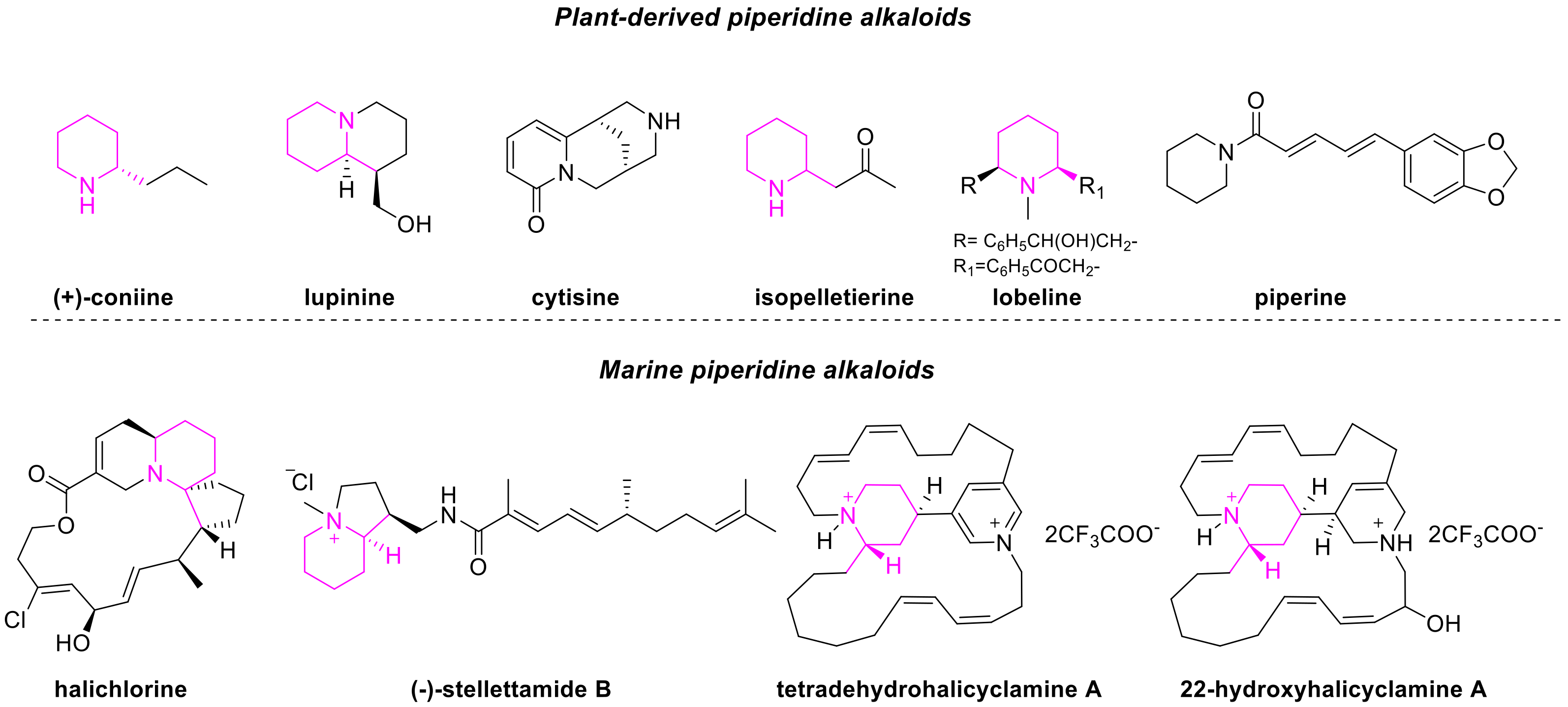
1. Introduction
Piperidine alkaloids are widespread natural products in the plant kingdom, but they are also present in the animal one, in particular in marine microorganisms [
1]. They are historically known as potent poisons and teratogens: some well-known examples include coniine (from hemlock), lupinine (from lupin flour) and cytisine, contained in
Laburnum spp. pods [
2]. These effects are mainly the result of the nicotinic acetylcholine receptors (nAChRs) desensitisation [
3]. However, piperidine alkaloids are characterised also by a wide range of interesting biological properties, including anthelminthic (e.g., isopelletierine), antioxidant and neuroprotective activities (e.g., lobeline) and the abilities of increasing drugs bioavailability and of contrasting multidrug resistance (e.g., piperine, lobeline) [
1]. The structures of the mentioned alkaloids are depicted in
Figure 1.
In the marine kingdom, some representative piperidine-containing bioactive compounds, isolated mainly from sponges, are halichlorine (anti-inflammatory, cytotoxic) [
4] and stellettamide B (antifungal and RNA-cleaving activities, from
Stelletta sp. [
5]) (
Figure 1).
Also several bis-piperidine alkaloids, in which the two heterocycles are embedded in complex macrocyclic systems, are diffused in marine microorganisms. These compounds proved to possess antifungal, antimicrobial properties and cytotoxic activity against different cancer cell lines. Some examples belonging to this class are tetradehydrohalicyclamine A and B, 22-hydroxyhalicyclamine A, haliclonacyclamine A and F, arenosclerins D and E, madangamine F and neopetrosiamine A [
6,
7,
8,
9,
10].
In this context, we realized that some of the cited natural products shared a common structural motif, constituted by a piperidine ring substituted in the 2-position, generating in this way a defined stereocenter (shown in pink in
Figure 1).
These features seemed quite familiar to us, because they characterised a library of compounds previously synthesised in our laboratory. In fact, the importance of the piperidine ring in bioactive natural products, prompted us to devise a diversity-oriented approach (DOS), aimed at the generation of a diversified library of derivatives containing this key heterocycle. In this project, the ideal starting point to achieve structural diversification proved to be the 2-piperidine ethanol (
1), a cheap and commercially available racemic product that incorporates the fundamental highlighted 2-substituted-piperidine moiety. Our library is composed by natural products (such as aloperine, coniine, boehmeriasin A and sedum alkaloids [
11,
12,
13,
14]), and synthetic derivatives [
15,
16,
17,
18], as depicted in
Figure 2.
We are currently trying to further enrich our library with new scaffolds. To this end, here we report the synthesis of (–)-anaferine starting from 1, as a further ramification of our DOS approach.
(–)-Anaferine is a bis-piperidine alkaloid, in which the two rings are connected through their C-2 atoms by a 2-propanone bridge. Being C2-symmetrical, this framework exists as two enantiomers and a meso-form. The diastereomeric mixture was isolated for the first time in 1962 [
19] from the root extract of
Withania somnifera, a plant widely exploited in the Ayurvedic medicine on the basis of several biological properties, including anti-inflammatory, anti-stress, immunomodulatory, adaptogenic, anticancer and neuroprotective activities [
20]. ORD (optical rotary dispersion) analysis led to the determination of the absolute configuration of the two enantiomers: (–)-anaferine proved to be the (
R,R)- enantiomer, and (+)-anaferine the (
S,S)-one [
21].
