Structural Features of Triethylammonium Acetate through Molecular Dynamics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
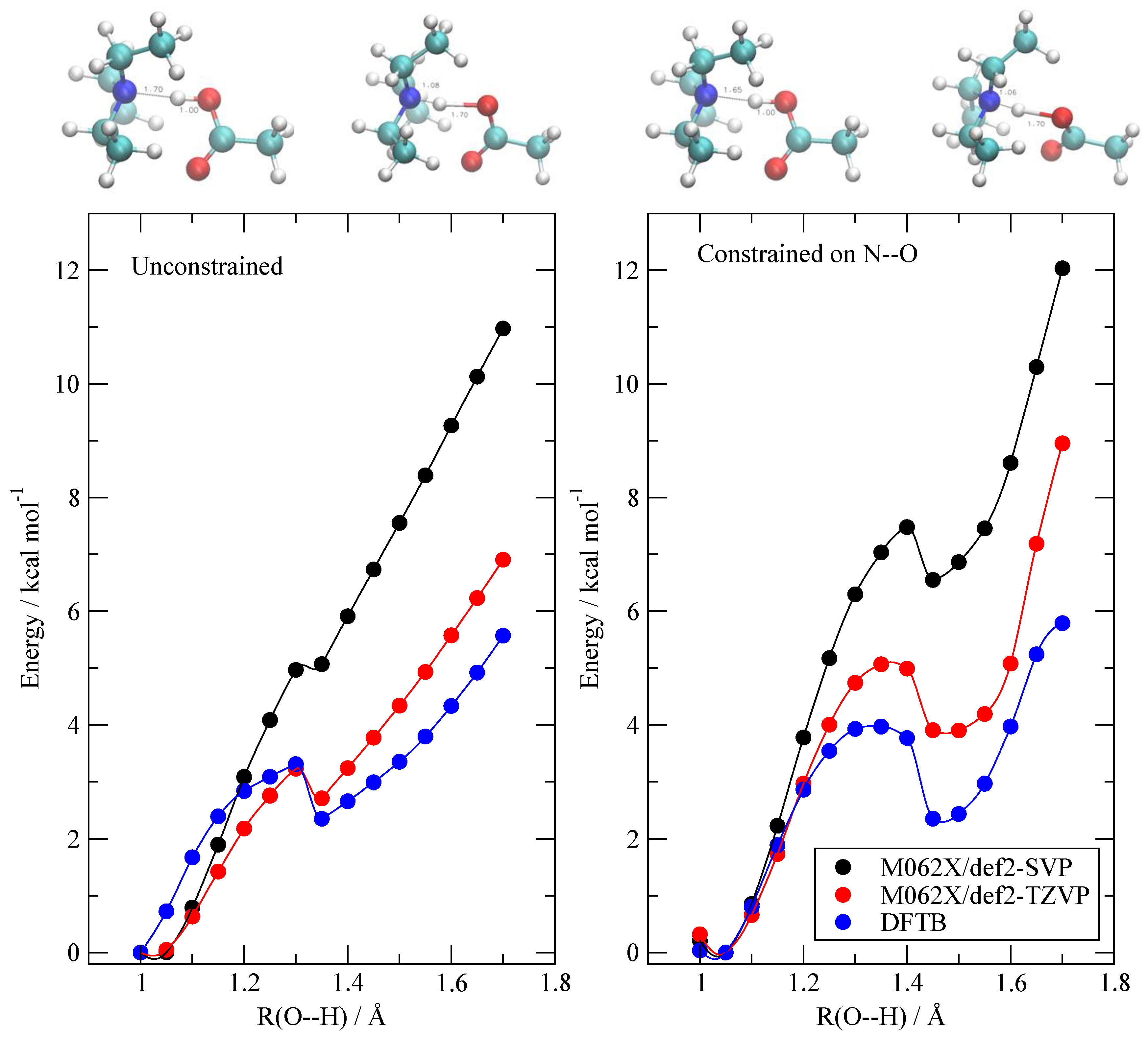
2. Computational Methods
- (1)
- 20:20, a simulation where a cubic cell with side length of 17.2 Å was initially made by 20 AcOH/TEA neutral pairs. NVT MD was run at a constant temperature of 300 K for a production time of 300 ps. The density was set to 1.04 g/cm3;
- (2)
- 10:10, a simulation where a cubic cell with 13.6 Å side length contained 10 AcOH/TEA initially neutral pairs. NVT MD was performed at 300 K for 600 ps. The density was set to 1.04 g/cm3;
- (3)
- 8:2, a simulation prepared with a 4:1 AcOH:TEA composition (to account for TEA separation/evaporation detected by experiments [27]) with two TEA molecules and eight AcOH in their neutral state. The cell side was set to 10.3 Å. Production time on this system was 950 ps at 300 K. The density was set to 1.02 g/cm3;
- (4)
- PI20:20, a simulation with a partially ionized composition [AcOH]3[TEA]3[AcOH]−[TEAH]+, where one fourth of the molecules are initially ionized. The cell size was set to 17.3 Å and the MD was performed for 300 ps of production time at 300 K. The density was set to 1.03 g/cm3;
- (5)
- I20:20, a simulation with a completely ionized equimolar composition [AcOH]−[TEAH]+, whose size is identical to the 20:20 one, but all TEA molecules are initially protonated and all AcOH are deprotonated. Production was 300 ps. This simulation was run at both 300 K and 360 K.
