Glucocorticoids Influencing Wnt/β-Catenin Pathway; Multiple Sites, Heterogeneous Effects

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Wnt/β-Catenin Pathway or Canonical Wnt-Signaling
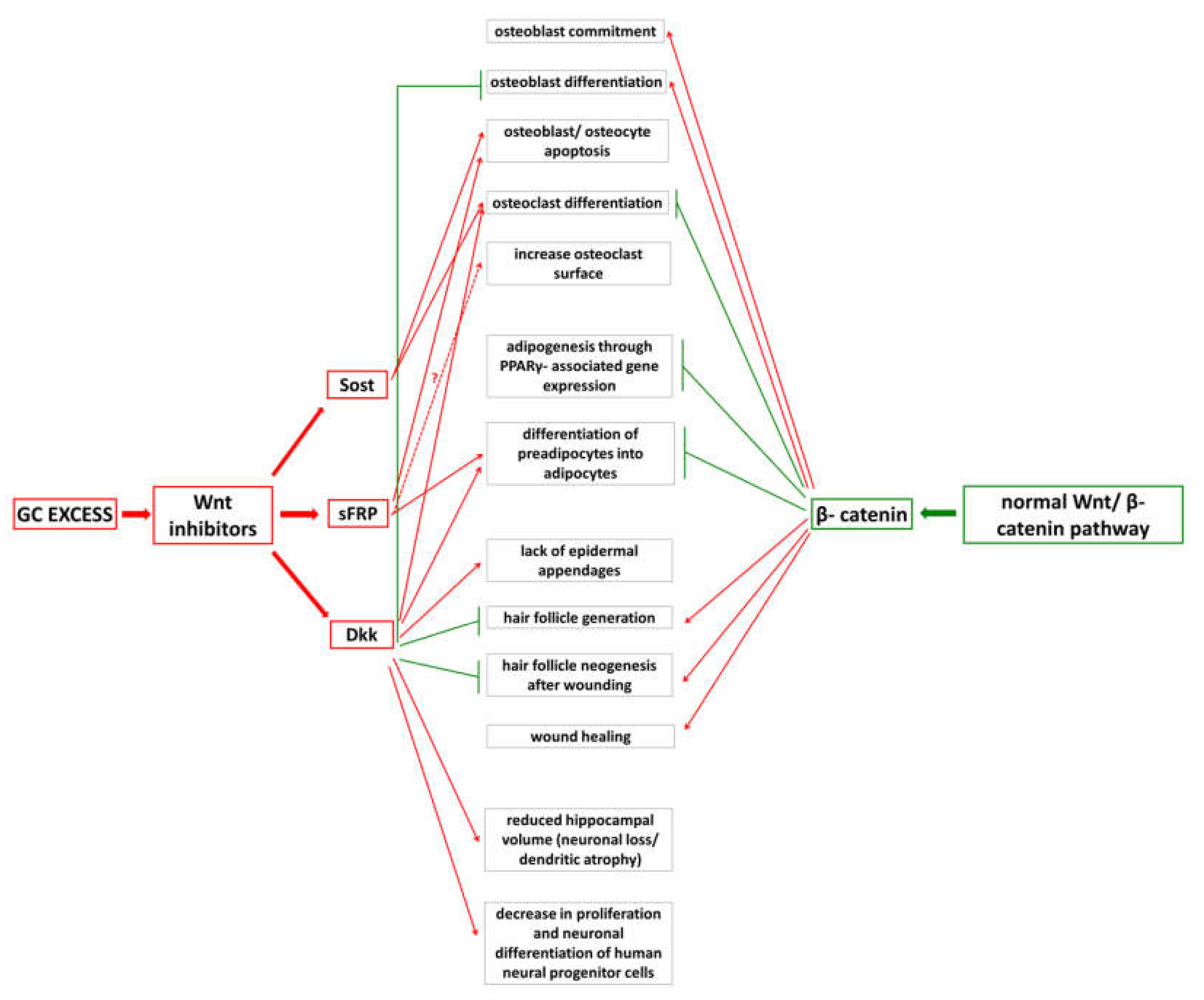
3. Wnt/β-catenin Pathway and Cushing’s Syndrome
4. Effects of GC Excess on Bone
4.1. Bone Homeostasis and Main Cytokines
4.2. Detrimental Effects of GC Excess
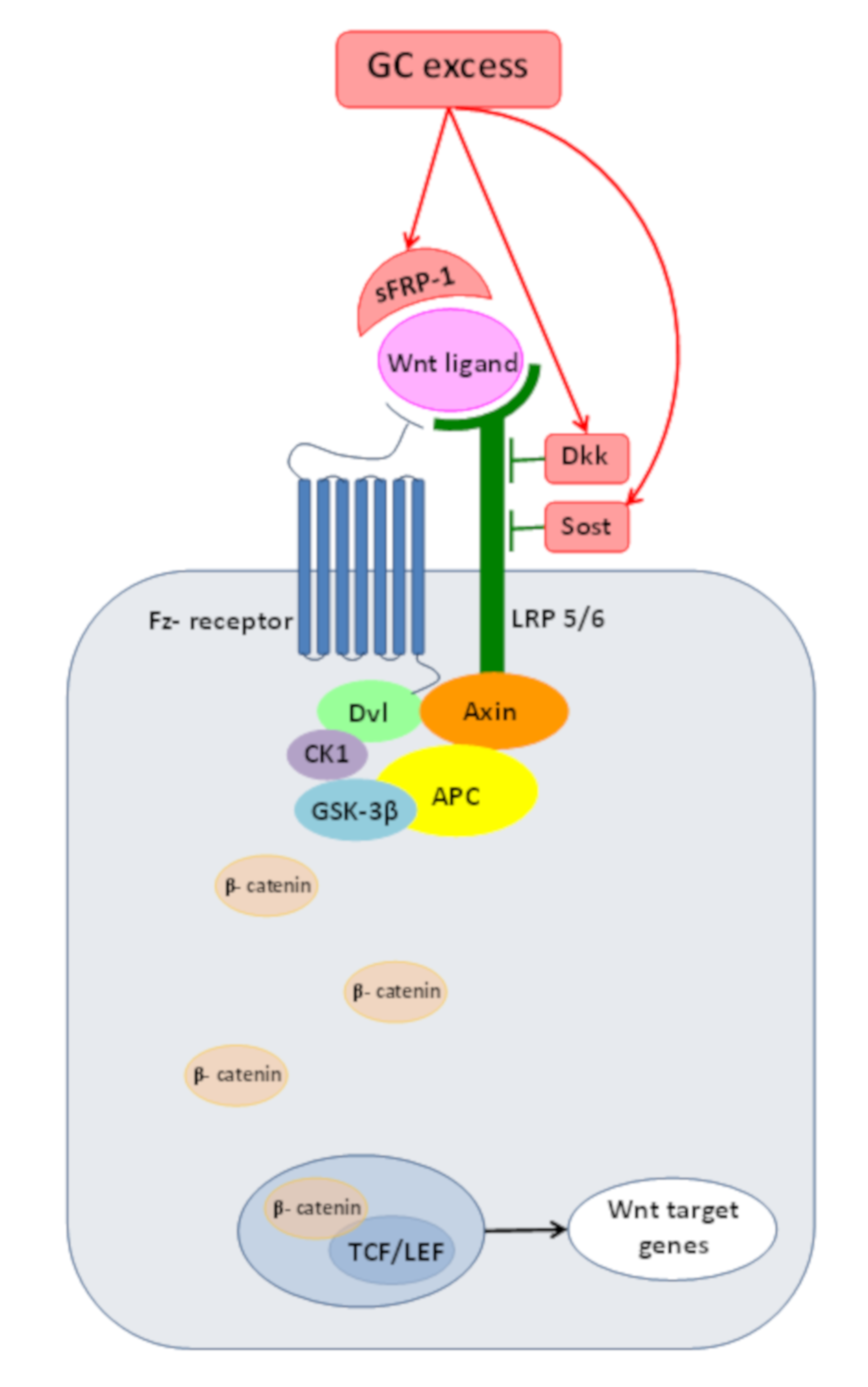
4.3. Inadequate Wnt/β-Catenin PathwayOperationdue to GC Excess
5. Effects of GC Excess on Adipose Tissue
5.1. Adipogenesis and Wnt-Signaling
5.2. Detrimental Effects of GC Excess
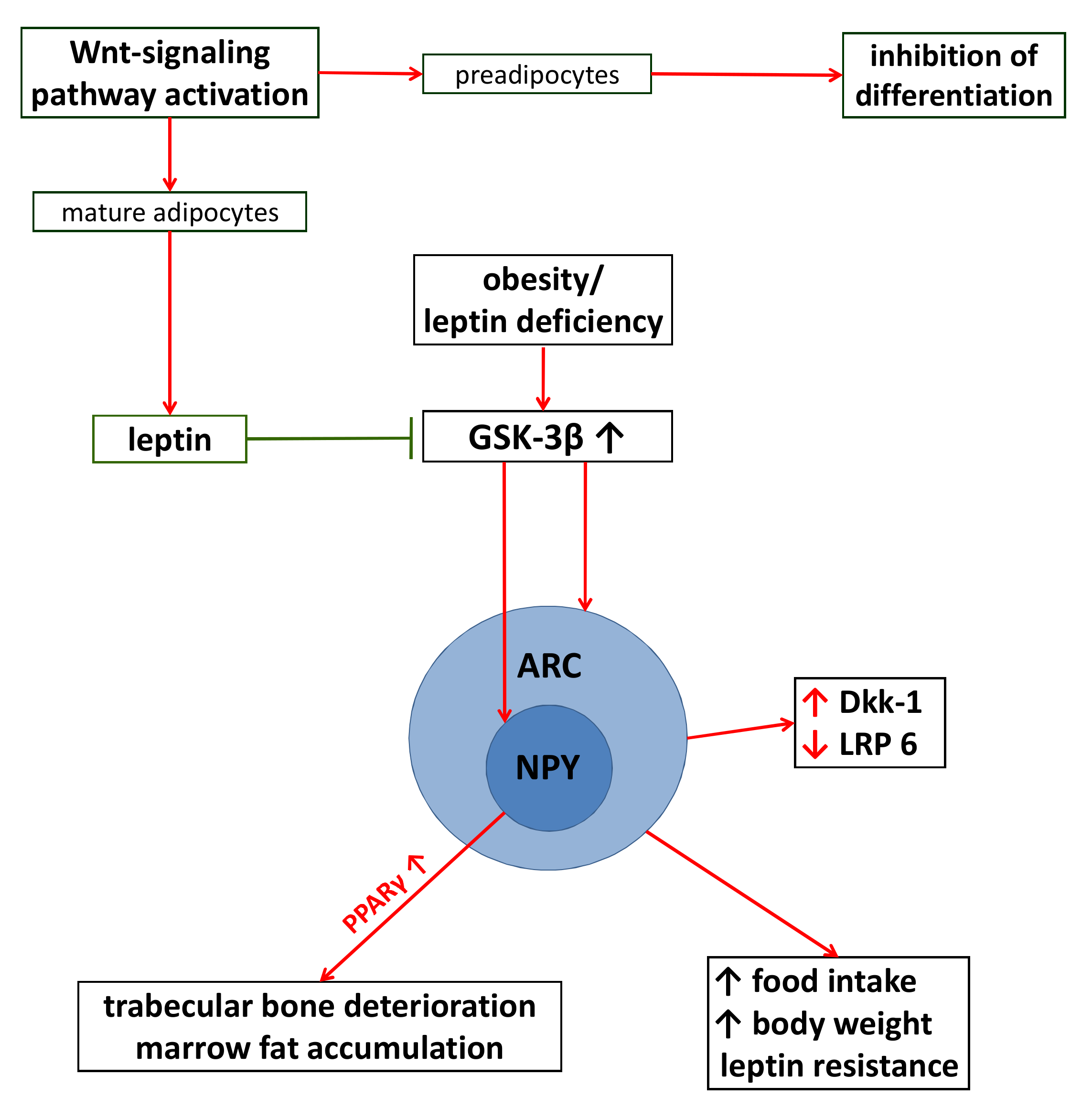
5.3. The Consequences of Adiposity on Wnt-Signaling
6. Effects of GC Excess on Brain
6.1. Neurogenesis and Wnt/β-Catenin Pathway
6.2. Detrimental Effects of GC Excess
6.3. GC Excess and Wnt-Signaling
7. Effects of GC Excess on Skin
7.1. Skin and Steroidogenesis
7.2. Detrimental Effects of GC Excess
7.3. Wnt-Pathway Alterations and Their Consequences
8. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nicolaides, N.C.; Charmandari, E.; George, P.; Chrousos, G.P.; Kino, T. Recent advances in the molecular mechanisms determining tissue sensitivity to glucocorticoids: Novel mutations, circadian rhythm and ligand- induced repression of the human glucocorticoid receptor. BMC Endocr. Disord. 2014, 14, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quax, R.A.; Manenschijn, L.; Koper, J.W.; Hazes, J.M.; Lamberts, S.W.J.; van Rossum, E.F.C.; Feelders, R.A. Glucocorticoid sensitivity in health and disease. Nat. Rev. Endocrinol. 2013, 9, 670–686. [Google Scholar] [CrossRef] [PubMed]
- Spies, C.M.; Strehl, C.; van der Goes, M.C.; Bijlsma, J.W.J.; Buttgereit, F. Glucocorticoids. Best Pract. Res. Clin. Rheumatol. 2011, 25, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, J.W.J.; Buttgereit, F. Adverse events of glucocorticoids during treatment of rheumatoid arthritis: Lessons from cohort and registry studies. Rheumatology 2016, 55, ii3–ii5. [Google Scholar] [CrossRef] [PubMed]
- Guaraldi, F.; Salvatori, R. Cushing syndrome: Maybe not so uncommon of an endocrine disease. J. Am. Board Fam. Med. 2012, 2, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, R.M.; Labad, J.; Sears, A.V.; Williamson, R.M.; Strachan, M.W.J.; Deary, I.J.; Lowe, G.D.O.; Price, J.F.; Walker, B.R. Glucocorticoid treatment and impaired mood, memory and metabolism in people with diabetes: The Edinburgh Type 2 Diabetes Study. Eur. J. Endocrinol. 2012, 166, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Overman, R.A.; Toliver, J.C.; Yeh, J.Y.; Gourlay, M.L.; Deal, C.L. United States adults meeting 2010 American College of Rheumatology criteria for treatment and prevention of glucocorticoid-induced osteoporosis. Arthritis Care Res. 2014, 66, 1644–1652. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/b-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [Green Version]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Cruciat, C.M.; Niehrs, C. Secreted and Transmembrane Wnt Inhibitors and Activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef] [Green Version]
- Schulze, J.; Seitz, S.; Saito, H.; Schneebauer, M.; Marshall, R.P.; Baranowsky, A.; Busse, B.; Schilling, A.F.; Friedrich, F.W.; Albers, J.; et al. Negative Regulation of Bone Formation by the Transmembrane Wnt Antagonist Kremen-2. PLoS ONE 2010, 5, e10309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin Binds to LRP5/6 and Antagonizes Canonical Wnt Signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, W.; Cheng, Z.; Shahnazari, M.; Dai, W.; Johnson, M.L.; Lane, N.E. Overexpression of secreted frizzled-related protein 1 inhibits bone formation and attenuates parathyroid hormone bone anabolic effects. J. Bone Miner. Res. 2010, 25, 190–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesa, P.; Fazio, V.W.; Church, J.M.; McGannon, E. Adrenal masses in patients with familial adenomatous polyposis. Dis. Colon Rectum 1997, 40, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Clark, S.K.; Katz, D.E.; Reznek, R.H.; Phillips, R.K. Adrenal masses are associated with familial adenomatous polyposis. Dis. Colon Rectum 2000, 43, 1739–1742. [Google Scholar] [CrossRef]