Most of the studies and the synthetic efforts on this topic, date back to the 70s, but in the last years this alkaloid has attracted once again the interest of the scientific community, thanks to
in silico studies that suggested possible applications in the treatment of neurodegenerative diseases [
22,
23].
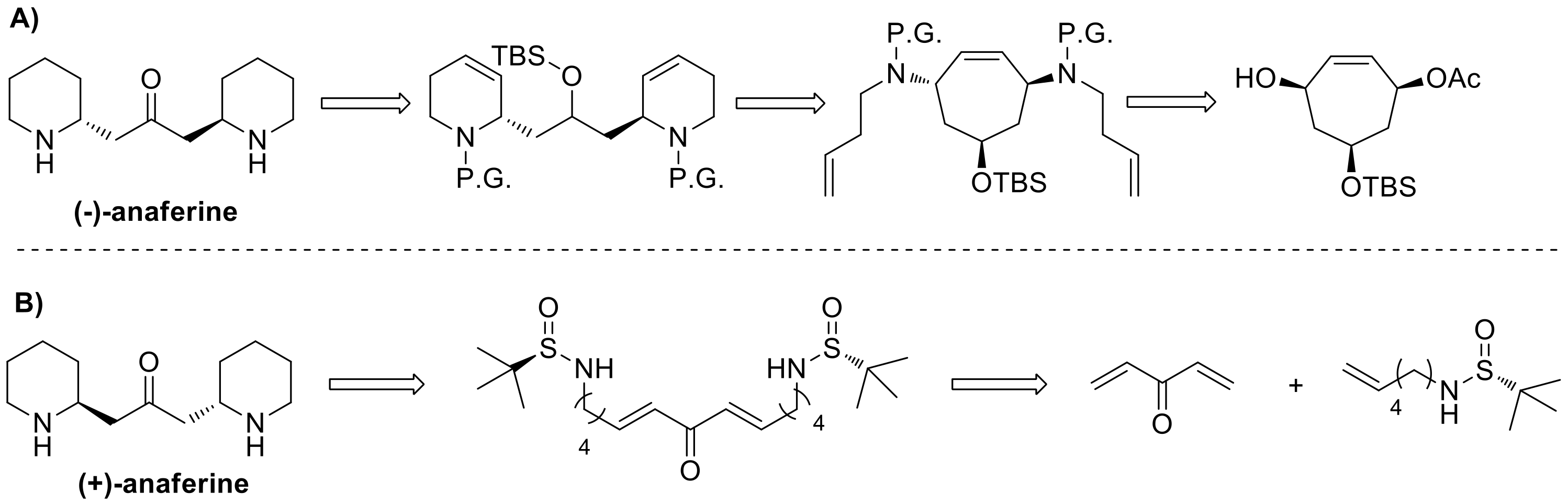
The last total synthesis of (–)-anaferine was accomplished in 2002 by Stapper and Blechert [
24], while del Pozo et al. recently reported the preparation of the (+)-enantiomer [
25] (
Figure 3).
Stapper and Blechert’s strategy took advantage of an enantiopure cycloheptene precursor, accessed in five steps from the commercially available tropone. Then, after its conversion into the corresponding diamine, a tandem ring rearrangement metathesis, consisting in a ring-opening metathesis, followed by two ring-closing metatheses, afforded the fundamental carbon skeleton, that was transformed into the final product upon reduction of the double bonds, cleavage of the protecting groups and oxidation of the secondary alcohol to ketone (
Figure 3A).
On the other hand, del Pozo’s approach involved the double use of
N-sulfinyl amines, as both chiral auxiliaries and nucleophiles in an intramolecular aza-Michael reaction, as appreciable in
Figure 3B. A bidirectional cross metathesis (CM), between the required sulfinyl amine and divinyl ketone gave an intermediate that underwent a double intramolecular aza-Michael reaction, forming the bis-piperidine core and generating at the same time the two stereocenters. Acidic cleavage of the sulfinyl groups led to (+)-anaferine synthesis completion.
2. Results and Discussion
