1. Introduction
The pursuit of environmental protection and the implementation of sustainability have become an important endeavour in current organic chemistry [
1,
2,
3]. However, in many cases, synthetic protocols utilize catalytic materials that rely on rare, expensive, toxic or harmful metal components [
4]. Truly sustainable and economically reliable synthetic methodologies must be free of such elements and should ideally rely on cost-efficient, nontoxic and readily available catalysts [
5]. In this manner, bismuth and its compounds are of significant current interest [
6,
7]. This is mainly due to the fact that, in contrast to other heavy metals, bismuth and its compounds are neither toxic nor harmful for the environment, while being stable to air, easy to handle and inexpensive [
8,
9,
10]. Importantly, bismuth(III) compounds are known to exhibit remarkable Lewis acid character, and they are capable of activating diverse substrates, such as alcohols, amines, olefins and alkynes [
11,
12,
13,
14,
15]. Recent examples for the catalytic utility of bismuth(III) compounds include oxidations, deprotections, acetal formations and, esterifications, as well as various carbon–carbon bond formations and heterocyclizations [
16,
17,
18,
19,
20,
21,
22,
23,
24]. Catalytic sources for such reactions are generally soluble salts, such as Bi(OTf)
3, BiBr
3, BiCl
3 and Bi(NO
3)
3·5H
2O [
16], whereas heterogeneous bismuth(III) catalysts are extremely scarce [
25]. Although most of the typical bismuth(III) salts are relatively cheap, the application of heterogeneous sources of the catalytic metal would involve obvious benefits, from environmental and practical aspects (e.g., facile product isolation and catalyst reusability).
The Markovnikov-type hydration of alkynes is a well-known process yielding valuable carbonyl compounds [
26,
27]. Its synthetic utility can be explained by the ease of introduction of the acetylene moiety, which can practically be regarded as a carbonyl equivalent upon unmasking by hydration [
28]. Traditional alkyne hydrations employ catalytic amounts of mercuric salts in strongly acidic media, which involves obvious environmental concerns [
27]. Recently, numerous transition metal salts and complexes were found applicable as catalysts for Markovnikov-type alkyne hydration—among these, gold, silver, platinum, palladium, rhodium and ruthenium compounds [
29,
30,
31,
32,
33,
34,
35,
36,
37,
38]. Although, these represent great progress compared to the classical methodologies, such reactions are generally promoted by soluble catalytic sources typically in the presence of various ligands. This fact, along with the high price of the catalytic metal, should be considered as a significant drawback.
Bismuth subnitrate (Bi
5O(OH)
9(NO
3)
4) is a commercially available bismuth(III) compound that bears significant medical uses (e.g., as an antidiarrheic agent) [
39]. It can readily be prepared by the controlled thermal decomposition of Bi(NO
3)
3·5H
2O [
40], and it is practically insoluble in most typical organic solvents. In spite of its beneficial properties, such as ready availability, nontoxicity and low price, it has scarcely been investigated as a heterogeneous bismuth catalyst in organic synthesis [
41,
42]. Earlier, we reported that various soluble bismuth salts are useful as homogeneous catalysts in alkyne hydration [
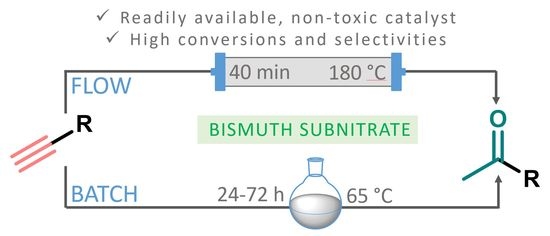
20]. We speculated that bismuth subnitrate may be applicable as a heterogeneous source for catalytic bismuth(III) and that it may prove useful as an efficient heterogeneous catalyst for Markovnikov-type alkyne hydrations. The application of heterogeneous catalysts in continuous flow systems have received an upsurge of interest, which is due to numerous benefits, such as facile catalyst handling, recycling and reuse, as well as simple product isolation [
43,
44,
45,
46,
47,
48,
49]. Additionally, in loaded catalyst columns, continuous substrate streams interact with a superstoichiometric amount of catalyst species, which enhances the reaction rates considerably [
50,
51], while the improved control over temperature and residence time ensures a high selectivity and low waste generation [
52,
53,
54,
55]. We therefore intended to study the reactions not only under the traditional batch conditions but, also, within a continuous flow packed-bed reactor environment. Our results are presented herein.
