Google Scholar]
- Costa, C.; Brandão, F.; Bessa, M.J.; Costa, S.; Valdiglesias, V.; Kiliç, G.; Fernández-Bertólez, N.; Quaresma, P.; Pereira, E.; Pásaro, E.; et al. In vitrocytotoxicity of superparamagnetic iron oxide nanoparticles on neuronal and glial cells. Evaluation of nanoparticle interference with viability tests. J. Appl.Toxicol. 2015, 36, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Patil, U.; Adireddy, S.; Jaiswal, A.; Mandava, S.; Lee, B.; Chrisey, D. In Vitro/In Vivo Toxicity Evaluation and Quantification of Iron Oxide Nanoparticles. Int. J. Mol. Sci. 2015, 16, 24417–24450. [Google Scholar] [CrossRef]
- Prodan, A.M.; Iconaru, S.L.; Ciobanu, C.S.; Chifiriuc, M.C.; Stoicea, M.; Predoi, D. Iron Oxide Magnetic Nanoparticles: Characterization and toxicity evaluation by in vitro and in vivo assays. J. Nanomat 2013, 1–10, article ID 587021. [Google Scholar] [CrossRef] [Green Version]
- Ngaboni Okassa, L.; Marchais, H.; Douziech-Eyrolles, L.; Cohen-Jonathan, S.; Soucé, M.; Dubois, P.; Chourpa, I. Development and characterization of sub-micron poly (d,l-lactide-co-glycolide) particles loaded with magnetite/maghemite nanoparticles. Int. J. Pharm 2005, 302, 187–196. [Google Scholar] [CrossRef]
- Li, H.; Wang, K.; Tuo, X.; Almásy, L.; Tian, Q.; Sun, G.; Henderson, M.J.; Li, Q.; Wacha, A.; Courtois, J.; et al. Thickness determination of ultrathin poly(acrylic acid) shell on γ-Fe2O3 nanocore via small-angle scattering. Mat. Chem. Phys. 2018, 204, 236–242. [Google Scholar] [CrossRef]
- Ramos Guivar, J.A.; Sadrollahi, E.; Menzel, D.; Ramos Fernandes, E.G.; López, E.O.; Torres, M.M.; Arsuaga, J.M.; Arencibia, A.; Litterst, F.J. Magnetic, structural and surface properties of functionalized maghemite nanoparticles for copper and lead adsorption. RSC Adv. 2017, 7, 28763–28779. [Google Scholar] [CrossRef] [Green Version]
- Woodard, L.E.; Dennis, C.L.; Borchers, J.A.; Attaluri, A.; Velarde, E. Nanoparticle architecture preserves magnetic properties during coating to enable robust multi-modal functionality. Sci. Rep. 2018, 8, 12706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadic, M.; Kralj, S.; Lalatonne, Y.; Motte, L. Iron oxide nanochains coated with silica: Synthesis, surface effects and magnetic properties. Appl. Surf. Sci. 2019, 476, 641–646. [Google Scholar] [CrossRef]
- Singh, N.; Jenkins, G.J.S.; Asadi, R.; Doak, S.H. Potential toxicity of superparamagnetic iron oxide nanoparticles (SPION). Nano Rev. 2010, 1, 5358. [Google Scholar] [CrossRef] [Green Version]
- Esqué-de los Ojos, D.; Pellicer, E.; Sort, J. The Influence of Pore Size on the Indentation Behavior of Metallic Nanoporous Materials: A Molecular Dynamics Study. Materials 2016, 9, 355. [Google Scholar] [CrossRef] [Green Version]
- Blechinger, J.; Bauer, A.T.; Torrano, A.A.; Gorzelanny, C.; Bräuchle, C.; Schneider, S.W. Uptake kinetics and nanotoxicity of silica nanoparticles are cell type dependent. Small 2013, 9, 3906, 3970–3980. [Google Scholar] [CrossRef]
