
Multivalent Ions as Reactive Crosslinkers for Biopolymers—A Review


Abstract
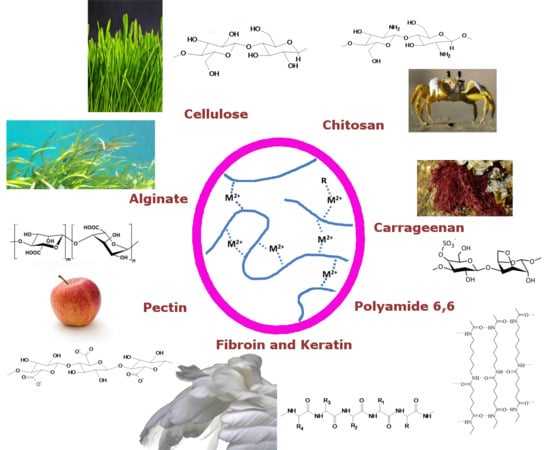
1. Introduction
2. Ion Hydration and Monosaccharide Complexation
2.1. The Hydrate Shell of Ions in Water
2.2. Complex Stability and Formation of Constants
- In analogy to polyhydroxycarboxylic acids also polysaccharides will be able to form a high number of complexes with multivalent ions, e.g., calcium and iron.
- The formation of complex species with 1:1:1 stoichiometry (e.g., [CaFeIIIH-3DGL]+ at pH 7) indicates that the presence of a carboxylic group may support formation of stable complexes at low pH, however participation of hydroxyl groups will also be contributing to complex formation. As an example, in highly alkaline aqueous solution the formation of iron-complexes with sorbitol complexes can be observed.
- Adsorption/complexation of metal ions e.g., Ca2+ and Fe3+ from aqueous solution into a solid polysaccharide matrix occurs,
- Dissolution of polysaccharides into the concentrated metal complex solutions can be achieved, e.g., dissolution of cellulose into alkaline Fe3+ tartaric complexes, and
- Gel-formation and precipitation of dissolved polysaccharides, e.g., alginates in presence of multi-valent ions (Ca2+), occurs due to formation of metal complexes with reduce solubility. As a result a polymer network is formed.
| Stoichiometry Ca2+:Fe3+:DGL 1:1:1 increasing pH | ||
| Ca2+ + Fe3+ + DGL↔[CaFeIIIDGL]4+ |  | (1) |
| Ca2+ + Fe3+ + DGL↔[CaFeIIIH-1DGL]3+ + H+ | (2) | |
| Ca2+ + Fe3+ + DGL↔[CaFeIIIH-2DGL]2+ + 2H+ | (3) | |
| Ca2+ + Fe3+ + DGL↔[CaFeIIIH-3DGL]+ + 3H+ | (4) | |
| Ca2+ + Fe3+ + DGL↔[CaFeIIIH-4DGL] + 4H+ | (5) | |
| Ca2+ + Fe3+ + DGL↔[CaFeIIIH-5DGL]− + 5H+ | (6) | |
3. Metal Ion Interactions with Polysaccharide Structures
3.1. Metal Complex as Structure Model
3.2. Ion-Uptake in Cellulose
3.3. Cellulose Solvents
- by derivatisation of the functional groups (hydroxyl groups C-2, C-3, and C-6) e.g., through xanthogenation, acetylation, or alkylation,
- formation of metal complexes, e.g., with iron-tartaric acid, copper-amino complexes, or the
- use of cellulose solvents, e.g., NMMO (N-methyl-morpholine-N-oxide) and ionic liquids.
4. Metal Ion Based Cross-Linking of Polysaccharides
- Formation of complexes at higher alkaline conditions with involvement of hydroxyl groups, e.g., the C-2 and C-3 groups [35].
4.1. Alginate
4.2. Carrageenans
4.2.1. κ -Carrageenan and Furcellaran
4.2.2. ι-Carrageenan
4.2.3. Aggregation and Gelation
4.3. Pectin
4.4. Xanthan and Other Polysaccharides
5. Metal-Ion Based Crosslinking on Fiber Surfaces and Interfaces
6. Ionic Interaction in Non- and Mixed-Polysaccharide Polymers
- expand multivalent ion complexing and interaction models
- refer to multivalent ion protein interaction and glycoproteins
6.1. Polyamide in CaCl2/Ethanol/Water Systems as the Model Compound for Polyamino Acid Structures
6.2. Ion-Rich Hydrate Shells in Protein Fiber Dissolution
6.3. Metal Complexes in Protein Fibers Forming Ionic
7. Synopsis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jarvis, M.C. Structure and properties of pectin gels in plant cell walls. Plant Cell Environ. 1984, 7, 153–164. [Google Scholar]
- Pilgrim, G.W.; Walter, R.H.; Oakenfull, D.G. CHAPTER 2 - Jams, Jellies, and Preserves. In The Chemistry and Technology of Pectin; Food Science and Technology; Walter, R.H., Ed.; Academic Press: San Diego, CA, USA, 1991; pp. 23–50. ISBN 978-0-08-092644-5. [Google Scholar]
- The Pectic Substances of Plants. Nature 1929, 124, 709. [CrossRef]
- Deniaud-Bouët, E.; Kervarec, N.; Michel, G.; Tonon, T.; Kloareg, B.; Herve, C. Chemical and enzymatic fractionation of cell walls from Fucales: Insights into the structure of the extracellular matrix of brown algae. Ann. Bot. 2014, 114, 1203–1216. [Google Scholar] [CrossRef] [PubMed]
- Rompp, W.; Axon, G.L.; Thompson, T. Sodium alginate: A textile printing thickener. Am. Dyest. Rep. 1983, 72, 31–32. [Google Scholar]
- Heinze, T.; Koschella, A. Solvents applied in the field of cellulose chemistry: A mini review. Polímeros Sci. Technol. 2005, 15, 84–90. [Google Scholar] [CrossRef]
- Henriksson, Å.; Gatenholm, P. Controlled Assembly of Glucuronoxylans onto Cellulose Fibres. Holzforschung 2001, 55, 494–502. [Google Scholar] [CrossRef]
