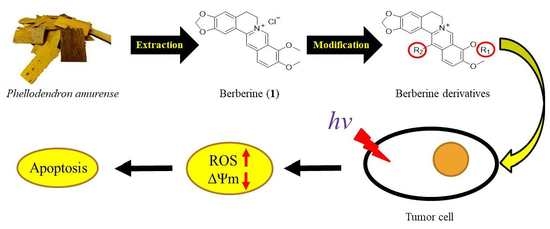
3. Materials and Methods
3.1. General Information
All chemical reagents and solvents in commercial quality were purchased from Sigma-Aldrich (St. Louis, MO, USA), Fluka (Roma, Italy), TCI (Tokyo, Japan), and Alpha Aesar without further purification. All chemical reagents in commercial quality were used as received (Sigma-Aldrich, St. Louis, MO, USA) and were used without further purification. Solvents were dried and the synthesized compounds were purified using standard techniques. Reactions-progression was monitored by TLC on aluminum plates coated with silica gel with a fluorescent indicator (Merck 60 F
254). Melting points of the synthesized compounds were determined by Fargo MP-2D apparatus and were uncorrected. The electronic absorption spectra were measured on a Shimadzu UV-1601 UV-vis spectrophotometer. Fluorescence spectra were taken on a Shimadzu RP-5301 spectrofluorometer. NMR spectra were recorded using TMS as an internal standard in CDCl
3 at 500 MHz for
1H and at 125 MHz for
13C (Bruker Biospin GmbH AVANCE III 500 MHz, Rheinstetten, Germany) (see
Supplementary Materials). Chemical shift (
δ) were reported in parts per million (ppm) measured relative to the internal standards (TMS), and the coupling constant (
J) were expressed in Hertz (Hz). Column chromatography was performed with silica gel Silia
Flash® G60 (60–200 μm) purchased from SiliCycle Inc. (Quebec City, QC, Canada). In general, the reactions were carried out under anhydrous conditions in dry solvent and nitrogen atmosphere. The purity of these compounds was based on the analysis of HPLC (Hitachi High-Technologies, Tokyo, Japan) equipped with a 280 nm detector and LiChroCART RP-C
18 column (4.6 mm i.d. × 250 mm, 5 μm, Merck, Darmstadt, Germany). The mobile phase was composed of MeOH-H
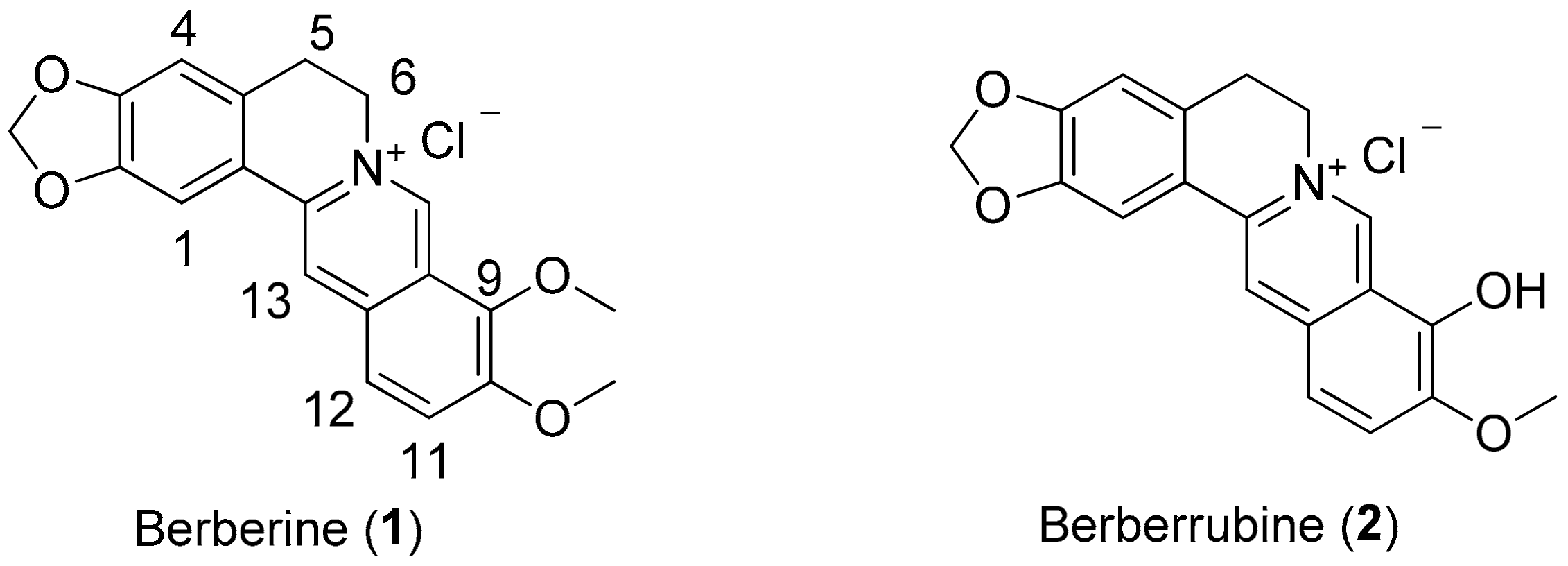
2O (0.05% TFA) (90:10) and the flow rate was 1.0 mL/min. The purity of all compounds was more than 98%. The mass spectra were acquired using a Thermo Finnigan model LXQ (Thermo Electron Co., Waltham, MA, USA) ion trap mass spectrometer equipped with ESI source interference and controlled by Xcalibur 2.06. The mass spectra were acquired in a positive ion mode or a negative ion mode. Berberrubine (
2), derivatives
3,
4,
5a,
5c, and
5e were synthesized according to previous methods [
27].
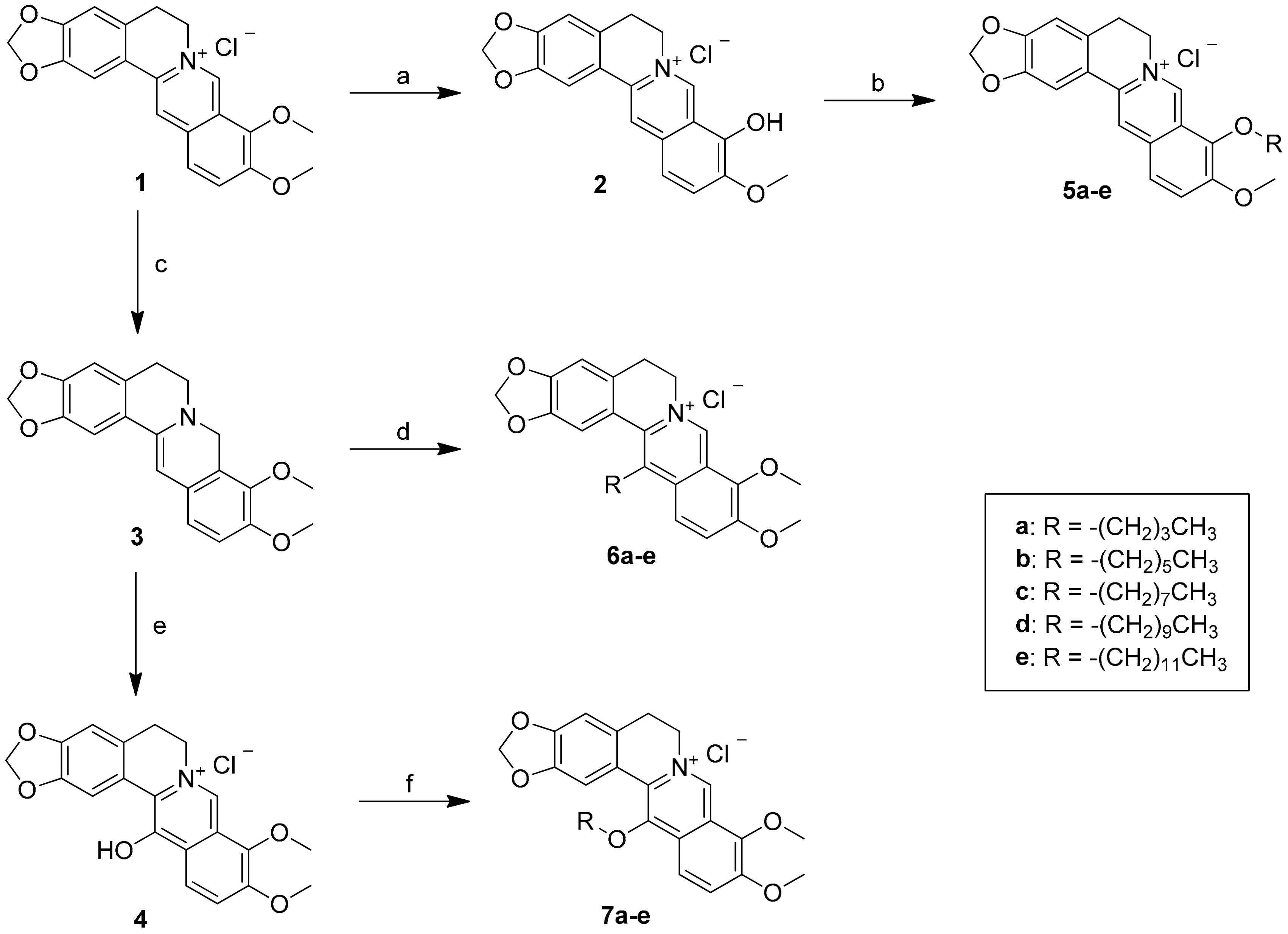
3.2. Synthesis of 9-O-Sunstituted Berberine Derivatives (5a–e)
To a solution of berberrubine 2 (1 mmol) in dry acetonitrile (10 mL) was added various n-alkyl bromide (1.2 mmol) and the reaction mixture was refluxed for 4–8 h. The progress of the reaction was monitored by TLC. Then, the reaction mixture was cooled to room temperature and the solvent was removed under reduced pressure. The crude product was chromatographed on a silica gel column and eluted with ethyl acetate/methanol (2/1) solvent to afford the desired berberine derivatives 5a–e.