- Yu, M.; Huang, S.; Yu, K.J.; Clyne, A.M. Dextran and polymer polyethylene glycol (PEG) coating reduce both 5 and 30 nm iron oxide nanoparticle cytotoxicity in 2D and 3D cell culture. Int. J. Mol. Sci. 2012, 13, 5554–5570. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Jadaun, A.; Arora, V.; Sinha, R.K.; Biyani, N.; Jain, V.K. In vitro toxicity assessment of chitosan oligosaccharide coated iron oxide nanoparticles. Toxicol. Rep. 2015, 2, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Ebrahiminezhad, A.; Rasoul-Amini, S.; Kouhpayeh, A.; Davaran, S.; Barar, J.; Ghasemi, Y. Impacts of amine functionalized iron oxide nanoparticles on HepG2 cell line. Curr. Nanosci. 2015, 11, 113–119. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Simchi, A.; Imani, M. Cytotoxicity of Uncoated and Polyvinyl Alcohol Coated Superparamagnetic Iron Oxide Nanoparticles. J. Phys. Chem. C 2009, 113, 9573–9580. [Google Scholar] [CrossRef]
- Souza, D.M.; Andrade, A.L.; Fabris, J.D.; Valério, P.; Góes, A.M.; Leite, M.F.; Domingues, R.Z. Synthesis and in vitro evaluation of toxicity of silica-coated magnetite nanoparticles. J. Non-Cryst. Solids 2008, 354, 4894–4897. [Google Scholar] [CrossRef]
- Gaharwar, U.S.; Rajamani, P. Iron Oxide Nanoparticles Induced Oxidative Damage in Peripheral Blood Cells of Rat. J. Biomed. Sci. Eng. 2015, 8, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Elias, A.; Tsourkas, A. Imaging circulating cells and lymphoid tissues with iron oxide nanoparticles. Hematol 2009, 720–726. [Google Scholar] [CrossRef] [Green Version]
- Roca, A.G.; Marco, J.F.; Morales, M.P.; Serna, C.J. Effect of Nature and Particle Size on Properties of Uniform Magnetite and Maghemite Nanoparticles. J. Phys. Chem. C 2007, 111, 18577–18584. [Google Scholar] [CrossRef]
- Baber, O.; Jang, M.; Barber, D.; Powers, K. Amorphous silica coatings on magnetic nanoparticles enhance stability and reduce toxicity to in vitro BEAS-2B cells. Inhal. Toxicol. 2011, 23, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Malvindi, M.A.; Greco, A.; Conversano, F.; Figuerola, A.; Corti, M.; Bonora, M.; Lascialfari, A.; Doumari, H.A.; Moscardini, M.; Cingolani, R.; et al. Magnetic/silica nanocomposites as dual-mode contrast agents for combined magnetic resonance imaging and ultrasonography. Adv. Funct. Mater. 2011, 21, 2548–2555. [Google Scholar] [CrossRef]
- Foglia, S.; Ledda, M.; Fioretti, D.; Iucci, G.; Papi, M.; Capellini, G.; Lolli, M.G.; Grimaldi, S.; Rinaldi, M.; Lisi, A. In vitro biocompatibility study of sub-5 nm silica-coated magnetic iron oxide fluorescent nanoparticles for potential biomedical application. Sci. Rep. 2017, 7, 46513. [Google Scholar] [CrossRef] [Green Version]
- Piccinetti, C.C.; Montis, C.; Bonini, M.; Laura, R.; Guerrera, M.C.; Radaelli, G.; Vianello, F.; Santinelli, V.; Maradonna, F.; Nozzi, V.; et al. Transfer of Silica-Coated Magnetic (Fe3O4) Nanoparticles Through Food: A Molecular and Morphological Study in Zebrafish. Zebrafish 2014, 11, 567–579. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Behera, B.; Sahu, S.K.; Ananthakrishnan, R.; Maiti, T.K.; Pramanik, P. Design of dual stimuli responsive polymer modified magnetic nanoparticles for targeted anti-cancer drug delivery and enhanced MR imaging. New J. Chem. 2016, 40, 545–557. [Google Scholar] [CrossRef]
- Moacă, E.A.; Farcas, C.; Coricovac, D.; Avram, S.; Mihali, C.V.; Drăghici, G.A.; Loghin, F.; Păcurariu, C.; Dehelean, C. Oleic acid double coated Fe3O4 nanoparticles as anti- melanoma compounds with a complex mechanism of activity-in vitro and in ovo assessment. J. Biomed. Nanotechnol. 2019, 15, 893–909. [Google Scholar] [CrossRef]
- He, X.; Liu, F.; Wang, K.; Ge, J.; Qin, D.; Gong, P.; Tan, W. Bioeffects of different functionalized silica nanoparticles on HaCaT cell line. Chinese Sci. Bull. 2006, 51, 1939–1946. [Google Scholar] [CrossRef]
