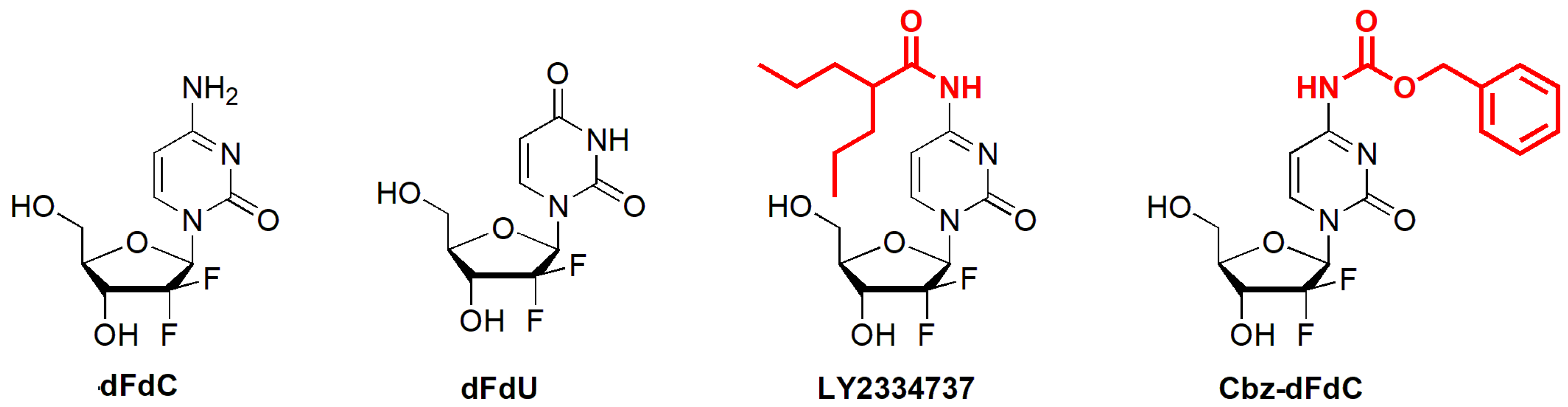
A Pharmacokinetic and Pharmacodynamic Evaluation of the Anti-Hepatocellular Carcinoma Compound 4-N-Carbobenzoxy-gemcitabine (Cbz-dFdC)


Abstract
:1. Introduction
2. Results
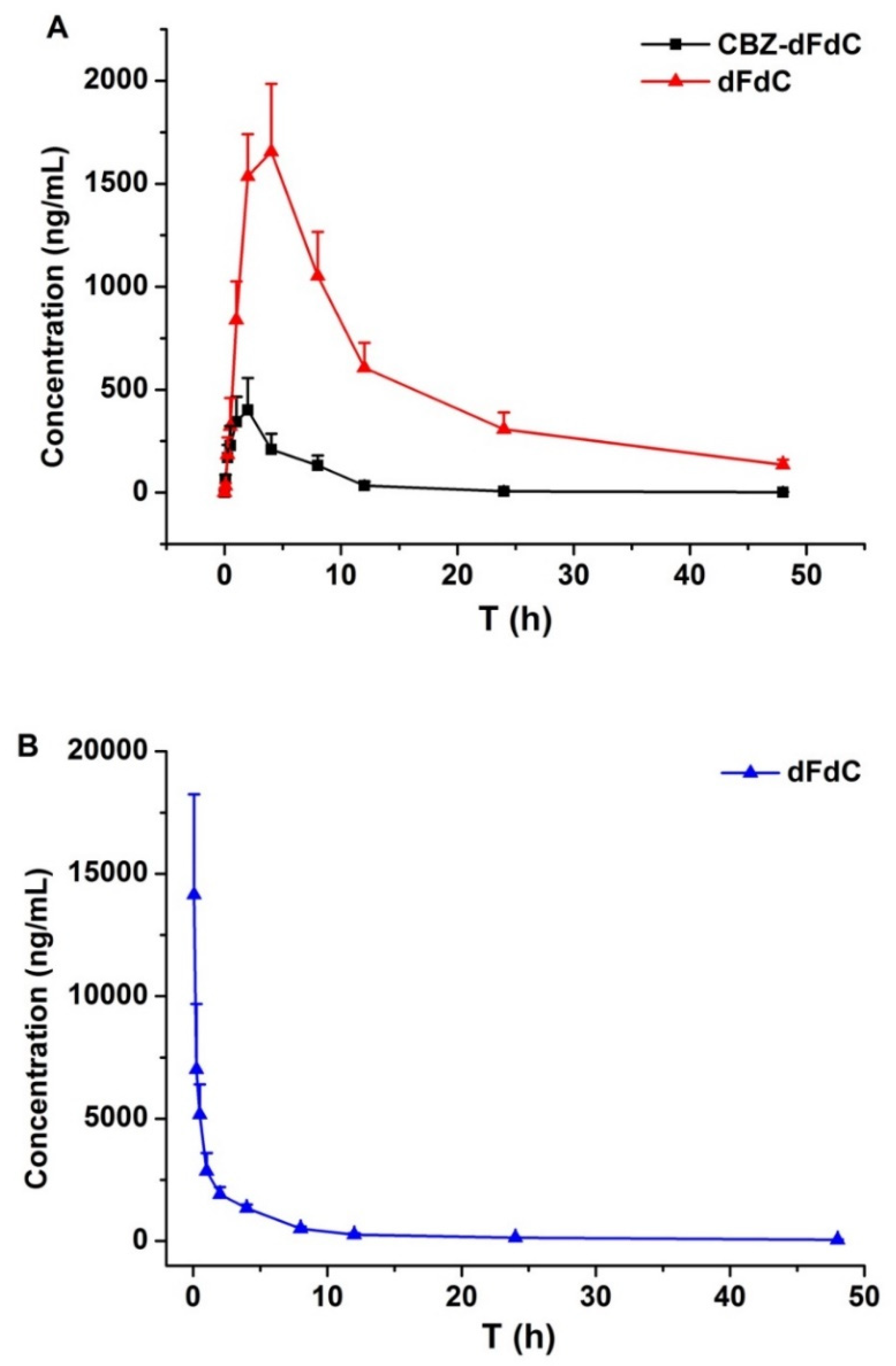
2.1. Plasma Pharmacokinetics In Vivo
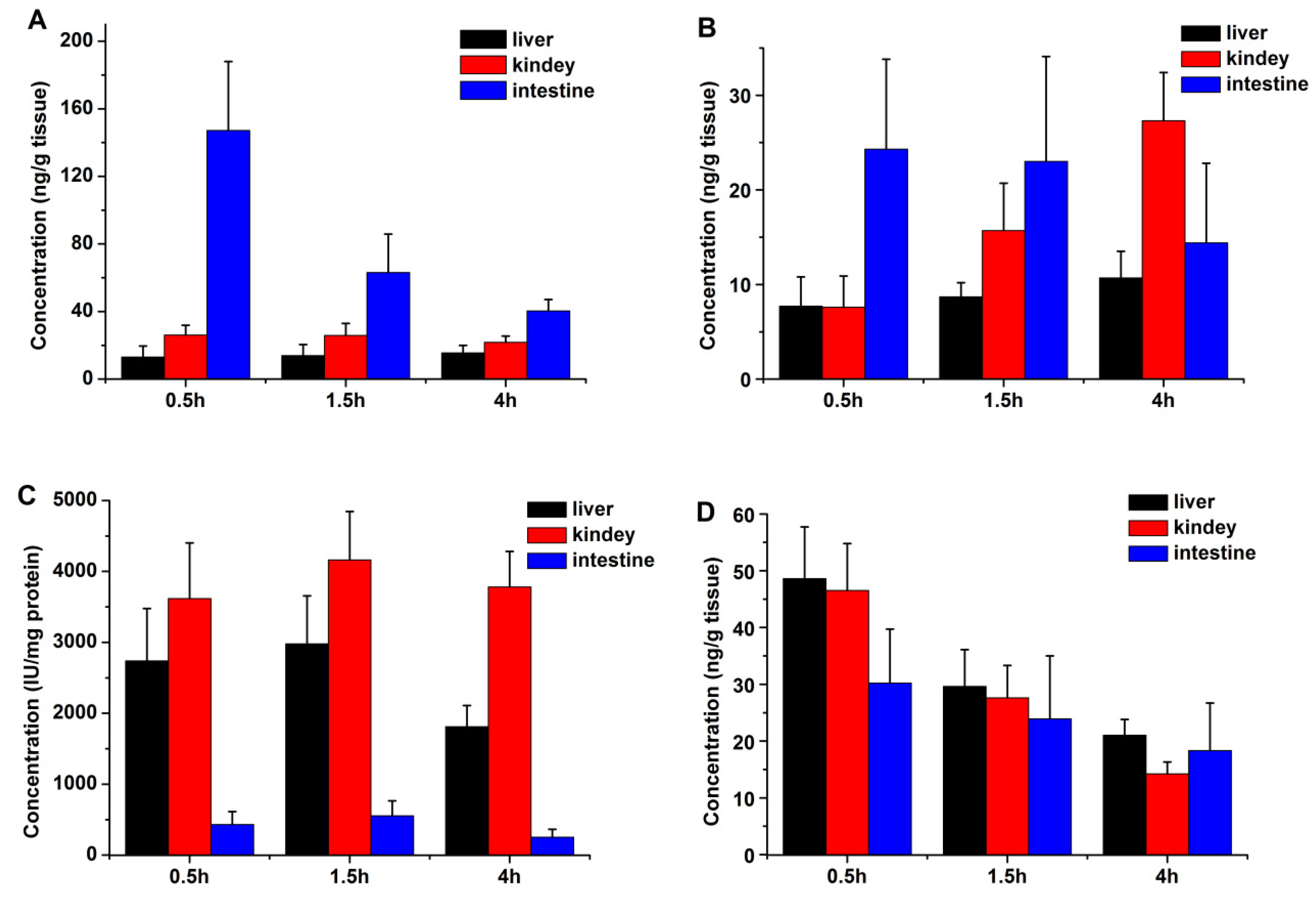
2.2. Tissue Distribution
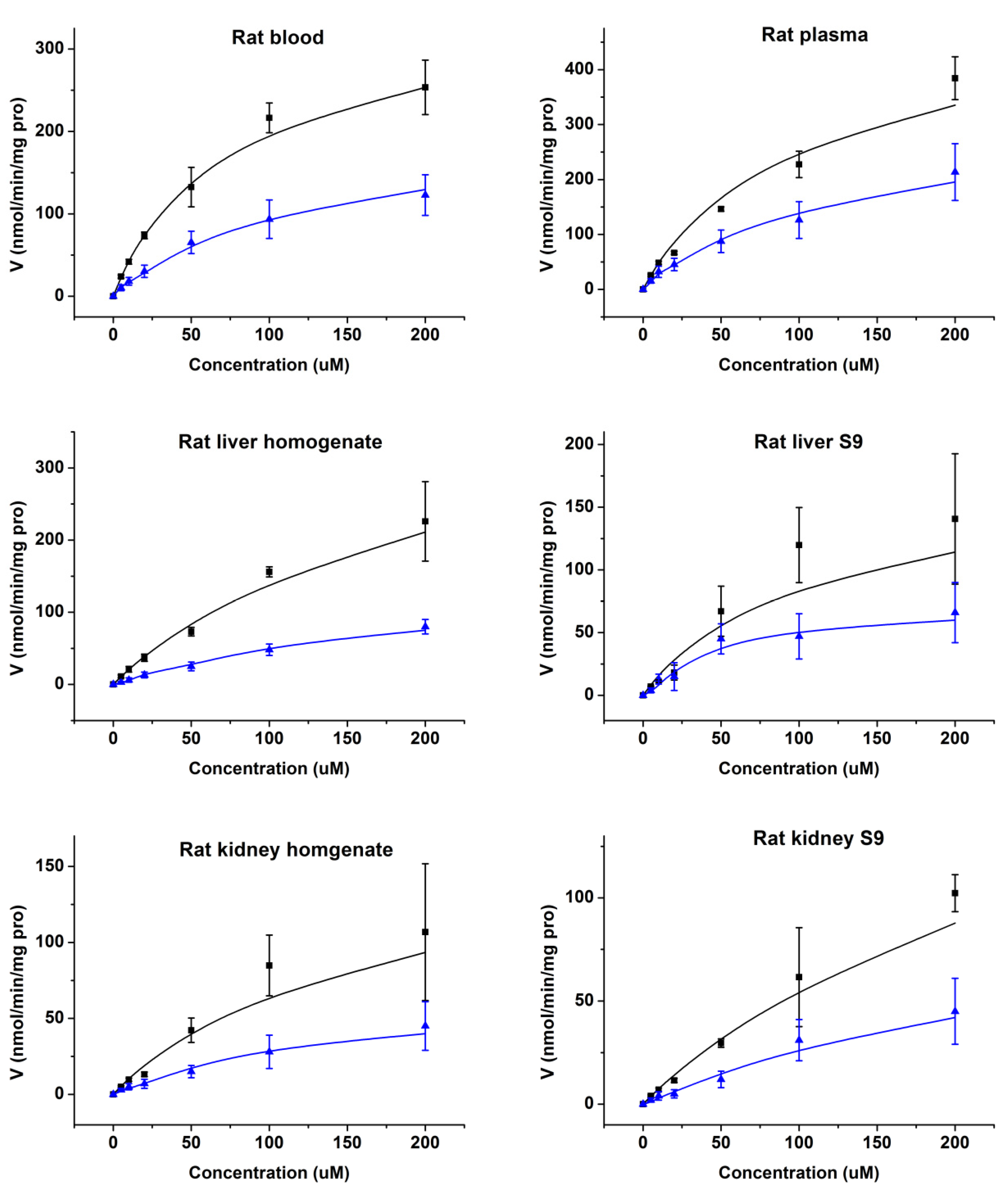
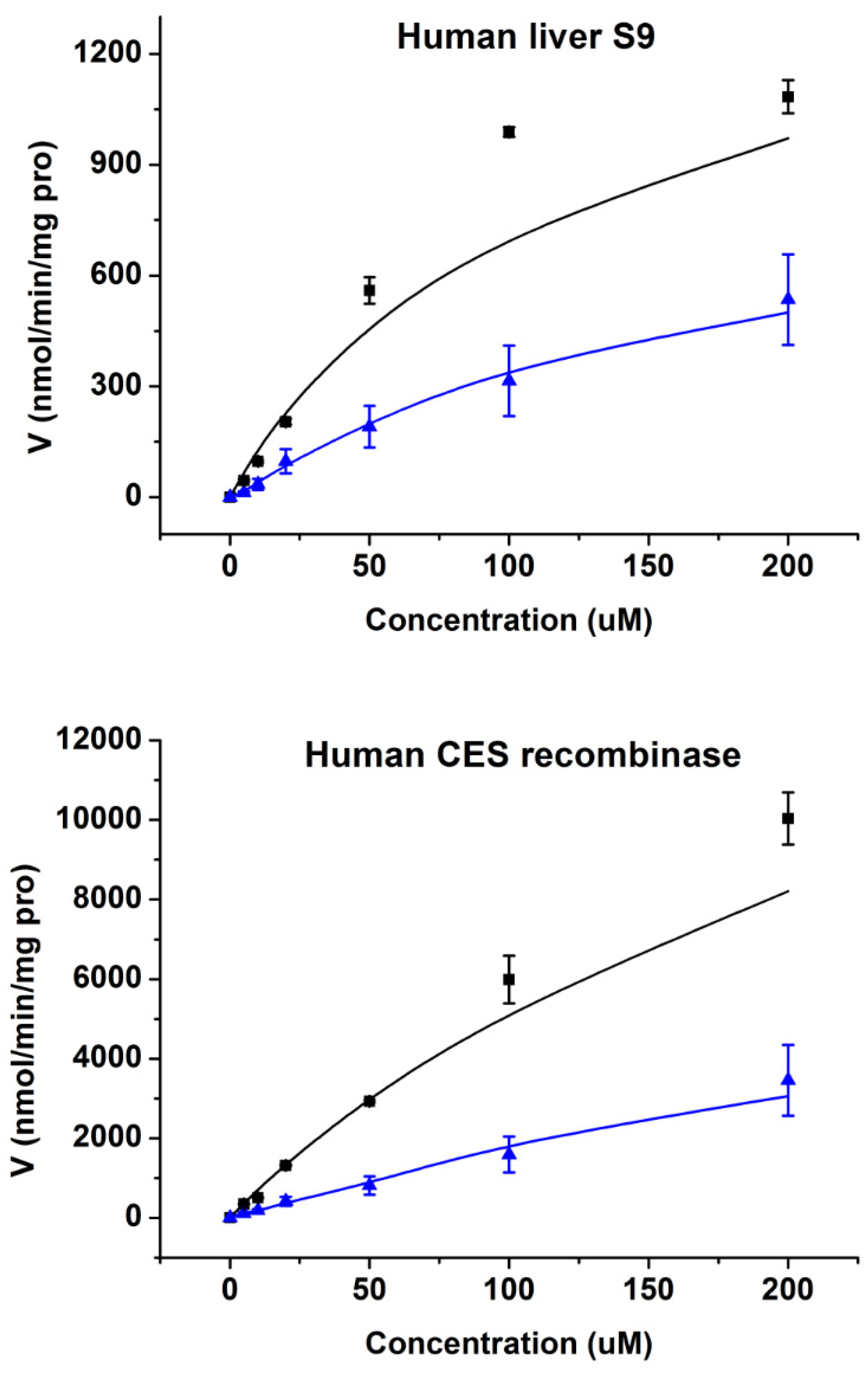
2.3. Enzymatic Kinetics In Vitro
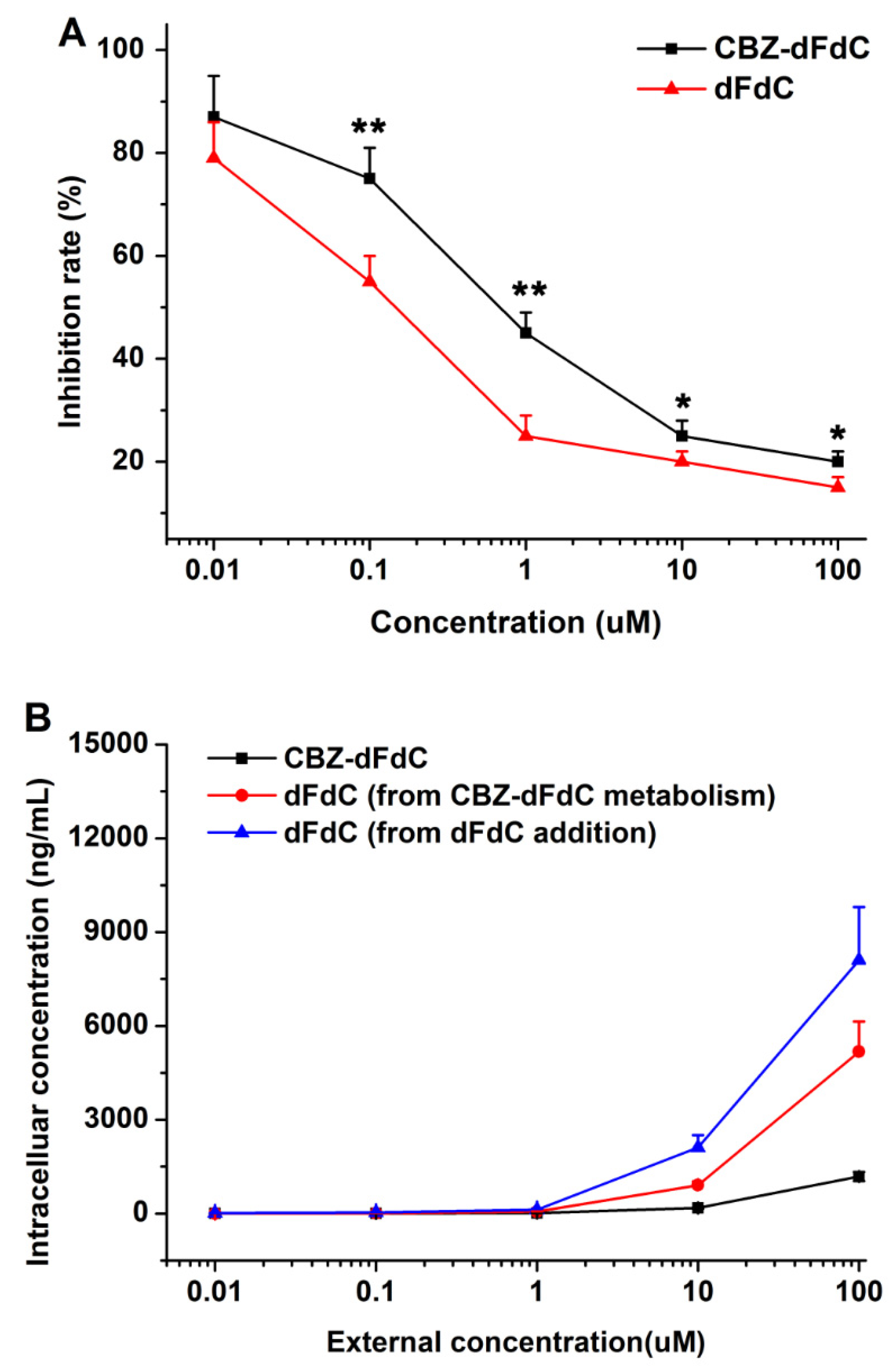
2.4. Cell Efficacy and Unptake
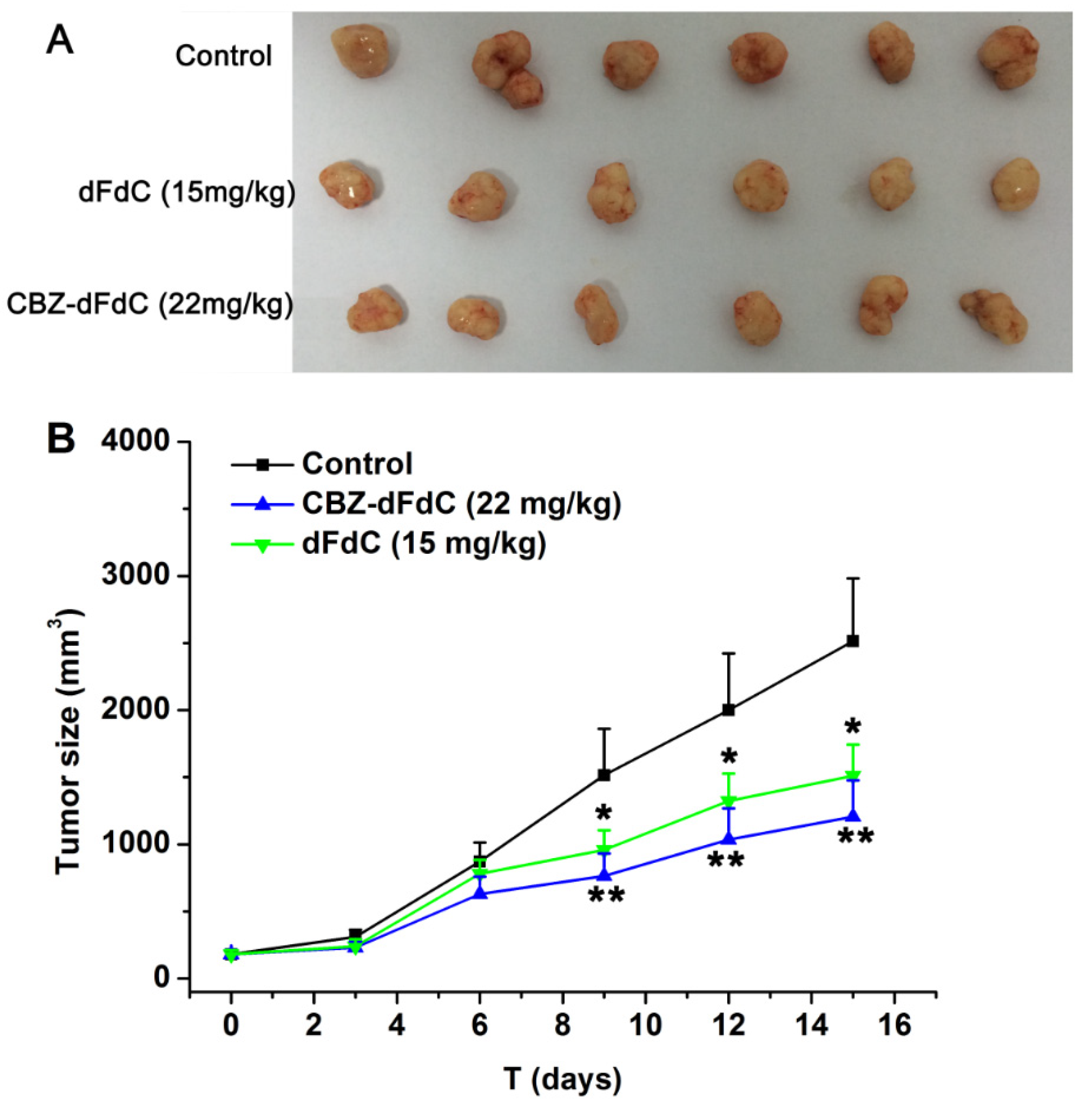
2.5. Antitumor Efficacy In Vivo
3. Discussion
4. Materials and Methods
4.1. Materials and Equipment
4.2. Animals
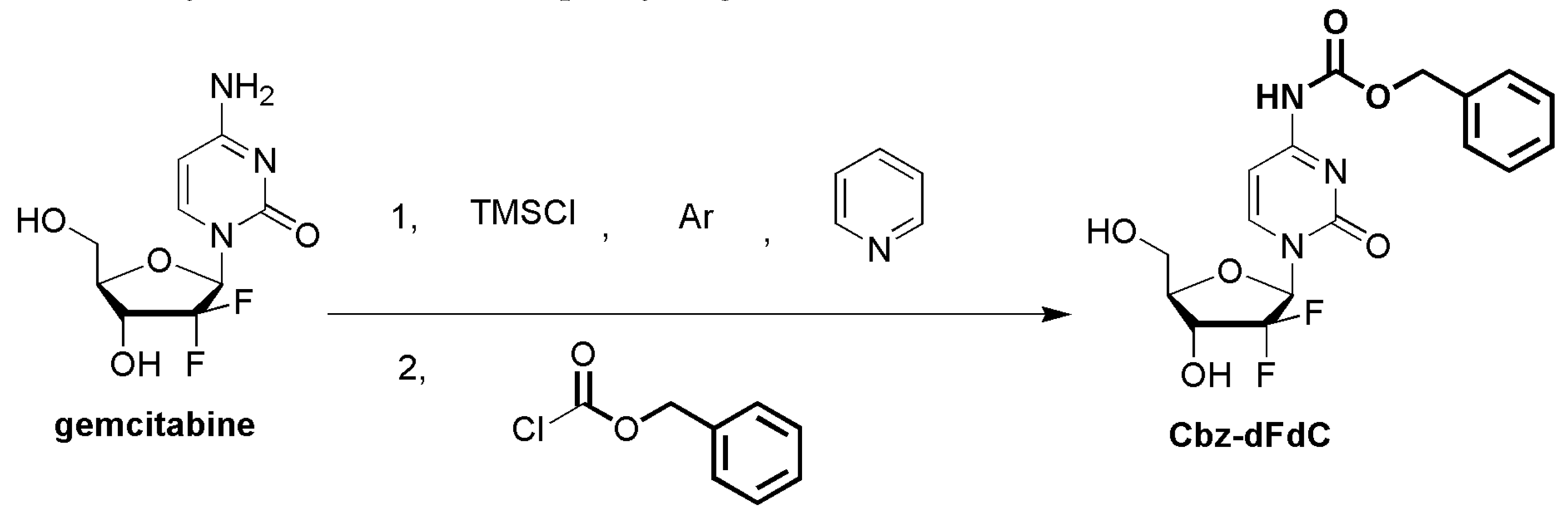
4.3. The Synthesis of Cbz-dFdC
4.4. Pharmacokinetics In Vivo
4.5. Tissue Distribution
4.6. Enzymatic Kinetics In Vitro
4.7. Antitumor Efficacy and Uptake In Vitro
4.8. Antitumor Efficacy In Vivo
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Cellular elimination of 2’,2’-difluorodeoxycytidine 5’-triphosphate: A mechanism of self-potentiation. Cancer Res. 1992, 52, 533–539. [Google Scholar] [PubMed]
- Symon, Z.; Davis, M.; Mcginn, C.J.; Zalupski, M.M.; Lawrence, T.S. Concurrent chemoradiotherapy with gemcitabine and cisplatin for pancreatic cancer: From the laboratory to the clinic. Int. J. Radiat. Oncol. 2002, 53, 140–145. [Google Scholar] [CrossRef]
- Heinemann, V. Gemcitabine in metastatic breast cancer. Expert Rev. Anticancer 2005, 5, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Couvreur, P.; Stella, B.; Reddy, L.H.; Hillaireau, H.; Dubernet, C.; Desmaele, D.; Lepetre-Mouelhi, S.; Rocco, F.; Dereuddre-Bosquet, N.; Clayette, P.; et al. Squalenoyl nanomedicines as potential therapeutics. Nano Lett. 2006, 6, 2544–2548. [Google Scholar] [CrossRef]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17, 7–12. [Google Scholar] [CrossRef]