- Tissier, F.; Cavard, C.; Groussin, L.; Perlemoine, K.; Fumey, G.; Hagneré, A.M.; René-Corail, F.; Jullian, E.; Gicquel, C.; Bertagna, X.; et al. Mutations of beta-catenin in adrenocortical tumors: Activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005, 65, 7622–7627. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. Wnt signaling and cancer. Genes Dev. 2000, 14, 1837–1851. [Google Scholar] [CrossRef] [Green Version]
- Tadjine, M.; Lampron, A.; Ouadi, L.; Bourdeau, I. Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin. Endocrinol. 2008, 68, 264–270. [Google Scholar] [CrossRef]
- Bonnet, S.; Gaujoux, S.; Launay, P.; Baudry, C.; Chokri, I.; Ragazzon, B.; Libé, R.; René-Corail, F.; Audebourg, A.; Vacher-Lavenu, M.C.; et al. Wnt/β-Catenin Pathway Activation in Adrenocortical Adenomas Is Frequently due to Somatic CTNNB1-Activating Mutations, Which Are Associated with Larger and Nonsecreting Tumors: A Study in Cortisol-Secreting and -Nonsecreting Tumors. J. Clin. Endocrinol. Metab. 2011, 96, E419–E426. [Google Scholar] [CrossRef] [Green Version]
- Rutkovskiy, A.; Stensløkken, K.O.; Vaage, I.J. Osteoblast Differentiation at a Glance. Med. Sci. Monit. Basic Res. 2016, 22, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Sapir-Koren, R.; Livshits, G. Osteocyte control of bone remodeling: Is sclerostin a key molecular coordinator of the balanced bone resorption– formation cycles? Osteoporos. Int. 2014, 25, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. The Great Beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 558, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Faccio, R.; Takeshita, S.; Zallone, A.; Ross, F.P.; Teitelbaum, S.L. c-Fms and the alphavbeta3 integrin collaborate during osteoclast differentiation. J. Clin. Investig. 2003, 111, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.C.; Choi, Y. Biology of the RANKL–RANK-OPG System in Immunity, Bone, and Beyond. Front. Immunol. 2014, 5, 511. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Janz, S. Attenuation of WNT signaling by DKK-1 and -2 regulates BMP2-induced osteoblast differentiation and expression of OPG, RANKL and M-CSF. Mol. Cancer 2007, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Muruganandan, S.; Sinal, C.J. The impact of bone marrow adipocytes on osteoblast and osteoclast differentiation. IUBMB Life 2014, 66, 147–155. [Google Scholar] [CrossRef]
- Tóth, M.; Grossman, A. Glucocorticoid- induced osteoporosis: Lessons from Cushing’s syndrome. Clin. Endocrinol. 2013, 79, 1–11. [Google Scholar] [CrossRef]
- Buehring, B.; Viswanathan, R.; Binkley, N.; Busse, W. Glucocorticoid-induced osteoporosis: An update on effects and management. J. Allergy Clin. Immunol. 2013, 132, 1019–1030. [Google Scholar] [CrossRef]
- Komori, T. Glucocorticoid Signaling and Bone Biology. Horm. Metab. Res. 2016, 48, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Turcott, E.; Skonier, J.E.; Winkler, D.G.; Latham, J.A. Sclerostin promotes the apoptosis of human osteoblastic cells: A novel regulation of bone formation. Bone 2004, 35, 828–835. [Google Scholar] [CrossRef]
- Bodine, P.V.; Billiard, J.; Moran, R.A.; Ponce-de-Leon, H.; McLarney, S.; Mangine, A.; Scrimo, M.J.; Bhat, R.A.; Stauffer, B.; Green, J.; et al. The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. J. Cell. Biochem. 2005, 96, 1212–1230. [Google Scholar] [CrossRef] [PubMed]