- Huang, X.; Teng, X.; Chen, D.; Tang, F.; He, J. The effect of the shape of mesoporous silica nanoparticles on cellular uptake and cell function. Biomaterials 2010, 31, 438–448. [Google Scholar] [CrossRef]
- Ali, D.; Verma, A.; Mujtaba, F.; Dwivedi, A.; Hans, R.K.; Ray, R.S. UVB-induced apoptosis and DNA damaging potential of chrysene via reactive oxygen species in human keratinocytes. Toxicol. Lett. 2011, 204, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Jin, C.; Tang, Y.; Wang, F.; Ma, C.; Yang, Y. Cytotoxicity of silica nanoparticles on HaCaT cells. J. Appl. Toxicol. 2014, 34, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Takakura, M.; Imamura, H.; Sugimoto, M.; Matsui, Y.; Miyoshi, H.; Nakayama, A.; Yoneda, M. Whole-body distribution of 14C-labeled silica nanoparticles and submicron particles after intravenous injection into Mice. J. Nanopart. Res. 2012, 14, 849. [Google Scholar] [CrossRef]
- Tao, K.; Liu, S.; Wang, L.; Qiu, H.; Li, B.; Zhang, M.; Guo, M.; Liu, H.; Zhang, X.; Liu, Y.; et al. Targeted multifunctional nanomaterials with MRI, chemotherapy and photothermal therapy for the diagnosis and treatment of bladder cancer. Biomater. Sci. 2020, 8, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Narayanaswamy, V.; Qurishi, Y.; Srivastava, C. GO-Fe3O4 Nanoparticle Composite for Selective Targeting of Cancer Cells. Nano Biomed. Eng. 2017, 9, 96–102. [Google Scholar] [CrossRef]
- Vairapperumal, T.; Saraswathy, A.; Ramapurath, J.S.; Janardhanan, S.K.; Unni, N.B. Catechin tuned magnetism of Gd- doped orthovanadate through morphology as T1-T2 MRI contrast agents. Sci. Rep. 2016, 6, 34976. [Google Scholar] [CrossRef] [Green Version]
- Leung, K. Cyclo(Arg-Gly-Asp- D-Try-Glu) Conjugated to Ultrasmall Superparamagnetic Iron Oxide Nanoparticles. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information (US) 2004–2013: Bethesda, MD, USA, 2008. Available online: https://www.ncbi.nlm.nih.gov/books/NBK23342/.
- Lanza, G.M.; Winter, P.M.; Caruthers, S.D.; Morawski, A.M.; Schmieder, A.H.; Crowder, K.C.; Wickline, S.A. Magnetic resonance molecular imaging with nanoparticles. J. Nucl. Cardiol. 2004, 11, 733–743. [Google Scholar] [CrossRef]
- Huang, X.; Zhuang, J.; Teng, X.; Li, L.; Chen, D.; Yan, X.; Tang, F. The promotion of human malignant melanoma growth by mesoporous silica nanoparticles through decreased reactive oxygen species. Biomaterials 2010, 31, 6142–6153. [Google Scholar] [CrossRef]
- Janko, C.; Ratschker, T.; Nguyen, K.; Zschiesche, L.; Tietze, R.; Lyer, S.; Alexiou, C. Functionalized Superparamagnetic Iron Oxide Nanoparticles (SPIONs) as Platform for the Targeted Multimodal Tumor Therapy. Front. Oncol. 2019, 9, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.M. The application of magnetic nanoparticles in the treatment and monitoring of cancer and infectious diseases. Biosci. Horizons Int. J. Student Res. 2017, 10, hzx009. [Google Scholar] [CrossRef] [Green Version]
- Lio, D.C.S.; Liu, C.; Oo, M.M.S.; Wiraja, C.; Teo, M.H.Y.; Zheng, M.; Chew, S.W.T.; Wang, X.; Xu, C. Transdermal delivery of small interfering RNAs with topically applied mesoporous silica nanoparticles for facile skin cancer treatment. Nanoscale 2019, 11, 17041–17051. [Google Scholar] [CrossRef] [PubMed]