9-O-Butylberberine bromide (5a). Berberrubine (2) was treated with n-butyl bromide according to the general procedure to give the desired bromide salt (5a) as a yellowish brown solid, yield: 86%; UV (MeOH) λmax (log ε): 431 (3.77), 350 (4.41), 267 (4.43), 231 (4.45), 205 (4.41) nm; 1H NMR (500 MHz, DMSO-d6): δ 9.75 (s, 1H, H-8), 8.94 (s, 1H, H-13), 8.20 (d, J = 9.1 Hz, H-12), 7.99 (d, J = 9.1 Hz, H-11), 7.80 (s, 1H, H-1), 7.09 (s, 1H, H-4), 6.17 (s, 2H, -OCH2O-), 4.95 (t, J = 6.3 Hz, 2H, H-6), 4.29 (t, J = 6.8 Hz, 2H, H-1’), 4.05 (s, 3H, -OCH3), 3.21 (t, J = 6.3 Hz, 2H, H-5), 1.86 (p, J = 7.2 Hz, 2H, H-2’), 1.52 (sex, J = 7.4 Hz, 2H, H-3’), 0.99 (t, J = 7.4 Hz, 3H, H-4’); 13C NMR (125 MHz, DMSO-d6): δ 150.4, 149.8, 147.7, 145.3, 142.9, 137.5, 133.0, 130.7, 126.7, 123.3, 121.7, 120.5, 120.2, 108.4, 105.4, 102.1, 73.9, 57.0, 55.3, 31.5, 26.3, 18.6, 13.8; LC-MS (ESI+, m/z) C23H24NO4+: 378.33 [M–Br]+; HRMS-ESI: m/z calculated for C23H22NO4+: 378.1705 [M − Br]+, found for 378.1708.
9-O-Hexylberberine bromide (5b). Berberrubine (2) was treated with n-hexyl bromide according to the general procedure to give the desired bromide salt (5b) as a yellowish brown solid, yield: 69%; UV (MeOH) λmax (log ε): 431.6 (3.92), 350.2 (4.54), 266.5 (4.60), 229.1 (4.67) nm; 1H NMR (500 MHz, DMSO-d6): δ 9.75 (s, 1H, H-8), 8.94 (s, 1H, H-13), 8.20 (d, J = 9.1 Hz, H-12), 7.99 (d, J = 9.1 Hz, H-11), 7.80 (s, 1H, H-1), 7.09 (s, 1H, H-4), 6.17 (s, 2H, -OCH2O-), 4.95 (t, J = 6.3 Hz, 2H, H-6), 4.28 (t, J = 6.8 Hz, 2H, H-1’), 4.05 (s, 3H, -OCH3), 3.21 (t, J = 6.3 Hz, 2H, H-5), 1.87 (p, J = 7.2 Hz, 2H, H-2’), 1.52 (p, J = 7.4 Hz, 2H, H-3’),1.37‒1.33 (m, 4H, H-4’, H-5’), 0.90 (t, J = 7.0 Hz, 3H, H-6’); 13C-NMR (125 MHz, DMSO-d6): δ 150.5, 149.8, 147.7, 145.3, 142.9, 137.5, 133.1, 130.7, 126.7, 123.4, 121.7, 120.5, 120.3, 108.5, 105.5, 102.1, 74.3, 57.1, 55.3, 31.1, 29.5, 26.4, 25.0, 22.1, 14.0; LC-MS (ESI+, m/z) C25H26NO4+: 406.33 [M–Br]+; HRMS-ESI: m/z calculated for C25H26NO4+: 406.2018 [M − Br]+, found for 406.2021.
9-O-Octylberberine bromide (5c). Berberrubine (2) was treated with n-octyl bromide according to the general procedure to give the desired bromide salt (5c) as a bright yellow solid, yield: 74%; UV (MeOH) λmax (log ε): 432 (3.79), 350 (4.42), 268 (4.44), 230 (4.47), 204 (4.48) nm; 1H NMR (500 MHz, DMSO-d6): δ 9.75 (s, 1H, H-8), 8.95 (s, 1H, H-13), 8.19 (d, J = 9.1 Hz, H-12), 7.99 (d, J = 9.1 Hz, H-11), 7.80 (s, 1H, H-1), 7.09 (s, 1H, H-4), 6.17 (s, 2H, -OCH2O-), 4.95 (t, J = 6.2 Hz, 2H, H-6), 4.28 (t, J = 6.8 Hz, 2H, H-1’), 4.06 (s, 3H, -OCH3), 3.21 (t, J = 6.2 Hz, 2H, H-5), 1.87 (p, J = 7.2 Hz, 2H, H-2’), 1.48 (p, J = 7.3 Hz, 2H, H-3’), 1.33 (m, 8H, H-4’–H-7’), 0.88 (t, J = 6.8 Hz, 3H, H-8’); 13C NMR (125 MHz, DMSO-d6): δ 150.4, 149.8, 147.7, 145.3, 142.9, 137.5, 133.0, 126.7, 123.3, 121.7, 120.5, 120.2, 108.4, 105.4, 102.1, 74.2, 57.0, 55.3, 31.2, 29.5, 28.8, 28.7, 26.3, 25.3, 22.1, 14.0; LC-MS (ESI+, m/z) C27H32NO4+: 434.41 [M–Br]+; HRMS-ESI: m/z calculated for C27H30NO4+: 434.2331 [M − Br]+, found for 434.2332.
9-O-Decylberberine bromide (5d). Berberrubine (2) was treated with n-decyl bromide according to the general procedure to give the desired bromide salt (5d) as a bright yellow solid, yield: 71%; UV (MeOH) λmax (log ε): 431.0 (3.68), 350.2 (4.31), 266.4 (4.35), 229.8 (4.38) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.75 (s, 1H, H-8), 8.95 (s, 1H, H-13), 8.19 (d, J = 9.1 Hz, H-12), 7.99 (d, J = 9.1 Hz, H-11), 7.80 (s, 1H, H-1), 7.09 (s, 1H, H-4), 6.17 (s, 2H, -OCH2O-), 4.95 (t, J = 6.2 Hz, 2H, H-6), 4.27 (t, J = 6.8 Hz, 2H, H-1’), 4.05 (s, 3H, -OCH3), 3.21 (t, J = 6.2 Hz, 2H, H-5), 1.87 (p, J = 7.2 Hz, 2H, H-2’), 1.48 (p, J = 7.3 Hz, 2H, H-3’), 1.40‒1.26 (m, 12H, H-4’–H-9’), 0.85 (t, J = 6.8 Hz, 3H, H-10’); 13C-NMR (125 MHz, DMSO-d6): δ 150.4, 149.8, 147.7, 145.3, 142.9, 137.5, 133.0, 130.7, 126.7, 123.3, 121.7, 120.5, 120.2, 108.4, 105.4, 102.1, 74.2, 57.0, 55.3, 31.3, 29.5, 29.0, 28.9, 28.8, 28.7, 26.3, 25.3, 22.1, 14.0; LC-MS (ESI+, m/z) C29H34NO4+: 462.43 [M–Br]+; HRMS-ESI: m/z calculated for C29H34NO4+: 462.2644 [M − Br]+, found for 462.2646.
9-O-Dodecylberberine bromide (5e). Berberrubine (2) was treated with n-dodecyl bromide according to the general procedure to give the desired bromide salt (5e) as a dark yellowish brown solid, yield: 61%; UV (MeOH) λmax (log ε): 431 (3.42), 351 (4.03), 267 (4.07), 230 (4.10), 202 (4.19) nm; 1H NMR (500 MHz, DMSO-d6): δ 9.77 (s, 1H, H-8), 8.98 (s, 1H, H-13), 8.21 (d, J = 9.1 Hz, H-12), 8.00 (d, J = 9.1 Hz, H-11), 7.82 (s, 1H, H-1), 7.10 (s, 1H, H-4), 6.18 (s, 2H, -OCH2O-), 4.96 (t, J = 6.1 Hz, 2H, H-6), 4.28 (t, J = 6.8 Hz, 2H, H-1’), 4.05 (s, 3H, -OCH3), 3.21 (t, J = 6.1 Hz, 2H, H-5), 1.88 (d, J = 7.4 Hz, H-2’), 1.47 (q, J = 7.5 Hz, 2H, H-3’), 1.29 (m, 16 H, H-4’–H-11’), 0.86 (t, J = 6.8 Hz, 3H, H-12’); 13C NMR (125 MHz, DMSO-d6): δ 150.4, 149.8, 147.7, 145.3, 142.9, 137.5, 133.0, 130.7, 126.7, 123.3, 121.7, 120.5, 120.3, 108.4, 105.5, 102.1, 74.2, 57.0, 55.3, 48.6, 31.3, 29.5, 29.08, 29.05, 28.9, 28.7, 26.3, 25.3, 22.1, 14.0; LC-MS (ESI+, m/z) C31H38NO4+: 490.50 [M − Br]+; HRMS-ESI: m/z calculated for C31H38NO4+: 490.2957 [M − Br]+, found for 490.2958.
3.3. Synthesis of 13-Sunstituted Berberine Derivatives (6a–e)
To a stirred solution of berberine (1) (3.71 g, 10 mmol) and K2CO3 (3.6 g, 30 mmol) in methanol (125 mL), 5% NaOH (5 mL) solution containing NaBH4 (0.30 g, 7.5 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 1 h and the precipitated product was filtered, washed with 30% ethanol (20 mL) and 80% ethanol (20 mL), and then recrystallized from absolute ethanol to provide dihydroberberine (3) (2.5 g, 74%) as a brown like solid, yield: 74%; UV (MeOH) λmax (log ε): 353.9 (3.96), 268 (4.01), 228.6 (4.09) nm;1H-NMR (500 MHz, DMSO-d6) δ 7.29 (s, 1H, H-13), 6.82 (d, J = 8.4 Hz, H-12), 6.76 (s, 1H, H-1), 6.69 (d, J = 8.4 Hz, 1H, H-11), 6.05 (s,1H, H-1), 6.00 (s, 2H, -OCH2-), 4.20 (s, 2H, H-8), 3.76 (s, 3H, -OCH3), 3.71 (s, 3H, -OCH3), 3.05 (t, J = 5.8 Hz, 2H, H-6), 2.79 (t, J = 5.8 Hz, 2H, H-5); 13C-NMR (125 MHz, DMSO-d6) δ 149.9, 146.9, 146.4, 143.9, 141.0, 128.6, 128.3, 123.9, 121.6, 118.4, 111.7, 107.9, 103.5, 101.0, 96.0, 60.2, 55.7, 48.8, 48.3, 28.9.