- Brabnikova Maresova, K.; Pavelka, K.; Stepan, J.J. Acute effects of glucocorticoids on serum markers of osteoclasts, osteoblast, and osteocytes. Calcif. Tissue Int. 2013, 92, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Van Lierop, A.H.; van der Eerden, A.W.; Hamdy, N.A.; Hermus, A.R.; den Heijer, M.; Papapoulos, S.E. Circulating sclerostin levels are decreased in patients with endogenous hypercortisolism and increase after treatment. J. Clin. Endocrinol. Metab. 2012, 97, E1953–E1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.; Han, L.; Ambrogini, E.; Weinstein, R.S.; Manolagas, S.C. Glucocorticoids and tumor necrosis factor α increase oxidative stress and suppress Wnt protein signaling in osteoblasts. J. Biol. Chem. 2011, 286, 44326–44335. [Google Scholar] [CrossRef] [Green Version]
- Hurson, C.J.; Butler, J.S.; Keating, D.T.; Murray, D.W.; Sadlier, D.M.; O’Byrne, J.M.; Doran, P.P. Gene expression analysis in human osteoblasts exposed to dexamethasone identifies altered developmental pathways as putative drivers of osteoporosis. BMC Musculoskelet. Disord. 2007, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hoogeboom, D.; Essers, M.A.; Polderman, P.E.; Voets, E.; Smits, L.M.; Burgering, B.M. Interaction of FOXO with beta-catenin inhibits beta-catenin/T cell factor activity. J. Biol. Chem. 2008, 283, 9224–9230. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.; Ambrogini, E.; Bartell, S.M.; Han, L.; Roberson, P.K.; de Cabo, R.; Jilka, R.L.; Weinstein, R.S.; O’Brien, C.A.; Manolagas, S.C.; et al. FOXOs attenuate bone formation by Wnt signaling. J. Clin. Investig. 2013, 123, 3409–3419. [Google Scholar] [CrossRef]
- Wang, F.S.; Lin, C.L.; Chen, Y.J.; Wang, C.J.; Yang, K.D.; Huang, Y.T.; Sun, Y.C.; Huang, H.C. Secreted frizzled- related protein 1 modulates glucocorticoid attenuation of osteogenic activities and bone mass. Endocrinology 2005, 146, 2415–2423. [Google Scholar] [CrossRef] [Green Version]
- Ohnaka, K.; Tanabe, M.; Kawate, H.; Nawata, H.; Takayanagi, R. Glucocorticoid suppresses the canonical Wnt signal in cultured human osteoblasts. Biochem. Biophys. Res. Commun. 2005, 329, 177–181. [Google Scholar] [CrossRef]
- Sato, A.Y.; Cregor, M.; Delgado-Calle, J.; Condon, K.W.; Allen, M.R.; Peacock, M.; Plotkin, L.I.; Bellido, T. Protection From Glucocorticoid-Induced Osteoporosis by Anti-Catabolic Signaling in the Absence of Sost/Sclerostin. J. Bone Miner. Res. 2016, 31, 1791–1802. [Google Scholar] [CrossRef] [Green Version]
- Morvan, F.; Boulukos, K.; Clément-Lacroix, P.; Roman Roman, S.; Suc-Royer, I.; Vayssière, B.; Ammann, P.; Martin, P.; Pinho, S.; Pognonec, P.; et al. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J. Bone Miner. Res. 2006, 21, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, M.; Xiong, J.; Fujiwara, Y.; Thostenson, J.D.; O’Brien, C.A. Cortical bone loss caused by glucocorticoid excess requires RANKL production by osteocytes and isassociated with reduced OPG expression in mice. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E587–E593. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.; Vayssière, B.; Garcia, T.; Newell, W.; Baron, R.; Roman-Roman, S.; Rawadi, G. Gene array analysis of Wnt-regulated genes in C3H10T1/2 cells. Bone 2005, 36, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Wijenayaka, A.R.; Kogawa, M.; Lim, H.P.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin stimulates osteocyte support of osteoclastactivity by a RANKL- dependent pathway. PLoS ONE 2011, 6, e25900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Wise, G.E. A DNA microarray analysis of chemokine and receptor genes in the rat dental follicle -role of secreted frizzled-related protein-1 in osteoclastogenesis. Bone 2007, 41, 266–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albers, J.; Keller, J.; Baranowsky, A.; Beil, F.T.; Catala-Lehnen, P.; Schulze, J.; Amling, M.; Schinke, T. Canonical Wnt signaling inhibits oteoclastogenesis independent of osteoprotegerin. J. Cell Biol. 2013, 200, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.D.; Sarraf, P.; Troy, A.E.; Bradwin, G.; Moore, K.; Milstone, D.S.; Spiegelman, B.M.; Mortensen, R.M. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol. Cell 1999, 4, 611–617. [Google Scholar] [CrossRef]
