
Aerogels from Cellulose Phosphates of Low Degree of Substitution: A TBAF·H2O/DMSO Based Approach

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
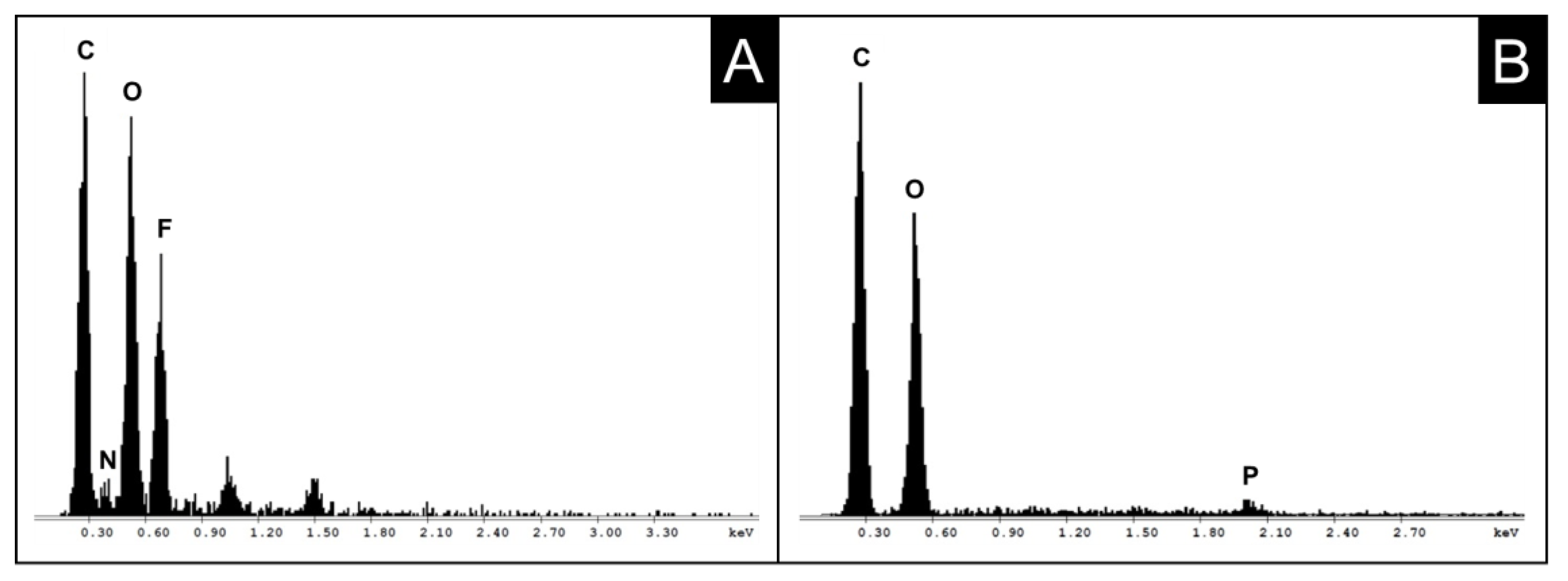
2.1. Phosphorylation
2.2. Chemical Integrity of Cellulose Phosphates during Dissolution
2.3. TBAF Content of Cellulose Phosphate Aerogels
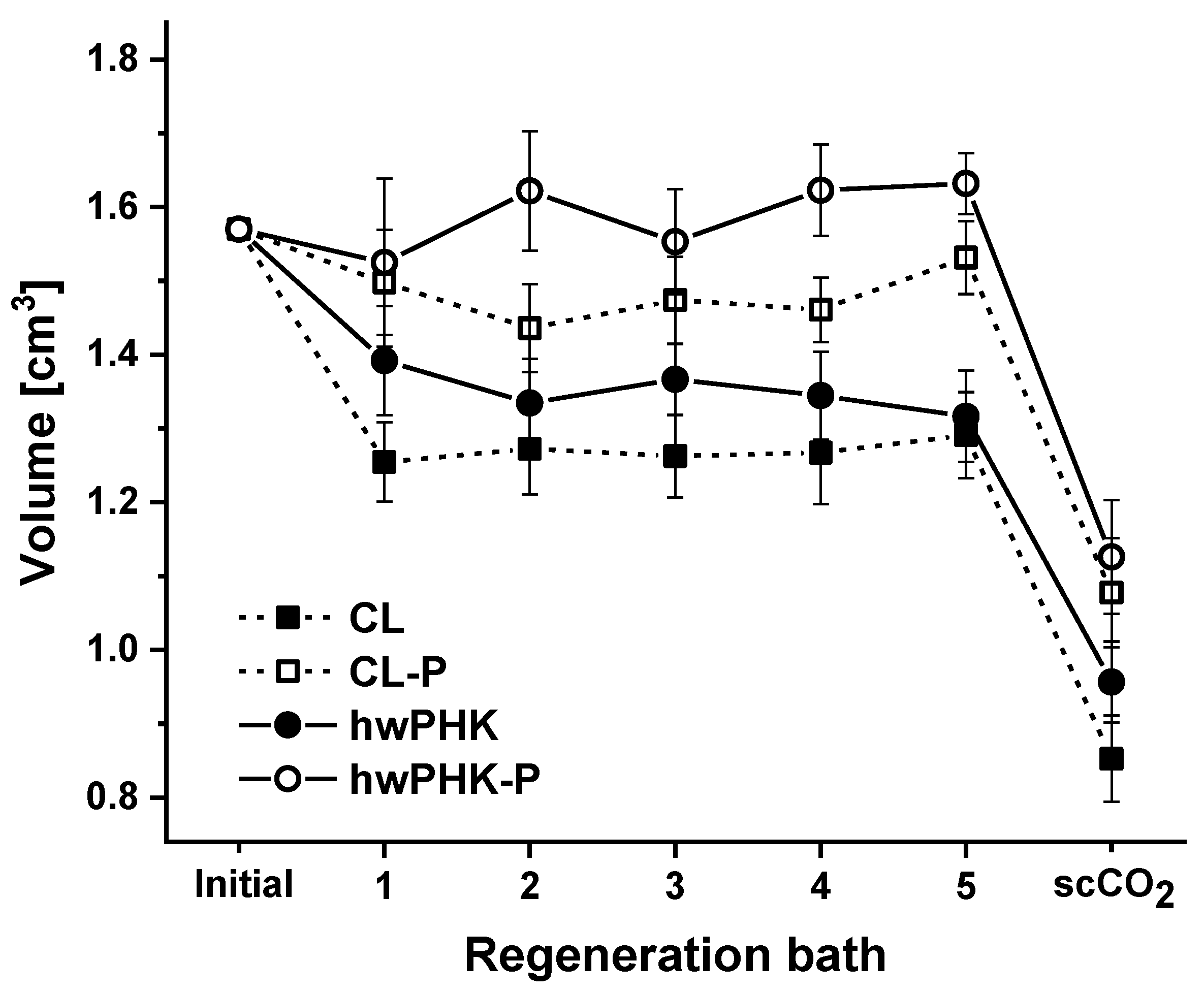
2.4. Aerogel Morphology
2.5. Compression Testing
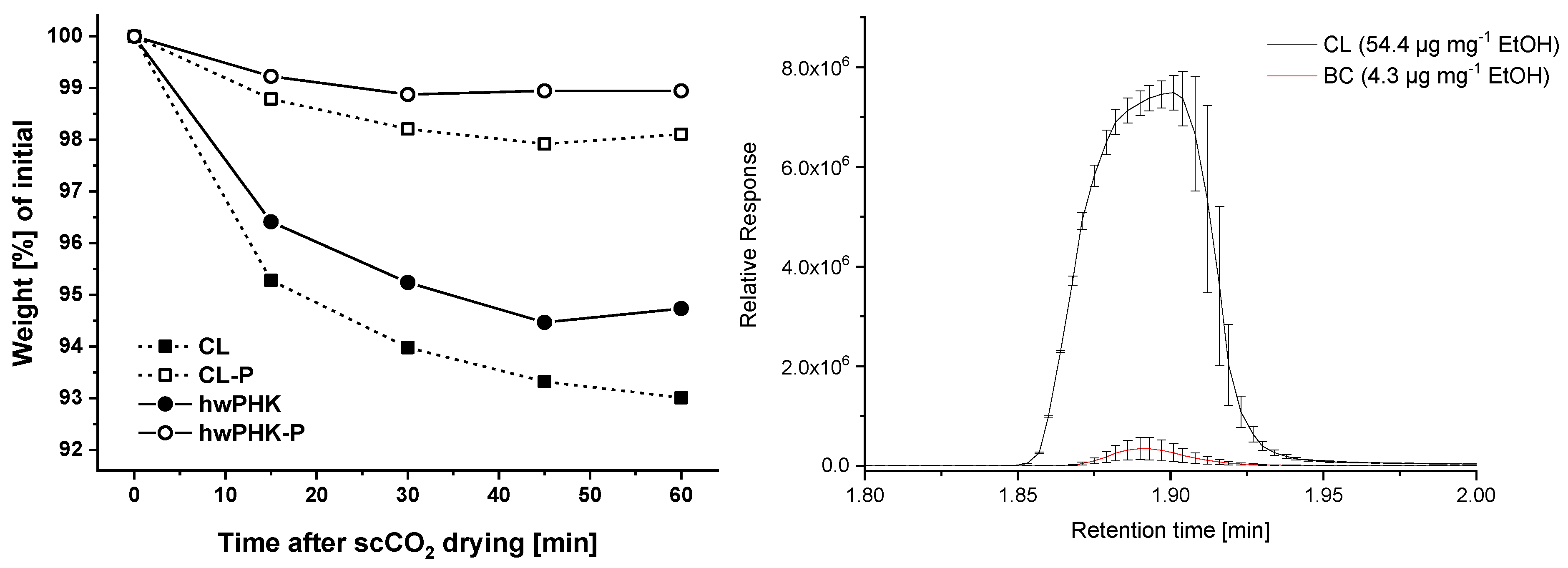
2.6. Aerogel Stability and Moisture Sorption during Storage
2.7. Thermostability
3. Experimental Section
- CL: MW 143.2 kg mol−1, 4.4 mmol g−1 C=O (fluorescence labelling of cellulose using carbazole-9-carbonyl-oxy-amine, CCOA;
- hwPHK: MW 154.8 kg mol−1, 8.1 mmol g−1 C=O;
3.1. Phosphorylation of Cellulose
3.2. Preparation of the Cellulose Solvent System TBAF (16.6 wt.%) / H2O (0.95 wt.%) / DMSO
3.3. Cellulose Dissolution, Casting, Coagulation, Solvent Exchange, and scCO2 Drying
4. Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Amaral, H.R.; Cipriano, D.F.; Santos, M.S.; Schettino, M.A.; Ferreti, J.V.; Meirelles, C.S.; Pereira, V.S.; Cunha, A.G.; Emmerich, F.G.; Freitas, J.C. Production of high-purity cellulose, cellulose acetate and cellulose-silica composite from babassu coconut shells. Carbohydr. Polym. 2019, 210, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hickey, R.J.; Pelling, A.E. Cellulose Biomaterials for Tissue Engineering. Front. Bioeng. Biotechnol. 2019, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Bao, C.; Hausmann, M.; Siqueira, G.; Zimmermann, T.; Kim, W.S. Electrochemical Sensors: 3D Printed Disposable Wireless Ion Sensors with Biocompatible Cellulose Composites. Adv. Electron. Mater. 2019, 5, 1800778. [Google Scholar] [CrossRef]
- Zhao, S.; Malfait, W.J.; Alburquerque, N.G.; Koebel, M.M.; Nyström, G. Biopolymer Aerogels and Foams: Chemistry, Properties, and Applications. Angew. Chem. Int. Ed. 2018, 57, 7580–7608. [Google Scholar] [CrossRef]
- García-González, C.A.; Alnaief, M.; Smirnova, I. Polysaccharide-based aerogels—Promising biodegradable carriers for drug delivery systems. Carbohydr. Polym. 2011, 86, 1425–1438. [Google Scholar] [CrossRef]