To a stirred solution of dihydroberberine (3) (170 mg, 5.0 mmol) in 80% ethanol (8 mL) and HOAc (2 mL), n-alkyl aldehyde (2 mL) was added. The reaction mixture was heated to 85–95 °C for 5 h. The solvent was removed by evaporation, and the residue was acidified by 2 N HCl (5 mL), and then stirred at room temperature for 1 h. The solid was collected by filtration and then purified by flash chromatography over silica gel, affording the title compounds 6a–e.
13-Butylberberine bromide (6a). Dihydroberberine (3) was treated with butanal according to the general procedure to give the desired chloride salt (6a) as a yellowish brown solid, yield: 68%; UV (MeOH) λmax (log ε): 420.3 (3.93), 341.9 (4.49), 265.1 (4.61), 231.9 (4.58) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.88 (s, 1H, H-8), 8.21 (d, J = 9.5 Hz, 1H, H-12), 8.18 (d, J = 9.5 Hz, 1H, H-11), 7.28 (s, 1H, H-1), 7.16 (s, 1H, H-4), 6.18 (s, 2H, -OCH2O-), 4.78 (t, J = 8.0 Hz, 2H, H-6), 4.09 (s, 3H, -OCH3), 4.08 (s, 3H, -OCH3), 3.33 (t, J = 8.0 Hz, 2H, H-5), 3.07 (t, J = 6.2 Hz, 2H, H-1’), 1.75 (p, J = 6.6 Hz, 2H, H-2’), 1.41 (hex, J = 7.2 Hz, 2H, H-3’), 0.91 (t, J = 6.9 Hz, 3H, H-4’); 13C-NMR (125 MHz, DMSO-d6): δ 150.3, 149.1, 146.6, 144.4, 144.3, 135.8, 134.2, 134.1, 132.3, 125.9, 121.5, 121.3, 109.2, 108.4, 102.2, 62.1, 57.1, 57.0, 32.6, 28.8, 27.4, 22.0, 13.5; LC-MS (ESI+, m/z) C24H26NO4+: 392.28 [M − Cl]+; HRMS-ESI: m/z calculated for C24H26NO4+: 392.1862 [M − Cl]+, found for 392.1861.
13-Hexylberberine bromide (6b). Dihydroberberine (3) was treated with hexanal according to the general procedure to give the desired chloride salt (6b) as a bright yellow solid, yield: 76%; UV (MeOH) λmax (log ε): 420.3 (3.73), 341.9 (4.29), 265.4 (4.40), 232 (4.37) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.91 (s, 1H, H-8), 8.22 (d, J = 9.7 Hz, 1H, H-12), 8.20 (d, J = 9.7 Hz, 1H, H-11), 7.30 (s, 1H, H-1), 7.18 (s, 1H, H-4), 6.20 (s, 2H, -OCH2O-), 4.80 (s, 2H, H-6), 4.10 (s, 3H, -OCH3), 4.10 (s, 3H, -OCH3), 3.33 (s,2H, H-5), 3.09 (t, J = 5.7 Hz, 2H, H-1’), 1.76 (s, 2H, H-2’), 1.40 (m, 2H, H-3’), 1.27‒1.26 (m, 4H, H-4’, H-5’), 0.86 (t, J = 6.9 Hz, 3H, H-6’); 13C-NMR (125 MHz, DMSO-d6): δ 150.2, 149.1, 146.6, 144.4, 144.2, 135.8, 134.2, 134.1, 132.2, 125.9, 121.5, 121.3, 120.3, 109.1, 108.4, 102.2, 62.0, 57.0, 30.7, 30.4, 28.9, 28.3, 27.4, 22.1, 13.9; LC-MS (ESI+, m/z) C26H30NO4+: 420.26 [M − Cl]+; HRMS-ESI: m/z calculated for C26H30NO4+: 420.2175 [M‒Cl]+, found for 420.2176.
13-Octylberberine chloride (6c). Dihydroberberine (3) was treated with octanal according to the general procedure to give the desired chloride salt (6c) as a bright yellow solid, yield: 65%; UV (MeOH) λmax (log ε): 420.1 (3.80), 341.4 (4.35), 265.3 (4.47), 232 (4.44) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.91 (s, 1H, H-8), 8.22 (d, J = 9.7 Hz, 1H, H-12), 8.20 (d, J = 9.7 Hz, 1H, H-11), 7.30 (s, 1H, H-1), 7.17 (s, 1H, H-4), 6.20 (s, 2H, -OCH2O-), 4.80 (s, 2H, H-6), 4.10 (s, 3H, -OCH3), 4.10 (s, 3H, -OCH3), 3.33 (s,2H, H-5), 3.09 (t, J = 5.7 Hz, 2H, H-1’), 1.76 (s, 2H, H-2’), 1.38‒1.37 (m, 2H, H-3’), 1.28‒1.23 (m, 8H, H-4’‒H-7’), 0.86 (t, J = 6.9 Hz, 3H, H-8’); 13C NMR (125 MHz, DMSO-d6): δ 150.2, 149.0, 146.6, 144.3, 144.2, 135.8, 134.2, 134.0, 132.2, 125.9, 121.4, 121.2, 120.3, 109.1, 108.3, 102.1, 62.0, 57.0, 31.2, 30.4, 28.8, 28.6, 28.4, 27.4, 22.0, 13.9; LC-MS (ESI+, m/z) C28H34NO4+: 448.28 [M–Cl]+; HRMS-ESI: m/z calculated for C28H34NO4+: 448.2488 [M − Cl]+, found for 448.2487.
13-Decylberberine chloride (6d). Dihydroberberine (3) was treated with decanal according to the general procedure to give the desired chloride salt (6d) as a bright yellow solid, yield: 43%; UV (MeOH) λmax (log ε): 420.3 (3.73), 341.9 (4.29), 265.3 (4.40), 231.7 (4.39) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.90 (s, 1H, H-8), 8.22 (d, J = 9.7 Hz, 1H, H-12), 8.20 (d, J = 9.7 Hz, 1H, H-11), 7.29 (s, 1H, H-1), 7.16 (s, 1H, H-4), 6.19 (s, 2H, -OCH2O-), 4.79 (s, 2H, H-6), 4.09 (s, 3H, -OCH3), 4.09 (s, 3H, -OCH3), 3.33 (s, 2H, H-5), 3.08 (t, J = 5.7 Hz, 2H, H-1’), 1.74 (s, 2H, H-2’), 1.36‒1.35 (m, 2H, H-3’), 1.27‒1.22 (m, 12H, H-4’‒H-9’), 0.86 (t, J = 6.9 Hz, 3H, H-10’); 13C-NMR (125 MHz, DMSO-d6): δ 150.2, 149.0, 146.6, 144.3, 144.2, 135.8, 134.2, 134.0, 132.2, 125.9, 121.4, 121.2, 120.3, 109.1, 108.3, 102.1, 62.0, 57.0, 31.3, 30.4, 28.9, 28.8, 28.6, 28.8, 28.4, 27.4, 22.1, 13.9; LC-MS (ESI+, m/z) C30H38NO4+: 476.40 [M–Cl]+; HRMS-ESI: m/z calculated for C30H38NO4+: 476.2801 [M − Cl]+, found for 476.2801.
13-Dodecylberberine chloride (6e). Dihydroberberine (3) was treated with dodecanal according to the general procedure to give the desired chloride salt (6e) as a bright yellow solid, yield: 29%; UV (MeOH) λmax (log ε): 420.6 (3.71), 341.8 (4.26), 265.3 (4.38), 232.0 (4.37) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.90 (s, 1H, H-8), 8.21 (d, J = 9.6 Hz, 1H, H-12), 8.19 (d, J = 9.6 Hz, 1H, H-11), 7.29 (s, 1H, H-1), 7.16 (s, 1H, H-4), 6.18 (s, 2H, -OCH2O-), 4.79 (s, 2H, H-6), 4.09 (s, 3H, -OCH3), 4.09 (s, 3H, -OCH3), 3.33 (s,2H, H-5), 3.08 (t, J = 5.7 Hz, 2H, H-1’), 1.74 (s, 2H, H-2’), 1.36‒1.35 (m, 2H, H-3’), 1.28‒1.22 (m, 16H, H-4’‒H-11’), 0.85 (t, J = 6.9 Hz, 3H, H-12’); 13C-NMR (125 MHz, DMSO-d6): δ 150.2, 149.0, 146.5, 144.3, 144.2, 135.8, 134.2, 134.0, 132.2, 125.9, 121.4, 121.2, 120.3, 109.1, 108.3, 102.1, 62.0, 57.0, 31.3, 30.4, 30.0, 28.9, 28.9, 28.8, 28.7, 28.6, 28.4, 27.4, 22.1, 13.9; LC-MS (ESI+, m/z) C32H42NO4+: 504.43 [M − Cl]+; HRMS-ESI: m/z calculated for C32H42NO4+: 504.3114 [M − Cl]+, found for 504.3115.
3.4. Synthesis of 13-O-Sunstituted Berberine Derivatives (7a–e)
Compound 3 (337 mg) was dissolved in 35 mL of CH2Cl2. Under nitrogen atmosphere, the temperature of the system was kept at ‒25 °C ~ ‒30 °C, and 8 mL of CH2Cl2 solution containing 258 mg MCPBA (1.5 mmol) was added slowly dropwise. After the addition, the temperature was kept and the reaction was carried out under agitation for 1 h. Then, the temperature was raised to 0 °C and 250 mg (2.0 mmol) of sodium sulfite was added therein, followed by being stirred at room temperature for 1 h. The reaction was stopped and the reaction mixture was filtered. The filtrate was evaporated to remove the solvent under reduced pressure and the residue was purified through silica gel column chromatography (CH2Cl2/CH3OH = 10:1) to obtain compound 4 (280 mg, 80 percent), yield: 80%;UV (MeOH) λmax (log ε): 444.5 (4.08), 367.9 (3.93), 305.3 (4.04), 235.0 (4.44), 212.1 (4.42) nm; 1H-NMR (500 MHz, DMSO-d6): δ 8.89 (s, 1H, H-8), 8.08 (d, J = 9.3 Hz, 1H, H-12), 8.06 (s, 1H, H-1), 7.52 (d, J = 9.3 Hz, 1H, H-11), 6.88 (s, 1H, H-4), 6.02 (s, 2H, -OCH2O-), 4.61 (t, J = 6.2 Hz, 2H, H-6), 3.95 (s, 3H, -OCH3), 3.93 (s, 3H, -OCH3), 3.01 (t, J = 6.2 Hz, 2H, H-5); 13C-NMR (125 MHz, DMSO-d6): δ 165.1, 150.2, 145.3, 145.2, 142.2, 130.2, 127.4, 123.9, 123.4, 122.3, 121.4, 117.6, 117.3, 107.5, 107.2, 100.8, 61.1, 56.4, 56.3, 27.6.