3. Results and Discussion
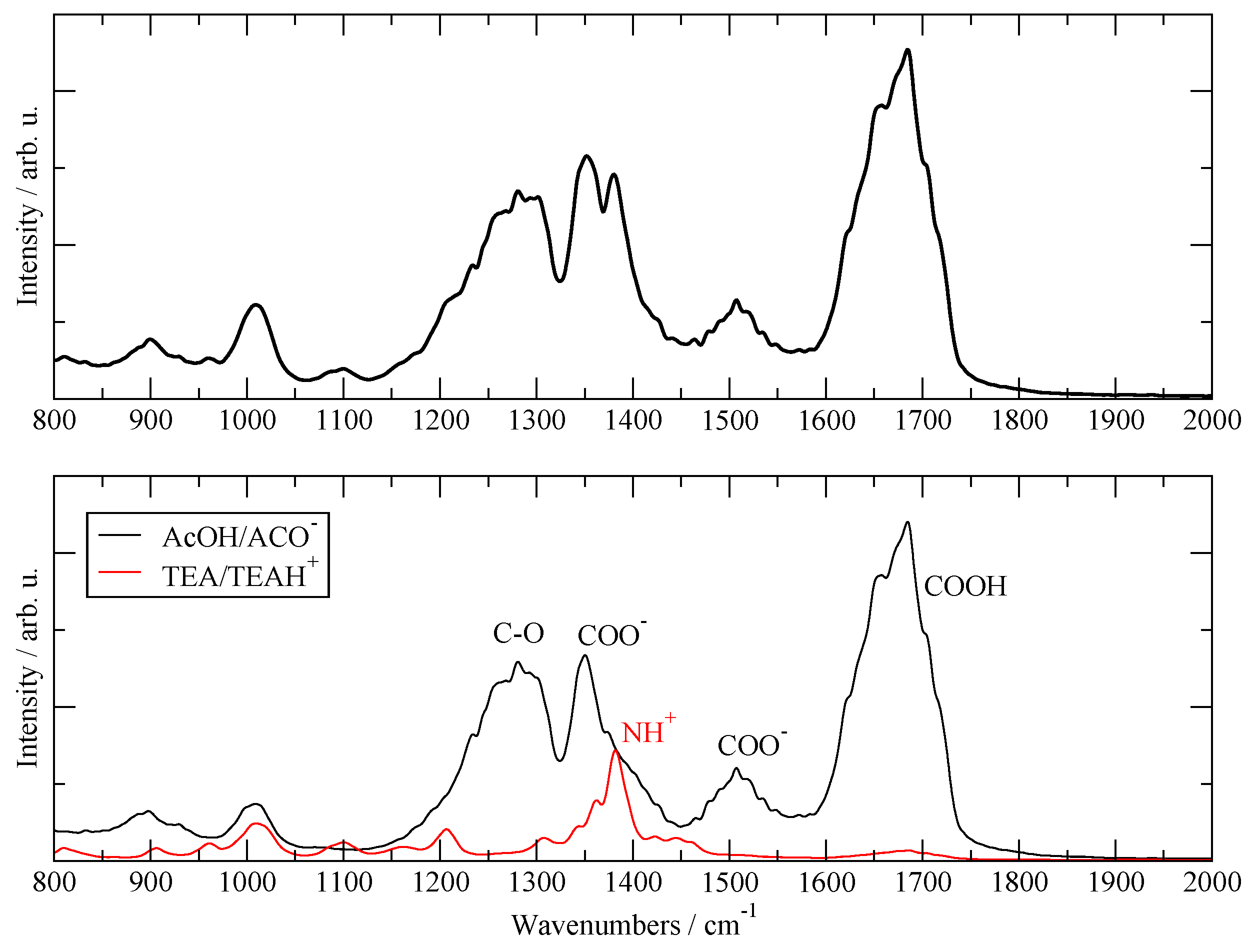
3.1. Validation with Available Data
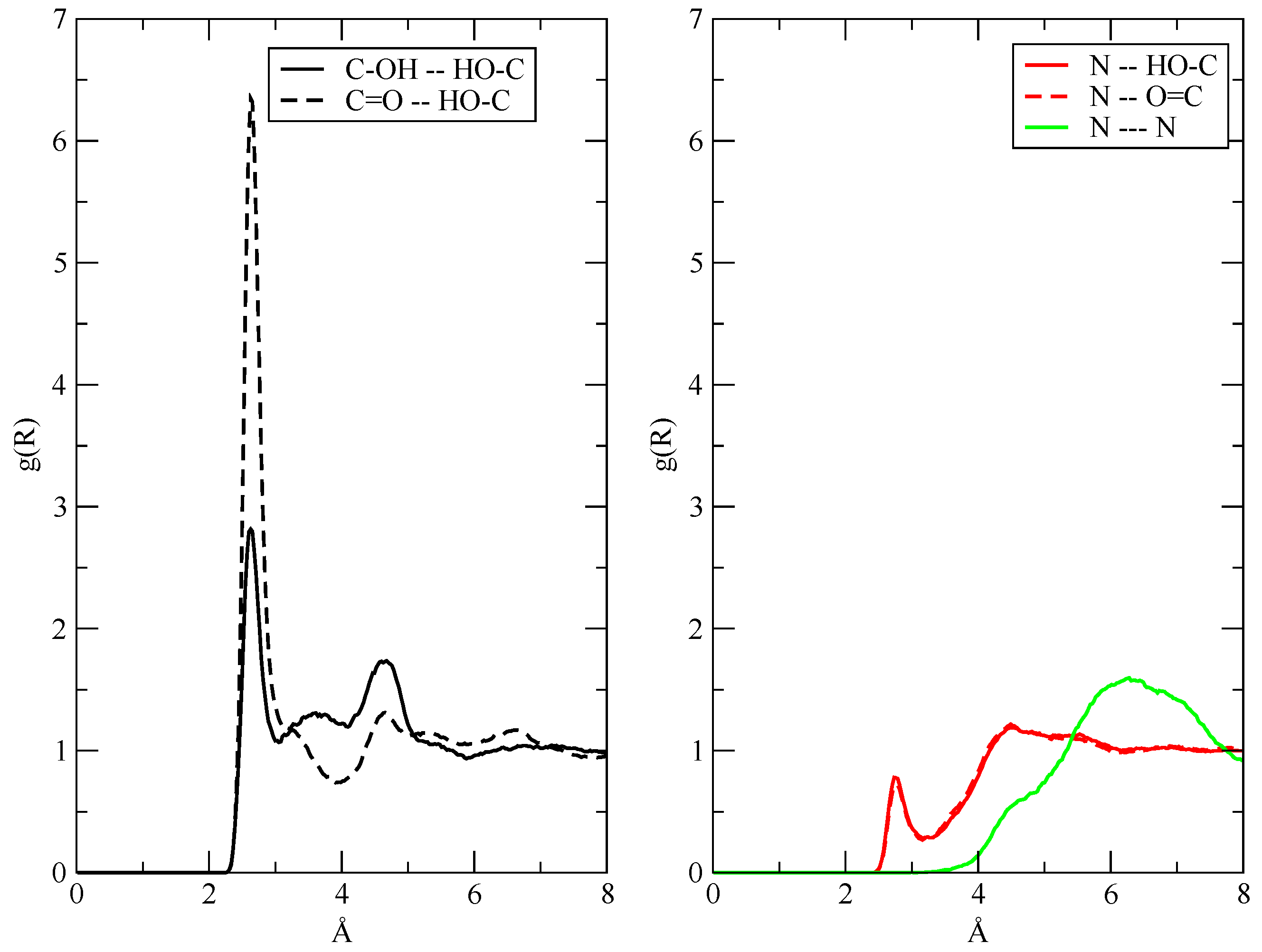
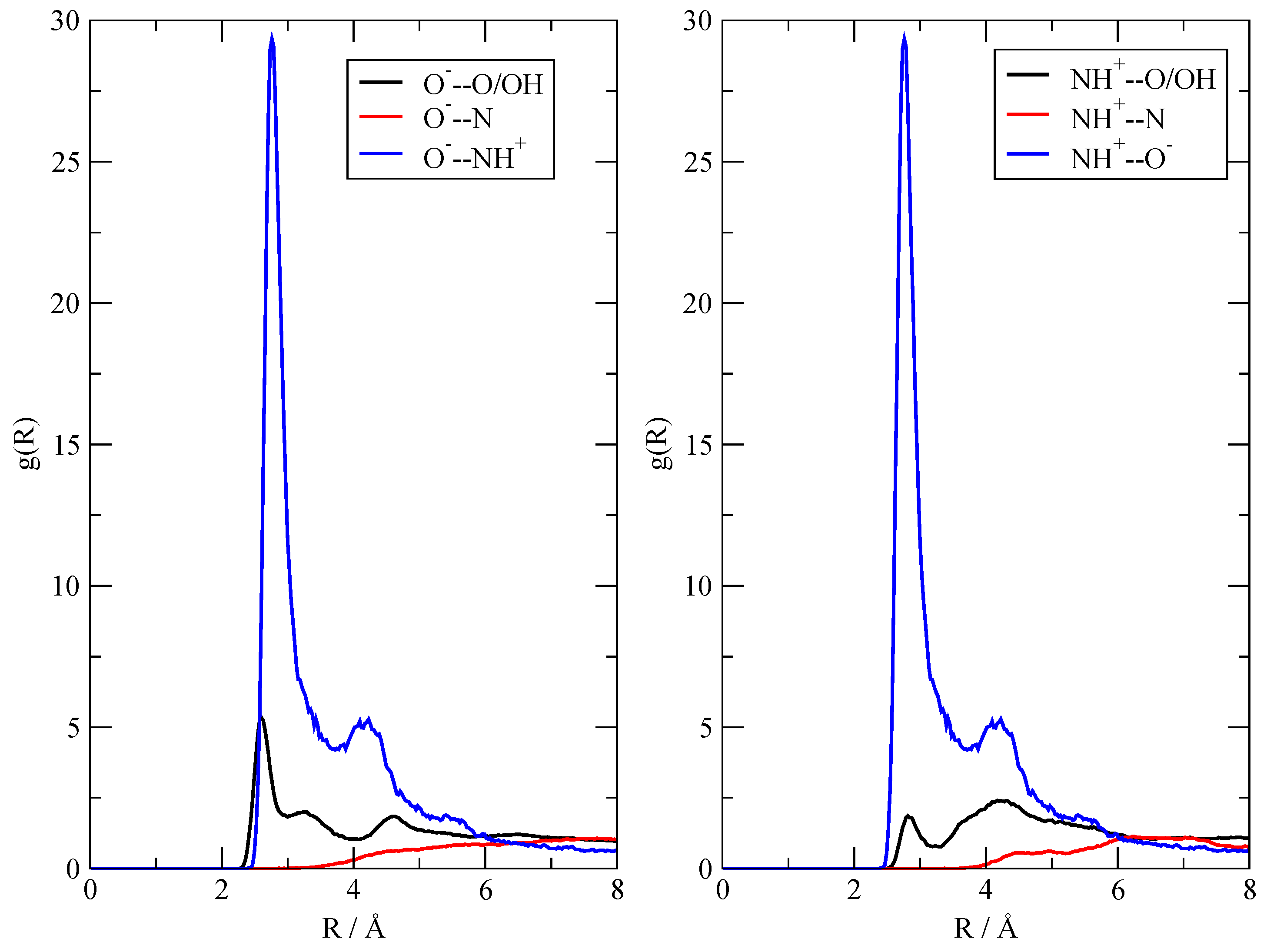
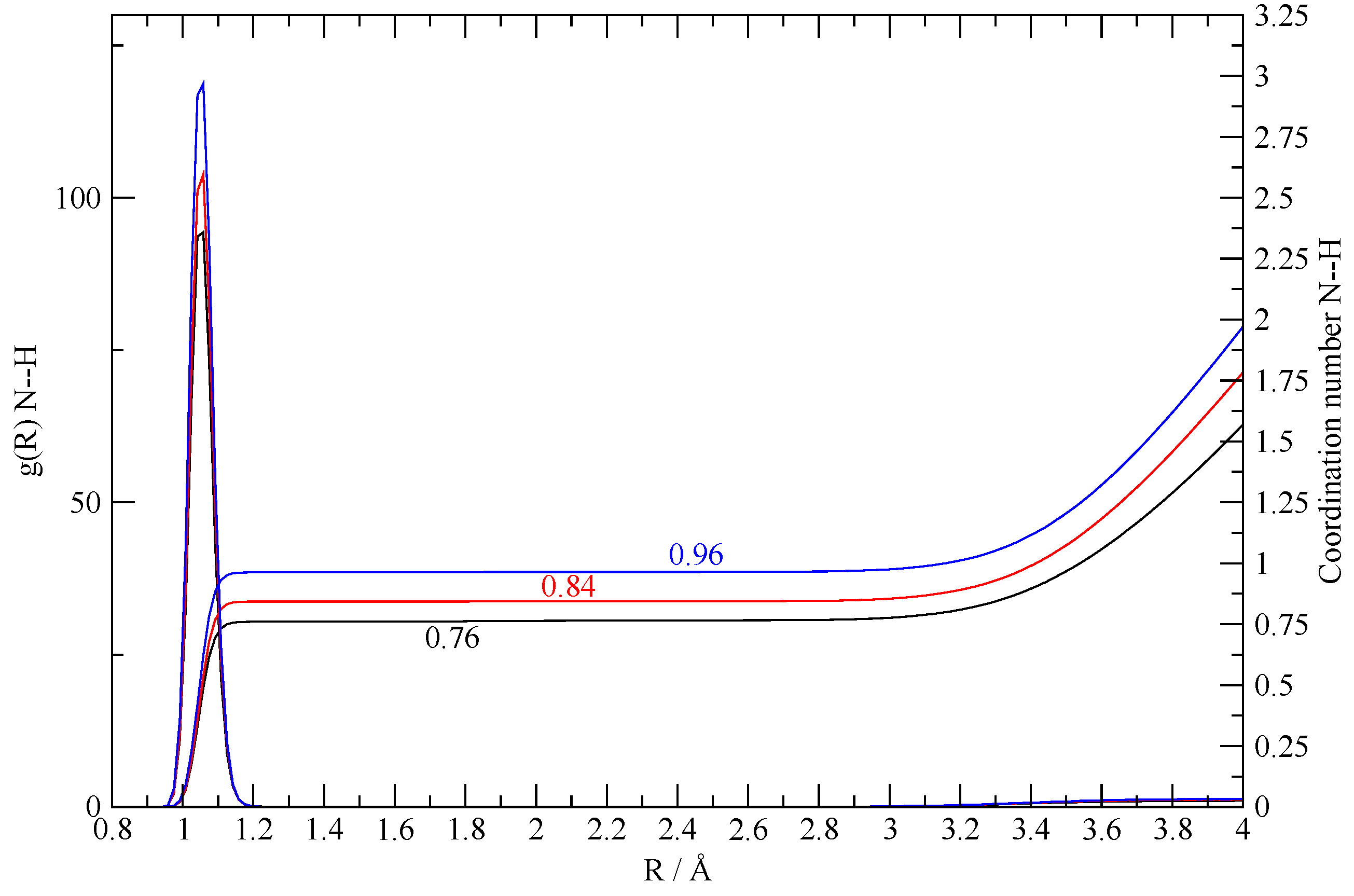
3.2. The Structure of the Fluid
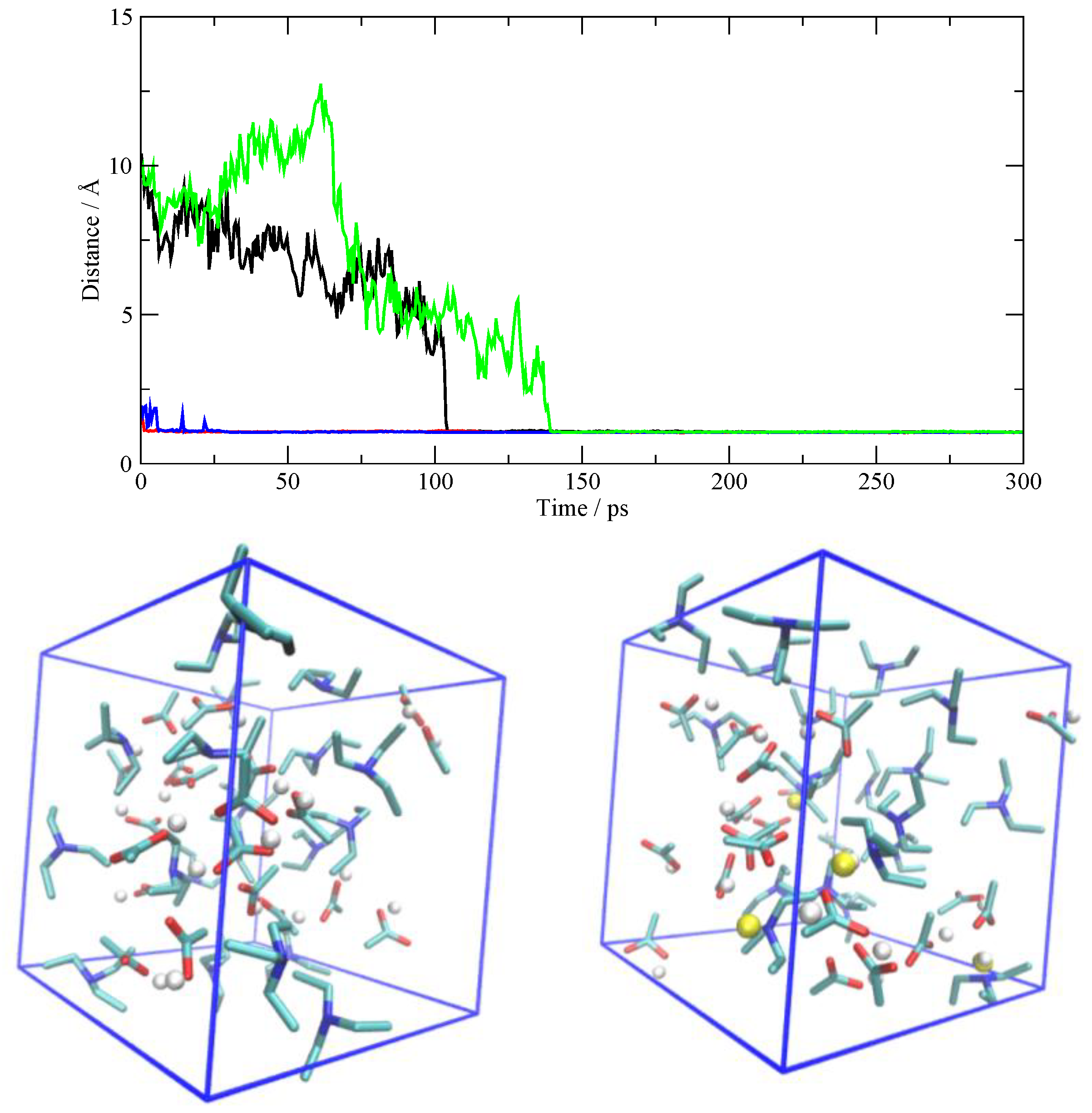
3.3. The Dynamics of the Fluid
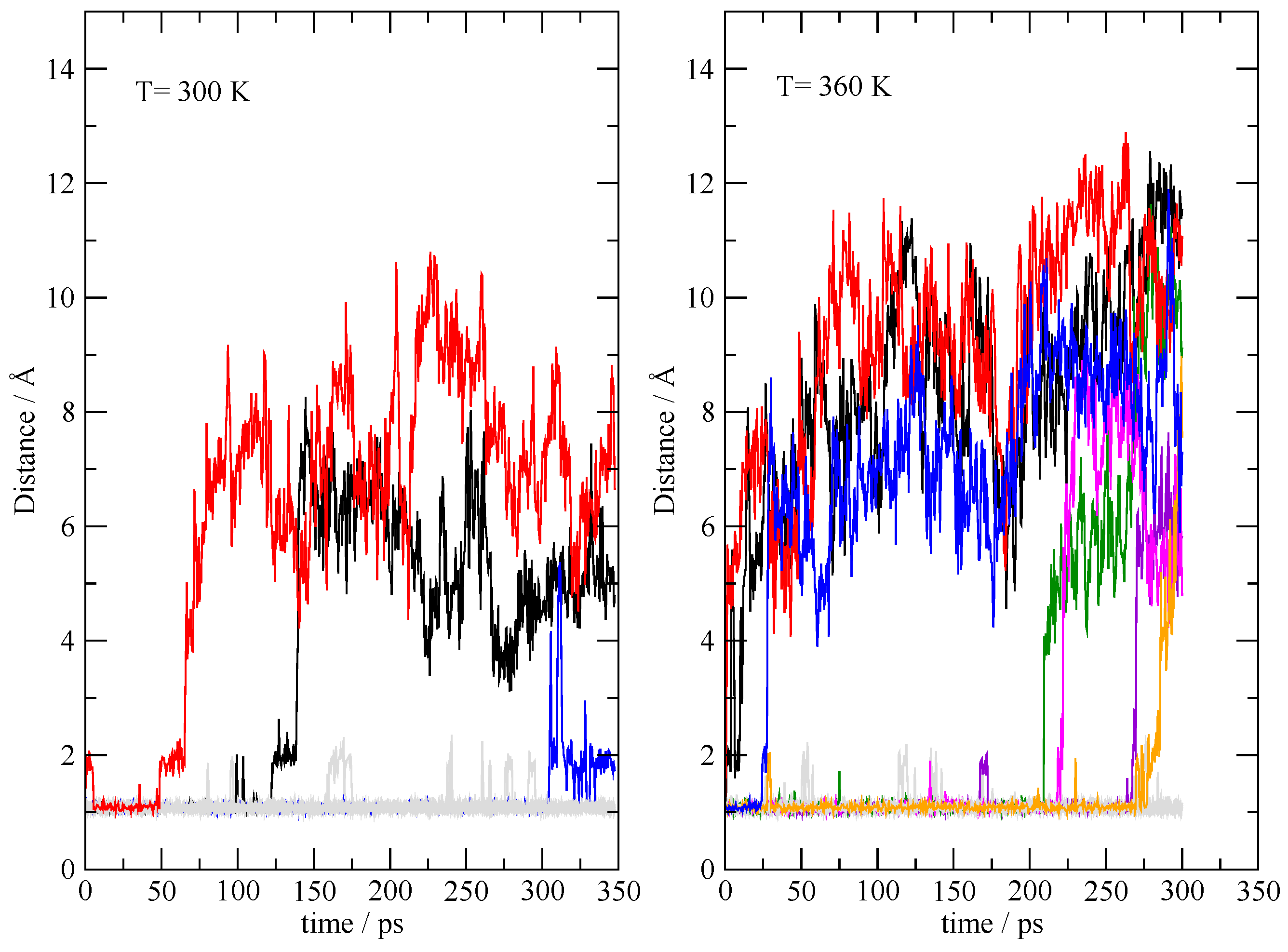
3.4. The Fate of the Ionic Pairs
3.5. The Other Simulations
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Evolving Structure–Property Relationships and Expanding Applications. Chem. Rev. 2015, 115, 11379–11448. [Google Scholar] [CrossRef] [PubMed]
- Belieres, J.-P.; Angell, C.A. Protic Ionic Liquids: Preparation, Characterization, and Proton Free Energy Level Representation†. J. Phys. Chem. B 2007, 111, 4926–4937. [Google Scholar] [CrossRef] [PubMed]
- Greaves, T.L.; Ha, K.; Howard, S.; Weerawardena, A.; Kirby, N.; Drummond, C.J.; Muir, B. Protic ionic liquids (PILs) nanostructure and physicochemical properties: Development of high-throughput methodology for PIL creation and property screens. Phys. Chem. Chem. Phys. 2015, 17, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Doi, H.; Song, X.; Minofar, B.; Kanzaki, R.; Takamuku, T.; Umebayashi, Y. A New Proton Conductive Liquid with No Ions: Pseudo-Protic Ionic Liquids. Chem. A Eur. J. 2013, 19, 11522–11526. [Google Scholar] [CrossRef]