- Annadurai, G.; Juang, R.-S.; Lee, D. Adsorption of heavy metals from water using banana and orange peels. Water Sci. Technol. 2003, 47, 185–190. [Google Scholar] [CrossRef]
- Diamond, C.; Wormmell, R.L. The Manufacture and Properties of “Casein Fibre”. J. Text. Inst. Proc. 1939, 30, P224–P228. [Google Scholar] [CrossRef]
- Miwa, Y.; Kurachi, J.; Kohbara, Y.; Kutsumizu, S. Dynamic ionic crosslinks enable high strength and ultrastretchability in a single elastomer. Commun. Chem. 2018, 1, 1–8. [Google Scholar] [CrossRef]
- Hossain, K.S.; Ochi, A.; Ooyama, E.; Magoshi, J.; Nemoto, N. Dynamic Light Scattering of Native Silk Fibroin Solution Extracted from Different Parts of the Middle Division of the Silk Gland of theBombyx moriSilkworm. Biomacromolecules 2003, 4, 350–359. [Google Scholar] [CrossRef]
- Ajisawa, A. Dissolution of silk fibroin with calciumchloride/ethanol aqueous solution. J. Seric. Sci. Jpn. 1998, 67, 91–94. [Google Scholar]
- The Chemistry of Hemoglobin and Myoglobin. Available online: https://chemed.chem.purdue.edu/genchem/topicreview/bp/1biochem/blood3.html (accessed on 7 April 2020).
- Chizhik, V.; Egorov, A.; Pavlova, M.S.; Eгopoвa, M.; Donets, A. Structure of hydration shell of calcium cation by NMR relaxation, Car-Parrinello molecular dynamics and quantum-chemical calculations. J. Mol. Liq. 2016, 224, 730–736. [Google Scholar] [CrossRef]
- Singh, R.P.; Yeboah, Y.D.; Pambid, E.R.; Debayle, P. Stability constant of the calcium-citrate(3-) ion pair complex. J. Chem. Eng. Data 1991, 36, 52–54. [Google Scholar] [CrossRef]
- Gácsi, A.; Kutus, B.; Csendes, Z.; Faragó, T.; Pálinkó, I.; Sipos, P.; Peintler, G. Calcium l -tartrate complex formation in neutral and in hyperalkaline aqueous solutions. Dalton Trans. 2016, 45, 17296–17303. [Google Scholar] [CrossRef] [PubMed]
- Swift, T.J.; Lo, H.H. The primary solvation number of magnesium(II) in liquid ammonia. J. Am. Chem. Soc. 1967, 89, 3988–3990. [Google Scholar] [CrossRef]
- Klockow, D. Hexacyanoferrat (III). Anal. Bioanal. Chem. 1965, 208, 135. [Google Scholar] [CrossRef]
- Martell, A.E.; Motekaitas, R.J. Determination and Use of Stability Constants; Inorganic Chemistry, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1992; ISBN 978-0-471-18817-9. [Google Scholar]
- Bechtold, T.; Burtscher, E.; Turcanu, A. Ca2+–Fe3+–d-gluconate-complexes in alkaline solution. Complex stabilities and electrochemical properties. J. Chem. Soc. Dalton Trans. 2002, 2683–2688. [Google Scholar] [CrossRef]
- Wyrzykowski, D.; Czupryniak, J.; Ossowski, T.; Chmurzyński, L. Thermodynamic interactions of the alkaline earth metal ions with citric acid. J. Therm. Anal. Calorim. 2010, 102, 149–154. [Google Scholar] [CrossRef]
- Meyer, J.L. Formation constants for interaction of citrate with calcium and magnesium lons. Anal. Biochem. 1974, 62, 295–300. [Google Scholar] [CrossRef]
- Gácsi, A.; Kutus, B.; Buckó, Á.; Csendes, Z.; Peintler, G.; Pálinkó, I.; Sipos, P. Some aspects of the aqueous solution chemistry of the Na+/Ca2+/OH−/Cit3− system: The structure of a new calcium citrate complex forming under hyperalkaline conditions. J. Mol. Struct. 2016, 1118, 110–116. [Google Scholar] [CrossRef]
- Mekmene, O.; Gaucheron, F. Determination of calcium-binding constants of caseins, phosphoserine, citrate and pyrophosphate: A modelling approach using free calcium measurement. Food Chem. 2011, 127, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Danilov, S.N.; Samsonova, T.I.; Bolotnikova, L.S. Investigation of Solutions of Cellulose. Russ. Chem. Rev. 1970, 39, 156–168. [Google Scholar] [CrossRef]
- Franke, W. Iron complexes of tartaric acid. Justus Liebigs Annalen der Chemie 1931, 242–284. [Google Scholar] [CrossRef]
- Ferrari, E.; Saladini, M. Iron(III) complexing ability of carbohydrate derivatives. J. Inorg. Biochem. 2004, 98, 1002–1008. [Google Scholar] [CrossRef] [PubMed]
- Jayme, G.; Bergmann, W. Simplified preparation of a cellulose solvent based on an iron-tartaric-acid-sodium complex. Reyon Zellwolle und Chemiefasern 1956, 34, 27–29. [Google Scholar]
- Fitz-Binder, C.; Bechtold, T. Ca2+ sorption on regenerated cellulose fibres. Carbohydr. Polym. 2012, 90, 937–942. [Google Scholar] [CrossRef]