2. Results and Discussion
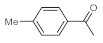
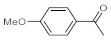
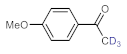
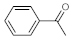
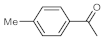
As the starting point of our study, the catalytic activity of different bismuth(III) compounds was compared using the Markovnikov-type hydration of
p-methoxyphenylacetylene as the model reaction (
Table 1). The reaction mixture containing the alkyne (1.0 M), together with 15 mol% of the selected catalyst, was refluxed for 24 h in MeOH as the solvent. Having confirmed the lack of conversion without any catalyst present (entry 1), we were delighted to find that, in the presence of bismuth subnitrate as the catalyst, the corresponding methyl ketone was formed in a quantitative and selective reaction (entry 2). Bi(OTf)
3 also furnished the quantitative conversion; however, dimethyl acetal
2 was detected as a side product to an extent of 12% (entry 3). According to the reaction mechanism suggested earlier [
56,
57],
2 is, in fact, an intermediary product, which is formed by the hydroalkoxylation of the alkyne with methanol;
2 is then hydrolyzed to yield methyl ketone
1 as the desired product. BiBr
3 proved less reactive as a catalyst and furnished only 45% conversion, along with some notable amount (8%) of
2 formed as a side product (entry 4). Bi(OAc)
3 and Bi
2O
3 proved inactive as a catalyst in the model reaction (entries 5 and 6).
Next, the effects of the most important reaction conditions were investigated carefully. As concerns the reaction time, it was found that 24 h is necessary for completion of the model reaction under reflux conditions in MeOH (further conditions: 15 mol% catalyst loading and 1 M substrate concentration). Shorter reaction times gave lower conversions—for example, 73% in the case of 12 h and 18% in the case of 3 h (
Table 2, entries 1–5). Upon investigating the effects of catalyst loading (entries 6–8), the best results were achieved with 15 mol%; however, with only 2 mol% of bismuth subnitrate catalyst present, an acceptable conversion of 62% could still be achieved. Importantly, in the cases of 2 and 5 mol% catalyst loading, dimethyl acetal
2 appeared as a side product to an extent of 29% and 12%, respectively. Heating at reflux temperature was found to be necessary for efficient alkyne hydration, since only traces of product formation occurred at room temperature (entry 9). Upon increasing the substrate concentration to 2 M, a notable decrease of the conversion and selectivity occurred (entry 10), and 1 M was therefore considered as an optimum value later on.
Besides MeOH, the model reaction was attempted using H
2O and different alcohols as the reaction medium (
Table 3). In EtOH and
iPrOH, a drastic reactivity decrease occurred that may be explained by the increased steric hindrance compared with MeOH during the hydroalkoxylation of the alkyne (entries 1–3) [
57]. The hydration process furnished methyl ketone
1 in H
2O also, but in this case, the conversion decreased to 69% (entry 4). In dry MeOH as the solvent, dimethyl acetal
2 was detected in the product solution to an extent of 8% (entry 5). This corroborates that trace amounts of water are necessary for effective hydrolysis of the dimethyl acetal intermediate into the desired methyl ketone product. Water traces may come from the moisture present in the air, from the solvent applied and/or from the catalyst itself. Additionally, it has been discussed earlier that bismuth(III) may bear some catalytic activity in the hydrolysis of the acetal intermediate [
20], thus explaining the appearance of
2 upon decreasing the catalyst loading to 2 or 5 mol% (
Table 2, entries 7 and 8).
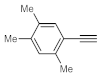
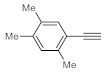
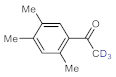
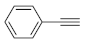
After acquiring sufficient data on the effects of the reaction conditions, the reactivity of various alkynes was next explored. Amongst aromatic alkynes, phenylacetylene and its
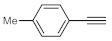
p-methyl-,
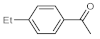
p-ethyl and
p-methoxy-substituted derivatives, as well as multi-methyl/methoxy-substituted phenylacetylenes, exhibited excellent reactivities with conversions in the range of 60–100% (
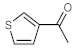
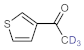
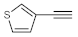
Table 4, entries 1–4, 6 and 7). Furthermore, 3-ethynylthiophene, ethynylferrocene and ethyl propiolate also proved as good substrates and gave conversions in the range of 74–100% (entries 8–10).
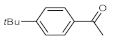
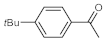
p-(tert-butyl)phenylacetylene gave only a moderate conversion, which is presumably due to steric effects (entry 5). In all the reactions investigated, the desired methyl ketones were formed selectively. Note that isolated yields were also determined in some representative instances.
