
Thiazole Ring—A Biologically Active Scaffold



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
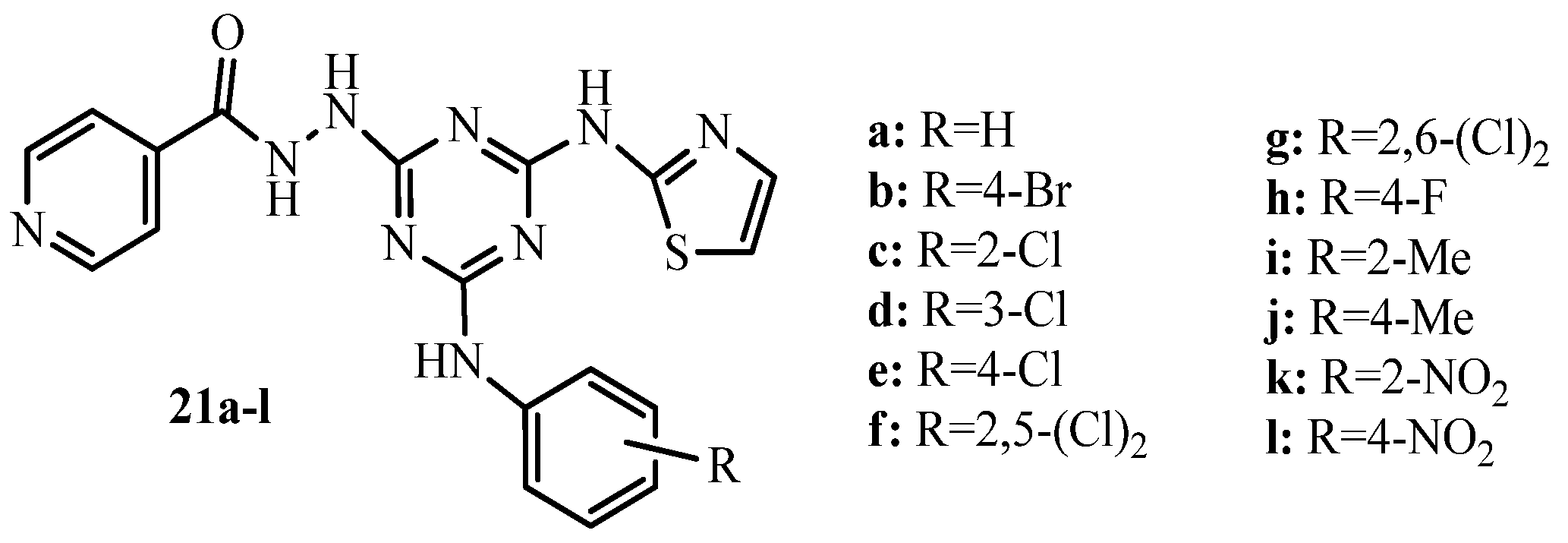