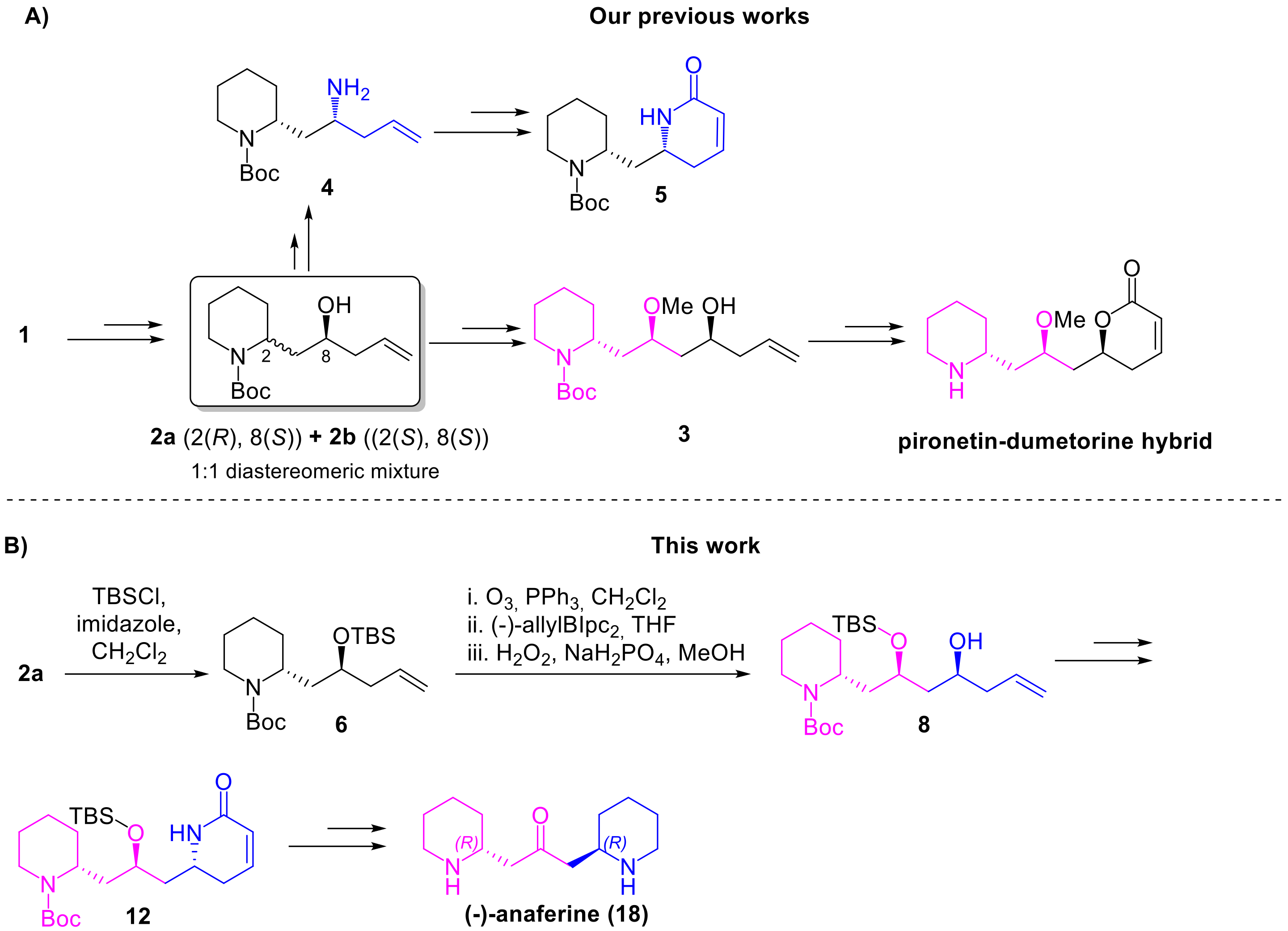
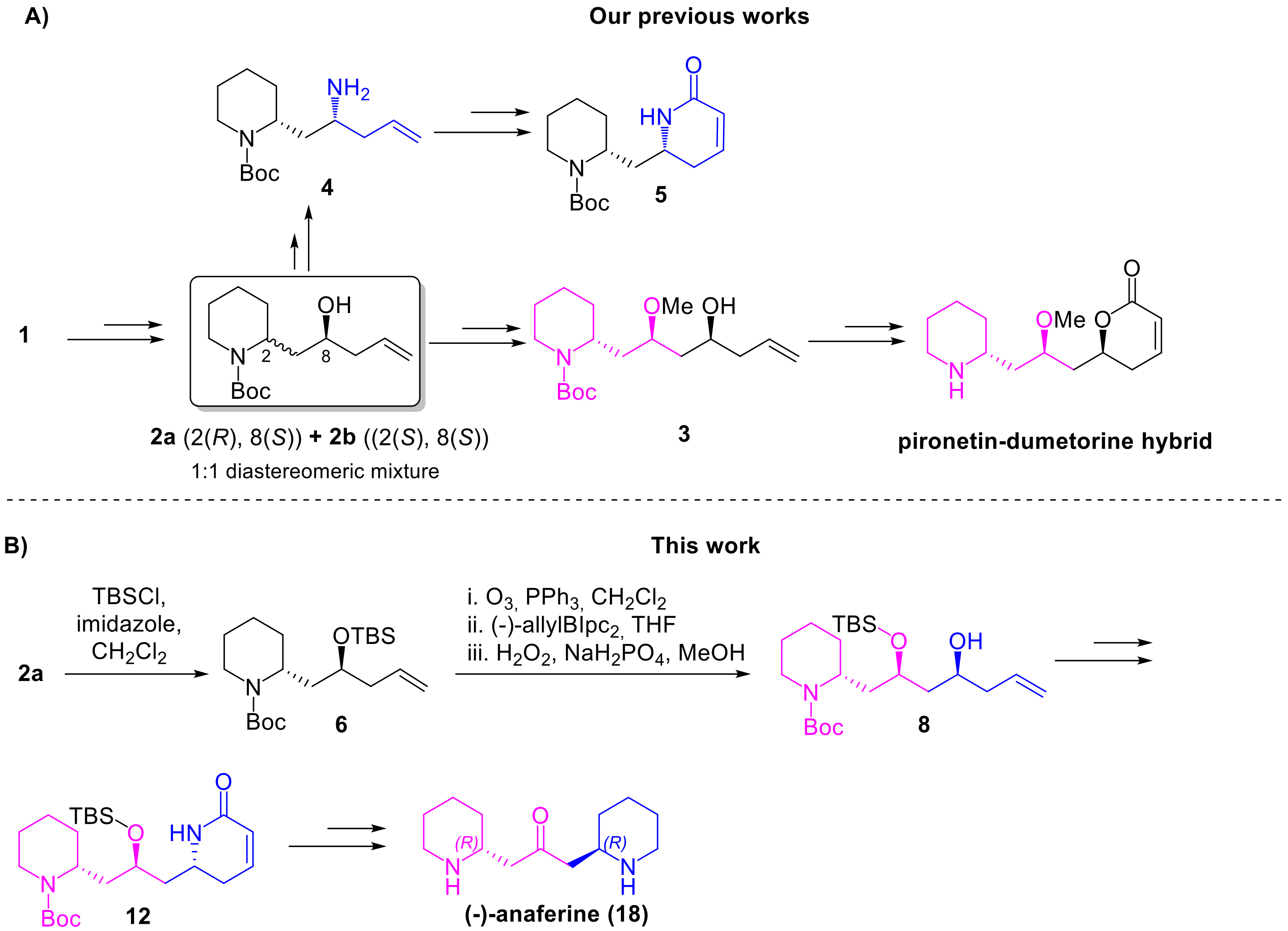
The idea of enriching our DOS library with (–)-anaferine was suggested by compound
3 (
Scheme 1A), previously obtained in our laboratory as an intermediate in the synthesis of the pironetin-dumetorine hybrids shown in
Figure 2 [
15]. Compound
3 is characterised by a piperidine core, deriving from the 2-piperidine ethanol, by a 2-methoxy propane bridge and by a homoallylic alcohol. The synthesis of
3, starting from 2-piperidine ethanol is summarized in
Scheme 1A, and involves as key step the conversion of
1 into the homoallylic alcohol
2a. Notably,
2a was recently exploited also for the synthesis of an α,β-unsaturated lactam (compound
5), as appreciable again in
Scheme 1A [
18]. Therefore, comparing the two synthetic routes, it’s possible to deduce the genesis of our synthetic plan. In fact, we reasoned that an analogue (
8) of compound
3, bearing an O-silylether instead of the methoxy group, could have been easily obtained starting from
2a, taking advantage of the same synthetic strategy. The homoallylic alcohol of
8 could be then converted into lactam
12, exploiting the same approach employed for the synthesis of
5 from
2a. The synthesis of (–)-anaferine could be completed reducing the
α,β-unsaturated lactam of
12 to the corresponding piperidine, and oxidizing the secondary alcohol to ketone, as depicted in the forward synthetic
Scheme 1B. Of course, the synthesis of (–)-anaferine requires a strict control over the stereochemistry, in order to access the correct
(R,R)-enantiomer. To this extent, two asymmetric Brown allylations were considered.