- Fitz-Binder, C.; Bechtold, T. One-sided surface modification of cellulose fabric by printing a modified TEMPO-mediated oxidant. Carbohydr. Polym. 2014, 106, 142–147. [Google Scholar] [CrossRef]
- Fitz-Binder, C.; Bechtold, T. Sorption of alkaline earth metal ions Ca2+ and Mg2+ on lyocell fibres. Carbohydr. Polym. 2009, 76, 123–128. [Google Scholar]
- Kongdee, A.; Bechtold, T. The complexation of Fe(III)-ions in cellulose fibres: A fundamental property. Carbohydr. Polym. 2004, 56, 47–53. [Google Scholar] [CrossRef]
- Imamura, T.; Hatanaka, C.; Doudou, T. Formation of a Copper-Sorbitol Complex in Copper Sulfate-Sorbitol Systems. J. Fac. Fish. Anim. Husb. Hiroshima Univ. 1978, 17, 107–116. [Google Scholar]
- Dale, B.E.; Tsao, G.T. A microcalorimetric study of complex formation between alkaline sodium tartrate and iron(III). J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 3163–3175. [Google Scholar] [CrossRef]
- Vu-Manh, H.; Öztürk, H.B.; Bechtold, T. Swelling and dissolution mechanism of lyocell fiber in aqueous alkaline solution containing ferric tartaric acid complex. Cellulose 2010, 17, 521–532. [Google Scholar] [CrossRef]
- Miyamoto, I.; Matsuoka, Y.; Matsui, T.; Okajima, K. Studies on Structure of Cuprammonium Cellulose II. Structural Change of Cellulose–Cuprammonium Complex as a Function of Hydroxyl Ion Concentration. Polym. J. 1995, 27, 1123–1131. [Google Scholar] [CrossRef][Green Version]
- Dong, M.; Xue, Z.; Wang, L.; Xia, Y. NaOH induced the complete dissolution of ι-carrageenan and the corresponding mechanism. Polymer 2018, 151, 334–339. [Google Scholar] [CrossRef]
- Wang, X.; Du, Y.; Fan, L.; Liu, H.; Hu, Y. Chitosan- metal complexes as antimicrobial agent: Synthesis, characterization and Structure-activity study. Polym. Bull. 2005, 55, 105–113. [Google Scholar] [CrossRef]
- Imeson, A. Food Stabilisers, Thickeners and Gelling Agents; Imeson, A., Ed.; Wiley-Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Smidsrød, O. Solution properties of alginate. Carbohydr. Res. 1970, 13, 359–372. [Google Scholar] [CrossRef]
- Smidsrød, O. Molecular basis for some physical properties of alginates in the gel state. Faraday Discuss. Chem. Soc. 1974, 57, 263–274. [Google Scholar] [CrossRef]
- McKee, J.W.A.; Kavalieris, L.; Brasch, D.J.; Brown, M.T.; Melton, L.D. Alginate content and composition ofMacrocystis pyrifera from New Zealand. Environ. Boil. Fishes 1992, 4, 357–369. [Google Scholar] [CrossRef]
- Grant, G.T.; Morris, E.R.; Rees, D.A.; Smith, P.J.; Thom, D. Biological interactions between polysaccharides and divalent cations: The egg-box model. FEBS Lett. 1973, 32, 195–198. [Google Scholar] [CrossRef]
- Morris, E.R.; Rees, D.A.; Thom, D.; Boyd, J. Chiroptical and stoichiometric evidence of a specific, primary dimerisation process in alginate gelation. Carbohydr. Res. 1978, 66, 145–154. [Google Scholar] [CrossRef]
- Braccini, I.; Pérez, S. Molecular Basis of Ca2+-Induced Gelation in Alginates and Pectins: The Egg-Box Model Revisited. Biomacromolecules 2001, 2, 1089–1096. [Google Scholar] [CrossRef]
- Stokke, B.T.; Draget, K.I.; Smidsrød, O.; Yuguchi, Y.; Urakawa, H.; Kajiwara, K. Small-Angle X-ray Scattering and Rheological Characterization of Alginate Gels. 1. Ca−Alginate Gels. Macromolecules 2000, 33, 1853–1863. [Google Scholar] [CrossRef]
- Kohn, R. Ion binding on polyuronates - alginate and pectin. Pure Appl. Chem. 1975, 42, 371–397. [Google Scholar] [CrossRef]
- Mørch, Y.; Donati, I.; Strand, B.L.; Skjåk-Bræk, G. Molecular Engineering as an Approach to Design New Functional Properties of Alginate. Biomacromolecules 2007, 8, 2809–2814. [Google Scholar] [CrossRef] [PubMed]
- Atkins, E.D.T.; Nieduszynski, I.A.; Mackie, W.; Parker, K.D.; Smolko, E.E. Structural components of alginic acid. II. The crystalline structure of poly-?-l-guluronic acid. Results of X-ray diffraction and polarized infrared studies. Biopolymers 1973, 12, 1879–1887. [Google Scholar] [CrossRef]
- Sikorski, P.; Mo, F.; Skjåk-Bræk, G.; Stokke, B.T. Evidence for Egg-Box-Compatible Interactions in Calcium−Alginate Gels from Fiber X-ray Diffraction. Biomacromolecules 2007, 8, 2098–2103. [Google Scholar] [CrossRef]