- Stoimenovski, J.; Izgorodina, E.I.; MacFarlane, D.R. Ionicity and proton transfer in protic ionic liquids Phys. Chem. Chem. Phys. 2010, 12, 10341–10347. [Google Scholar] [CrossRef]
- Kelley, S.P.; Narita, A.; Holbrey, J.D.; Green, K.D.; Reichert, W.M.; Rogers, R.D. Understanding the Effects of Ionicity in Salts, Solvates, Co-Crystals, Ionic Co-Crystals, and Ionic Liquids, Rather than Nomenclature, Is Critical to Understanding Their Behavior. Cryst. Growth Des. 2013, 13, 965–975. [Google Scholar] [CrossRef]
- Bodo, E.; Mangialardo, S.; Capitani, F.; Gontrani, L.; Leonelli, F.; Postorino, P. Interaction of a long alkyl chain protic ionic liquid and water. J. Chem. Phys. 2014, 140, 204503. [Google Scholar] [CrossRef] [Green Version]
- Campetella, M.; Gontrani, L.; Leonelli, F.; Bencivenni, L.; Caminiti, R. Two different models to predict ionic-liquid diffraction patterns: Fixed-charge versus polarizable potentials. Chem. Phys. Chem. 2015, 16, 197–203. [Google Scholar] [CrossRef]
- Fumino, K.; Fossog, V.; Stange, P.; Paschek, D.; Hempelmann, R.; Ludwig, R. Controlling the subtle energy balance in protic ionic liquids: Dispersion forces compete with hydrogen bonds. Angew. Chem. Int. Ed. 2015, 54, 2792–2795. [Google Scholar] [CrossRef]
- Campetella, M.; Montagna, M.; Gontrani, L.; Scarpellini, E.; Bodo, E. Unexpected proton mobility in the bulk phase of cholinium-based ionic liquids: New insights from theoretical calculations. Phys. Chem. Chem. Phys. 2017, 19, 11869–11880. [Google Scholar] [CrossRef]