- Cardea, S.; Reverchon, E. Supercritical Fluid Processing of Polymers. Polymers 2019, 11, 1551. [Google Scholar] [CrossRef] [Green Version]
- Liebner, F.; Pircher, N.; Schimper, C.; Haimer, E.; Rosenau, T. Cellulose-based aerogels. In Encyclopedia of Biomedical Polymers and Polymeric Biomaterials; CRC Press: New York, NY, USA; Taylor & Francis Group: New York, NY, USA, 2015. [Google Scholar]
- Tabernero, A.; Baldino, L.; Cardea, S.; Del Valle, E.M.M.; Reverchon, E. A Phenomenological Approach to Study Mechanical Properties of Polymeric Porous Structures Processed Using Supercritical CO2. Polymers 2019, 11, 485. [Google Scholar] [CrossRef] [Green Version]
- Plappert, S.; Nedelec, J.-M.; Rennhofer, H.; Lichtenegger, H.; Liebner, F.W. Strain Hardening and Pore Size Harmonization by Uniaxial Densification: A Facile Approach toward Superinsulating Aerogels from Nematic Nanofibrillated 2,3-Dicarboxyl Cellulose. Chem. Mater. 2017, 29, 6630–6641. [Google Scholar] [CrossRef]
- Fauziyah, M.; Widiyastuti, W.; Balgis, R.; Setyawan, H. Production of cellulose aerogels from coir fibers via an alkali–urea method for sorption applications. Cellulose 2019, 26, 9583–9598. [Google Scholar] [CrossRef]
- Kanomata, K.; Tatebayashi, N.; Habaki, X.; Kitaoka, T. Cooperative catalysis of cellulose nanofiber and organocatalyst in direct aldol reactions. Sci. Rep. 2018, 8, 4098. [Google Scholar] [CrossRef] [Green Version]
- Pircher, N.; Veigel, S.; Aigner, N.; Nedelec, J.-M.; Rosenau, T.; Liebner, F.W. Reinforcement of bacterial cellulose aerogels with biocompatible polymers. Carbohydr. Polym. 2014, 111, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, H.; Hu, Y.; Chen, Z.; Zhong, L. Cellulose carbon aerogel/PPy composites for high-performance supercapacitor. Carbohydr. Polym. 2019, 215, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Hakkarainen, T.; Koivuniemi, R.; Kosonen, M.; Escobedo-Lucea, C.; Sanz-García, A.; Vuola, J.; Valtonen, J.; Tammela, P.; Mäkitie, A.A.; Luukko, K.; et al. Nanofibrillar cellulose wound dressing in skin graft donor site treatment. J. Control. Release 2016, 244, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Sulaeva, I.; Henniges, U.; Rosenau, T.; Potthast, A. Bacterial cellulose as a material for wound treatment: Properties and modifications. A review. Biotechnol. Adv. 2015, 33, 1547–1571. [Google Scholar] [CrossRef]