- Braccini, I.; Grasso, R.P.; Pérez, S. Conformational and configurational features of acidic polysaccharides and their interactions with calcium ions: A molecular modeling investigation. Carbohydr. Res. 1999, 317, 119–130. [Google Scholar] [CrossRef]
- Angyal, S.J. Complex fomation between sugars and metal ions. Pure Appl. Chem. 1973, 35, 131–146. [Google Scholar] [CrossRef]
- Donati, I.; Holtan, S.; Mørch, Y.; Borgogna, M.; Dentini, M. New Hypothesis on the Role of Alternating Sequences in Calcium−Alginate Gels. Biomacromolecules 2005, 6, 1031–1040. [Google Scholar] [CrossRef]
- Higgs, P.G.; Ball, R.C. Some ideas concerning the elasticity of biopolymer networks. Macromolecules 1989, 22, 2432–2437. [Google Scholar] [CrossRef]
- Fang, Y.; Al-Assaf, S.; Phillips, G.O.; Nishinari, K.; Funami, T.; Williams, P.A.; Li, L. Multiple Steps and Critical Behaviors of the Binding of Calcium to Alginate. J. Phys. Chem. B 2007, 111, 2456–2462. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.; Formo, K.; Strand, B.L.; Alsberg, E.; Christensen, B.E. Chapter 9. Alginates as biomaterials in tissue engineering. Carbohydr. Chem. 2011, 37, 227–258. [Google Scholar] [CrossRef]
- Sun, J.; Tan, H. Alginate-Based Biomaterials for Regenerative Medicine Applications. Matererials 2013, 6, 1285–1309. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xie, Y.-J.; He, W. Research progress on chemical modification of alginate: A review. Carbohydr. Polym. 2011, 84, 33–39. [Google Scholar] [CrossRef]
- Qin, Y.; Jiang, J.; Zhao, L.; Zhang, J.; Wang, F. Chapter 13—Applications of Alginate as a Functional Food Ingredient. In Biopolymers for Food Design; Handbook of Food Bioengineering; Grumezescu, A.M., Holban, A.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 409–429. ISBN 978-0-12-811449-0. [Google Scholar]
- Klein, J.; Stock, J.; Vorlop, K.-D. Pore size and properties of spherical Ca-alginate biocatalysts. Appl. Microbiol. Biotechnol. 1983, 18, 86–91. [Google Scholar] [CrossRef]
- Bidarra, S.; Barrias, C.C.; Granja, P.L. Injectable alginate hydrogels for cell delivery in tissue engineering. Acta Biomater. 2014, 10, 1646–1662. [Google Scholar] [CrossRef]
- Draget, K.I.; Skjåk-Bræk, G.; Smidsrød, O. Alginate based new materials. Int. J. Boil. Macromol. 1997, 21, 47–55. [Google Scholar] [CrossRef]
- Lee, K.Y.; Mooney, D. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef]
- Larsen, B.E.; Bjørnstad, J.; Pettersen, E.O.; Tønnesen, H.H.; Melvik, J.E. Rheological characterization of an injectable alginate gel system. BMC Biotechnol. 2015, 15, 29. [Google Scholar] [CrossRef]
- FAO Alginate. Available online: http://www.fao.org/docrep/006/y4765e/y4765e08.htm (accessed on 6 February 2019).
- Orive, G.; Ponce, S.; Bayat, A.; Gascón, A.R.; Igartua, M.; Pedraz, J.L. Biocompatibility of microcapsules for cell immobilization elaborated with different type of alginates. Biomaterials 2002, 23, 3825–3831. [Google Scholar] [CrossRef]
- Paredes-Juarez, G.A.; Spasojevic, M.; Faas, M.M.; De Vos, P. Immunological and Technical Considerations in Application of Alginate-Based Microencapsulation Systems. Front. Bioeng. Biotechnol. 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Piculell, L.; Nilsson, S.; Muhrbeck, P. Effects of small amounts of kappa-carrageenan on the rheology of aqueous iota-carrageenan. Carbohydr. Polym. 1992, 18, 199–208. [Google Scholar] [CrossRef]
- Piculell, L. Gelling Carrageenans. In Food Polysaccharides and Their Applications; Stephen, A.M., Phillips, G.O., Williams, P.A., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 239–288. ISBN 978-0-8247-5922-3. [Google Scholar]
- Knutsen, S.H.; Grasdalen, H. Characterization of Water-extractable Polysaccharides from Norwegian Furcellaria lumbricalis (Huds.) Lamour. (Gigartinales, Rhodophyceae) by IR and NMR Spectroscopy. Bot. Mar. 1987, 30, 497–506. [Google Scholar] [CrossRef]
- Tuvikene, R.; Truus, K.; Robal, M.; Volobujeva, O.; Mellikov, E.; Pehk, T.; Kollist, A.; Kailas, T.; Vaher, M. The extraction, structure, and gelling properties of hybrid galactan from the red alga Furcellaria lumbricalis (Baltic Sea, Estonia). Environ. Boil. Fishes 2009, 22, 51–63. [Google Scholar] [CrossRef]