- Low, K.; Tan, S.Y.S.; Izgorodina, E.I. An ab initio Study of the Structure and Energetics of Hydrogen Bonding in Ionic Liquids. Front. Chem. 2019, 7, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, P. Quantum Chemical Modeling of Hydrogen Bonding in Ionic Liquids. Top. Curr. Chem. 2017, 375, 59. [Google Scholar] [CrossRef] [PubMed]
- Zentel, T.; Overbeck, V.; Michalik, D.; Kühn, O.; Ludwig, R. Hydrogen bonding in protic ionic liquids: Structural correlations, vibrational spectroscopy, and rotational dynamics of liquid ethylammonium nitrate. J. Phys. B 2018, 51, 034002. [Google Scholar] [CrossRef] [Green Version]
- Campetella, M.; Le Donne, A.; Daniele, M.; Gontrani, L.; Lupi, S.; Bodo, E.; Leonelli, F. Hydrogen Bonding Features in Cholinium-Based Protic Ionic Liquids from Molecular Dynamics Simulations. J. Phys. Chem. B 2018, 122, 2635–2645. [Google Scholar] [CrossRef] [PubMed]
- Le Donne, A.; Adenusi, H.; Porcelli, F.; Bodo, E. Structural Features of Cholinium Based Protic Ionic Liquids through Molecular Dynamics. J. Phys. Chem. B 2019, 123, 5568–5576. [Google Scholar] [CrossRef]
- Zhao, Z.; Ueno, K.; Angell, C.A. High Conductivity, and “Dry” Proton Motion, in Guanidinium Salt Melts and Binary Solutions. J. Phys. Chem. B 2011, 115, 13467–13472. [Google Scholar] [CrossRef]