- Sulaeva, I.; Hettegger, H.; Bergen, A.; Rohrer, C.; Kostic, M.; Konnerth, J.; Rosenau, T.; Potthast, A. Fabrication of bacterial cellulose-based wound dressings with improved performance by impregnation with alginate. Mater. Sci. Eng. C 2020, 110, 110619. [Google Scholar] [CrossRef]
- Plappert, S.; Liebner, F.W.; Konnerth, J.; Nedelec, J.-M. Anisotropic nanocellulose gel–membranes for drug delivery: Tailoring structure and interface by sequential periodate–chlorite oxidation. Carbohydr. Polym. 2019, 226, 115306. [Google Scholar] [CrossRef]
- Hujaya, S.D.; Lorite, G.S.; Vainio, S.J.; Liimatainen, H. Polyion complex hydrogels from chemically modified cellulose nanofibrils: Structure-function relationship and potential for controlled and pH-responsive release of doxorubicin. Acta Biomater. 2018, 75, 346–357. [Google Scholar] [CrossRef]
- Zaborowska, M.; Bodin, A.; Bäckdahl, H.; Popp, J.; Goldstein, A.; Gatenholm, P. Microporous bacterial cellulose as a potential scaffold for bone regeneration. Acta Biomater. 2010, 6, 2540–2547. [Google Scholar] [CrossRef]
- Bonilla, M.R.; Lopez-Sanchez, P.; Gidley, M.; Stokes, J.R. Micromechanical model of biphasic biomaterials with internal adhesion: Application to nanocellulose hydrogel composites. Acta Biomater. 2016, 29, 149–160. [Google Scholar] [CrossRef]
- Osorio, D.A.; Lee, B.E.J.; Kwiecien, J.M.; Wang, X.; Shahid, I.; Hurley, A.L.; Cranston, E.D.; Grandfield, K. Cross-linked cellulose nanocrystal aerogels as viable bone tissue scaffolds. Acta Biomater. 2019, 87, 152–165. [Google Scholar] [CrossRef]
- Budtova, T. Bio-based Aerogels: A New Generation of Thermal Superinsulating Materials, In Cellulose Science and Technology: Chemistry, Analysis, and Applications; John Wiley & Sons: Chichester, UK, 2018; pp. 371–392. [Google Scholar]