- Greer, C.W.; Yaphe, W. Characterization of Hybrid (Beta-Kappa-Gamma) Carrageenan from Eucheuma gelatinae J. Agardh (Rhodophyta, Solieriaceae) Using Carrageenases, Infrared and 13C-Nuclear Magnetic Resonance Spectroscopy. Bot. Mar. 1984, 27, 473–478. [Google Scholar] [CrossRef]
- Morris, E.R.; Rees, D.A.; Robinson, G. Cation-specific aggregation of carrageenan helices: Domain model of polymer gel structure. J. Mol. Boil. 1980, 138, 349–362. [Google Scholar] [CrossRef]
- Viebke, C.; Borgström, J.; Piculell, L. Characterisation of kappa- and iota-carrageenan coils and helices by MALLS/GPC. Carbohydr. Polym. 1995, 27, 145–154. [Google Scholar] [CrossRef]
- Millane, R.P.; Chandrasekaran, R.; Arnott, S.; Dea, I.C. The molecular structure of kappa-carrageenan and comparison with iota-carrageenan. Carbohydr. Res. 1988, 182, 1–17. [Google Scholar] [CrossRef]
- Cairns, P.; Atkins, E.; Miles, M.; Morris, V. Molecular transforms of kappa carrageenan and furcellaran from mixed gel systems. Int. J. Boil. Macromol. 1991, 13, 65–68. [Google Scholar] [CrossRef]
- Almutairi, F.M.; Adams, G.G.; Kök, M.S.; Lawson, C.J.; Gähler, R.; Wood, S.; Foster, T.; Rowe, A.J.; Harding, S.E. An analytical ultracentrifugation based study on the conformation of lambda carrageenan in aqueous solution. Carbohydr. Polym. 2013, 97, 203–209. [Google Scholar] [CrossRef]
- Running, C.; Falshaw, R.; Janaswamy, S. Trivalent iron induced gelation in lambda-carrageenan. Carbohydr. Polym. 2012, 87, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, S.; Piculell, L. Helix-coil transitions of ionic polysaccharides analyzed within the Poisson-Boltzmann cell model. 4. Effects of site-specific counterion binding. Macromolecules 1991, 24, 3804–3811. [Google Scholar] [CrossRef]
- Zhang, W.; Piculell, L.; Nilsson, S. Salt dependence and ion specificity of the coil-helix transition of furcellaran. Biopolymers 1991, 31, 1727–1736. [Google Scholar] [CrossRef]
- Zhang, W.; Piculell, L.; Nilsson, S.; Knutsen, S.H. Cation specificity and cation binding to low sulfated carrageenans. Carbohydr. Polym. 1994, 23, 105–110. [Google Scholar] [CrossRef]
- Michel, A.S.; Mestdagh, M.M.; Axelos, M. Physico-chemical properties of carrageenan gels in presence of various cations. Int. J. Boil. Macromol. 1997, 21, 195–200. [Google Scholar] [CrossRef]
- Nilsson, S.; Piculell, L.; Joensson, B. Helix-coil transitions of ionic polysaccharides analyzed within the Poisson-Boltzmann cell model. 1. Effects of polyion concentration and counterion valency. Macromolecules 1989, 22, 2367–2375. [Google Scholar] [CrossRef]
- Nilsson, S.; Piculell, L. Helix-coil transitions of ionic polysaccharides analyzed within the Poisson-Boltzmann cell model. 2. Effects of salt concentration on the thermal transition. Macromolecules 1989, 22, 3011–3017. [Google Scholar] [CrossRef]
- Belton, P.S.; Chilvers, G.R.; Morris, V.J.; Tanner, S.F. Effects of group I cations on the gelation of iota carrageenan. Int. J. Boil. Macromol. 1984, 6, 303–308. [Google Scholar] [CrossRef]
- Picullel, L.; Håkansson, C.; Nilsson, S. Cation specificity of the order—Disorder transition in iota carrageenan: Effects of kappa carrageenan impurities. Int. J. Boil. Macromol. 1987, 9, 297–301. [Google Scholar] [CrossRef]
- Borgström, J.; Piculell, L.; Viebke, C.; Talmon, Y. On the structure of aggregated kappa-carrageenan helices. A study by cryo-TEM, optical rotation and viscometry. Int. J. Boil. Macromol. 1996, 18, 223–229. [Google Scholar] [CrossRef]
- Yuguchi, Y.; Thuy, T.T.T.; Urakawa, H.; Kajiwara, K. Structural characteristics of carrageenan gels: Temperature and concentration dependence. Food Hydrocoll. 2002, 16, 515–522. [Google Scholar] [CrossRef]
- Ikeda, S.; Morris, V.J.; Nishinari, K. Microstructure of Aggregated and Nonaggregated κ-Carrageenan Helices Visualized by Atomic Force Microscopy. Biomacromolecules 2001, 2, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, W.R.; Harpell, A.R. Carrageenan. In Food Stabilisers, Thickeners and Gelling Agents; Imeson, A., Ed.; Wiley-Blackwell Publishing Ltd.: Hoboken, NJ, USA, 2010; pp. 73–94. [Google Scholar]
- Stanley, N. FAO - chapter 3 - Production, Properties and Uses of Carrageenan. Available online: http://www.fao.org/3/x5822e/x5822e05.htm (accessed on 20 February 2019).