- Hansen, S.W. Gemcitabine in the treatment of ovarian cancer. Int. J. Gynecol. Cancer 2010, 11, 39–41. [Google Scholar] [CrossRef]
- Favaretto, A.G. Non-platinum combination of gemcitabine in NSCLC. Ann. Oncol. 2006, 17, 82–85. [Google Scholar] [CrossRef]
- Pectasides, D.; Pectasides, M.; Economopoulos, T. Systemic chemotherapy in locally advanced and/or metastatic bladder cancer. Cancer Treat. Rev. 2006, 32, 456–470. [Google Scholar] [CrossRef]
- Dent, S.; Messersmith, H.; Trudeau, M. Gemcitabine in the management of metastatic breast cancer: A systematic review. Breast Cancer Res. Treat. 2008, 108, 319–331. [Google Scholar] [CrossRef]
- Hilbig, A.; Oettle, H. Gemcitabine in the treatment of metastatic pancreatic cancer. Expert Rev. Anticancer 2008, 8, 511–523. [Google Scholar] [CrossRef]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2’,2’-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar] [PubMed]
- Grunewald, R.; Kantarjian, H.; Du, M.; Faucher, K.; Tarassoff, P.; Plunkett, W. Gemcitabine in leukemia: A phase I clinical, plasma, and cellular pharmacology study. J. Clin. Oncol. 1992, 10, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Ruiz van Haperen, V.W.; Veerman, G.; Eriksson, S.; Boven, E.; Stegmann, A.P.; Hermsen, M.; Vermorken, J.B.; Pinedo, H.M.; Peters, G.J. Development and molecular characterization of a 2’,2’-difluorodeoxycytidine -resistant variant of the human ovarian carcinoma cell line A2780. Cancer Res. 1994, 54, 4138–4143. [Google Scholar] [PubMed]