- Zhao, C.; Burrell, G.; Torriero, A.A.J.; Separovic, F.; Dunlop, N.F.; Macfarlane, D.R.; Bond, A.M. Electrochemistry of Room Temperature Protic Ionic Liquids. J. Phys. Chem. B 2008, 112, 6923–6936. [Google Scholar] [CrossRef]
- Watanabe, H.; Umecky, T.; Arai, N.; Nazet, A.; Takamuku, T.; Harris, K.R.; Kameda, Y.; Buchner, R.; Umebayashi, Y. Possible Proton Conduction Mechanism in Pseudo-Protic Ionic Liquids: A Concept of Specific Proton Conduction. J. Phys. Chem. B 2019, 123, 6244–6252. [Google Scholar] [CrossRef]
- Knorr, A.; Stange, P.; Fumino, K.; Weinhold, F.; Ludwig, R. Spectroscopic Evidence for Clusters of Like-Charged Ions in Ionic Liquids Stabilized by Cooperative Hydrogen Bonding. ChemPhysChem 2016, 17, 458–462. [Google Scholar] [CrossRef] [Green Version]
- Menges, F.S.; Zeng, H.J.; Kelleher, P.; Gorlova, O.; Johnson, M.A.; Niemann, T.; Strate, A.; Ludwig, R. Structural Motifs in Cold Ternary Ion Complexes of Hydroxyl-Functionalized Ionic Liquids: Isolating the Role of Cation–Cation Interactions. J. Phys. Chem. Lett. 2018, 9, 2979–2984. [Google Scholar] [CrossRef]
- Niemann, T.; Stange, P.; Strate, A.; Ludwig, R. Like-likes-Like: Cooperative Hydrogen Bonding Overcomes Coulomb Repulsion in Cationic Clusters with Net Charges up to Q=+6e. ChemPhysChem 2018, 19, 1691–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Donne, A.; Adenusi, H.; Porcelli, F.; Bodo, E. Hydrogen Bonding as a Clustering Agent in Protic Ionic Liquids: Like-Charge vs