- Hollister, S. Porous scaffold design for tissue engineering. Nat. Mater. 2005, 4, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, W. Designing porous scaffolds for tissue engineering. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2005, 364, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Pircher, N.; Fischhuber, D.; Carbajal, L.; Strauß, C.; Nedelec, J.-M.; Kasper, C.; Rosenau, T.; Liebner, F.W. Preparation and Reinforcement of Dual-Porous Biocompatible Cellulose Scaffolds for Tissue Engineering. Macromol. Mater. Eng. 2015, 300, 911–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puppi, D.; Chiellini, F.; Piras, A.M.; Chiellini, E. Polymeric materials for bone and cartilage repair. Prog. Polym. Sci. 2010, 35, 403–440. [Google Scholar] [CrossRef]
- Mi, Q.-Y.; Ma, S.-R.; Yu, J.; He, J.-S.; Zhang, J. Flexible and Transparent Cellulose Aerogels with Uniform Nanoporous Structure by a Controlled Regeneration Process. ACS Sustain. Chem. Eng. 2016, 4, 656–660. [Google Scholar] [CrossRef]
- Plappert, S.; Nedelec, J.-M.; Rennhofer, H.; Lichtenegger, H.; Bernstorff, S.; Liebner, F.W. Self-Assembly of Cellulose in Super-Cooled Ionic Liquid under the Impact of Decelerated Antisolvent Infusion: An Approach toward Anisotropic Gels and Aerogels. Biomacromolecules 2018, 19, 4411–4422. [Google Scholar] [CrossRef]
- Ebner, G.; Schiehser, S.; Potthast, A.; Rosenau, T. Side reaction of cellulose with common 1-alkyl-3-methylimidazolium-based ionic liquids. Tetrahedron Lett. 2008, 49, 7322–7324. [Google Scholar] [CrossRef]
- Liebner, F.W.; Haimer, E.; Potthast, A.; Loidl, D.; Tschegg, S.; Neouze, M.-A.; Wendland, M.; Rosenau, T. Cellulosic aerogels as ultra-lightweight materials. Part 2: Synthesis and properties 2nd ICC 2007, Tokyo, Japan, October 25–29, 2007. Holzforschung 2009, 63, 3–11. [Google Scholar] [CrossRef]
- Onwukamike, K.N.; Grelier, S.; Grau, E.; Cramail, H.; Meier, M.A.R. Critical Review on Sustainable Homogeneous Cellulose Modification: Why Renewability Is Not Enough. ACS Sustain. Chem. Eng. 2018, 7, 1826–1840. [Google Scholar] [CrossRef] [Green Version]