- Campo, V.L.; Kawano, D.F.; Braz da Silva, D.; Ivone, C. Carrageenans: Biological properties, chemical modifications and structural analysis—A review. Carbohydr. Polym. 2009, 77, 167–180. [Google Scholar] [CrossRef]
- BeMiller, J.N. An Introduction to Pectins: Structure and Properties. In Chemistry and Function of Pectins; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1986; Volume 310, pp. 2–12. ISBN 978-0-8412-0974-9. [Google Scholar]
- De Vries, J.A.; Voragen, A.G.J.; Rombouts, F.M.; Pilnik, W. Structural Studies of Apple Pectins with Pectolytic Enzymes. In Chemistry and Function of Pectins; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1986; Volume 310, pp. 38–48. ISBN 978-0-8412-0974-9. [Google Scholar]
- Thibault, J.-F.; Renard, C.M.; Axelos, M.A.; Roger, P.; Crépeau, M.-J. Studies of the length of homogalacturonic regions in pectins by acid hydrolysis. Carbohydr. Res. 1993, 238, 271–286. [Google Scholar] [CrossRef]
- Devries, J.; Denuijl, C.; Voragen, A.; Rombouts, F.; Pilnik, W. Structural features of the neutral sugar side chains of apple pectic substances. Carbohydr. Polym. 1983, 3, 193–205. [Google Scholar] [CrossRef]
- Lopes da Silva, J.A.; Rao, M.A. Pectins: Structure, Functionality and Uses. In Food Polysaccharides and Their Applications; Stephen, A.M., Phillips, G.O., Williams, P.A., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 353–411. ISBN 978-0-8247-5922-3. [Google Scholar]
- Mohnen, D. Pectin structure and biosynthesis. Curr. Opin. Plant Boil. 2008, 11, 266–277. [Google Scholar] [CrossRef]
- Garnier, C.; Axelos, M.A.; Thibault, J.-F. Selectivity and cooperativity in the binding of calcium ions by pectins. Carbohydr. Res. 1994, 256, 71–81. [Google Scholar] [CrossRef]
- Morris, E.R.; A Powell, D.; Gidley, M.J.; A Rees, D. Conformations and interactions of pectins. I. Polymorphism between gel and solid states of calcium polygalacturonate. J. Mol. Boil. 1982, 155, 507–516. [Google Scholar] [CrossRef]
- Rees, D.A.; Wight, A.W. Polysaccharide conformation. Part VII. Model building computations for ?-1,4 galacturonan and the kinking function of L-rhamnose residues in pectic substances. J. Chem. Soc. B 1971, 1366. [Google Scholar] [CrossRef]
- Cros, S.; Garnier, C.; Axelos, M.A.; Imberty, A.; Pérez, S. Solution conformations of pectin polysaccharides: determination of chain characteristics by small angle neutron scattering, viscometry, and molecular modeling. Biopolymers 1996, 39, 339–351. [Google Scholar] [CrossRef]
- Berth, G.; Anger, H.; Plashchina, I.; Braudo, E.; Tolstoguzov, V. Structural study of the solutions of acidic polysaccharides. II. Study of some thermodynamic properties of the dilute pectin solutions with different degrees of esterification. Carbohydr. Polym. 1982, 2, 1–8. [Google Scholar] [CrossRef]
- Anger, H.; Berth, G. Gel permeation chromatography and the Mark-Houwink relation for pectins with different degrees of esterification. Carbohydr. Polym. 1986, 6, 193–202. [Google Scholar] [CrossRef]
- Smidsrød, O.; Haug, A. Estimation of the relative stiffness of the molecular chain in polyelectrolytes from measurements of viscosity at different ionic strengths. Biopolymers 1971, 10, 1213–1227. [Google Scholar] [CrossRef] [PubMed]
- Deckers, H.; Olieman, C.; Rombouts, F.; Pilnik, W. Calibration and application of high-performance size exclusion columns for molecular weight distribution of pectins. Carbohydr. Polym. 1986, 6, 361–378. [Google Scholar] [CrossRef]
- Morris, G.A.; Foster, T.; Harding, S.E. The effect of the degree of esterification on the hydrodynamic properties of citrus pectin. Food Hydrocoll. 2000, 14, 227–235. [Google Scholar] [CrossRef]
- Walkinshaw, M.D.; Arnott, S. Conformations and interactions of pectins. II. Models for junction zones in pectinic acid and calcium pectate gels. J. Mol. Boil. 1981, 153, 1075–1085. [Google Scholar] [CrossRef]
- Oakenfull, D.; Scott, A. Hydrophobic Interaction in the Gelation of High Methoxyl Pectins. J. Food Sci. 1984, 49, 1093–1098. [Google Scholar] [CrossRef]
- Thakur, B.R.; Singh, R.K.; Handa, A.K.; Rao, D.M.A. Chemistry and uses of pectin—A review. Crit. Rev. Food Sci. Nutr. 1997, 37, 47–73. [Google Scholar] [CrossRef]
- Sanderson, G.R. Applications of Xanthan gum. Br. Polym. J. 1981, 13, 71–75. [Google Scholar] [CrossRef]
- Nolte, H.; John, S.; Smidsrød, O.; Stokke, B.T. Gelation of xanthan with trivalent metal ions. Carbohydr. Polym. 1992, 18, 243–251. [Google Scholar] [CrossRef]
- Bergmann, D.; Furth, G.; Mayer, C. Binding of bivalent cations by xanthan in aqueous solution. Int. J. Boil. Macromol. 2008, 43, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.J. Bacterial Polysaccharides. In Food Polysaccharides and Their Applications; Stephen, A.M., Phillips, G.O., Williams, P.A., Eds.; CRC Press: Boca Raton, FL, USA, 2006; pp. 413–454. ISBN 978-0-8247-5922-3. [Google Scholar]