Compound 4 (1.0 g) and potassium carbonate (200 mg) were dissolved in 20 mL of acetone followed by adding 0.25 mL of alkyl bromide. The obtained reaction solution was heated and refluxed for 3 h. Then, the reaction mixture was filtered and the filtrate was evaporated under reduced pressure. The residual was purified through silica gel column (CH2Cl2/CH3OH = 20: 1) to obtain compounds 7a–e.
13-O-Butylberberine bromide (7a). Compound (4) was treated with n-butyl bromide according to the general procedure to give the desired bromide salt (7a) as a yellowish brown solid, yield: 66%; UV (MeOH) λmax (log ε): 427.8 (3.46), 349.7 (3.97), 265.5 (4.07), 232.6 (4.17) nm;1H-NMR (500 MHz, DMSO-d6): δ 9.82 (s, 1H, H-8), 8.21 (d, J = 9.3 Hz, 1H, H-12), 8.09 (d, J = 9.3 Hz, 1H, H-11), 7.90 (s, 1H, H-1), 7.13 (s, 1H, H-4), 6.17 (s, 2H, -OCH2O-), 4.88 (t, J = 5.8 Hz, 2H, H-6), 4.10 (s, 3H, -OCH3), 4.09 (s, 3H, -OCH3), 3.84 (t, J = 6.4 Hz, 2H, H-1’), 3.17 (t, J = 4.7 Hz, 2H,H-5), 1.80‒1.74 (m, 2H, H-2’), 1.53‒1.45 (m, 2H, H-3’), 0.91 (t, J = 7.4 Hz, 3H, H-4’); 13C-NMR (125 MHz, DMSO-d6): δ 150.9, 150.0, 149.2, 146.8, 144.2, 142.0, 132.5, 130.8, 128.8, 126.1, 122.1, 118.4, 108.4, 107.9, 102.1, 75.0, 62.0, 57.1, 56.4, 31.5, 26.9, 18.6, 13.6; HRMS-ESI: m/z calculated for C24H26NO5+: 408.1811 [M − Br]+, found for 408.1812.
13-O-Hexylberberine bromide (7b). Compound (4) was treated with n-hexyl bromide according to the general procedure to give the desired bromide salt (7b) as a bright yellow solid, yield: 76%; UV (MeOH) λmax (log ε): 428.0 (3.75), 350.3 (4.26), 266.5 (4.32), 229.0 (4.45) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.81 (s, 1H, H-8), 8.21 (d, J = 9.4 Hz, 1H, H-12), 8.09 (d, J = 9.4 Hz, 1H, H-11), 7.90 (s, 1H, H-1), 7.13 (s, 1H, H-4), 6.17 (s, 2H, -OCH2O-), 4.87 (t, J = 5.9 Hz, 2H, H-6), 4.10 (s, 3H, -OCH3), 4.09 (s, 3H, -OCH3), 3.84 (t, J = 6.4 Hz, 2H, H-1’), 3.16 (t, J = 5.9 Hz, 2H, H-5), 1.78 (p, J = 7.0 Hz, 2H, H-2’), 1.45 (p, J = 7.5 Hz,2H, H-3’), 1.33‒1.23 (m, 4H, H-4’, H-5’), 0.86 (t, J = 6.9 Hz, 3H, H-6’); 13C-NMR (125 MHz, DMSO-d6): δ 150.9, 150.1, 149.2, 146.8, 144.2, 141.9, 132.5, 130.8, 128.8, 126.1, 122.1, 118.5, 118.4, 108.3, 108.0, 102.1, 75.4, 62.0, 57.1, 30.9, 29.4, 25.1, 22.0, 13.9; HRMS-ESI: m/z calculated for C26H30NO5+: 436.2124 [M − Br]+, found for 436.2122.
13-O-Octylberberine chloride (7c). Compound (4) was treated with n-octyl bromide according to the general procedure to give the desired bromide salt (7c) as a bright yellow solid, yield: 61%; UV (MeOH) λmax (log ε): 430.6 (3.73), 349.2 (4.23), 265.7 (4.32), 234.0 (4.42) nm; 1H-NMR (500 MHz, CDCl3): δ 10.33 (s, 1H, H-8), 7.98 (d, J = 9.2 Hz, 1H, H-12), 7.94 (s, 1H, H-1), 7.83 (d, J = 9.2 Hz, 1H, H-11), 6.85 (s, 1H, H-4), 6.09 (s, 2H, -OCH2O-), 5.24 (d, J = 5.8 Hz, 1H, H-6), 4.34 (s, 3H, -OCH3), 4.08 (s, 3H, -OCH3), 3.83 (d, J = 6.5 Hz, 2H, H-1’), 3.29 (t, J = 5.8 Hz, 2H, H-5), 1.83 (p, J = 7.1 Hz, 2H, H-2’), 1.50 (p, J = 7.2 Hz, 2H, H-3’), 1.33‒1.26 (m, 8H, H-4’‒H-7’), 0.90 (t, J = 6.8 Hz, 3H, H-8’); 13C NMR (125 MHz, CDCl3): δ 152.3, 150.7, 149.9, 147.1, 146.2, 143.4, 132.1, 130.9, 129.9, 125.4, 123.0, 118.5, 117.9, 108.5, 108.4, 102.0, 75.9, 63.2, 57.1, 31.8, 30.2, 29.3,29.2, 28.2, 26.0, 22.6, 14.1; LC-MS (ESI+, m/z) C28H34NO5+: 464.58 [M − Br]+; HRMS-ESI: m/z calculated for C28H34NO5+: 464.2437 [M‒Br]+, found for 464.2436.
13-O-Decylberberine chloride (7d). Compound (4) was treated with n-decyl bromide according to the general procedure to give the desired bromide salt (7d) as a bright yellow solid, yield: 58%; UV (MeOH) λmax (log ε): 428.3 (3.70), 349.6 (4.19), 265.4 (4.28), 223.8 (4.47) nm; 1H-NMR (500 MHz, CDCl3): δ 10.50 (s, 1H, H-8), 7.97 (d, J = 9.2 Hz, 1H, H-12), 7.94 (s, 1H, H-1), 7.81 (d, J = 9.2 Hz, 1H, H-11), 6.86 (s, 1H, H-4), 6.09 (s, 2H, -OCH2O-), 5.31 (d, J = 5.8 Hz, 1H, H-6), 4.34 (s, 3H, -OCH3), 4.09 (s, 3H, -OCH3), 3.82 (d, J = 6.5 Hz, 2H, H-1’), 3.28 (t, J = 5.8 Hz, 2H, H-5), 1.83 (p, J = 7.2 Hz, 2H, H-2’), 1.50 (d, J = 7.2 Hz, 2H, H-3’), 1.32‒1.23 (m, 12H, H-4’‒H-9’), 0.90 (t, J = 6.9 Hz, 3H, H-10’); 13C-NMR (125 MHz, CDCl3): δ 151.3, 150.6, 149.9, 147.5, 146.4, 143.9, 132.2, 130.8, 129.8, 125.3, 123.1, 118.5, 117.8, 108.4, 102.0, 75.8, 63.2, 57.0, 56.7, 31.9, 30.1,29.5, 29.33, 29.29, 28.2, 26.0, 22.7, 14.1; LC-MS (ESI+, m/z) C30H38NO5+: 492.62 [M − Br]+; HRMS-ESI: m/z calculated for C30H38NO5+: 492.2750 [M − Br]+, found for 492.2751.
13-O-Dodecylberberine chloride (7e). Compound (4) was treated with n-dodecyl bromide according to the general procedure to give the desired bromide salt (7e) as a bright yellow solid, yield: 40%; UV (MeOH) λmax (log ε): 427.5 (3.63), 350 (4.13), 266.4 (4.20), 225.4 (4.37) nm; 1H-NMR (500 MHz, DMSO-d6): δ 9.80 (s, 1H, H-8), 8.20 (d, J = 9.4 Hz, 1H, H-12), 8.09 (d, J = 9.4 Hz, 1H, H-11), 7.89 (s, 1H, H-1), 7.13 (s, 1H, H-4), 6.16 (s, 2H, -OCH2O-), 4.86 (t, J = 5.9 Hz, 2H, H-6), 4.09 (s, 3H, -OCH3), 4.08 (s, 3H, -OCH3), 3.84 (t, J = 6.4 Hz, 2H, H-1’), 3.16 (t, J = 5.9 Hz, 2H, H-5),1.77 (p, J = 7.0 Hz, 2H, H-2’), 1.43(m, 2H, H-3’), 1.24 (m, 16H, H-4’‒H-11’), 0.85 (t, J = 6.9 Hz, 3H, H-12’); 13C-NMR (125 MHz, DMSO-d6): δ 150.9, 150.1, 149.2, 146.8, 144.2, 142.0, 132.5, 130.9, 128.8, 126.1, 122.2, 118.5, 118.4, 108.3, 108.0, 102.1, 75.4, 62.0, 57.1, 56.4, 31.3, 29.4, 29.04, 29.02, 28.96, 28.89, 28.75, 28.7, 26.9, 25.4, 22.1, 14.0; LC-MS (ESI+, m/z) C32H42NO5+: 520.68 [M‒Br]+; HRMS-ESI: m/z calculated for C32H42NO5+: 520.3063 [M − Br]+, found for 520.3066.