- Pircher, N.; Carbajal, L.; Schimper, C.; Bacher, M.; Rennhofer, H.; Nedelec, J.-M.; Lichtenegger, H.; Rosenau, T.; Liebner, F.W. Impact of selected solvent systems on the pore and solid structure of cellulose aerogels. Cellulose 2016, 23, 1949–1966. [Google Scholar] [CrossRef] [Green Version]
- Sala, M.R.; Peng, C.; Skalli, O.; Sabri, F. Tunable neuronal scaffold biomaterials through plasmonic photo-patterning of aerogels. MRS Commun. 2019, 9, 1249–1255. [Google Scholar] [CrossRef]
- Lynch, K.; Skalli, O.; Sabri, F. Growing Neural PC-12 Cell on Crosslinked Silica Aerogels Increases Neurite Extension in the Presence of an Electric Field. J. Funct. Biomater. 2018, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sala, M.R.; Lynch, K.; Chandrasekaran, S.; Skalli, O.; Worsley, M.; Sabri, F. PC-12 cells adhesion and differentiation on carbon aerogel scaffolds. MRS Commun. 2018, 8, 1426–1432. [Google Scholar] [CrossRef]
- Schimper, C.B.; Pachschwoell, P.; Wendland, M.; Smid, E.; Neouze, M.-A.; Nedelec, J.-M.; Henniges, U.; Rosenau, T.; Liebner, F.W. Fine-fibrous cellulose II aerogels of high specific surface from pulp solutions in TBAF·H2O/DMSO. Holzforschung 2018, 73, 65–81. [Google Scholar] [CrossRef]
- Sharma, R.; Fry, J.L. Instability of anhydrous tetra-n-alkylammonium fluorides. J. Org. Chem. 1983, 48, 2112–2114. [Google Scholar] [CrossRef]
- Heinze, T.; Köhler, S. Dimethyl Sulfoxide and Ammonium Fluorides—Novel Cellulose Solvents. In Chemistry Student Success: A Field-Tested, Evidence-Based Guide; American Chemical Society: Washington, DC, USA, 2010; Volume 1033, pp. 103–118. [Google Scholar]
- Liebner, F.W.; Dunareanu, R.; Opietnik, M.; Haimer, E.; Wendland, M.; Werner, C.; Maitz, M.; Seib, P.; Neouze, M.-A.; Potthast, A.; et al. Shaped hemocompatible aerogels from cellulose phosphates: Preparation and properties. Holzforschung 2012, 66, 317–321. [Google Scholar] [CrossRef]
- Schimper, C.; Haimer, E.; Wendland, M.; Potthast, A.; Rosenau, T.; Liebner, F. The effects of different process parameters on the properties of cellulose aerogels obtained via the Lyocell route. Lenzing. Ber. 2011, 89, 109–117. [Google Scholar]