- Liu, J.; Farmer, S.R. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J. Biol. Chem. 2004, 279, 45020–45027. [Google Scholar] [CrossRef] [Green Version]
- Moldes, M.; Zuo, Y.; Morrison, R.F.; Silva, D.; Park, B.H.; Liu, J.; Farmer, S.R. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta- catenin signalling during adipogenesis. Biochem. J. 2003, 376, 607–613. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, H.; Zuo, Y.; Farmer, S.R. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol. Cell. Biol. 2006, 26, 5827–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christodoulides, C.; Scarda, A.; Granzotto, M.; Milan, G.; Dalla Nora, E.; Keogh, J.; De Pergola, G.; Stirling, H.; Pannacciulli, N.; Sethi, J.K.; et al. WNT10B mutations in human obesity. Diabetologia 2006, 49, 678–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieman, L.K. Cushing’s syndrome: Update on signs, symptoms and biochemical screening. Eur. J. Endocrinol. 2015, 173, M33–M38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, R.T.; Lee, M.J.; Karastergiou, K.; Gower, A.; Fried, S.K. Depot dependent effects of dexamethasone on gene expression in human omental and abdominal subcutaneous adipose tissues from obese women. PLoS ONE 2016, 11, e0167337. [Google Scholar] [CrossRef]
- Xiao, X.; Li, H.; Yang, J.; Qi, X.; Zu, X.; Yang, J.; Zhong, J.; Cao, R.; Liu, J.; Wen, G. Wnt/β-catenin signaling pathway and lipolysis enzymes participate in methylprednisolone induced fat differential distribution between subcutaneous and visceral adipose tissue. Steroids 2014, 84, 30–35. [Google Scholar] [CrossRef]
- Rebuffé-Scrive, M.; Krotkiewski, M.; Elfverson, J.; Björntorp, P. Muscle and adipose tissue morphology and metabolism in Cushing’s syndrome. J. Clin. Endocrinol. Metab. 1988, 67, 1122–1128. [Google Scholar] [CrossRef]
- Ehrlund, A.; Mejhert, N.; Lorente-Cebrián, S.; Aström, G.; Dahlman, I.; Laurencikiene, J.; Rydén, M. Characterization of the Wnt inhibitors secreted frizzled- related proteins (SFRPs) in human adipose tissue. J. Clin. Endocrinol. Metab. 2013, 98, E503–E508. [Google Scholar] [CrossRef]
- Lagathu, C.; Christodoulides, C.; Tan, C.Y.; Virtue, S.; Laudes, M.; Campbell, M.; Ishikawa, K.; Ortega, F.; Tinahones, F.J.; Fernández-Real, J.M.; et al. Secreted frizzled-related protein 1 regulates adipose tissue expansion and is dysregulated in severe obesity. Int. J. Obes. 2010, 34, 1695–1705. [Google Scholar] [CrossRef] [Green Version]
- Abdallah, B.M. Marrow adipocytes inhibit the differentiation of mesenchymal stem cells into osteoblasts via suppressing BMP- signaling. J. Biomed. Sci. 2017, 24, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Blüher, M.; Mantzoros, C.S. From leptin to other adipokines in health and disease: Facts and expectations at the beginning of the 21st century. Metabolism 2015, 64, 131–145. [Google Scholar] [CrossRef]
- Chen, Z.L.; Shao, W.J.; Xu, F.; Liu, L.; Lin, B.S.; Wei, X.H.; Song, Z.L.; Lu, H.G.; Fantus, I.G.; Weng, J.P.; et al. Acute Wnt pathway activation positively regulates leptin gene expression in mature adipocytes. Cell Signal. 