{kind=link}
{kind=link}
{kind=link}
{kind=link}
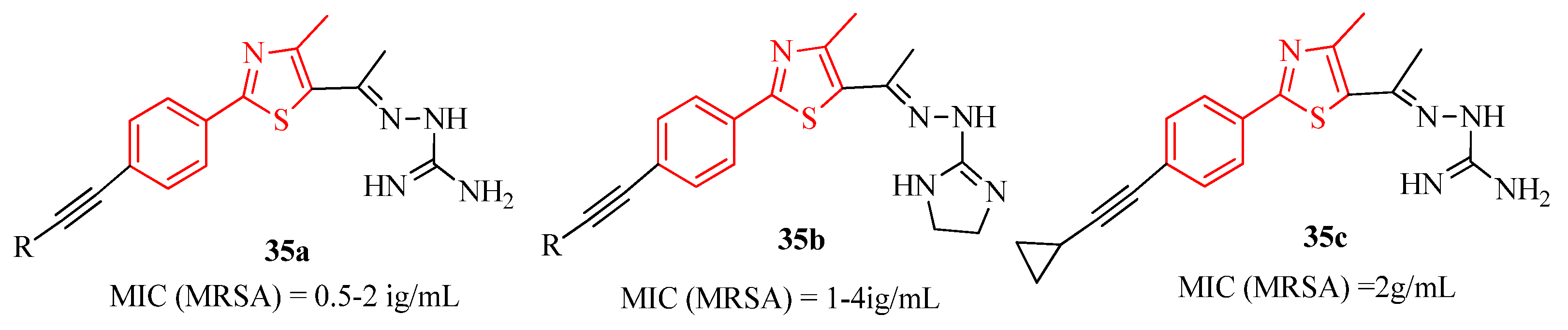{kind=link}
{kind=link}