3.5. Biological Activity
3.5.1. Cell Lines and Cell Culture Conditions
All cells were obtained from the Bioresource Collection and Research Center (Hsinchu, Taiwan). Human colon cancer cells (HT-29) was cultured in RPMI 1640 (HyClone, South Logan, UT, USA) medium, supplemented with 10% fetal bovine serum (FBS, Gibco, Life Technologies, Carlsbad, CA, USA) and antibiotics (100 mg/mL streptomycin and 100 U/mL penicillin, Gibco, Life Technologies, USA). Human bladder cancer cells (BFTC 905) and human liver carcinoma (HepG2) cell lines were cultured in Dulbecco’s modified Eagle medium (DMEM, HyClone, USA) medium, supplemented with 10% fetal bovine serum (FBS) and antibiotics. Human cancer cells were maintained in humidified atmosphere with 5% CO2 and 95% air at 37 °C in a carbon dioxide incubator (SANYO, CO2 incubator, Osaka, Japan).
3.5.2. Cell Viability Inhibition Assay
Cell viability was assessed by staining with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT, Bionovas Biotechnology Co., Ltd., Toronto, Ontario, Canada) [
32]. Briefly, the cancer cells were plated at 5 × 10
3 – 1 × 10
4 cells into 96-well plates incubated at 37 °C in a humidified 5% CO2 incubator for 24 h. After medium removal, 100 μL of culture medium with 0.1% DMSO containing the test compounds at different concentrations was added to each well and incubated at 37 °C for another 24 h. At the end of treatment, final concentration of 0.5 mg/mL MTT was added, and cells were incubated for a further 1.5 h. The absorbance was recorded on an ELISA plate reader (SpectraMax 340PC
384, Molecular Devices, Orleans, CA, USA) at a 540 nm wavelength. Inhibition ratio (%) was calculated using the following equation:
A
treated and A
control are the average absorbance of three independent experiments from treated and control groups, respectively.
Cell viability was determined the test compound concentration required to inhibit tumor cell proliferation by 50% (IC50) from the dose-response curves. All data average values from triplicate samples and the experiments were repeated at least three times.
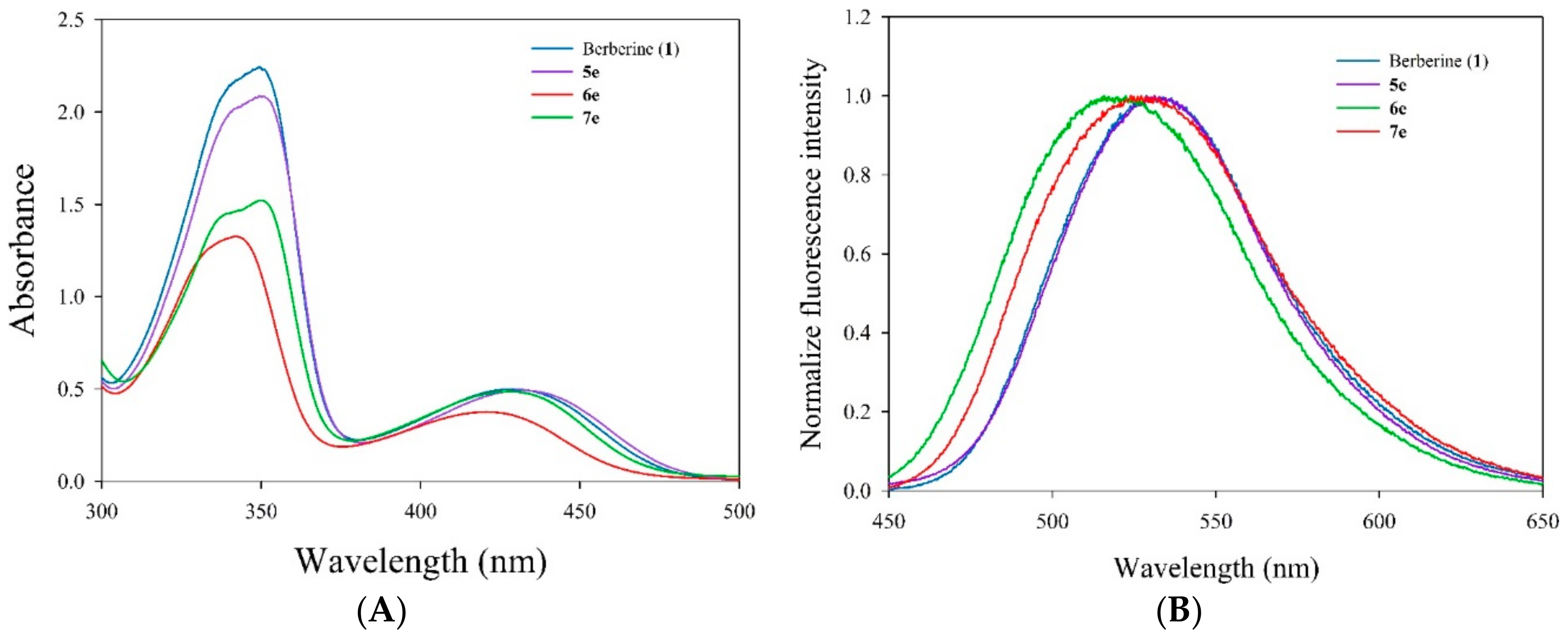
3.5.3. Measurement of Absorption and Fluorescence Spectra
Absorption spectrum (250–500 nm) of berberine and its derivatives (5e, 6e, and 7e) were recorded on a UV–vis spectrophotometer (UV-1601, Shimadzu, Kyoto, Japan) with a quartz cuvette (path length 1 cm, accuracy ± 0.2 nm). Fluorescence emission spectrum were recorded on a fluorescence spectrophotometer (RF-5301PC, Shimadzu, Kyoto, Japan) with excitation at 420 nm and a measure of emission from 450 to 700 nm (slit 3/3 nm) in a 1 cm quartz cuvette. A final concentration of berberine and its derivatives 5e, 6e, and 7e were 10 μM diluted using methanol. Appropriate blanks corresponding to the methanol were subtracted to correct the background.
3.5.4. Light Source
The UVA light box consisting of 8 UVA lamps (Blacklight blue, F8T5BLB, Sankyo Denki Co. Ltd., Osaka, Japan) was custom made [
37]. The light spectral irradiance of the light box was determined using a NIST-Traceable radiometer (ILT1400 Portable radiometer, International Light Technologies Inc., MA, USA). The light dose of the UVA light box, with the maximum emission at 420 nm, was routinely measured using a photochemical reactor (PR-1000, Panchum Scientific Corp., Kaohsiung, Taiwan). In the study, the light-irradiation doses were determined at 1.8, 3.6, 7.2 J/cm