- Bergman, A.M.; Pinedo, H.M.; Peters, G.J. Determinants of resistance to 2’,2’-difluorodeoxycytidine (gemcitabine). Drug Resist. Updat. 2002, 5, 19–33. [Google Scholar] [CrossRef]
- Baker, J.A.; Wickremsinhe, E.R.; Li, C.H.; Oluyedun, O.A.; Dantzig, A.H.; Hall, S.D.; Qian, Y.W.; Ring, B.J.; Wrighton, S.A.; Guo, Y. Pharmacogenomics of gemcitabine metabolism: Functional analysis of genetic variants in cytidine deaminase and deoxycytidine kinase. Drug Metab. Dispos. 2013, 41, 541–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giusti, G.; Mangoni, C.; De Petrocellis, B.; Scarano, E. Deoxycytidylate deaminase and deoxycytidine deaminase in normal and neoplastic human tissues. Enzym. Biol. Clin. 1970, 11, 375–383. [Google Scholar] [CrossRef]
- Abbruzzese, J.L.; Grunewald, R.; Weeks, E.A.; Gravel, D.; Adams, T.; Nowak, B.; Mineishi, S.; Tarassoff, P.; Satterlee, W.; Raber, M.N. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498. [Google Scholar] [CrossRef]
- Reddy, L.H.; Khoury, H.; Paci, A.; Deroussent, A.; Ferreira, H.; Dubernet, C.; Declèves, X.; Besnard, M.; Chacun, H.; Lepêtre-Mouelhi, S. Squalenoylation favorably modifies the in vivo pharmacokinetics and biodistribution of gemcitabine in mice. Drug Metab. Dispos. 2008, 36, 1570–1577. [Google Scholar] [CrossRef] [Green Version]
- Bender, D.M.; Bao, J.; Dantzig, A.H.; Diseroad, W.D.; Law, K.L.; Magnus, N.A.; Peterson, J.A.; Perkins, E.J.; Pu, Y.J.; Reutzel-Edens, S.M.; et al. Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J. Med. Chem. 2009, 52, 6958–6961. [Google Scholar] [CrossRef]