Deuterium-labeled compounds have numerous applications—for example, as analytical standards or for the evaluation of the metabolic pathways or in tracer studies to investigate pharmacokinetics, catalytic cycles and reaction pathways [
58,
59,
60]. The incorporation of deuterium into various organic compounds has therefore gained significant importance in medicinal, analytical and pharmaceutical chemistry [
61]. In most cases, deuteration methods involve halogen/D exchange and are typically mediated by strong bases, which severely limits the functional group tolerance [
62]. Moreover, catalytic H/D exchange reactions are also known; however, these often involve selectivity and/or environmental issues [
63,
64,
65]. Inspired by these limitations, we explored a facile methodology to prepare valuable trideuteromethyl ketones by simply switching the reaction medium from MeOH to MeOH-
d4 in bismuth subnitrate-catalyzed alkyne hydrations. As shown in
Table 5, high conversion and 100% chemoselectivity were achieved in all the reactions investigated, and deuteration was highly favored over incidental hydrogen incorporation, as demonstrated by deuterium contents of >99%.
With convincing results under batch conditions in our hands, we next attempted to translate the bismuth subnitrate-catalyzed alkyne hydration into a practical continuous flow process. Therefore, a simple setup was assembled that consisted of a stainless steel HPLC column as a catalyst bed and a 10-bar backpressure regulator (BPR), which enabled the safe overheating of the reaction mixture. The column encompassed 0.9 g of bismuth subnitrate as catalyst, and the 0.2-M alkyne solution in MeOH was continuously pumped by using an HPLC pump. Similarly, as in the batch experiments, the Markovnikov-type hydration of
p-methoxyphenylacetylene was chosen as a model reaction. Firstly, the effect of the reaction temperature was investigated at a fixed flow rate of 50 µL min
−1 (24-min residence time). A gradual improvement of conversion was found from 37% to 88%, with increasing temperatures in the range 120–180 °C (
Figure 1A). Considering that in batch, a reaction time of 24 h was required for a quantitative reaction, 88% conversion within 12 min residence time should be regarded as a significant improvement. Next, the effects of different residence times were explored by employing different flow rates at 180 °C reaction temperature (
Figure 1B). To our delight, upon decreasing the flow rate to 30 µL min
−1 (40 min residence time), quantitative and selective hydration occurred into the desired acetophenone derivative. Shorter residence times gave lower conversions—for example, 49% in case of 6 min (200 µL min
−1 flow rate). Gratifyingly, the formation of intermediary dimethyl acetal was not detected in any of these experiments.
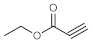
After getting familiar with the effects of the key flow conditions, a range of alkynes were next submitted to bismuth subnitrate-catalyzed hydration at 180 °C and 30 µL min
−1 flow rate (
Table 6). Despite the fact that the 40 min residence time on the catalyst bed was reasonably shorter than the batch reaction times applied earlier (24–72 h), the flow reactions worked comparably well to the corresponding batch experiments. Excellent conversion (71–100%) and chemoselective acetophenone formation were observed in reactions of phenylacetylene and most of its mono- and multisubstituted derivatives investigated (entries 1–6). Similarly as in batch, only
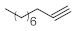
p-(tert-butyl)phenylacetylene gave moderate conversion among phenylacetylene derivatives investigated (entry 4). Gratifyingly, ethynylferrocene, 3-ethynylthiophene and ethyl propiolate, as well as dec-1-yne, were also tolerated well by the flow process, and their hydration furnished excellent conversions (90–100%) and selective formation of the desired methyl ketones (entries 7–10).
Finally, the stability of the packed-bed system was tested. For this, the Markovnikov-type hydration of
p-methoxyphenylacetylene was performed continuously for 15 h under optimum flow conditions (180 °C, 30 µL min
−1 flow rate). During this period, five samples were taken and analyzed separately. To our delight, conversion was steady around 95% in the first 12 h of the experiment, and only a small decrease to 87% occurred in the last sample collected after 15 h (
Figure 2). Importantly, the chemoselectivity towards acetophenone
1 was 100% in all samples investigated. As the result of this experiment, 725 mg of
1 was isolated, which corresponded to a yield of 89%. The bismuth content of the catalyst used during scale-out (including all washing cycles) showed a 3.1% decrease as compared with an unused batch of bismuth subnitrate, indicating the presence of some metal leaching under the comparatively harsh reaction conditions applied.






{kind=link}
{kind=link}
{kind=link}