2 for 420 nm (dose rate 5.9 mW/cm
2), which were obtained by approximately 5, 10, and 20 minutes of exposure, respectively. To determine visible light dose using the following equation:
where
E stands for visible light energy,
t represents time expressed in second and, finally,
P is lamp potency.
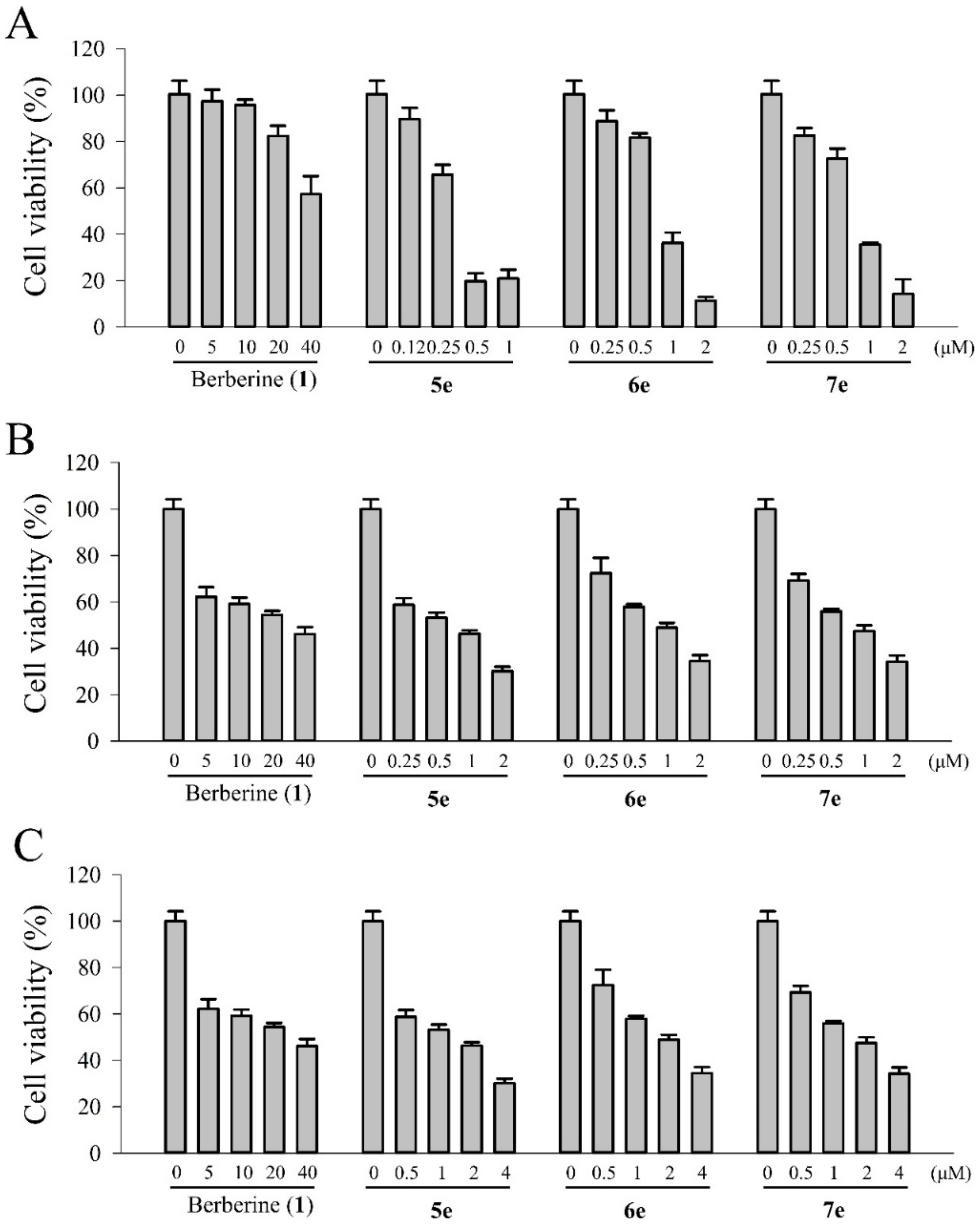
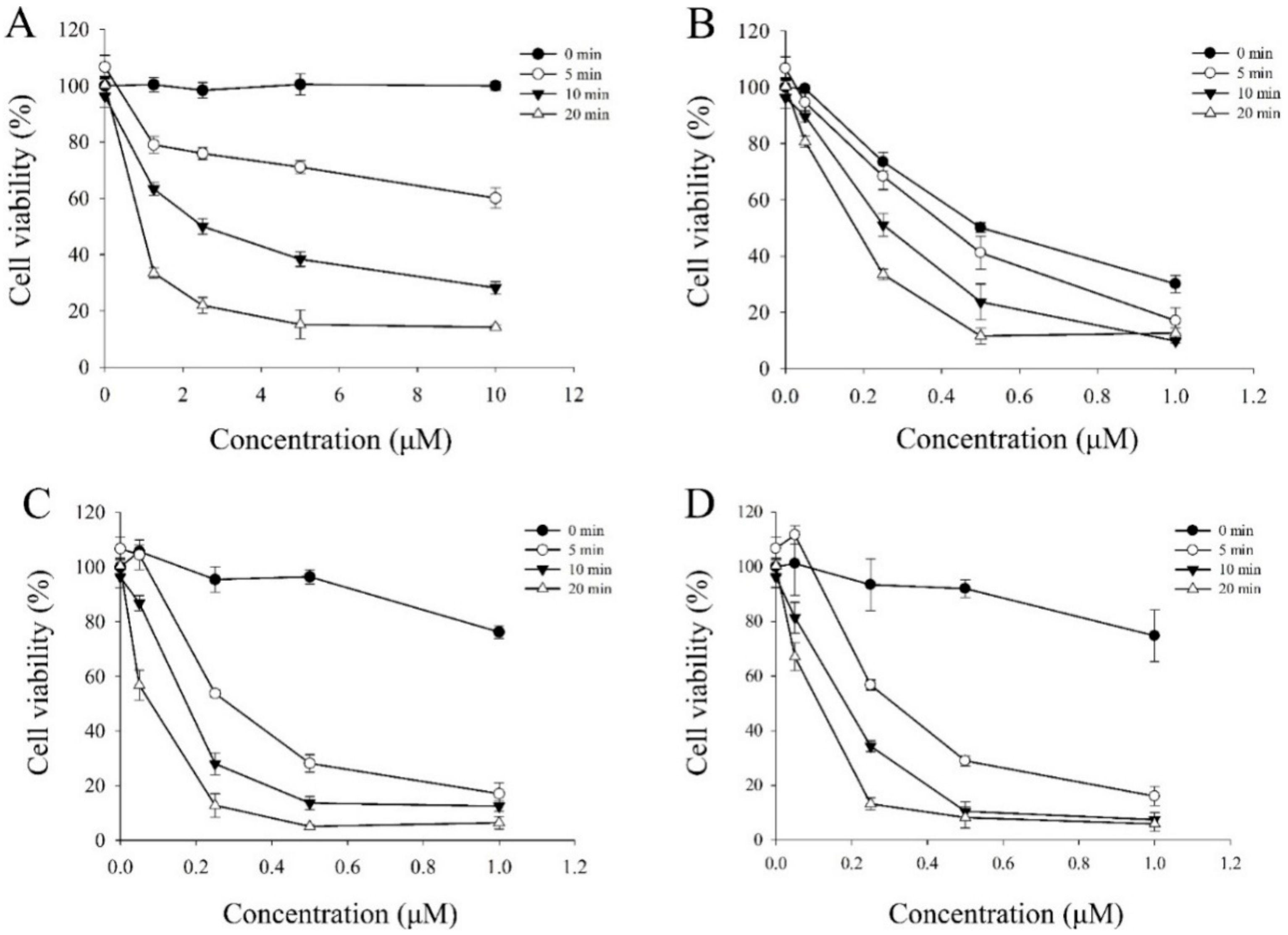
3.5.5. Photocytotoxic Assay
The photocytotoxicity of berberine (
1) and its derivatives (
5e,
6e, and
7e) were studied by using the MTT colorimetric assay with quantification of formazan formed by cleavage of the tetrazolium rings of MTT in DMSO by spectral measurement [
32]. Various concentrations of the berberine (
1) and berberine derivatives (
5e,
6e, and
7e) dissolved in 1% DMSO were added to the cells and incubation was continued for 4 h in the dark, followed by photo-irradiation with visible light of 420 nm (1.8, 3.6 and 7.2 J/cm
2 for 5, 10 and 20 min of irradiation, respectively) by using a photochemical reactor (PR-1000, Panchum Scientific Corp., Kaohsiung, Taiwan) filled with 8 fluorescent black tubes lamps (Blacklight blue, F8T5BLB, Sankyo Denki Co. Ltd., Osaka, Japan). For these experiments, the cells were seeded in 96-well microliter plates (5000 cells/well) and grown for at least 24 h in 100 μL DMEM containing 10% FBS. The growth medium was replaced with fresh medium containing 3% FBS and berberine (5, 10, 20, and 40 μM) and its derivatives (0.05, 0.25, 0.5, and 1.0 μM) and the cells were incubated for 4 h at 37 °C. At the end of the incubation, the drug-containing medium was removed, and the cells were washed with fresh medium and irradiated for 5, 10, and 20 min with visible light (420 nm). After the incubation period for 20 h, 10 μL MTT (5 mg/mL) was added to each well and incubated for an additional 1.5 h at 37 °C, 100 μL DMSO was added to dissolve the formazan crystals. The absorbance of the samples was measured using an ELISA microplate reader (SpectraMax 340PC
384, Molecular Devices, Orleans, CA, USA) and selecting 540 nm as the lest wavelengths. The survival of the treated cells was calculated as the percentage of their absorbance referred to the absorbance of the untreated (no drug, no light) cells taken as 100% viability. The IC
50 values were obtained by nonlinear regression analysis (Sigma 11.0 software).
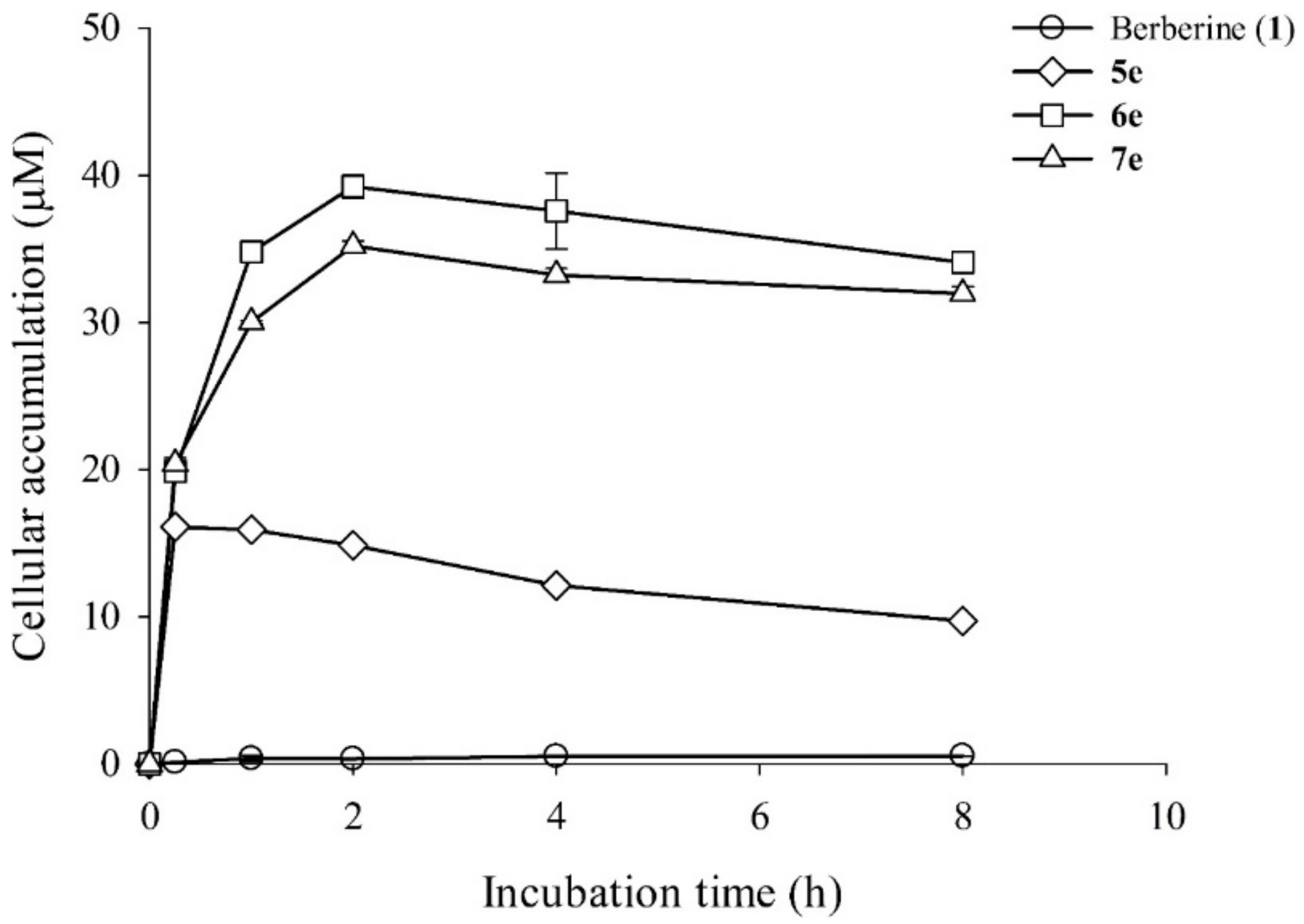
3.5.6. Intracellular Uptake Profiling of Berberine and its Derivatives in HepG2 Cells
In order to establish a time profile for intracellular accumulation of these berberine derivatives, an HPLC analysis was carried out. The HepG2 cells treated with different berberine (1) and its derivatives (5e, 6e, and 7e) were allowed to grow in 5 mL liquid culture. The cultures were incubated in a shaking incubator in the dark. After 0 h, 0.25 h, 1 h, 2 h, 4h, and 8 h, aliquots of cells were harvested by centrifugation at 6000 rpm for 5 min. The supernatant liquid was discarded and the cell pellet was washed thrice with 1 mL of 2×PBS in order to remove the uninternalized berberine and its derivatives. After this, the cells were gently resuspended in 0.2 mL methanol and then were filtered with a 0.22 μm filter into HPLC analysis. The HPLC analysis was carried out by exciting the sample at 350 nm and emission at 530 nm.