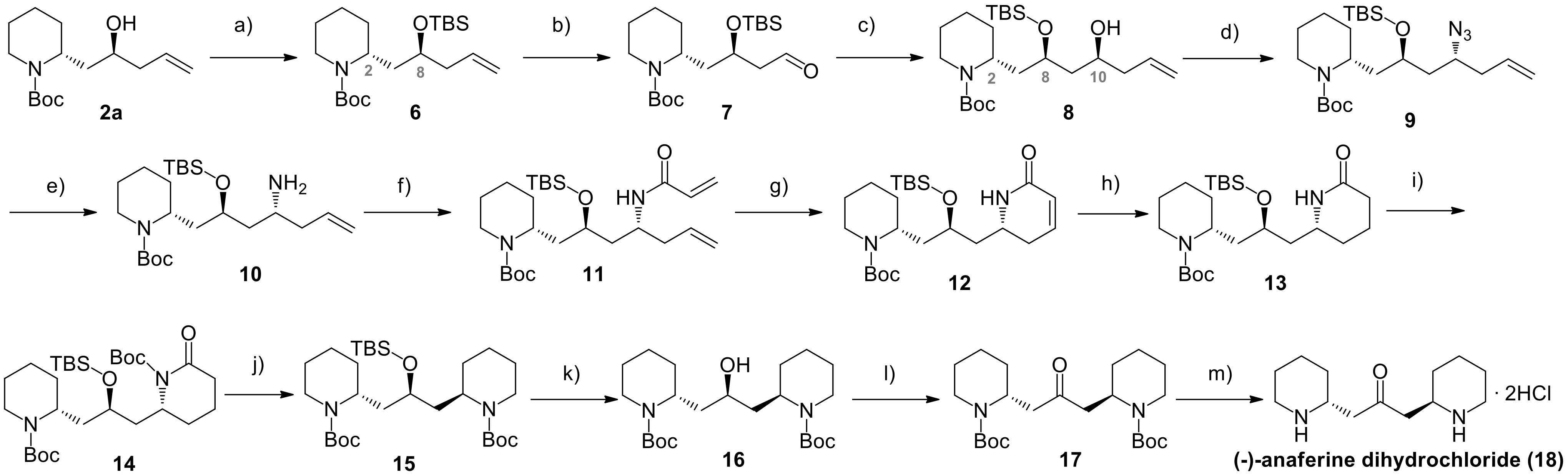
The proposed synthetic strategy started from the homoallylic alcohol
2a, previously obtained in our research group from
1 in three steps, involving the protection of the piperidinic nitrogen, oxidation of the alcohol to aldehyde and its asymmetric allylation under Brown’s conditions, leading to two diastereomeric alcohols
2a and
2b [
18] (
Scheme 1A). The relative
anti-configuration of
2a was established by X-ray analysis, while the desired absolute (
R,S)-configuration resulted from the use of the proper allylating agent enantiomer (in this case the (–)-B-allyl diisopinocampheylborane). In this way,
2a was obtained with an
e.e. = 84% (
Supplementary Information, Figure S1) and was then protected at the hydroxyl group as
tert-butylsilyl ether (
6).
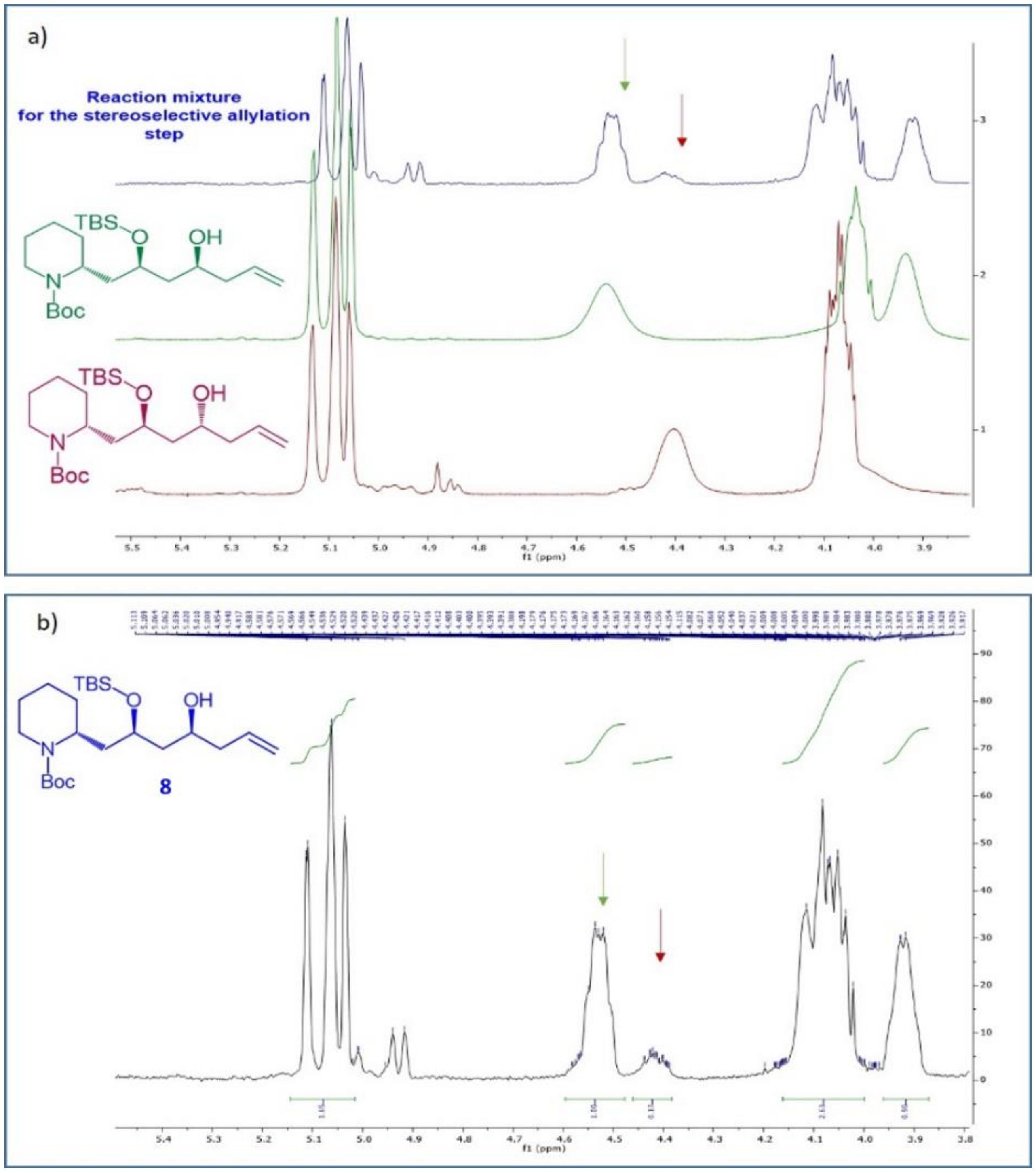
Oxidative cleavage of the double bond afforded aldehyde
7, that was allylated again exploiting a Brown allylation. Also in this case, the stereocontrol was exerted mainly by the chirality of the Ipc (isopinocampheyl) substituents on the borane, rather than by the stereocenters already present in the substrate. The use of (–)-B-allyl diisopinocampheylborane, secured the formation of the desired stereoisomer of compound
8, that was accessed with a
d.r.: 85:15. The diastereomeric ratio was determined through
1H-NMR analysis. In fact, the two possible diastereomers
8 and
8b, characterized by an opposite configuration at the C-10 stereocenter, were independently synthesised. Their
1H-NMR spectra in CDCl
3 were almost completely superimposable, rendering impossible the determination of the diastereomeric composition of the reaction mixture obtained under asymmetric conditions. However, we realized that using deuterated benzene a satisfactory signals separation for the two single diastereomers was achieved (
Figure 4a, green and red spectra). In this way, once identified the signals of each diastereomers, the
d.r. of the asymmetric reaction (blue spectrum in
Figure 4a) was calculated on the basis of signals integrals, as reported in
Figure 4b.