2015, 27, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Benzler, J.; Ganjam, G.K.; Krüger, M.; Pinkenburg, O.; Kutschke, M.; Stöhr, S.; Steger, J.; Koch, C.E.; Ölkrug, R.; Schwartz, M.W.; et al. Hypothalamic glycogen synthase kinase 3β has a central role in the regulation of food intake and glucose metabolism. Biochem. J. 2012, 447, 175–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzler, J.; Andrews, Z.B.; Pracht, C.; Stöhr, S.; Shepherd, P.R.; Grattan, D.R.; Tups, A. Hypothalamic WNT signalling is impaired during obesity and reinstated by leptin treatment in male mice. Endocrinology 2013, 154, 4737–4745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.S.; Lian, W.S.; Weng, W.T.; Sun, Y.C.; Ke, H.J.; Chen, Y.S.; Ko, J.Y. Neuropeptide Y mediates glucocorticoid-induced osteoporosis and marrow adiposity in mice. Osteoporos. Int. 2016, 27, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Lang, J.; Sohn, J.; Hammond, E.; Chang, M.; Pleasure, D. Canonical Wnt signaling in the oligodendroglial lineage-puzzles remain. Glia 2015, 63, 1671–1693. [Google Scholar] [CrossRef] [Green Version]
- Tatomir, A.; Micu, C.; Crivii, C. The impact of stress and glucocorticoids on memory. Clujul Med. 2014, 87, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Andela, C.D.; van Haalen, F.M.; Ragnarsson, O.; Papakokkinou, E.; Johannsson, G.; Santos, A.; Webb, S.M.; Biermasz, N.R.; van der Wee, N.J.; Pereira, A.M. Cushing’s syndrome causes irreversible effects on the human brain: A systematic review of structural and functional magnetic resonance imaging studies. Eur. J. Endocrinol. 2015, 173, R1–R14. [Google Scholar] [CrossRef] [Green Version]
- Matrisciano, F.; Busceti, C.L.; Bucci, D.; Orlando, R.; Caruso, A.; Molinaro, G.; Cappuccio, I.; Riozzi, B.; Gradini, R.; Motolese, M.; et al. Induction of the Wnt antagonist Dickkopf-1 is involved in stress-induced hippocampal damage. PLoS ONE 2011, 1, e16447. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, N.; Machida, T.; Takahashi, T.; Takatsu, H.; Shinkai, T.; Abe, K.; Urano, S. Elevation by oxidative stress and aging of hypothalamic-pituitary-adrenal activity in rats and its prevention by vitamin E. J. Clin. Biochem. Nutr. 2009, 45, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.P.; Sandman, C.A.; Buss, C.; Wing, D.A.; Head, K. Fetal glucocorticoid exposure is associated with preadolescent brain development. Biol. Psychiatry 2013, 74, 647–655. [Google Scholar] [CrossRef] [Green Version]
- Bose, R.; Moors, M.; Tofighi, R.; Cascante, A.; Hermanson, O.; Ceccatelli, S. Glucocorticoids induce long-lasting effects in neural stem cells resulting in senescence-related alterations. Cell Death Dis. 2010, 1, e92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crudo, A.; Suderman, M.; Moisiadis, V.G.; Petropoulos, S.; Kostaki, A.; Hallett, M.; Szyf, M.; Matthews, S.G. Glucocorticoid programming of the fetal male hippocampal epigenome. Endocrinology 2013, 154, 1168–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boku, S.; Nakagawa, S.; Masuda, T.; Nishikawa, H.; Kato, A.; Takamura, N.; Omiya, Y.; Kitaichi, Y.; Inoue, T.; Kusumi, I. Valproate recovers the inhibitory effect of dexamethasone on the proliferation of the adult dentate gyrus-derived neural precursor cells via GSK-3β and β-catenin pathway. Eur. J. Pharmacol. 2014, 723, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Boku, S.; Nakagawa, S.; Masuda, T.; Nishikawa, H.; Kato, A.; Kitaichi, Y.; Inoue, T.; Koyama, T. Glucocorticoids and lithium reciprocally regulate the proliferation of adult dentate gyrus-derived neural precursor cells through GSK-3b and b-catenin/TCF pathway. Neuropsychopharmacology 2009, 34, 805–815. [Google Scholar] [CrossRef] [Green Version]
- Chenn, A.; Walsh, C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297, 365–369. [Google Scholar] [CrossRef] [Green Version]
- Moors, M.; Bose, R.; Johansson-Haque, K.; Edoff, K.; Okret, S.; Ceccatelli, S. Dickkopf 1 mediates glucocorticoid-induced changes in human neural progenitor cell proliferation and differentiation. Toxicol. Sci. 2012, 125, 488–495. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, L.J.; Sapolsky, R.M. Glucocorticoids increase the accumulation of reactive oxygen species and enhance adriamycin-induced toxicity in neuronal culture. Exp. Neurol. 1996, 141, 201–206. [Google Scholar] [CrossRef]