- Zhang, M.; Dou, M.; Wang, M.; Yu, Y. Study on the solubility parameter of supercritical carbon dioxide system by molecular dynamics simulation. J. Mol. Liq. 2017, 248, 322–329. [Google Scholar] [CrossRef]
- Granja, P.L.; Pouysegu, L.; Petraud, M.; De Jeso, B.; Baquey, C.; Barbosa, M.A. Cellulose phosphates as biomaterials. I. Synthesis and characterisation of highly phosphory-lated cellulose gels. J. Appl. Polym. Sci. 2001, 82, 3341–3353. [Google Scholar] [CrossRef]
- Gavillon, R.; Budtova, T. Aerocellulose: New Highly Porous Cellulose Prepared from Cellulose—NaOH Aqueous Solutions. Biomacromolecules 2008, 9, 269–277. [Google Scholar] [CrossRef]
- Sescousse, R.; Gavillon, R.; Budtova, T. Aerocellulose from cellulose–ionic liquid solutions: Preparation, properties and comparison with cellulose–NaOH and cellulose–NMMO routes. Carbohydr. Polym. 2011, 83, 1766–1774. [Google Scholar] [CrossRef]
- Abu-Rous, M.; Varga, K.; Bechtold, T.; Schuster, K.C. A new method to visualize and characterize the pore structure of TENCEL® (Lyocell) and other man-made cellulosic fibres using a fluorescent dye molecular probe. J. Appl. Polym. Sci. 2007, 106, 2083–2091. [Google Scholar] [CrossRef]
- Mao, Y.; Zhou, J.; Cai, J.; Zhang, L. Effects of coagulants on porous structure of membranes prepared from cellulose in NaOH/urea aqueous solution. J. Membr. Sci. 2006, 279, 246–255. [Google Scholar] [CrossRef]
- Fink, H.-P.; Weigel, P.; Purz, H.; Ganster, J. Structure formation of regenerated cellulose materials from NMMO-solutions. Prog. Polym. Sci. 2001, 26, 1473–1524. [Google Scholar] [CrossRef]
- Ioelovich, M.; Leykin, A. Vapor sorption by cellulose. BioResources 2011, 6, 178–195. [Google Scholar]
- Rouquerol, F.; Rouquerol, J.; King, S. Adsorption by Powders and Porous Solids: Principles. In Methodology and Applications, 1st ed.; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Lampke, T. Beitrag zur Charakterisierung naturfaserverstärkter Verbundwerkstoffe mit hochpolymerer Matrix, in Fakultät für Maschinenbau und Verfahrenstechnik. Available online: https://monarch.qucosa.de/api/qucosa%3A17757/attachment/ATT-0/ (accessed on 1 March 2020).