With the correct stereoisomer of compound
8 in our hands, we proceeded with the following step, that was a Mitsunobu reaction in the presence of diphenylphosphorylazide, that converted the alcohol into the corresponding azide
9, with inversion of configuration. In this way, the correct (
R)-configuration of C-10 stereocenter was accessed. Azide reduction under mild Staudinger conditions afforded homoallylic amine
10. Acylation with the commercially available acryloyl chloride, followed by a ring closing metathesis, led to the formation of the desired α,β-unsaturated lactam
12. As reported in our previous studies, Umicore M73 SIMes catalyst proved to be the most efficient to favour the ring enclosure [
18,
26]. In this way, the fundamental (–)-anaferine carbon skeleton, characterised by two nitrogen-containing hexacycles, connected by the 2-hydroxypropane linker and maintaining the (
R,R)-configuration of the two key stereocenters, was secured. The next goal was the reduction of the α,β-unsaturated lactam into the corresponding piperidine. First of all, the double bond was selectively reduced through catalytic hydrogenation in the presence of Pd/C. Then, the amidic nitrogen of
13 was protected as Boc [
27] and the piperidone was reduced to piperidine
15, using BH
3·SMe
2. This mild reducing agent was preferred to the most common LiAlH
4, to avoid the conversion of the two Boc protecting groups into methyl amines [
28]. The TBS protecting group was selectively cleaved using TBAF, affording the known intermediate
16, reported in Blechert and Stapper’s synthesis of (–)-anaferine [
24].
The use of TBAF instead of acid conditions was fundamental to preserve the two Boc protecting groups: this was a mandatory requirement, because two basic nitrogen atoms would have interfered with the following oxidation of the hydroxyl group to ketone. Dess-Martin periodinane (DMP) was employed instead of pyridinium chlorochromate, that was the oxidizing agent of choice in the previously reported synthesis. DMP is a less toxic reagent, easier to handle and that gave, after a basic work-up, the desired product
17 in quantitative yield and without need of further purification. Finally, the cleavage of the two Boc protecting groups in acid environment, led to the desired (-)-anaferine dihydrochloride (
18), whose identity was confirmed comparing analytical and spectroscopic properties with data reported in literature. The whole synthetic route is depicted in
Scheme 2.
In conclusion, 2-piperidine ethanol proved to be once again a flexible synthon for the obtainment of diversified piperidine-containing compounds. In this work, the collection of derivatives previously synthesised in our research group was enriched with (–)-anaferine, that was accessed as a new branching of our diversity-oriented approach, with an overall 9% yield over 13 steps. Notably, this strategy could open the possibility of affording interesting analogues of piperidine-based marine and plant-deriving natural products, characterised by a polyketide side chain in C-2 position.
3. Experimental Section
3.1. General Remarks
Unless otherwise stated, reagents and solvents were purchased from Sigma Aldrich (St. Louis, MO, USA), Fluorochem (Zentek, Milano, Italy) or TCI (Zentek, Milano, Italy) and used without further purification. Unless otherwise stated, all reactions were carried out in oven-dried glassware and dry solvents, under nitrogen atmosphere and were monitored by thin layer chromatography (TLC) on silica gel (precoated 60F254 plates, Merck, (Darmstadt, Germany, with detection by UV light (254 nm) or by solutions of potassium permanganate stain, or ninhydrin, or p-anisaldehyde stain. Flash chromatography was performed using silica gel (240−400 mesh, Merck) as stationary phase.
1H-NMR spectra were recorded on an Avance (400 MHz) spectrometer (Bruker, Billerica, MA, USA) and are reported relative to residual CDCl3 or CD3OD. 13C-NMR spectra were recorded on the same instruments (100 MHz) and are reported relative to residual CDCl3 or CD3OD. All 1D and 2D NMR spectra (Bruker, Billerica, MA, USA) were collected using the standard pulse sequences available with Bruker Topspin 1.3. Chemical shifts (δ) for proton and carbon resonances are quoted in parts per million (ppm) relative to tetramethylsilane (TMS), used as an internal standard. Data for 1H-NMR are reported as follows: chemical shift (δ/ppm) (multiplicity, coupling constant (Hz), integration). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, m = multiplet, bs = broad singlet. Data for 13C-NMR are reported in terms of chemical shift (δ/ppm).
Mass spectra were registered exploiting the electrospray ionisation (ESI) technique on a Q-Tof micro mass spectrometer (Waters, Milford, MA, USA).
Specific rotation values were measured on a P-1030 polarimeter (Jasco, Europe, Cremella, LC, Italy), using 1 mL cells, with path length of 10 cm. Measures were collected at 20–25 °C, using sodium D line wavelength λ = 589 nm.
3.2. Synthesis of tert-butyl (2R)-2-[(2S)-2-[(tert-butyldimethylsilyl)oxy]pent-4-en-1-yl]piperidine- 1-carboxylate (6)
TBSCl (0.574 g, 3.81 mmol) was added to a solution of 2a (0.790 g, 2.93 mmol) and imidazole (0.400 g, 5.86 mmol) in anhydrous CH2Cl2 (15 mL), previously cooled at 0 °C. The reaction mixture was then diluted with CH2Cl2 and washed with brine. The aqueous layer was extracted with CH2Cl2 and the collected organic phases were dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (hexane: EtOAc = 9:1), to give 6 (1.010 g, 90%) as a yellow oil. [α]D20: +16.9 (c = 0.62, CHCl3). 1H-NMR (CDCl3) δ 5.95−5.65 (m, 1H), 5.14–4.94 (m, 2H), 4.36–4.16 (m, 1H), 4.07−3.87 (m, 1H), 3.67 (dq, J = 7.6, 5.2 Hz, 1H), 2.86–2.63 (m, 1H), 2.50–2.31 (m, 1H), 2.31–2.16 (m, 1H), 1.83 (m, 1H), 1.75–1.47 (m, 7H), 1.46 (s, 9H), 0.98−0.79 (m, 9H), 0.07 (s, 3H), 0.06 (s, 3H). 13C-NMR (CDCl3) δ 155.0, 135.3, 117.1, 79.4, 69.5, 47.5, 41.2, 39.0, 36.1, 36.8, 29.1, 28.7 (3 CH3), 26.0, 25.8 (3 CH3), 19.3, −4.3 (2 CH3). MS (ESI) m/z 406.31 [M + Na]+ (calcd. for C21H41NO3SiNa, 406.27).