- Wickremsinhe, E.R.; Bao, J.; Smith, R.; Burton, R.; Dow, S.; Perkins, E. Preclinical absorption, distribution, metabolism, and excretion of an oral amide prodrug of gemcitabine designed to deliver prolonged systemic exposure. Pharmaceutics 2013, 5, 261–276. [Google Scholar] [CrossRef]
- Wickremsinhe, E.R.; Lee, L.B.; Schmalz, C.A.; Torchia, J.; Ruterbories, K.J. High sensitive assay employing column switching chromatography to enable simultaneous quantification of an amide prodrug of gemcitabine (LY2334737), gemcitabine, and its metabolite dFdU in human plasma by LC–MS/MS. J. Chromatogr. B 2013, 932, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Miwa, M.; Ura, M.; Nishida, M.; Sawada, N.; Ishikawa, T.; Mori, K.; Shimma, N.; Umeda, I.; Ishitsuka, H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur. J. Cancer 1998, 34, 1274–1281. [Google Scholar] [CrossRef]
- Immordino, M.L.; Brusa, P.; Rocco, F.; Arpicco, S.; Ceruti, M.; Cattel, L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing lipophilic gemcitabine prodrugs. J. Control. Release 2004, 100, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Rejiba, S.; Reddy, L.H.; Bigand, C.; Parmentier, C.; Couvreur, P.; Hajri, A. Squalenoyl gemcitabine nanomedicine overcomes the low efficacy of gemcitabine therapy in pancreatic cancer. Nanomedicine 2011, 7, 841–849. [Google Scholar] [CrossRef]
- Wang, G.; Chen, H.; Zhao, D.; Ding, D.; Sun, M.; Kou, L.; Luo, C.; Zhang, D.; Yi, X.; Dong, J.; et al. Combination of l-Carnitine with Lipophilic Linkage-Donating Gemcitabine Derivatives as Intestinal Novel Organic Cation Transporter 2-Targeting Oral Prodrugs. J. Med. Chem. 2017, 60, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zhen, L.; Peng, Y.; Wang, J.; Fei, F.; Aa, L.; Jiang, W.; Pei, X.; Lu, L.; Liu, J.; et al. Simultaneous determination of gemcitabine prodrug, gemcitabine and its major metabolite 2’,2’-difluorodeoxyuridine in rat plasma by UFLC–MS/MS. J. Chromatogr. B 2018, 1084, 4–13. [Google Scholar] [CrossRef]
- Munger, J.S.; Shi, G.P.; Mark, E.A.; Chin, D.T.; Gerard, C.; Chapman, H.A. A serine esterase released by human alveolar macrophages is closely related to liver microsomal carboxylesterases. J. Biol. Chem. 1991, 266, 18832–18838. [Google Scholar]
- Satoh, T.; Hosokawa, M. Molecular aspects of carboxylesterase isoforms in comparison with other esterases. Toxicol. Letts. 1995, 82, 439–445. [Google Scholar] [CrossRef]
- Sanghani, S.P.; Quinney, S.K.; Fredenburg, T.B.; Sun, Z.; Davis, W.I.; Murry, D.J.; Cummings, O.W.; Seitz, D.E.; Bosron, W.F. Carboxylesterases expressed in human colon tumor tissue and their role in CPT-11 hydrolysis. Clin. Cancer Res. 2003, 9, 4983–4991. [Google Scholar]
- Imai, T. Human Carboxylesterase Isozymes: Catalytic Properties and Rational Drug Design. Drug Metab. Pharm. 2006, 21, 173–185. [Google Scholar] [CrossRef]
- Takai, S.; Matsuda, A.; Usami, Y.; Adachi, T.; Sugiyama, T.; Katagiri, Y.; Tatematsu, M.; Hirano, K. Hydrolytic profile for ester- or amide-linkage by carboxylesterases pI 5.3 and 4.5 from human liver. Biol. Pharm. Bull. 1993, 20, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Pearce, R.E.; Wang, X.; Gaedigk, R.; Wan, Y.J.; Yan, B. Human carboxylesterases HCE1 and HCE2: Ontogenic expression, inter-individual variability and differential hydrolysis of oseltamivir, aspirin, deltamethrin and permethrin. Biochem. Pharm. 