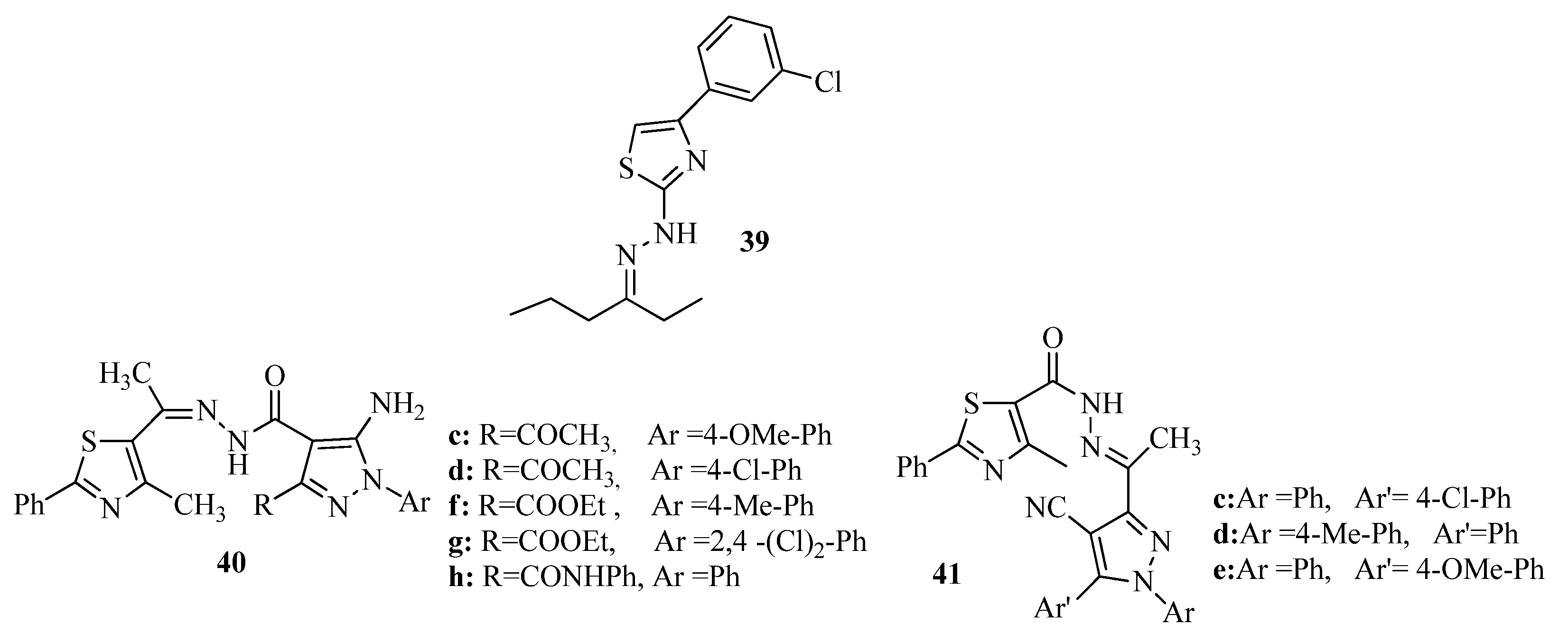{kind=link}
{kind=link}
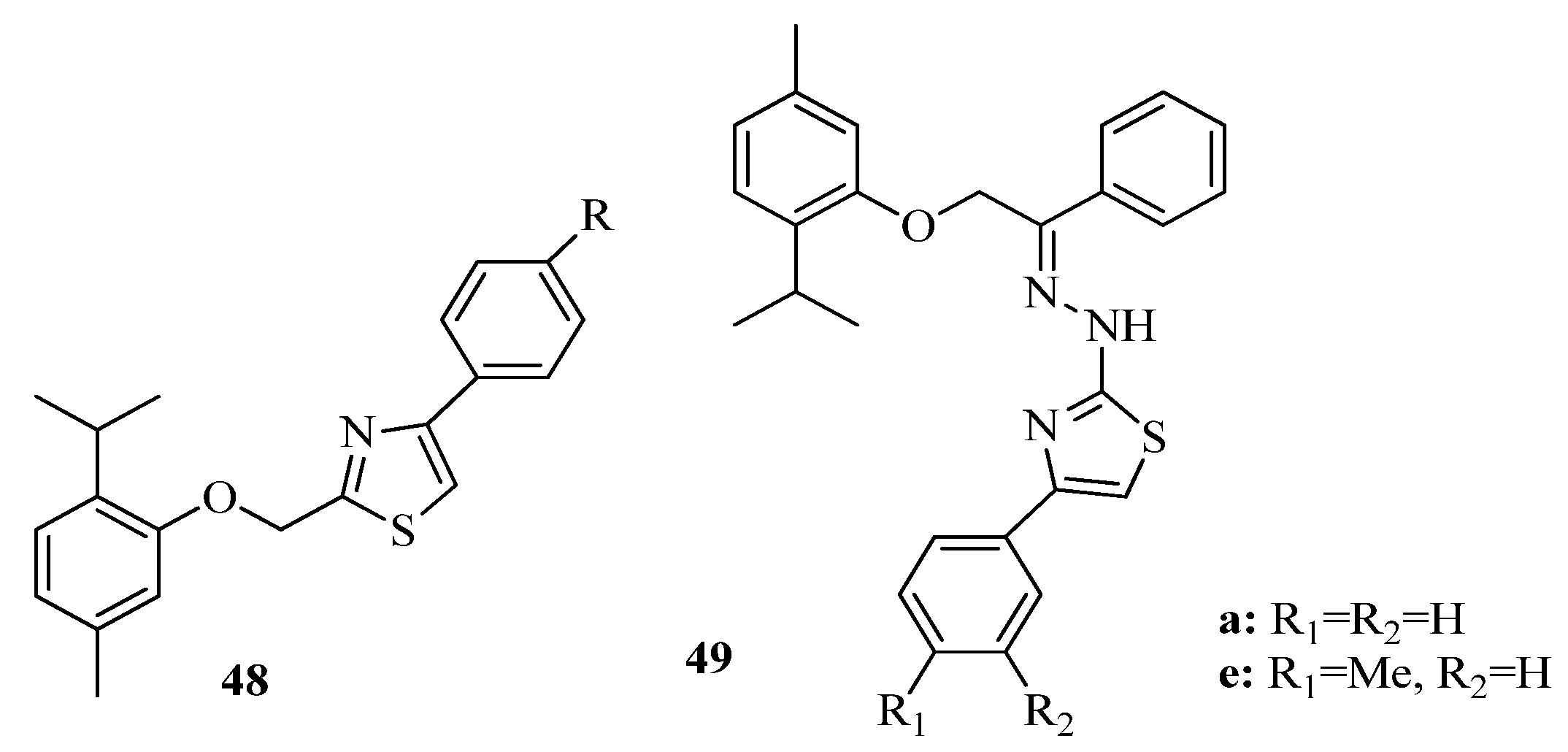{