3.3. Synthesis of tert-butyl (2R)-2-[(2R)-2-[(tert-butyldimethylsilyl)oxy]-4-oxobutyl]piperidine- 1-carboxylate (7)
Ozone was fluxed through a solution of 6 (1.000 g, 2.61 mmol) in CH2Cl2 (30 mL), cooled at −78 °C. After 1 h, the residual ozone was removed from the solution fluxing nitrogen for 10 min. PPh3 (1.026 g, 3.91 mmol) was added at −78 °C, then the reaction mixture was stirred for 2 h at r.t.. The solvent was removed under vacuum and the residue was purified by column chromatography on silica gel (hexane: EtOAc = 9:1), affording 7 (0.603 g, 60%) as a yellow oil. [α]D20: +22.2 (c = 0.52, CHCl3). 1H-NMR (CDCl3) δ 9.77 (s, 1H), 4.36−4.23 (m, 1H), 4.13 (d. quint., J = 11.5, 4.2 Hz, 1H), 4.07−3.95 (m, 1H), 2.76 (t, J = 12.2 Hz, 2H), 2.53 (ddd, J = 15.6, 7.0, 3.2 Hz, 1H), 2.14−1.97 (m, 1H), 1.56 (dd, J = 29.8, 11.2 Hz, 6H), 1.44 (s, 9H), 1.41−1.32 (m, 1H), 0.86 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H). 13C- NMR (CDCl3) δ 202.3, 155.0, 79.7, 66.2, 50.4, 47.4, 41.5, 38.2, 29.5, 28.6 (3 CH3), 25.9 (3 CH3), 25.8, 19.4, 18.1, −4.19, −4.60. MS (ESI) m/z 408.38 [M + Na]+ (calcd. for C20H39NO4SiNa, 408.25).
3.4. Synthesis of (R)-tert-butyl 2-((2R,4S)-2-((tert-butyldimethylsilyl)oxy)-4-hydroxyhept-6-en-1-yl) piperidine-1-carboxylate (8)
(-)-DIP-Cl (1.292 g, 4.03 mmol) was dissolved in anhydrous THF (16 mL) and cooled at −78 °C. Allylmagnesium bromide (1.0 M solution, 4.14 mL, 4.14 mmol) was added dropwise. The reaction mixture was stirred for 1 h at −5 °C and then the white solid was allowed to precipitate. 7 (0.914 g, 2.37 mmol) was dissolved in anhydrous THF (8 mL) and added to the supernatant solution of the previous mixture, cooled at −78 °C. After 1 h, the reaction was warmed to r.t. and stirred overnight. The mixture was concentrated and a saturated solution of NaH2PO4 (14mL) was added, followed by MeOH (14 mL) and H2O2 35% (7mL). The mixture was stirred for 30 min; a saturated solution of NaHCO3 was added. The layers were separated and the aqueous one was extracted with Et2O. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (hexane: EtOAc = 8:2, second column CH2Cl2, then CH2Cl2: MeOH = 95:5), to give 8 (0.932 g, 92%) as light yellow oil. [α]D20: +3.1 (c = 0.63, CHCl3). 1H-NMR (CDCl3) δ 5.86 (m, 1H), 5.17−5.02 (m, 2H), 4.36−4.20 (m, 1H), 4.15−3.91 (m, 1H), 3.91−3.76 (m, 2H), 2.78 (t, J = 12.7 Hz, 1H), 2.23 (m, 2H), 2.12−1.98 (m, 1H), 1.98−1.77 (m, 1H), 1.72−1.48 (m, 7H), 1.45 (s, 9H), 1.43−1.38 (m, 1H), 0.89 (s, 9H), 0.10 (s, 6H). 13C-NMR (CDCl3) δ 155.1, 135.2, 117.4, 79.6, 70.7, 70.1, 47.4, 43.7, 42.4, 39.2 (2 CH2), 30.0, 28.7 (3CH3), 26.0 (3CH3), 25.8, 19.4, 18.4, −3.9, −4.5. MS (ESI) m/z 450.79 [M + Na]+ (calcd. for C23H45NO4SiNa, 450.30).
3.5. Synthesis of tert-butyl(R)-2-((2R,4R)-4-azido-2-((tert-butyldimethylsilyl)oxy)hept-6-en-1-yl) piperidine-1-carboxylate (9)
8 (0.932 g, 2.18 mmol) and PPh3 (0.687 g, 2.62 mmol) were dissolved in anhydrous THF (18 mL) and cooled at 0 °C. DIAD (0.52 mL, 2.62 mmol) was added dropwise. The reaction mixture was stirred for 10′ at 0 °C and then DPPA (0.57 mL, 2.62 mmol) was added dropwise. The reaction was warmed to r.t. and stirred overnight. The mixture was concentrated, and the crude product was purified by column chromatography on silica gel (hexane: EtOAc = 97:3), to give 9 (0.839 g, 85%) as light yellow oil. [α]D20: −29.7 (c = 0.54, CHCl3). 1H-NMR (CDCl3) δ 5.82 (ddt, J = 17.1, 10.1, 7.0 Hz, 1H), 5.21−5.08 (m, 2H), 4.36−4.22 (m, 1H), 4.09−3.94 (m, 1H), 3.80 (m, 1H), 3.66−3.54 (m, 1H), 2.77 (t, J = 13.1 Hz, 1H), 2.35 (m, 2H), 2.10−1.98 (m, 1H), 1.80−1.66 (m, 1H), 1.64−1.53 (m, 5H), 1.51 (m, 1H), 1.47 (s, 9H), 1.44−1.36 (m, 2H), 0.89 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H). 13C-NMR (CDCl3) 154.1, 134.1, 118.4, 79.4, 67.2, 58.6, 47.3, 41.1, 39.6, 38.6 (2 CH2), 29.8, 28.6 (3 CH3), 26.1 (3 CH3), 25.8, 19.4, 18.2, −3.82, −4.69. MS (ESI) m/z 475.31 [M + Na]+ (calcd. for C23H44N4O3SiNa, 475.31).
3.6. Synthesis of (R)-tert-butyl 2-((2S,4R)-4-amino-2-((tert-butyldimethylsilyl)oxy)hept-6-en-1-yl) piperidine-1-carboxylate (10)
Compound 9 (0.821 g, 1.81 mmol) and PPh3 (0.951 g, 3.63 mmol) were dissolved in a mixture of THF and H2O (10:1, 25 mL) and heated at 40 °C for 8 h. At that point, water was added (10.4 mL). The layers were separated and the aqueous one was extracted with Et2O. The combined organic layers were washed with brine, dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (CH2Cl2: MeOH = 95:5), to give 10 (0.601 g, 78%) as light yellow oil. [α]D20: +6.8 (c = 0.46, CHCl3). 1H-NMR (CDCl3) δ 5.79 (ddt, J = 17.2, 10.2, 7.1 Hz, 1H), 5.31−4.94 (m, 2H), 4.28–4.12 (m, 1H), 4.08–3.90 (m, 1H), 3.87–3.80 (m, 1H), 3.10–3.05 (m, 1H), 2.90−2.59 (m, 1H), 2.40–2.20 (m, 2H), 2.09−2.01 (m, 1H), 1.76−1.48 (m, 7H), 1.45 (s, 9H), 1.40−1.30 (m, 2H), 0.96−0.75 (m, 9H), 0.10–−0.15 (m, 6H). 13C-NMR (CDCl3) δ 155.3, 135.3, 117.9, 79.6, 68.4 (2 CH), 47.8, 42.5 (2 CH2), 39.00, 37.8, 29.8, 28.7 (3 CH3), 26.0 (3 CH3), 25.8, 19.4, 18.1, −4.17, −4.47. MS (ESI) m/z 427.63 [M + H]+ (calcd. for C23H47N2O3Si, 427.33).