2009, 77, 238–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.J.; Appel, D.I.; Johnson, J.A.; Chavin, K.D.; Markowitz, J.S. Role of carboxylesterase 1 and impact of natural genetic variants on the hydrolysis of trandolapril. Biochem. Pharm. 2009, 77, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Masakiyo, H. Structure and catalytic properties of carboxylesterase isozymes involved in metabolic activation of prodrugs. Molecules 2008, 13, 412–431. [Google Scholar]
- Heinrich, M.; Edwards, S.; Moerman, D.E.; Leonti, M. Ethnopharmacological field studies: A critical assessment of their conceptual basis and methods. J. Ethnopharmacol. 2009, 124, 1–17. [Google Scholar] [CrossRef]
- Huang, J.; Robertson, J.M.; Ye, H.; Margolis, J.; Nadeau, L.; Yan, D. Dose-volume analysis of predictors for gastrointestinal toxicity after concurrent full-dose gemcitabine and radiotherapy for locally advanced pancreatic adenocarcinoma. Int. J. Radiat Oncol Biol Phys. 2012, 83, 1120–1125. [Google Scholar] [CrossRef]
- Malatesta, L.; Cosco, D.; Paolino, D.; Cilurzo, F.; Costa, N.; Di Tullio, A.; Fresta, M.; Celia, C.; Di Marzio, L.; Locatelli, M. Simultaneous quantification of Gemcitabine and Irinotecan hydrochloride in rat plasma by using high performance liquid chromatography-diode array detector. J. Pharm. Biomed. Anal. 2018, 159, 192–199. [Google Scholar] [CrossRef]
- Kim, T.H.; Shin, S.; Kim, S.; Bulitta, J.B.; Weon, K.Y.; Joo, S.H.; Ma, E.; Yoo, S.D.; Park, G.Y.; Kwon, D.R.; et al. Alterations in Pharmacokinetics of Gemcitabine and Erlotinib by Concurrent Administration of Hyangsayukgunja-Tang, a Gastroprotective Herbal Medicine. Molecules 2017, 22, 1515. [Google Scholar] [CrossRef] [Green Version]