- Portilho, F.L.; Helal-Neto, E.; Cabezas, S.S.; Pinto, S.R.; Nascimento dos Santos, S.; Pozzo, L.; Sancenón, F.; Martínez-Máñez, R.; Santos-Oliveira, R. Magnetic core mesoporous silica nanoparticles doped with dacarbazine and labelled with 99mTc for early and differential detection of metastatic melanoma by single photon emission computed tomography. Artif. Cells. Nanomed. Biotechnol. 2018, 46 (Suppl. 1), 1080–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avram, S.; Coricovac, D.E.; Pavel, I.Z.; Pinzaru, I.; Ghiulai, R.; Baderca, F.; Soica, C.; Muntean, D.; Branisteanu, D.E.; Spandidos, D.A.; et al. Standardization of A375 human melanoma models on chicken embryo chorioallantoic membrane and Balb/c nude mice. Oncol Rep. 2017, 38, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianasi, C.; Costisor, O.; Putz, A.-M.; Plocek, J.; Săcărescu, L.; Nižňanský, D.; Savii, C. Superparamagnetic γ-Fe2O3-SiO2 Nanocomposites from Fe2O3-SiO2-PVA Hybrid Xerogels. Characterization and MRI Preliminary Testing. Curr. Org. Chem. 2017, 21, 2783–2791. [Google Scholar] [CrossRef]
- Ianasi, C.; Costisor, O.; Putz, A.-M.; Lazau, R.; Negrea, A.; Niznansky, D.; Sacarescu, L.; Savii, C. Low temperature superparamagnetic nanocomposites obtained by Fe(Acac)3-SiO2-PVA hybrid xerogel thermolysis. Process. Appl. Ceram. 2016, 10, 265–275. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Wang, D. Pore surface fractal dimension of sol–gel derived Al2O3–SiO2 membranes. Mater. Lett. 2003, 57, 2015–2020. [Google Scholar]
- Kline, S.R. Reduction and analysis of SANS and USANS data using IGOR Pro. J. Appl. Cryst. 2006, 39, 895–900. [Google Scholar] [CrossRef]
- Beaucage, G.J. Approximation leading to a unified exponential/power-law approach to small-angle scattering. J. Appl. Cryst. 1995, 28, 717–728. [Google Scholar] [CrossRef]
- Dudás, Z.; Fagadar-Cosma, E.; Len, A.; Románszki, L.; Almásy, L.; Vlad-Oros, B.; Dascălu, D.; Krajnc, A.; Kriechbaum, M.; Kuncser, A. Improved Optical and Morphological Properties of Vinyl-Substituted Hybrid Silica Materials Incorporating a Zn-Metalloporphyrin. Materials 2018, 11, 565. [Google Scholar] [CrossRef] [Green Version]
- Gubanova, N.N.; Baranchikov, A.Y.; Kopitsa, G.P.; Almásy, L.; Angelov, B.; Yapryntsev, A.D.; Rosta, L.; Ivanov, V.K. Combined SANS and SAXS study of the action of ultrasound on the structure of amorphous zirconia gels. Ultrason. Sonochem. 2015, 24, 230–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putz, A.-M.; Savii, C.; Ianăşi, C.; Dudás, Z.; Székely, K.N.; Plocek, J.; Sfârloagă, P.; Săcărescu, L.; Almásy, L. Pore ordering in mesoporous matrices induced by different directing agents. J. Porous Mater. 2015, 22, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.W. Small-angle scattering studies of disordered, porous and fractal systems. J. Appl. Cryst. 1991, 24, 414–435. [Google Scholar] [CrossRef]
- Borcan, F.; Soica, C.M.; Dehelean, C.A.; Ganta, S.; Amiji, M.M. Size and Stability Optimization for Polyurethane Nanostructures used as Transdermal Drug Vehicle. Rev. Chim. Bucharest 2012, 63, 1164–1166. [Google Scholar]
- Heghes, A.; Soica, C.M.; Ardelean, S.; Ambrus, R.; Muntean, D.; Galuscan, A.; Dragos, D.; Ionescu, D.; Borcan, F. Influence of emulsifiers on the characteristics of polyurethane structures used as drug carrier. Chem. Cent. J. 2013, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Balas, M.; Dumitrache, F.; Badea, M.A.; Fleaca, C.; Badoi, A.; Tanasa, E.; Dinischiotu, A. Coating dependent in vitro biocompatibility of new Fe-Si Nanoparticles. Nanomaterials 2018, 8, 495. [Google Scholar] [CrossRef] [Green Version]