3.7. Synthesis of (R)-tert-butyl 2-((2S,4R)-4-acrylamido-2-((tert-butyldimethylsilyl)oxy) hept-6-en- 1-yl) piperidine-1-carboxylate (11)
Compound 10 (0.558 g, 1.31 mmol) was dissolved in anhydrous CH2Cl2 (5.5 mL) and cooled at 0 °C. Anhydrous TEA (547 μL, 3.92 mmol) was added and the reaction was stirred for 10 min. Acryloyl chloride (160 μL, 1.96 mmol) was added dropwise; the mixture was warmed to r.t. and stirred for 5 h. At that point, cold water (0 °C) was added. The layers were separated and the aqueous one was extracted with CH2Cl2. The combined organic layers were washed twice with a saturated solution of NH4Cl, dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (CH2Cl2: MeOH = 95:5), to give 11 (0.548 g, 88%) as a brown oil. [α]D20: −8.1 (c = 0.42, CHCl3). 1H-NMR (CDCl3) δ 6.42 (bs, 1H), 6.29−6.03 (m, 2H), 5.81 (ddt, J = 19.2, 9.6, 7.2 Hz, 1H), 5.65−5.51 (m, 1H), 5.16−4.94 (m, 2H), 4.22−4.09 (m, 1H), 4.05−3.91 (m, 2H), 3.77−3.60 (m, 1H), 2.86 (m, 1H), 2.53−2.30 (m, 1H), 2.14−2.02 (m, 1H), 1.80 (t, J = 12.8 Hz, 1H), 1.66−1.53 (m, 6H), 1.45 (m, 11H), 1.31 (m, 1H), 0.91 (s, 9H), 0.02 (s, 6H). 13C-NMR (CDCl3) δ 165.3, 134.6, 131.7, 125.4, 117.4, 79.6, 66.8, 46.6, 39.3 (3CH2), 31.4, 30.5 (2CH2), 28.6 (3CH3), 25.9 (3CH3), 25.6, 19.3 (2 CH2), 17.9, −4.5 (2CH3). MS (ESI) m/z 503.71 [M + Na]+ (calcd. for C26H48N2O4SiNa, 503.33).
3.8. Synthesis of (R)-tert-butyl 2-((S)-2-((tert-butyldimethylsilyl)oxy)-3-((R)-6-oxo- 1,2,3,6- tetrahydro-pyridin -2-yl)propyl)piperidine-1-carboxylate (12)
Compound 11 (0.540 g, 1.12 mmol) was dissolved in anhydrous degassed CH2Cl2 (47 mL). A solution of Umicore catalyst 14% wt (59.0 mg, 0.01 mmol) in degassed CH2Cl2 (16.2 mL) was added dropwise and the reaction was stirred for 5 h at 50 °C. The reaction mixture was concentrated under vacuum. The crude product was purified by column chromatography on silica gel (CH2Cl2: MeOH = 98:2), to give 12 (0.364 g, 71%) as a light yellow oil. [α]D20: +46.2 (c = 0.66, CHCl3). 1H-NMR (CDCl3) δ6.67 (bs, 1H), 6.60−6.51 (m, 1H), 5.91−5.83 (m, 1H), 4.19 (m, 1H), 3.99−3.94 (m, 1H), 3.80−3.76 (m, 2H), 2.81−2.73 (m, 1H), 2.17−2.16 (m, 2H), 1.84-1.75 (m, 2H), 1.54−1.46 (m, 6H), 1.45 (s, 9H), 1.36−1.28 (m, 2H), 0.89 (s, 9H), 0.06 (s, 6H). 13C-NMR (CDCl3) δ 166.0, 155.5, 140.4, 125.2, 79.9, 68.2, 48.0 (2 CH), 31.5 (2 CH2), 30.0, 29.8, 28.6 (3 CH3), 26.0 (3 CH3), 25.9, 25.7, 19.3, 18.1, −4.3, −4.8. MS (ESI) m/z 475.52 [M + Na]+ (calcd. for C24H44N2O4SiNa, 475.30).
3.9. Synthesis of (R)-tert-butyl 2-((S)-2-((tert-butyldimethylsilyl)oxy)-3-((R)-6- oxopiperidin -2-yl)propyl) piperidine-1-carboxylate (13)
Compound 12 (0.205 g, 0.45 mmol) was dissolved in MeOH (10 mL); Pd/C 10% wt (0.205 g) was added, and the reaction mixture was put under H2 atmosphere and stirred overnight. The mixture was filtered over Celite, washed with MeOH and CH2Cl2 and concentrated to give 13 (0.206 g, quant.) as a dark yellow oil. [α]D20: +1.4 (c = 0.70, CHCl3). 1H-NMR (CDCl3) δ 6.73 (s, 1H), 4.23−4.19 (m, 1H), 4.00−3.96 (m, 1H), 3.82−3.77 (m, 1H), 3.59−3.54 (m, 1H), 2.78 (t, J = 13.2 Hz, 1H), 2.44−2.32 (m, 1H), 2.25 (ddd, J = 17.7, 11.1, 6.5 Hz, 1H), 2.07 (ddd, J = 14.6, 11.4, 3.7 Hz, 1H), 1.88 (m, 1H), 1.84−1.66 (m, 4H), 1.66−1.48 (m, 6H), 1.45 (s, 9H), 1.37 (m, 2H), 0.91 (s, 9H), 0.08 (s, 6H). 13C-NMR (CDCl3) δ 171.6, 155.2, 79.8, 68.6, 49.9, 47.7, 41.5, 39.2, 31.3, 30.3, 30.0, 29.9, 28.7 (3 CH3), 26.1 (3 CH3), 25.8, 20.6, 19.4, 18.2, −4.59, −4.91. MS (ESI) m/z 477.91 [M + Na]+ (calcd. for C24H46N2O4SiNa, 477.31).