- Tabata, T.; Katoh, M.; Tokudome, S.; Nakajima, M.; Yokoi, T. Identification of the cytosolic carboxylesterase catalyzing the 5’-deoxy-5-fluorocytidine formation from capecitabine in human liver. Drug Metab. Dispos. 2004, 32, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Hosokawa, M. The mammalian carboxylesterases: From molecules to functions. Annu. Rev. Pharm. Toxicol. 1998, 38, 257–288. [Google Scholar] [CrossRef]
- Hosokawa, M. Structure, function and regulation of carboxylesterases. Chem. Biol. Interact. 2006, 162, 195–211. [Google Scholar]
- Sanghani, S.P.; Sanghani, P.C.; Schiel, M.A.; Bosron, W.F. Human carboxylesterases: An update on CES1, CES2 and CES3. Protein Pept. Lett. 2009, 16, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Taylor, P.; Bosron, W.F.; Sanghani, S.P.; Hosokawa, M.; Du, B.N.L. Current Progress on Esterases: From Molecular Structure to Function. Drug Metab. Dispos. 2002, 30, 488–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redinbo, M.R.; Potter, P.M. Keynote review: Mammalian carboxylesterases: From drug targets to protein therapeutics. Drug Discov. Today 2005, 10, 313–325. [Google Scholar] [CrossRef]
- Ross, M.K.; Crow, J.A. Human Carboxylesterases and Their Role in Xenobiotic and Endobiotic Metabolism. J. Biochem. Mol. Toxicol. 2010, 21, 187–196. [Google Scholar] [CrossRef]
Sample Availability: All Samples are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Cbz-dFdC (i.g. 22 mg/kg) | dFdC (i.v. 15 mg/kg) | |
|---|---|---|---|
| Cbz-dFdC | dFdC | dFdC | |
| t1/2 (h) | 9.91 ± 1.87 | 10.43 ± 0.91 | 10.56 ± 1.86 |