- Sato, H.; Takahashi, T.; Sumitani, K.; Takatsu, H.; Urano, S. Glucocorticoidcgenerates ROS to induce oxidative injury in the hippocampus, leading to impairment of cognitive function of rats. J. Clin. Biochem. Nutr. 2010, 47, 224–232. [Google Scholar] [CrossRef] [Green Version]
- Camm, E.J.; Tijsseling, D.; Richter, H.G.; Adler, A.; Hansell, J.A.; Derks, J.B.; Cross, C.M.; Giussani, D.A. Oxidative stress in the developing brain: Effects of postnatal glucocorticoid therapy and antioxidants in the rat. PLoS ONE 2011, 6, e21142. [Google Scholar] [CrossRef]
- Paik, J.H.; Ding, Z.; Narurkar, R.; Ramkissoon, S.; Muller, F.; Kamoun, W.S.; Chae, S.S.; Zheng, H.; Ying, H.; Mahoney, J.; et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell 2009, 5, 540–553. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A. A nervous breakdown in the skin: Stress and the epidermal barrier. J. Clin. Investig. 2007, 117, 3166–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vukelic, S.; Stojadinovic, O.; Pastar, I.; Rabach, M.; Krzyzanowska, A.; Lebrun, E.; Davis, S.C.; Resnik, S.; Brem, H.; Tomic-Canic, M. Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J. Biol. Chem. 2011, 286, 10265–10275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jozic, I.; Stojadinovic, O.; Kirsner, R.S.; Tomic-Canic, M. Stressing the steroids in skin: Paradox or fine-tuning? J. Investig. Dermatol. 2014, 134, 2869–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.; Zbytek, B.; Nikolakis, G.; Manna, P.R.; Skobowiat, C.; Zmijewski, M.; Li, W.; Janjetovic, Z.; Postlethwaite, A.; Zouboulis, C.C.; et al. Steroidogenesis in the skin: Implications for local immune functions. J. Steroid Biochem. Mol. Biol. 2013, 137, 107–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccato, F.; Boscaro, M. Cushing’s syndrome: Screening and diagnosis. High Blood Press. Cardiovasc. Prev. 2016, 23, 209–215. [Google Scholar] [CrossRef]
- Schoepe, S.; Schäcke, H.; May, E.; Asadullah, K. Glucocorticoid therapy- induced skin atrophy. Exp. Dermatol. 2006, 15, 406–420. [Google Scholar] [CrossRef]
- Stojadinovic, O.; Lee, B.; Vouthounis, C.; Vukelic, S.; Pastar, I.; Blumenberg, M.; Brem, H.; Tomic-Canic, M. Novel genomic effects of glucocorticoids in epidermal keratinocytes: Inhibition of apoptosis, interferon-gamma pathway, and wound healing along with promotion of terminal differentiation. J. Biol. Chem. 2007, 282, 4021–4034. [Google Scholar] [CrossRef] [Green Version]
- Driskell, R.R.; Jahoda, C.A.; Chuong, C.M.; Watt, F.M.; Horsley, V. Defining dermal adipose tissue. Exp. Dermatol. 2014, 23, 629–631. [Google Scholar] [CrossRef] [Green Version]
- Walker, G.E.; Verti, B.; Marzullo, P.; Savia, G.; Mencarelli, M.; Zurleni, F.; Liuzzi, A.; Di Blasio, A.M. Deep subcutaneous adipose tissue: A distinct abdominal adipose depot. Obesity 2007, 15, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Sbarbati, A.; Accorsi, D.; Benati, D.; Marchetti, L.; Orsini, G.; Rigotti, G.; Panettiere, P. Subcutaneous adipose tissue classification. Eur. J. Histochem. 2010, 54, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Baida, G.; Bhalla, P.; Yemelyanov, A.; Stechschulte, L.A.; Shou, W.; Readhead, B.; Dudley, J.T.; Sánchez, E.R.; Budunova, I. Deletion of the glucocorticoid receptor chaperone FKBP51 prevents glucocorticoid-induced skin atrophy. Oncotarget 2018, 9, 34772–34783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valassi, E.; Santos, A.; Yaneva, M.; Tóth, M.; Strasburger, C.J.; Chanson, P.; Wass, J.A.; Chabre, O.; Pfeifer, M.; Feelders, R.A.; et al. The European Registry on Cushing’s syndrome: 2-year experience. Baseline demographic and clinical characteristics. Eur. J. Endocrinol. 