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
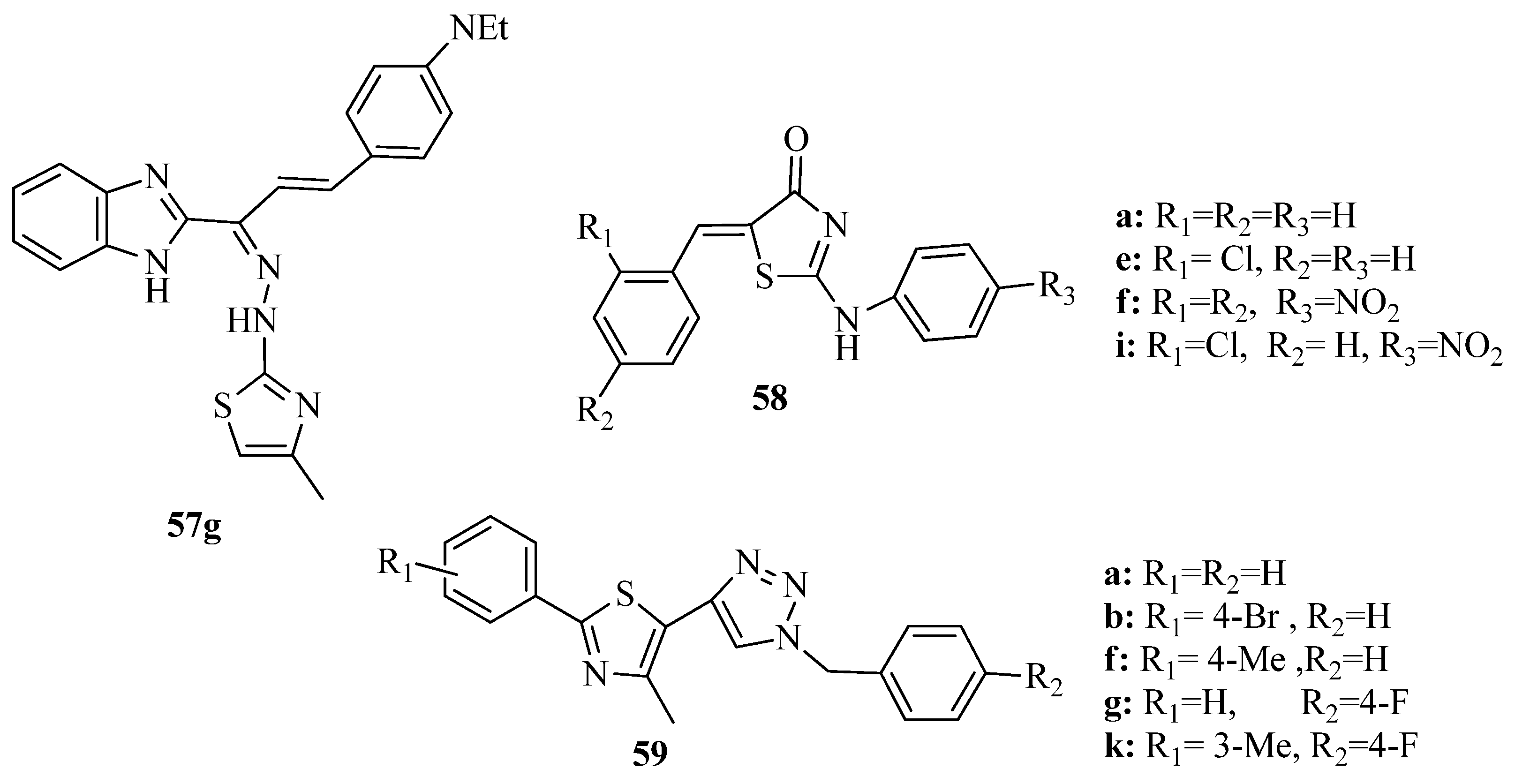{kind=link}
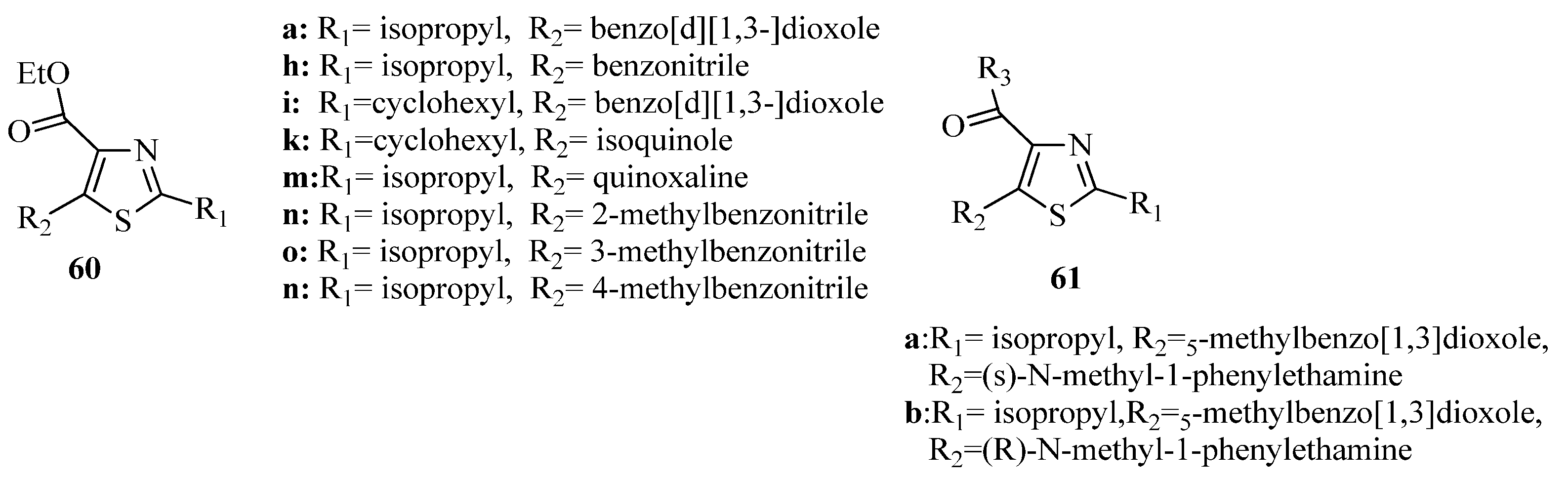