3.10. Synthesis of (R)-tert-butyl 2-((R)-3-((R)-1-(tert-butoxy carbonyl)piperidin-2-yl)-2- ((tert-butyl-dimethylsilyl)oxy)propyl)-6-oxopiperidine-1-carboxylate (14)
Compound 13 (0.206 g, 0.45 mmol) was dissolved in anhydrous CH2Cl2 (1.5 mL) and cooled to 0 °C. TEA (190 μL, 1.36 mmol), DMAP (0.012 g, 0.09 mmol) and Boc2O (0.298 g, 1.36 mmol) were added. The mixture was stirred at r.t. for 48 h and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (hexane: EtOAc = 7:3), to give 14 (0.204 g, 82%) as a dark yellow oil. [α]D20: +23.2 (c = 0.82, CHCl3). 1H-NMR (CDCl3) δ 4.48−4.33 (m, 1H), 4.23 (m, 1H), 4.08−3.92 (m, 1H), 3.78 (quint., J = 6.2 Hz, 1H), 2.77 (t, J = 12.7 Hz, 1H), 2.55−2.38 (m, 2H), 1.96−1.70 (m, 7H), 1.88- 1.56 (m, 5H), 1.52 (s, 9H), 1.44 (s, 9H), 1.31−1.19 (m, 2H), 0.88 (s, 9H), 0.07 (s, 6H). 13C-NMR (CDCl3) δ 171.3, 154.8, 152.8, 82.9, 79.2, 68.5, 53.7, 47.3, 41.6, 39.2, 36.7, 34.1, 29.7, 28.6 (3 CH3), 28.0 (3 CH3), 26.7, 25.9 (3 CH3), 25.6, 19.2, 17.9, 17.1, −4.3 (2 CH3). MS (ESI) m/z 577.48 [M + Na]+ (calcd. for C29H54N2O6SiNa: 577.36).
3.11. Synthesis of (2R,2′R)-di-tert-butyl 2,2′-(2-((tert-butyldimethylsilyl)oxy)propane- 1,3-diyl) bis (piperidine-1-carboxylate) (15)
Compound 14 (0.170 g, 0.31 mmol) was dissolved in anhydrous THF (7.5 mL) and cooled to 0 °C. BH3∙SMe2 (68 μL, 0.67 mmol), was added dropwise. The mixture was stirred at r.t. for 5h and quenched with a saturated solution of NH4Cl. The layers were separated, and the organic one was extracted with Et2O. The combined organic layers were dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (hexane: EtOAc = 8:2), to give 15 (0.126 g, 76%) as a yellow oil. [α]D20: +28.0 (c = 0.48, CHCl3). 1H-NMR (CDCl3) δ 4.40–4.24 (m, 2H), 4.00–3.87 (m, 2H), 3.72–3.61 (m, 1H), 2.84–2.71 (m, 2H), 1.94–1.63 (m, 6H), 1.60–1.51 (m, 6H), 1.45 (s, 18H), 1.39–1.34 (m, 4H), 0.89 (s, 9H), 0.06 (s, 6H). 13C-NMR (CDCl3) δ 154.9 (2 Cq), 79.1 (2 Cq), 68.7, 47.4 (2 CH), 39.5 (2 CH2), 37.6 (2 CH2), 29.68 (2 CH2), 28.5 (6 CH3), 25.7 (3 CH3), 25.6 (2 CH2), 19.1 (2 CH2), 18.01, −4.2, −4.4. MS (ESI) m/z 563.99 [M + Na]+ (calcd. for C29H56N2O5SiNa, 563.38).
3.12. Synthesis of (2R,2′R)-di-tert-butyl 2,2′-(2-hydroxypropane-1,3- diyl)bis(piperidine-1-carboxylate) (16)
Compound 15 (0.112g, 0.21 mmol) was dissolved in anhydrous THF (1.5 mL) and cooled to −15 °C. TBAF 1.0 M in THF (620 μL, 0.62 mmol), was added dropwise. The mixture was stirred for 16h, slowly warming to 0 °C. The mixture was quenched with a saturated solution of NH4Cl. The layers were separated, and the organic one was extracted with Et2O. The combined organic layers were dried over Na2SO4, filtered and concentrated under vacuum. The crude product was purified by column chromatography on silica gel (hexane: EtOAc = 85:15), to give 16 (0.090 g, 71%) as a white solid. [α]D20: +43.3 (c = 0.53, CHCl3). Ref: [α]D20: +41.4 (c = 0.72, CHCl3). 1H-NMR (CDCl3) δ = 4.28−4.52 (m, 2 H), 3.70−3.98 (m, 2 H), 3.08−3.34 (m, 1 H), 2.60−2.92 (m, 2 H), 1.80−2.38 (m, 2 H), 1.00−1.74 (m, 14 H), 1.42 (s, 18 H). 13C-NMR (CDCl3) δ = 156.5, 155.0, 79.9, 79.0, 64.7, 47.4, 46.3, 39.3, 39.0, 37.0, 36.7, 29.7, 29.4, 28.5 (3 CH3), 28.3 (3 CH3), 25.6, 25.5, 19.1 (detected signals). MS (ESI) m/z 449.71 [M + Na]+ (calcd. for C23H42N2O5Na, 449.30).
3.13. Synthesis of (2R,2′R)-di-tert-butyl 2,2′-(2-oxopropane-1,3-diyl)bis(piperidine-1- carboxylate) (17)
Compound 16 (0.043 g, 0.10 mmol) was dissolved in anhydrous CH2Cl2 (1 mL) at 0 °C, and DMP (50.7 mg, 0.12 mmol) was added. The mixture was stirred for 16h at r.t. The mixture was concentrated; a 10% solution of NaOH was added and the aqueous phase was extracted with Et2O. The combined organic layers were washed with NaOH (10%) and H2O. The combined organic layers were dried over Na2SO4, filtered and concentrated under vacuum, to give 17 (0.042 g, quant.) as a colourless solid. [α]D20: +19.9 (c = 0.77, CHCl3). 1H-NMR (CDCl3) δ = 4.60−4.76 (m, 2 H), 3.82−4.02 (m, 2H), 2.50−2.86 (m, 6 H), 1.20−1.71 (m, 12 H), 1.41 (s, 18 H). 13C-NMR (CDCl3) δ 207.2, 154.8 (2 Cq), 79.6 (2 Cq), 47.3 (2 CH), 43.4 (2 CH2), 39.6 (2 CH2), 28.5 (6 CH3), 28.4 (2 CH2), 25.4 (2 CH2), 19.1 (2 CH2). MS (ESI) m/z 447.81 [M + Na]+ (calcd. for C23H40N2O5Na, 447.28).
3.14. Synthesis of 1,3-di((R)-piperidin-2-yl)propan-2-one (18)
Compound 17 (0.030 g, 0.07 mmol) was dissolved in CH2Cl2 (2 mL) and cooled to 0 °C. A 4M solution of HCl (75 μL, 0.21 mmol) in dioxane was added dropwise. The mixture was stirred for 16h at r.t. The mixture was concentrated to give (–)-anaferine dihydrochloride (18, 0.021 g, quant.) as a colourless solid, without need of further purification. [α]D20: −47.9 (c = 0.65, MeOH/H2O = 1:1). 1H NMR (CD3OD) δ = 3.50−3.61 (m, 2 H), 3.32−3.40 (m, 2 H), 2.88−3.05 (m, 6 H), 1.82−1.95 (m, 6 H), 1.00−1.74 (m, 6 H). 13C-NMR (CD3OD) δ = 205.0, 52.3 (2 CH), 44.8 (2CH2), 44.7 (2CH2), 28.3 (2CH2), 22.0 (2CH2), 21.6 (2CH2). MS (ESI) m/z 224.31 [C13H24N2O]+ (calcd. for C13H24N2O, 224.19).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}