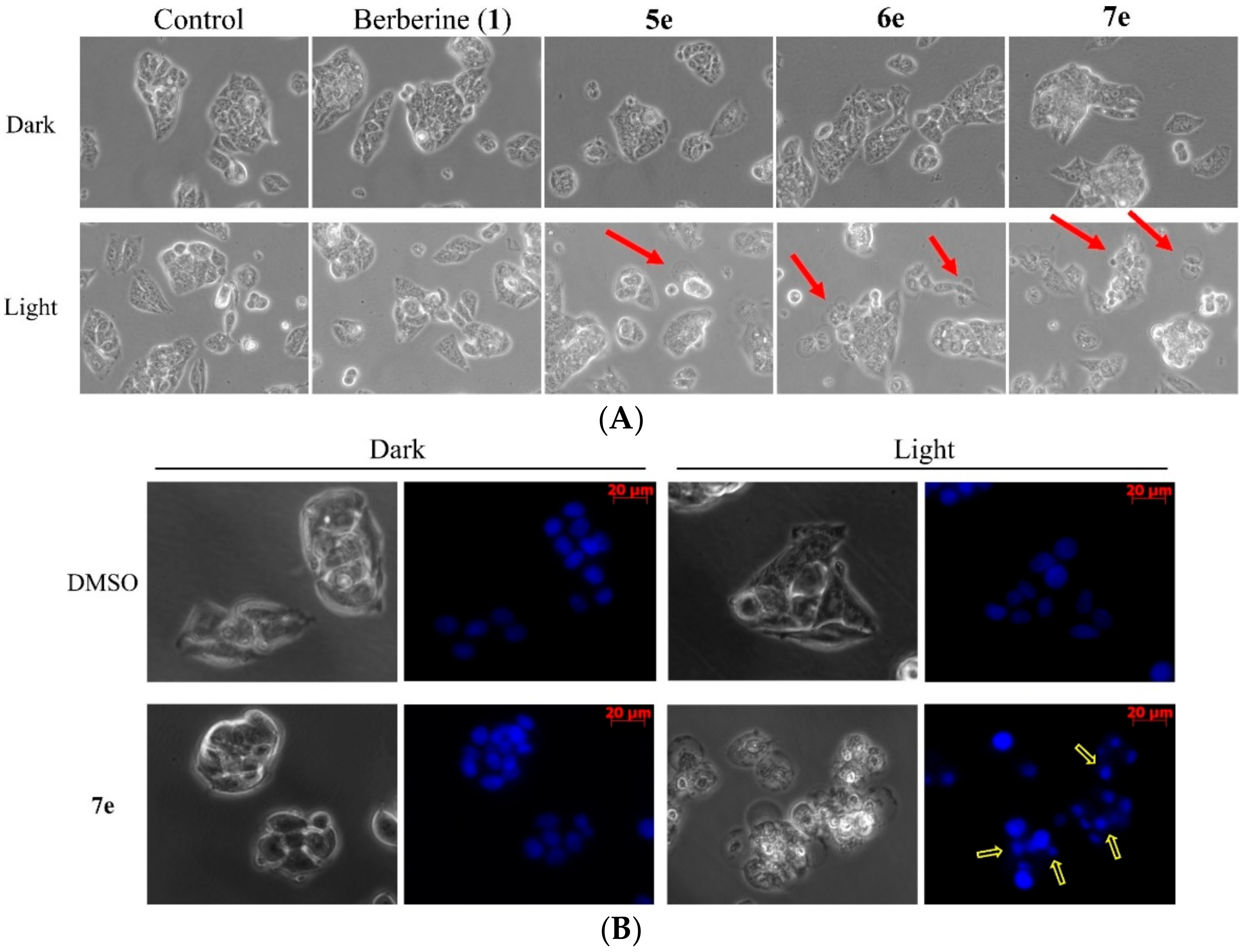
3.5.7. Cell Morphological Assessment
To detect morphological evidence of apoptosis, cells were visualized following DNA staining with the fluorescent dye Hoechst 33258 [
34]. The HepG2 cells were plated in a 6 cm dish at a density of 7×10
5 cells/well and were incubated overnight. The cells were treated with 0.5 μM concentrations berberine (
1), compounds
5e,
6e, and
7e for 4 h in the dark and treated with visible light (420 nm) for 10 min. Additionally, the HepG2 cells of compound
7e treatment were stained with 10 μM Hoechst 33258 (Sigma-Aldrich, St. Louis, MO, USA). After Cells were incubated in a dark room for 5 min, the cells were washed with PBS and then were observed by a fluorescent microscope (Zeiss, Axio Observer A1, Japan).
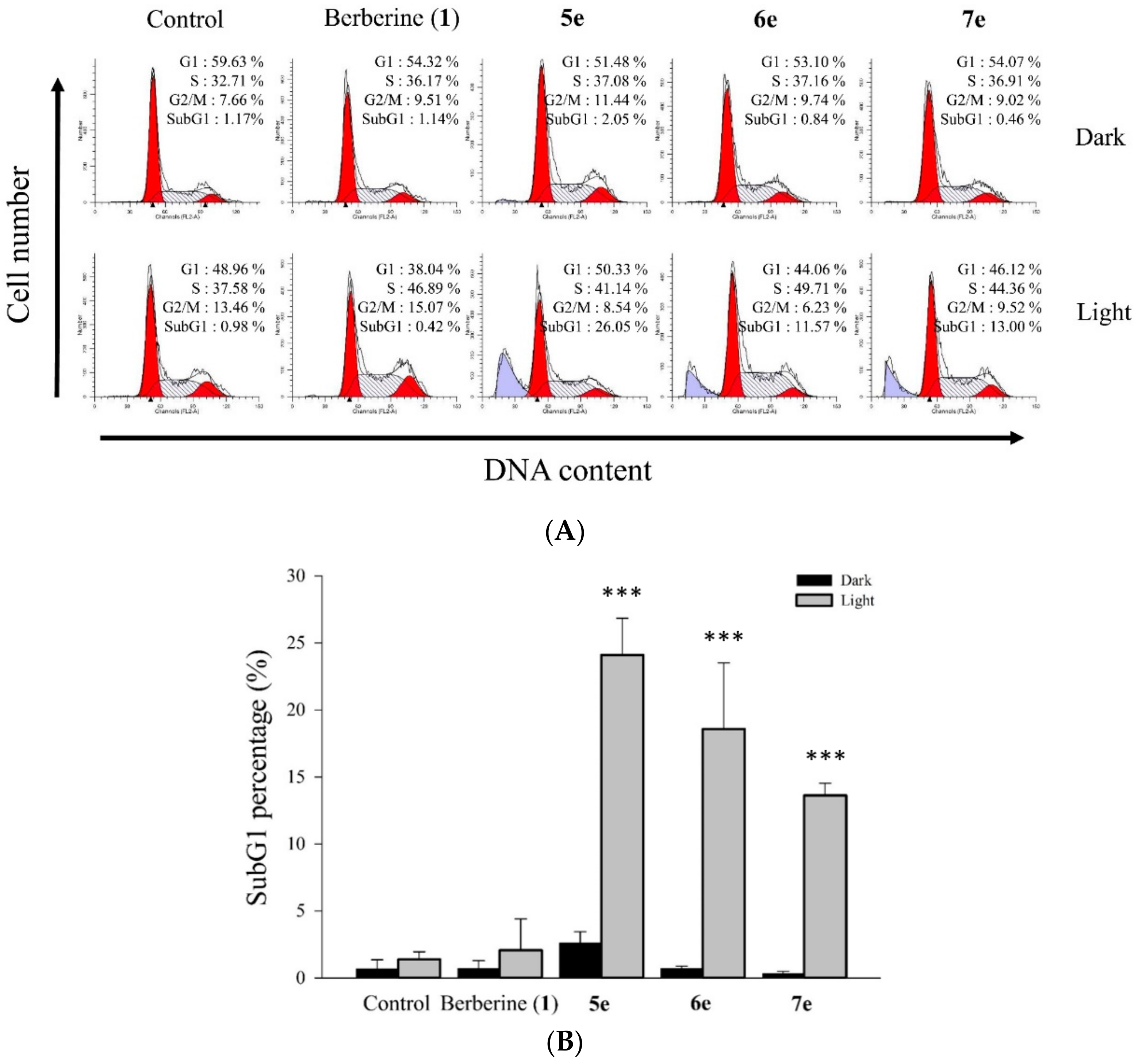
3.5.8. Cell Cycle Distribution Analysis
Flow cytometry was used to obtain the cell cycle distribution and the apoptotic rate [
35]. The HepG2 cells were plated at 1×10
6 cells per 6 cm dish and cultured overnight. Then, various concentrations of 0.5 μM berberine (
1), compounds
5e,
6e, and
7e were added for 2 h. At the end of the incubation, the drug-containing medium was removed, and the cells were washed with fresh medium and irradiated for 10 min with visible light (420 nm). The cells of each dish were harvested, washed once with PBS, and fixed with methanol at 4 °C. After 24 h, the cells of each dish were harvested and washed once with PBS. The cells were suspended in 473 μL of PBS containing 40 μg/mL propidium iodide (PI) and 40 μg/mL RNase. Cells were incubated in a dark room for 30 min at room temperature then subjected to cell cycle analysis using a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA) and analyzed using the ModFit 3.0 software (Verity Software House, ME, USA).