Opposite-Charge Dimer Formation. ACS Omega 2018, 3, 10589–10600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Donne, A.; Bodo, E. Isomerization patterns and proton transfer in ionic liquids constituents as probed by ab-initio computation. J. Mol. Liq. 2018, 249, 1075–1082. [Google Scholar] [CrossRef]
- Knorr, A.; Fumino, K.; Bonsa, A.; Ludwig, R. Spectroscopic evidence of ‘jumping and pecking’ of cholinium and H-bond enhanced cation–cation interaction in ionic liquids. Phys. Chem. Chem. Phys. 2015, 17, 30978–30982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Guo, Y.; Luo, X.; Li, H. Infrared spectroscopic study on chemical and phase equilibrium in triethylammonium acetate. Sci. China Chem. 2012, 55, 1688–1694. [Google Scholar] [CrossRef]
- Berton, P.; Kelley, S.P.; Wang, H.; Rogers, R.D. Elucidating the triethylammonium acetate system: Is it molecular or is it ionic? J. Mol. Liq. 2018, 269, 126–131. [Google Scholar] [CrossRef]
- Borodin, O. Polarizable Force Field Development and Molecular Dynamics Simulations of Ionic Liquids. J. Phys. Chem. B 2009, 113, 11463–11478. [Google Scholar] [CrossRef]
- Brehm, M.; Weber, H.; Pensado, A.S.; Stark, A.; Kirchner, B. Proton transfer and polarity changes in ionic liquid–water mixtures: A perspective on hydrogen bonds from ab initio molecular dynamics at the example of 1-ethyl-3-methylimidazolium acetate–water mixtures—Part 1. Phys. Chem. Chem. Phys. 2012, 14, 5030–5044. [Google Scholar] [CrossRef]