| AUC0–∞ (ng·h/mL) | 2691.3 ± 456.8 | 27,498.0 ± 5653.3 | 22,685 ± 4136.2 |
| Tmax (h) | 2.00 ± 0.00 | 4.00 ± 0.00 | 0.083 ± 0.00 |
| Cmax (ng/mL) | 451.1 ± 106.7 | 1656.3 ± 431.5 | 14,145 ± 4115 |
| Parameters | Disappearance of Cbz-dFdC | Appearance of dFdC | ||||
|---|---|---|---|---|---|---|
| Vmax (nmol/min/mg pro) | Km (μM) | Clint (mL/min/mg) | Vmax (nmol/min/mg pro) | Km (μM) | Clint (mL/min/mg) | |
| Rat Blood | 341.1 | 69.8 | 4.89 | 178.5 | 73.2 | 2.44 |
| Rat Plasma | 486.2 | 90.3 | 5.38 | 230.4 | 101.2 | 2.28 |
| Rat Liver Homogenate | 411.6 | 189.6 | 2.17 | 86.8 | 96.4 | 0.90 |
| Rat Liver S9 | 169.6 | 96.3 | 1.76 | 49.5 | 41.2 | 1.20 |
| Rat Kidney Homogenate | 162.7 | 147.5 | 1.10 | 44.3 | 48.9 | 0.91 |
| Rat Kidney S9 | 206.5 | 269.3 | 0.77 | 50.2 | 78.7 | 0.64 |
| Human Liver S9 | 1491.3 | 107.8 | 13.83 | 480.6 | 65.7 | 7.32 |
| Human CES1 Recombinase | 18,802.3 | 258.6 | 72.71 | 3910.4 | 107.2 | 36.48 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Wang, J.; Hao, K. A Pharmacokinetic and Pharmacodynamic Evaluation of the Anti-Hepatocellular Carcinoma Compound 4-N-Carbobenzoxy-gemcitabine (Cbz-dFdC). Molecules 2020, 25, 2218. https://doi.org/10.3390/molecules25092218
Sun Y, Wang J, Hao K. A Pharmacokinetic and Pharmacodynamic Evaluation of the Anti-Hepatocellular Carcinoma Compound 4-N-Carbobenzoxy-gemcitabine (Cbz-dFdC). Molecules. 2020; 25(9):2218. https://doi.org/10.3390/molecules25092218
Chicago/Turabian StyleSun, Yilin, Jiankun Wang, and Kun Hao. 2020. "A Pharmacokinetic and Pharmacodynamic Evaluation of the Anti-Hepatocellular Carcinoma Compound 4-N-Carbobenzoxy-gemcitabine (Cbz-dFdC)" Molecules 25, no. 9: 2218. https://doi.org/10.3390/molecules25092218