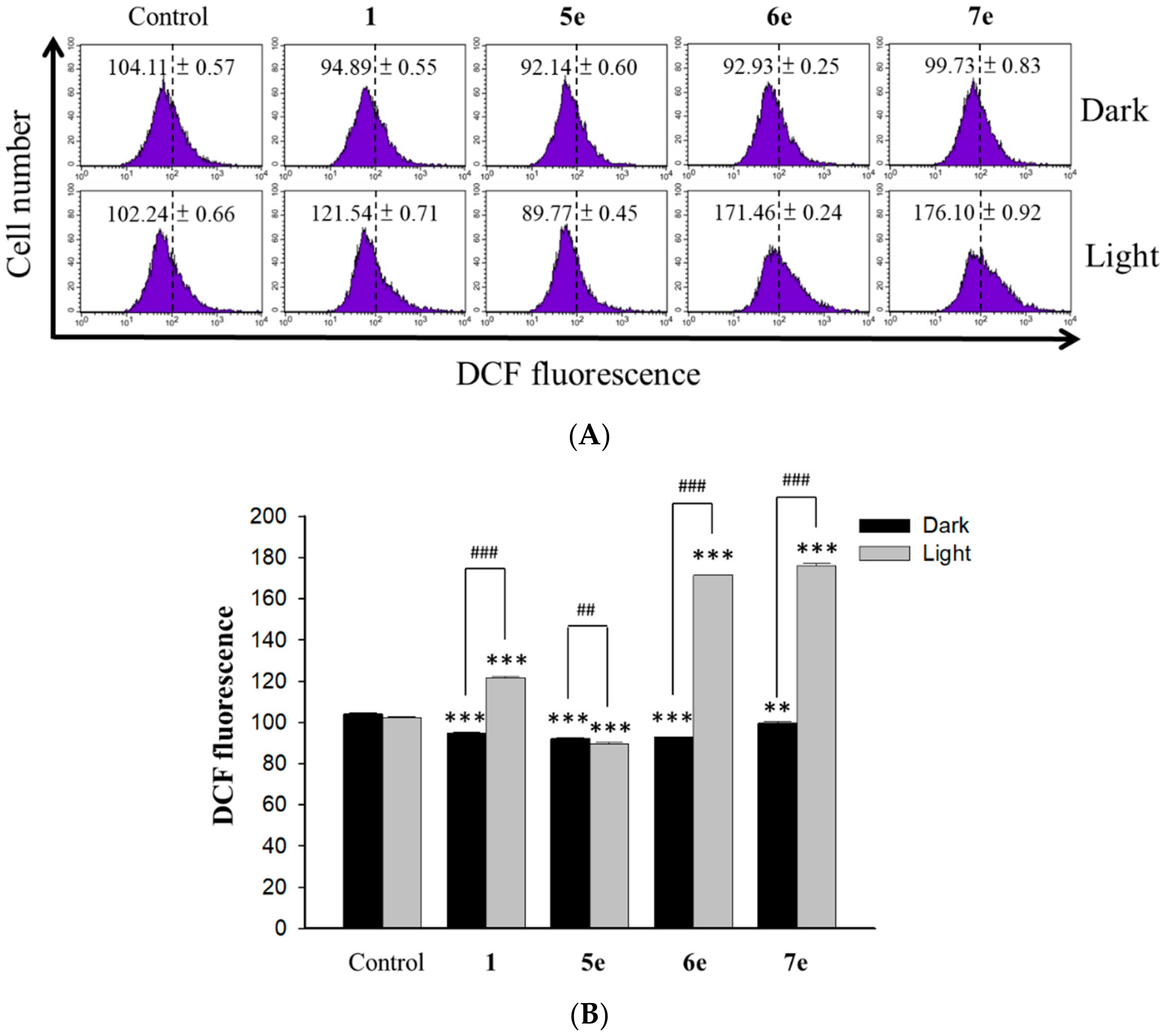
3.5.9. Measurement of Intracellular Singlet Oxygen Production
Intracellular oxidant stress was monitored by measuring changes in fluorescence resulting from intracellular probe oxidation. Intracellular production of ROS, namely singlet oxygen (
1O
2), was measured using the DCFH-DA (Molecular Probes, USA) probe, respectively [
36]. The level of intracellular singlet oxygen was detected using fluorescent dye DCF sensitivity. Briefly, the HepG2 (4×10
4 cells/ml) cells were pretreated with 0.5 μM of berberine (
1) and 0.25 μM of its derivative (
5e,
6e and
7e) for 2 h. At the end of the incubation, the drug-containing medium was removed, and the cells were washed with fresh medium and irradiated for 10 min with visible light (420 nm). After the incubation period for 24 h, intracellular peroxide level was detected by DCFH-DA as an intracellular fluorescence probe. After incubation with 20 μM DCFH-DA for 1 h, the cells were harvested and washed with PBS twice. A flow cytometer (Becton Dickinson, San Jose, CA, USA) was used to detect dichlorofluorescein (DCF). Data were collected and analyzed by ModFit 3.0 software (Verity Software House, ME, USA).
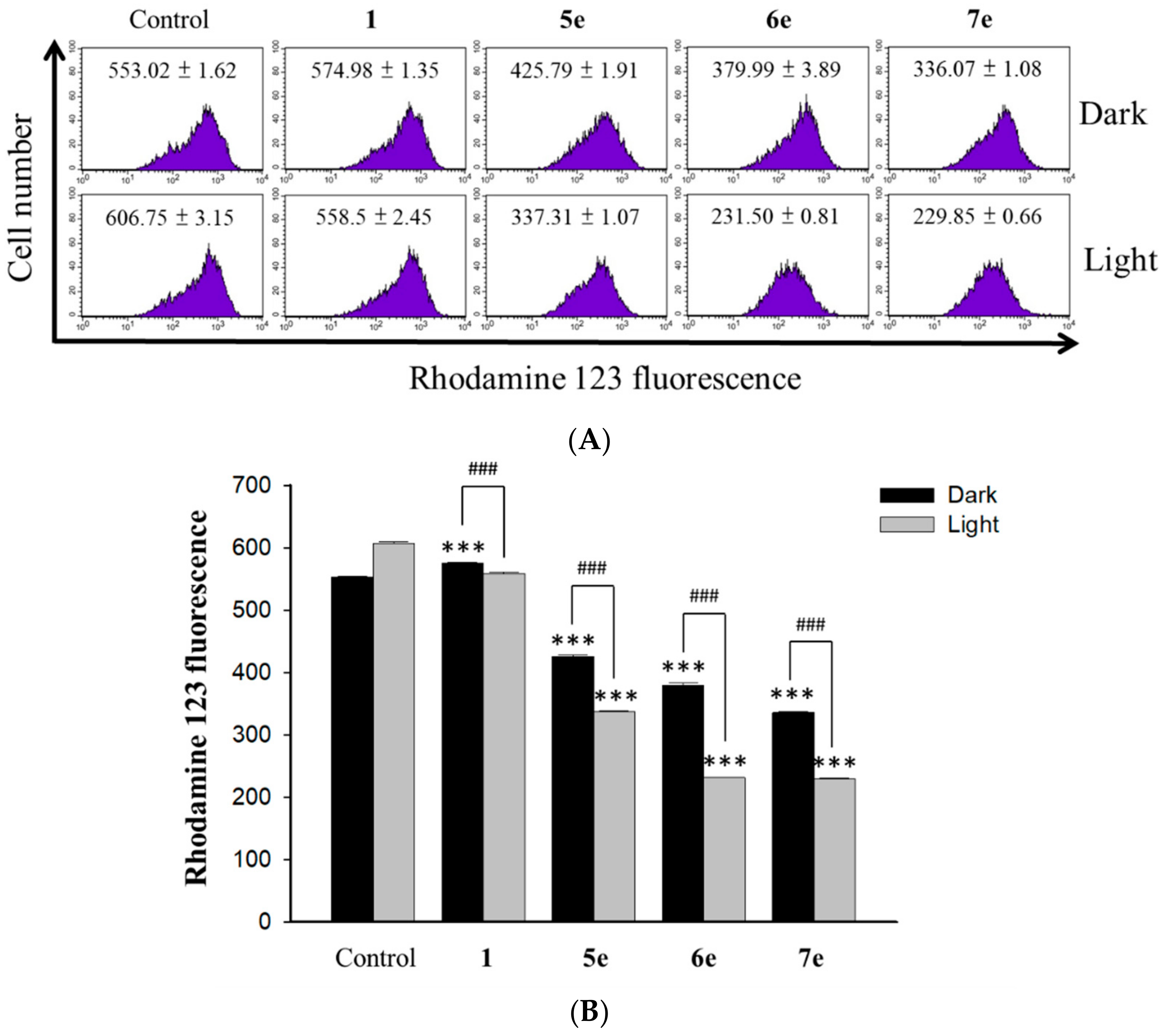
3.5.10. Measurement of the Mitochondrial Membrane Potential
The changes in the mitochondrial membrane potential (MMP) were determined by using the fluorescent probe rhodamine 123 (Sigma, St Louis, USA). The dye Rh123 selectively enters mitochondria without influencing the membrane potential and is retained inside the mitochondria [
37]. Once the MMP is lost, rhodamine 123 is subsequently washed out of the cells, leading to the decline of the fluorescence signal. HepG2 cells (1×10
5 cells/mL) were incubated with concentrations of 0.5 μM berberine (
1) and 0.25 μM of its derivative (
5e,
6e, and
7e) for 2 h. At the end of the incubation, the drug-containing medium was removed, and the cells were washed with fresh medium and irradiated for 10 min with visible light (420 nm) for 24 h. Then, stained with 2 mg/mL of rhodamine 123 for 20 min under gentle shaking. After washing with PBS, cells were immediately analyzed by flow cytometer (Becton Dickinson, San Jose, CA, USA) and data were analyzed using the ModFit 3.0 software (Verity Software House, ME, USA).
3.5.11. Statistical Analysis
The values shown are mean ± SD of three independent experiments. Data are statistically evaluated by Student’s t-test of SigmaPlot 11.0 and shown significantly different in * p < 0.05, ** p < 0.01, and *** p < 0.001.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}