- Aradi, B.; Hourahine, B.; Frauenheim, T.; Frauenheim, T. DFTB+, a Sparse Matrix-Based Implementation of the DFTB Method†. J. Phys. Chem. A 2007, 111, 5678–5684. [Google Scholar] [CrossRef]
- Gaus, M.; Goe, A.; Elstner, M. Parametrization and benchmark of dftb3 for organic molecules. J. Chem. Theory Comput. 2013, 9, 338–354. [Google Scholar] [CrossRef]
- Řezáč, J. Empirical Self-Consistent Correction for the Description of Hydrogen Bonds in DFTB3. J. Chem. Theory Comput. 2017, 13, 4804–4817. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 73–78. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Addicoat, M.A.; Stefanovic, R.; Webber, G.B.; Atkin, R.; Page, A.J. Assessment of the Density Functional Tight Binding Method for Protic Ionic Liquids. J. Chem. Theory Comput. 2014, 10, 4633–4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, P.; Qian, H.-J.; Irle, S.; Lu, X.; Roston, D.; Mori, T.; Elstner, M.; Cui, Q. Molecular Simulation of Water and Hydration Effects in Different Environments: Challenges and Developments for DFTB Based Models. J. Phys. Chem. B 2014, 118, 11007–11027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theor. Chem. Accounts 2005, 116, 316–325. [Google Scholar] [CrossRef]