{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
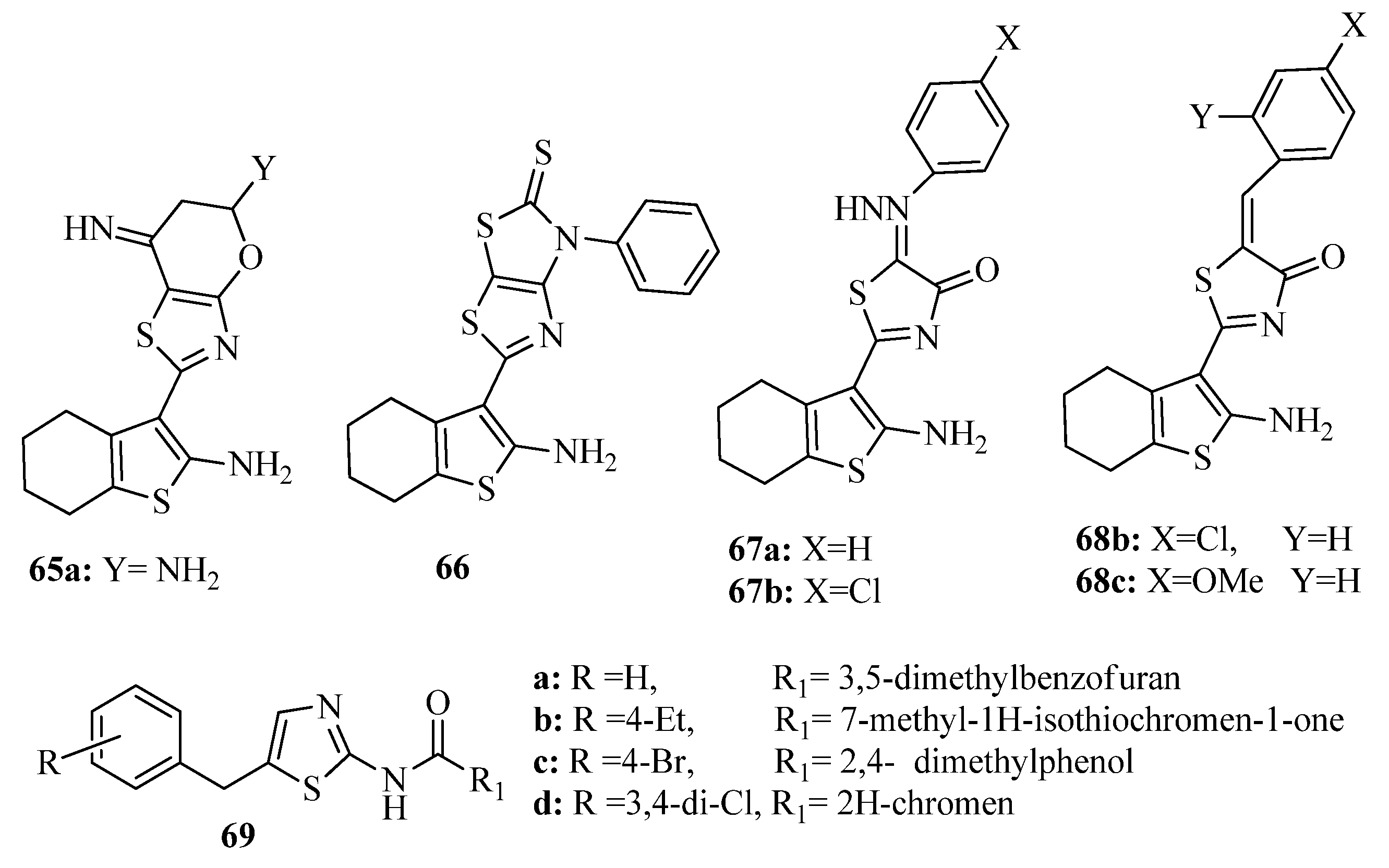