- Tang, M.; Bacon, R. 125. Carbonization of cellulose fibers. I. Low temperature pyrolysis. Carbon 1964, 1, 390. [Google Scholar] [CrossRef]
- Bradbury, A.G.W.; Sakai, Y.; Shafizadeh, F. A kinetic model for pyrolysis of cellulose. J. Appl. Polym. Sci. 1979, 23, 3271–3280. [Google Scholar] [CrossRef]
- Hosoya, T.; Kawamoto, H.; Saka, S. Pyrolysis behaviors of wood and its constituent polymers at gasification temperature. J. Anal. Appl. Pyrolysis 2007, 78, 328–336. [Google Scholar] [CrossRef] [Green Version]
- Röhrling, J.; Potthast, A.; Rosenau, T.; Lange, T.; Ebner, G.; Sixta, H.; Kosma, P. A Novel Method for the Determination of Carbonyl Groups in Cellulosics by Fluorescence Labeling. 1. Method Development. Biomacromolecules 2002, 3, 959–968. [Google Scholar] [CrossRef]
- Patton, H.W.; Touey, G.P. Gas Chromatographic Determination of Some Hydrocarbons in Cigarette Smoke. Anal. Chem. 1956, 28, 1685–1688. [Google Scholar] [CrossRef]
- Suflet, D.M.; Chitanu, G.C.; Popa, V.I. Phosphorylation of polysaccharides: New results on synthesis and characterisation of phosphorylated cellulose. React. Funct. Polym. 2006, 66, 1240–1249. [Google Scholar] [CrossRef]
- Liebner, F.W.; Haimer, E.; Wendland, M.; Neouze, M.-A.; Schlufter, K.; Miethe, P.; Heinze, T.; Potthast, A.; Rosenau, T. Aerogels from Unaltered Bacterial Cellulose: Application of scCO2Drying for the Preparation of Shaped, Ultra-Lightweight Cellulosic Aerogels. Macromol. Biosci. 2010, 10, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, M.; Rao, B.S. Modeling of supercritical drying of ethanol-soaked silica aerogels with carbon dioxide. J. Chem. Technol. Biotechnol. 2008, 83, 1101–1109. [Google Scholar] [CrossRef]
- Fischer, F.; Rigacci, A.; Pirard, R.; Berthon-Fabry, S.; Achard, P. Cellulose-based aerogels. Polymer 2006, 47, 7636–7645. [Google Scholar] [CrossRef]
- International Organization for Standardization. Determination of the Specific Surface Area of Solids by Gas Adsorption Using the BET Method (ISO Standard No. 9277); International Organization for Standardization: Geneva, Switzerland, 2010. [Google Scholar]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The determination of pore volume and area distributions in porous substances. I. Computations from nitrogen isotherms. J. Am. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | P Content [wt.%] | DSP | Shrinking (−) / Swelling (+) | ||
|---|---|---|---|---|---|
| Regeneration (EtOH, before Drying) [%] | After scCO2 Drying [%] | Apparent Density [mg cm−3] | |||
| hwPHK | - | - | −16.2 ± 4.9 | −39.1 ± 5.5 | 58.1 ± 3.5 |
| hwPHK-P | 4.91 | 0.24 | +4.0 ± 2.5 | −28.3 ± 6.8 | 47.0 ± 5.8 |
| CL | - | - | −17.8 ± 4.5 | −45.7 ± 6.8 | 71.2 ± 5.0 |
| CL-P | 3.34 | 0.18 | −2.4 ± 2.8 | −31.4 ± 6.7 | 50.0 ± 4.0 |
| Extraction Medium | Extraction Time [h] | Final DSP | [%] of Initial DSP | |
|---|---|---|---|---|
| DMSO (control) | 4 | 0.149 | 71 | ±0.0 |
| TBAF/DMSO | 2 | 0.094 | 45 | ±1.9 |
| 4 | 0.093 | 44 | ±1.2 | |
| 8 | 0.096 | 46 | ±0.0 | |
| 8 (60 °C) | 0.096 | 46 | ±0.0 | |