2011, 165, 383–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broder, M.S.; Chang, E.; Cherepanov, D.; Neary, M.P.; Ludlam, W.H. Identification of potential markers for Cushing disease. Endocr. Pract. 2016, 22, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Lefkowitz, E.G.; Cossman, J.P.; Fournier, J.B. A case report of Cushing’s disease presenting as hair loss. Case Rep. Dermatol. 2017, 9, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT signals are required for the initiation of hair follicle development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Choi, Y.S.; Zhang, Y.; Xu, M.; Yang, Y.; Ito, M.; Peng, T.; Cui, Z.; Nagy, A.; Hadjantonakis, A.K.; Lang, R.A.; et al. Distinct functions for Wnt/β-catenin in hair follicle stem cell proliferation and survival and interfollicular epidermal homeostasis. Cell Stem Cell 2013, 13, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Hawkshaw, N.J.; Hardman, J.A.; Haslam, I.S.; Shahmalak, A.; Gilhar, A.; Lim, X.; Paus, R. Identifying novel strategies for treating human hair loss disorders: Cyclosporine A suppress the Wnt inhibitor, SFRP1, int he dermal papillaof human scalp hair follicles. PLoS Biol. 2018, 16, e2003705. [Google Scholar] [CrossRef] [Green Version]
- Närhi, K.; Järvinen, E.; Birchmeier, W.; Taketo, M.M.; Mikkola, M.L.; Thesleff, I. Sustained epithelial beta-catenin activity induces precocious hair development but disrupts hair follicle down-growth and hair shaft formation. Development 2008, 135, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.Y.; Yamada, S.; Izumi, H.; Tsukamoto, M.; Nakashima, T.; Tasaki, T.; Guo, X.; Uramoto, H.; Sasaguri, Y.; Kohno, K. Critical in vivo roles of WNT10A in wound healing by regulating collagen expression/synthesis in WNT10A- deficient mice. PLoS ONE 2018, 13, e0195156. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Yang, Z.; Andl, T.; Cui, C.; Kim, N.; Millar, S.E.; Cotsarelis, G. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 2007, 447, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Stojadinovic, O.; Brem, H.; Vouthounis, C.; Lee, B.; Fallon, J.; Stallcup, M.; Merchant, A.; Galiano, R.D.; Tomic-Canic, M. Molecular pathogenesis of chronic wounds: The role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am. J. Pathol. 2005, 167, 59–69. [Google Scholar] [CrossRef]
- Stojadinovic, O.; Pastar, I.; Nusbaum, A.G.; Vukelic, S.; Krzyzanowska, A.; Tomic-Canic, M. Deregulation of epidermal stem cell niche contributes to pathogenesis of nonhealing venous ulcers. Wound Repair Regen. 2014, 22, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jozic, I.; Vukelic, S.; Stojadinovic, O.; Liang, L.; Ramirez, H.A.; Pastar, I.; Tomic-Canic, M. Stress Signals, Mediated by Membranous Glucocorticoid Receptor, Activate PLC/PKC/GSK-3β/β-catenin Pathway to Inhibit Wound Closure. J. Investig. Dermatol. 2017, 137, 1144–1154. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Chesi, M.; Bergsagel, P.L. Bone lesions in molecular subtypes of multiple myeloma. N. Engl. J. Med. 2004, 351, 197–198. [Google Scholar] [CrossRef]
- Oyajobi, B.O.; Garrett, I.R.; Gupta, A.; Flores, A.; Esparza, J.; Muñoz, S.; Zhao, M.; Mundy, G.R. Stimulation of new bone formation by the proteasome inhibitor, bortezomib: Implications for myeloma bone disease. Br. J. Haematol. 2007, 139, 434–438. [Google Scholar] [CrossRef]
- Qiang, Y.W.; Hu, B.; Chen, Y.; Zhong, Y.; Shi, B.; Barlogie, B.; Shaughnessy, J.D., Jr. Bortezomib induces osteoblast differentiation via Wnt-independent activation of beta-catenin/TCF signaling. Blood 2009, 113, 4319–4330. [Google Scholar] [CrossRef] [Green Version]
- Bodine, P.V.; Stauffer, B.; Ponce-de-Leon, H.; Bhat, R.A.; Mangine, A.; Seestaller-Wehr, L.M.; Moran, R.A.; Billiard, J.; Fukayama, S.; Komm, B.S.; et al. A small molecule inhibitor of the Wnt antagonist secreted frizzled-related protein-1 stimulates bone formation. Bone 2009, 44, 1063–1068. [Google Scholar] [CrossRef]