- Dong, C.; Zhang, F.; Ji, H.; Yang, G. Efficient and selective adsorption of multi-metal ions using sulfonated cellulose as adsorbent. Carbohydr. Polym. 2016, 151, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Paul, U.; Manian, A.; Siroka, B.; Duelli, H.; Bechtold, T. Sorption of anionic polysaccharides by cellulose. Carbohydr. Polym. 2012, 87, 695–700. [Google Scholar] [CrossRef]
- Paul, U.; Manian, A.; Široká, B.; Duelli, H.; Bechtold, T. Sorption of iron(III)–alginate complexes on cellulose fibres. Cellulose 2013, 20, 2481–2490. [Google Scholar] [CrossRef]
- Chen, K.L.; Mylon, S.E.; Elimelech, M. Aggregation Kinetics of Alginate-Coated Hematite Nanoparticles in Monovalent and Divalent Electrolytes. Environ. Sci. Technol. 2006, 40, 1516–1523. [Google Scholar] [CrossRef]
- Dong, H.; Snyder, J.F.; Williams, K.S.; Andzelm, J.W. Cation-Induced Hydrogels of Cellulose Nanofibrils with Tunable Moduli. Biomacromolecules 2013, 14, 3338–3345. [Google Scholar] [CrossRef]
- Liu, M.; Yue, X.; Dai, Z.; Ma, Y.; Xing, L.; Zha, Z.; Liu, S.; Li, Y. Novel Thrombo-Resistant Coating Based on Iron−Polysaccharide Complex Multilayers. ACS Appl. Mater. Interfaces 2008, 1, 113–123. [Google Scholar] [CrossRef]
- Machida-Sano, I.; Matsuda, Y.; Namiki, H. In vitro adhesion of human dermal fibroblasts on iron cross-linked alginate films. Biomed. Mater. 2009, 4, 025008. [Google Scholar] [CrossRef]
- Synhaivska, O.; Mermoud, Y.; Baghernejad, M.; Alshanski, I.; Hurevich, M.; Yitzchaik, S.; Wipf, M.; Calame, M. Detection of Cu2+ Ions with GGH Peptide Realized with Si-Nanoribbon ISFET. Sensors 2019, 19, 4022. [Google Scholar] [CrossRef]
- Schuur, B.; Verkuijl, B.J.V.; Minnaard, A.J.; De Vries, J.G.; Heeres, H.J.; Feringa, B.L. Chiral separation by enantioselective liquid–liquid extraction. Org. Biomol. Chem. 2011, 9, 36–51. [Google Scholar] [CrossRef]
- Smith, E.R. The Isolation and Quantitation of Fetuin-A-Containing Calciprotein Particles from Biological Fluids. In Kidney Research: Experimental Protocols; Methods in Molecular Biology; Hewitson, T.D., Smith, E.R., Holt, S.G., Eds.; Springer: New York, NY, USA, 2016; pp. 221–240. ISBN 978-1-4939-3353-2. [Google Scholar]
- Bauer, C.; Reutter, W.; Gerok, W. Neue Ergebnisse zur biologischen und medizinischen Bedeutung von Glycoproteinen. J. Mol. Med. 1979, 57, 199–214. [Google Scholar] [CrossRef]
- Burger, K.; Nagy, L.; Gyuresik, B. The structure of metal complexes of small models of glycoproteins in aqueous solution. J. Mol. Liq. 1995, 65, 213–219. [Google Scholar] [CrossRef]
- Polák, B.; Aunter, B.D. Seminal plasma biochemistry. IV: Enzymes involved in the liquefaction of human seminal plasma. Int. J. Androl. 1989, 12, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Hassan, I.; Kumar, V.; Singh, T.P.; Yadav, S. Purification and characterization of zinc α2-glycoprotein-Prolactin inducible protein complex from human seminal plasma. J. Sep. Sci. 2008, 31, 2318–2324. [Google Scholar] [CrossRef]
- Roberts, M.F.; Jenekhe, S.A. Lewis acid complexation of polymers: Gallium chloride complex of nylon 6. Chem. Mater. 1990, 2, 224–226. [Google Scholar] [CrossRef]
- Roberts, M.F.; Jenekhe, S.A. Site-specific reversible scission of hydrogen bonds in polymers: An investigation of polyamides and their Lewis acid-base complexes by infrared spectroscopy. Macromolecules 1991, 24, 3142–3146. [Google Scholar] [CrossRef]
- Vasanthan, N.; Kotek, R.; Jung, D.-W.; Shin, D.; E Tonelli, A.; Salem, D.R. Lewis acid–base complexation of polyamide 66 to control hydrogen bonding, extensibility and crystallinity. Polymer 2004, 45, 4077–4085. [Google Scholar] [CrossRef]
- Sun, B. Study on the mechanism of nylon 6.6 dissolving process using CaCl₂/MeOH as the solvent. Chin. J. Polym. Sci. 1994, 12, 57–65. [Google Scholar]
- Rietzler, B.; Bechtold, T.; Pham, T. Controlled Surface Modification of Polyamide 6.6 Fibres Using CaCl2/H2O/EtOH Solutions. Polymer 2018, 10, 207. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, C.; Proniewicz, E.; Proniewicz, L.M.; Kim, Y.; Liu, J.; Zhao, Y.; Xu, Y.; Wu, J. Crystalline transition and morphology variation of polyamide 6/CaCl2 composite during the decomplexation process. Spectrochim. Acta Mol. Biomol. Spectrosc. 2013, 115, 783–788. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, Y.; Wang, D.; Zhao, Y.; Weng, S.; Xu, D.; Wu, J. FT-IR spectroscopic investigation on the interaction between nylon 66 and lithium salts. J. Appl. Polym. Sci. 2004, 91, 2869–2875. [Google Scholar] [CrossRef]
- Afshari, M.; Gupta, A.; Jung, D.; Kotek, R.; Tonelli, A.; Vasanthan, N. Properties of films and fibers obtained from Lewis acid–base complexed nylon 6,6. Polymer 2008, 49, 1297–1304. [Google Scholar] [CrossRef]
- Hattori, M.; Saito, M. Thermal Gelation of the Nylon 6,6-Calcium Chloride-Methanol System. Polym. J. 1996, 28, 139–144. [Google Scholar] [CrossRef]