| 16 | 0.093 | 44 | ±0.5 | |
| Sample | E [MPa] | Eρ [MPa cm3 g−1] | σy [MPa] | εy [%] |
|---|---|---|---|---|
| CL (TBAF/DMSO) | 11.9 | 167 | 5.3 | 3.4 |
| ([EMIM][OAc]/DMSO) a | 1.121 a | 20 a | 2.1 a | n.d. b |
| NMMO⋅H2O a | 4.26 a | 68 a | 2.9 a | n.d. b |
| CL-P | 10.8 | 216 | 3.6 | 5.1 |
| hwPHK | 5.9 | 102 | 5.7 | 7.6 |
| hwPHK-P | 3.2 | 68 | 1.9 | 3.4 |
| Nitrogen Sorption Experiments | Thermoporosimetry | |||||
|---|---|---|---|---|---|---|
| Sample | Calculated Porosity [%] | Specific Surface [m2 g−1] | Sorbed Volume [cm3 g−1] | C Constant | Vp PSD [cm3 g−1] | Rp [nm] max PSD |
| CL | 96.28 | 355 (43) | 0.80 (0.20) | 101 (1.0) | 6.06 | 19.19 |
| CL-P | 96.99 | 311 (6.9) | 0.55 (0.10) | 48 (2.0) | 9.27 | 21.23 |
| hwPHK | 95.46 | 366 (2.0) | 0.7 (0) | 104 (2.0) | 7.32 | 19.74 |
| hwPHK-P | 96.80 | 270 (20) | 0.5 (0) | 45 (5.9) | 10.60 | 24.49 |
| Sample | Conditioning [% RH] | 2 d | 84 d | ||
|---|---|---|---|---|---|
| Volume [%] | Weight [%] | Volume [%] | Weight [%] | ||
| CL | 0 | 78.7 (1.3) | −4.2 (0.48) | 68.0 (1.6) | −6.7 (0.25) |
| 30 | 72.0 (4.8) | −1.4 (0.06) | 62.6 (5.2) | −1.5 (0.29) | |
| 65 | 30.5 (3.0) | 5.0 (1.0) | 24.7 (2.9) | 5.2 (0.63) | |
| 98 | 10.0 (3.0) | 23.6 (0.53) | 10.4 (5.5) | 25.8 (0.05) | |
| CL-P | 0 | 88.6 (1,4) | −1.7 (0.04) | 78.9 (5.1) | −3.1 (0.13) |
| 65 | 27.4 (1.1) | 6.6 (0.39) | 19.7 (1.7) | 5.8 (0.21) | |
| hwPHK | 0 | 85.6 (11) | −4.8 (0.37) | 76.9 (8.4) | −4.9 (0.36) |
| 30 | 76.0 (6.5) | −0.3 (0.02) | 68.7 (6.2) | −0.7 (0.13) | |
| 65 | 36.3 (0.48) | 5.5 (0.54) | 27.2 (0.13) | 5.4 (0.81) | |
| 98 | 7.1 (0.72) | 24.2 (0.01) | 6.5 (0.08) | 25.0 (0.07) | |
| hwPHK-P | 0 | 95.6 (5.2) | −0.6 (0.01) | 87.6 (4.9) | −1.8 (0.01) |
| 65 | 30.9 (3.3) | 6.1 (0.24) | 21.4 (3.4) | 4.8 (0.17) | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schimper, C.B.; Pachschwoell, P.S.; Hettegger, H.; Neouze, M.-A.; Nedelec, J.-M.; Wendland, M.; Rosenau, T.; Liebner, F. Aerogels from Cellulose Phosphates of Low Degree of Substitution: A TBAF·H2O/DMSO Based Approach. Molecules 2020, 25, 1695. https://doi.org/10.3390/molecules25071695
Schimper CB, Pachschwoell PS, Hettegger H, Neouze M-A, Nedelec J-M, Wendland M, Rosenau T, Liebner F. Aerogels from Cellulose Phosphates of Low Degree of Substitution: A TBAF·H2O/DMSO Based Approach. Molecules. 2020; 25(7):1695. https://doi.org/10.3390/molecules25071695
Chicago/Turabian StyleSchimper, Christian B., Paul S. Pachschwoell, Hubert Hettegger, Marie-Alexandra Neouze, Jean-Marie Nedelec, Martin Wendland, Thomas Rosenau, and Falk Liebner. 2020. "Aerogels from Cellulose Phosphates of Low Degree of Substitution: A TBAF·H2O/DMSO Based Approach" Molecules 25, no. 7: 1695. https://doi.org/10.3390/molecules25071695
APA StyleSchimper, C. B., Pachschwoell, P. S., Hettegger, H., Neouze, M.-A., Nedelec, J.-M., Wendland, M., Rosenau, T., & Liebner, F. (2020). Aerogels from Cellulose Phosphates of Low Degree of Substitution: A TBAF·H2O/DMSO Based Approach. Molecules, 25(7), 1695. https://doi.org/10.3390/molecules25071695