- Pannone, G.; Bufo, P.; Santoro, A.; Franco, R.; Aquino, G.; Longo, F.; Botti, G.; Serpico, R.; Cafarelli, B.; Abbruzzese, A.; et al. WNT pathway in oral cancer: Epigenetic inactivation of WNT-inhibitors. Oncol. Rep. 2010, 24, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.T.; Li, J.; Jang, E.R.; Gulhati, P.; Rychahou, P.G.; Napier, D.L.; Wang, C.; Weiss, H.L.; Lee, E.Y.; Anthony, L.; et al. Deregulation of Wnt/β-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis 2013, 34, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Bimonte, V.M.; Fittipaldi, S.; Marocco, C.; Emerenziani, G.P.; Fornari, R.; Guidetti, L.; Poggiogalle, E.; Nicolai, E.; Di Luigi, L.; Donini, L.M.; et al. Physical activity and hypocaloric diet recovers osteoblasts homeostasis in women affected by abdominal obesity. Endocrine 2017, 58, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voorzanger-Rousselot, N.; Journe, F.; Doriath, V.; Body, J.J.; Garnero, P. Assessment of circulating Dickkopf-1 with a new two-site immunoassay in healthy subjects and women with breast cancer and bone metastases. Calcif. Tissue Int. 2009, 84, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Chang, J.S.; Park, K.S.; Park, J.; Kim, N.; Lee, J.I.; Kong, I.D. Effects of exercise training on circulating levels of Dickkpof-1 and secreted frizzled-related protein-1 in breast cancer survivors: A pilot single-blind randomized controlled trial. PLoS ONE 2017, 12, e0171771. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J. Bone Miner. Res. 2009, 24, 1651–1661. [Google Scholar] [CrossRef]
- Spatz, J.M.; Fields, E.E.; Yu, E.W.; DivietiPajevic, P.; Bouxsein, M.L.; Sibonga, J.D.; Zwart, S.R.; Smith, S.M. Serum sclerostin increases in healthy adult men during bed rest. J. Clin. Endocrinol. Metab. 2012, 97, E1736–E1740. [Google Scholar] [CrossRef]
- Chen, C.; Nakagawa, S.; An, Y.; Ito, K.; Kitaichi, Y.; Kusumi, I. The exercise-glucocorticoid paradox: How exercise is beneficial to cognition, mood, and the brain while increasing glucocorticoid levels. Front. Neuroendocrinol. 2017, 44, 83–102. [Google Scholar] [CrossRef]
- Ueda, S.; Ichiseki, T.; Yoshitomi, Y.; Yonekura, H.; Ueda, Y.; Kaneuji, A.; Matsumoto, T. Osteocytic cell necrosis is caused by a combination of glucocorticoid-induced Dickkopf-1 and hypoxia. Med. Mol. Morphol. 2015, 48, 69–75. [Google Scholar] [CrossRef]
- Noguchi, T.; Ebina, K.; Hirao, M.; Morimoto, T.; Koizumi, K.; Kitaguchi, K.; Matsuoka, H.; Iwahashi, T.; Yoshikawa, H. Oxygen ultra-fine bubbles water administration prevents bone loss of glucocorticoid-induced osteoporosis in mice by suppressing osteoclast differentiation. Osteoporos. Int. 2017, 28, 1063–1075. [Google Scholar] [CrossRef]
- Liu, X.X.; Zhu, X.M.; Miao, Q.; Ye, H.Y.; Zhang, Z.Y.; Li, Y.M. Hyperglycemia induced by glucocorticoids in nondiabetic patients: A meta-analysis. Ann. Nutr. Metab. 2014, 65, 324–332. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meszaros, K.; Patocs, A. Glucocorticoids Influencing Wnt/β-Catenin Pathway; Multiple Sites, Heterogeneous Effects. Molecules 2020, 25, 1489. https://doi.org/10.3390/molecules25071489
Meszaros K, Patocs A. Glucocorticoids Influencing Wnt/β-Catenin Pathway; Multiple Sites, Heterogeneous Effects. Molecules. 2020; 25(7):1489. https://doi.org/10.3390/molecules25071489
Chicago/Turabian StyleMeszaros, Katalin, and Attila Patocs. 2020. "Glucocorticoids Influencing Wnt/β-Catenin Pathway; Multiple Sites, Heterogeneous Effects" Molecules 25, no. 7: 1489. https://doi.org/10.3390/molecules25071489
APA StyleMeszaros, K., & Patocs, A. (2020). Glucocorticoids Influencing Wnt/β-Catenin Pathway; Multiple Sites, Heterogeneous Effects. Molecules, 25(7), 1489. https://doi.org/10.3390/molecules25071489