- Ruusuvuori, K.; Kurtén, T.; Ortega, I.K.; Faust, J.; Vehkamäki, H. Proton affinities of candidates for positively charged ambient ions in boreal forests. Atmospheric Chem. Phys. Discuss. 2013, 13, 10397–10404. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Brehm, M.; Fligg, R.; Vöhringer, P.; Kirchner, B. Computing vibrational spectra from ab initio molecular dynamics. Phys. Chem. Chem. Phys. 2013, 15, 6608–6622. [Google Scholar] [CrossRef]
- Heimer, N.E.; Del Sesto, R.E.; Meng, Z.; Wilkes, J.S.; Carper, W.R. Vibrational spectra of imidazolium tetrafluoroborate ionic liquids. J. Mol. Liq. 2006, 124, 84–95. [Google Scholar] [CrossRef]
- Ojha, D.; Chandra, A. Temperature dependence of the ultrafast vibrational echo spectroscopy of OD modes in liquid water from first principles simulations. Phys. Chem. Chem. Phys. 2019, 21, 6485–6498. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bodo, E. Structural Features of Triethylammonium Acetate through Molecular Dynamics. Molecules 2020, 25, 1432. https://doi.org/10.3390/molecules25061432
Bodo E. Structural Features of Triethylammonium Acetate through Molecular Dynamics. Molecules. 2020; 25(6):1432. https://doi.org/10.3390/molecules25061432
Chicago/Turabian StyleBodo, Enrico. 2020. "Structural Features of Triethylammonium Acetate through Molecular Dynamics" Molecules 25, no. 6: 1432. https://doi.org/10.3390/molecules25061432
APA StyleBodo, E. (2020). Structural Features of Triethylammonium Acetate through Molecular Dynamics. Molecules, 25(6), 1432. https://doi.org/10.3390/molecules25061432