- Danciu, C.; Muntean, D.; Alexa, E.; Farcas, C.; Oprean, C.; Zupko, I.; Bor, A.; Minda, D.; Proks, M.; Buda, V.; et al. Phytochemical Characterization and Evaluation of the Antimicrobial, Antiproliferative and Pro- Apoptotic Potential of Ephedraalata Decne. Hydroalcoholic Extract against the MCF-7 Breast Cancer Cell Line. Molecules 2019, 24, 13. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: The synthetized samples from this study are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samp. | SBET*1 [m2/g] | S L*2 [m2/g] | S t-plot*3 [m2/g] | dDFT*4 [nm] | dBJH*5 [nm] | TPV *6 [cm3/g] | Alpha-S*7 [cm3/g] | dfs FHH ads*8 | dfs FHH des*8 |
|---|---|---|---|---|---|---|---|---|---|
| P-120 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| P-220 | 99 | 158 | 64 | 2.7 | 3.7 | 0.055 | 0.038 | 2.69/2.89 | 2.77/2.92 |
| P-260 | 344 | 519 | 246 | 2.6 | 3.1 | 0.183 | 0.131 | 2.77/2.92 | 2.57/2.85 |
| P-300 | 468 | 640 | 349 | 2.1 | 3.4 | 0.227 | 0.170 | 2.85/2.95 | 2.68/2.89 |
| P-400 | 382 | 539 | 258 | 2.4 | 3.1 | 0.194 | 0.130 | 2.82/2.94 | 2.55/2.85 |
| ΔT, [°C] | Tmax, [°C] | Weight Loss | Thermic Effect | ||
|---|---|---|---|---|---|
| ΔG/ΔT [%] | Cumulativ ΔG, [%] | ΔG/ΔT [mg] | |||
| 25–125 | 71 | 4.16 | 4.16 | 0.83 | endothermic |
| 125–215 | 193 | 8.27 | 12.43 | 1.65 | exothermic |
| 215–275 | 257 | 16.69 | 29.27 | 3.34 | exothermic |
| 275–800 | 361 | 22.31 | 51.44 | 4.46 | exothermic |
| Sample Name | Calcination Temperature | Rg (nm) | p | Fractal Dimension |
|---|---|---|---|---|
| P-120 | 120 °C | - | 3.82 ± 0.03 | Dsurface = 2.18 |
| P-220 | 220 °C | 10.01 ± 0.09 | 2.42 ± 0.01 | Dvolume = 2.42 |
| P-260 | 260 °C | 7.54 ± 0.02 | 2.21 ± 0.01 | Dvolume = 2.21 |
| P-300 | 300 °C | 7.79 ± 0.02 | 2.26 ± 0.01 | Dvolume = 2.26 |
| P-400 | 400 °C | 9.59 ± 0.03 | 2.18 ± 0.01 | Dvolume =2.18 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Putz, A.-M.; Ianăși, C.; Dudás, Z.; Coricovac, D.; Watz, C.; Len, A.; Almásy, L.; Sacarescu, L.; Dehelean, C. SiO2-PVA-Fe(acac)3 Hybrid Based Superparamagnetic Nanocomposites for Nanomedicine: Morpho-textural Evaluation and In Vitro Cytotoxicity Assay. Molecules 2020, 25, 653. https://doi.org/10.3390/molecules25030653
Putz A-M, Ianăși C, Dudás Z, Coricovac D, Watz C, Len A, Almásy L, Sacarescu L, Dehelean C. SiO2-PVA-Fe(acac)3 Hybrid Based Superparamagnetic Nanocomposites for Nanomedicine: Morpho-textural Evaluation and In Vitro Cytotoxicity Assay. Molecules. 2020; 25(3):653. https://doi.org/10.3390/molecules25030653
Chicago/Turabian StylePutz, Ana-Maria, Cătălin Ianăși, Zoltán Dudás, Dorina Coricovac, Claudia (Farcas) Watz, Adél Len, László Almásy, Liviu Sacarescu, and Cristina Dehelean. 2020. "SiO2-PVA-Fe(acac)3 Hybrid Based Superparamagnetic Nanocomposites for Nanomedicine: Morpho-textural Evaluation and In Vitro Cytotoxicity Assay" Molecules 25, no. 3: 653. https://doi.org/10.3390/molecules25030653
APA StylePutz, A.-M., Ianăși, C., Dudás, Z., Coricovac, D., Watz, C., Len, A., Almásy, L., Sacarescu, L., & Dehelean, C. (2020). SiO2-PVA-Fe(acac)3 Hybrid Based Superparamagnetic Nanocomposites for Nanomedicine: Morpho-textural Evaluation and In Vitro Cytotoxicity Assay. Molecules, 25(3), 653. https://doi.org/10.3390/molecules25030653