- Yang, Z.; Yin, H.; Li, X.; Liu, Z.; Jia, Q. Study on dry spinning and structure of low mole ratio complex of calcium chloride-polyamide 6. J. Appl. Polym. Sci. 2010, 118, 1996–2004. [Google Scholar] [CrossRef]
- Jia, Q.; Xiong, Z.-J.; Shi, C.-M.; Zhao, X.; Wang, X.-N. Preparation and properties of polyamide 6 fibers prepared by the gel spinning method. J. Appl. Polym. Sci. 2011, 5165–5171. [Google Scholar] [CrossRef]
- Wei, W.; Qiu, L.; Wang, X.L.; Chen, H.P.; Lai, Y.C.; Tsai, F.C.; Zhu, P.; Yeh, J.T. Drawing and tensile properties of polyamide 6/calcium chloride composite fibers. J. Polym. Res. 2011, 18, 1841–1850. [Google Scholar] [CrossRef]
- Zhang, H.; Shi, C.; He, M.; Jia, Q.; Zhao, X. Structure and property changes of polyamide 6 during the gel-spinning process. J. Appl. Polym. Sci. 2013, 130, 4449–4456. [Google Scholar] [CrossRef]
- Mathur, A.; Tonelli, A.; Rathke, T.; Hudson, S. The dissolution and characterization of Bombyx mori silk fibroin in calcium nitrate-methanol solution and the regeneration of films. Biopolymers 1997, 42, 61–74. [Google Scholar] [CrossRef]
- Matsumoto, A.; Lindsay, A.; Abedian, B.; Kaplan, D.L. Silk Fibroin Solution Properties Related to Assembly and Structure. Macromol. Biosci. 2008, 8, 1006–1018. [Google Scholar] [CrossRef]
- Sun, Y.; Shao, Z.; Ma, M.; Hu, P.; Liu, Y.; Yu, T. Acrylic polymer-silk fibroin blend fibers. J. Appl. Polym. Sci. 1997, 65, 959–966. [Google Scholar] [CrossRef]
- Kaplan, D.L.; Mello, C.M.; Arcidiacono, S.; Fossey, S.; Senecal, K.; Muller, W. Silk. In Protein-Based Materials; Bioengineering of Materials; McGrath, K., Kaplan, D., Eds.; Birkhäuser Basel: Basel, Switzerland, 1997; pp. 103–131. ISBN 978-0-8176-3848-1. [Google Scholar]
- Ngo, H.-T.; Bechtold, T. Analysis of the Fibroin Solution State in Calcium Chloride/Water/Ethanol for Improved Understanding of the Regeneration Process. Fibres Text. East. Eur. 2018, 26, 43–50. [Google Scholar] [CrossRef]
- Rey, F.; Antelo, J.; Arce, F.; Penedo, F. Equilibrium constants of metal amino acid complexes. Polyhedron 1990, 9, 665–668. [Google Scholar] [CrossRef]
- Fox, S.; Büsching, I.; Barklage, W.; Strasdeit, H. Coordination of Biologically Important α-Amino Acids to Calcium(II) at High pH: Insights from Crystal Structures of Calcium α-Aminocarboxylates. Inorg. Chem. 2007, 46, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Halpern, J.; Kauffman, G.B. Ligands and chelates. In Encyclopaedia Britannica; Encyclopaedia Britannica, Inc.: Chicago, IL, USA, 2018. [Google Scholar]
- Bechtold, T.; Fitz-Binder, C.; Krüger, M. Process for Production of Leather Substitutes. Austrian Patent Application no.: A51050/2017, 2017. [Google Scholar]
- Hoshino, M.; Song, J.C.; Yoshida, T.; Uryu, T. Carbon-13 NMR Studies of Keratin Hydrolysate-, Polylysine-, and Polyornithine-Metal Ion Complexes. Kobunshi Ronbunshu 1991, 48, 341–346. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; He, Y.; Song, P.; Wang, R.-M. Preparation of Keratin-Glycine Metal Complexes and Their Scavenging Activity for Superoxide Anion Radicals. Int. J. Polym. Sci. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Hemrajani, S.N.; Narwani, C.S. Polarographic study of metal ion complexes with keratin fibers (wool) at pH 4-5 and at 30°. I. Chlorides and nitrated of mercury(II), copper(II), cadmium(II), and lead(II). J. Indian Chem. Soc. 1967, 44, 704–709. [Google Scholar]
- Khandker, H.S. Association behavior of native and regenerated silk fibroin in aqueous solution. Foods Food Ingred. J. Jpn. 2008, 213, 526–536. [Google Scholar]
- Chen, W.X.; Lu, S.F.; Yao, Y.Y.; Pan, Y.; Shen, Z.Q. Copper(II)-Silk Fibroin Complex Fibers as Air-Purifying Materials for Removing Ammonia. Text. Res. J. 2005, 75, 326–330. [Google Scholar] [CrossRef]
- Shirai, H.; Hojo, N. Metal complexes of silk fibroin. VI. Formation of nickel(II) silk fibroin (Bombyx mori) complexes and their secondary structures. Nippon Kagaku Kaishi 1975, 12, 2233–2237. [Google Scholar] [CrossRef]
Sample Availability: Since the manuscript is a review, no specific samples are available. We offer discussion and support with materials if possible, upon contact. |
























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wurm, F.; Rietzler, B.; Pham, T.; Bechtold, T. Multivalent Ions as Reactive Crosslinkers for Biopolymers—A Review. Molecules 2020, 25, 1840. https://doi.org/10.3390/molecules25081840
Wurm F, Rietzler B, Pham T, Bechtold T. Multivalent Ions as Reactive Crosslinkers for Biopolymers—A Review. Molecules. 2020; 25(8):1840. https://doi.org/10.3390/molecules25081840
Chicago/Turabian StyleWurm, Florian, Barbara Rietzler, Tung Pham, and Thomas Bechtold. 2020. "Multivalent Ions as Reactive Crosslinkers for Biopolymers—A Review" Molecules 25, no. 8: 1840. https://doi.org/10.3390/molecules25081840
APA StyleWurm, F., Rietzler, B., Pham, T., & Bechtold, T. (2020). Multivalent Ions as Reactive Crosslinkers for Biopolymers—A Review. Molecules, 25(8), 1840. https://doi.org/10.3390/molecules25081840