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
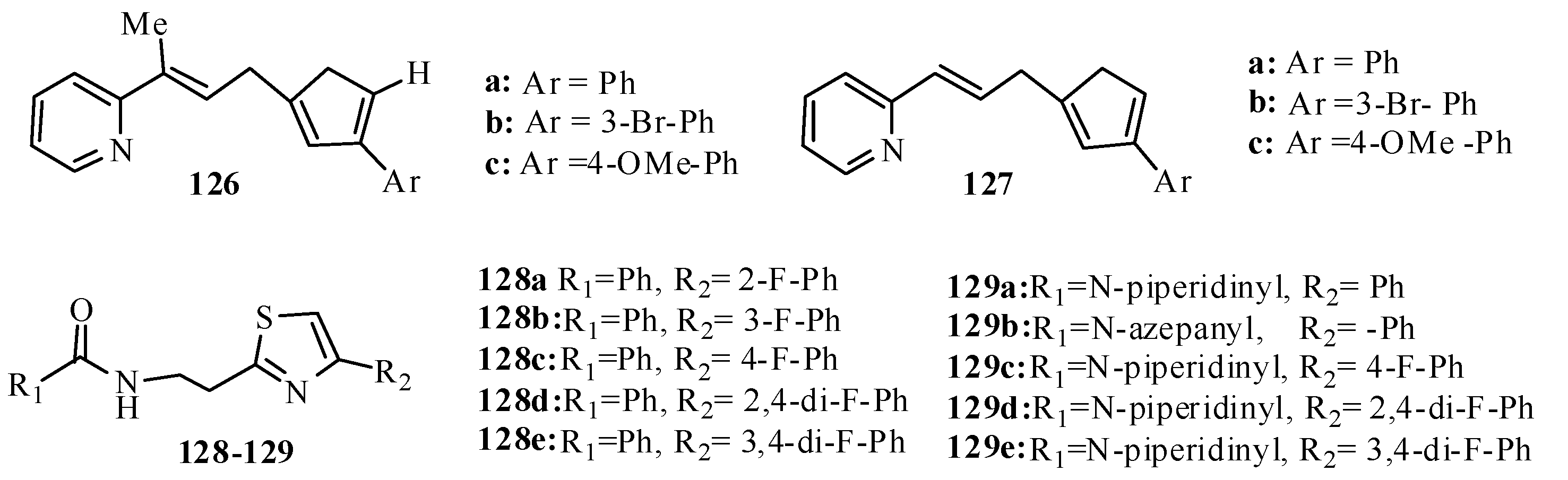{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
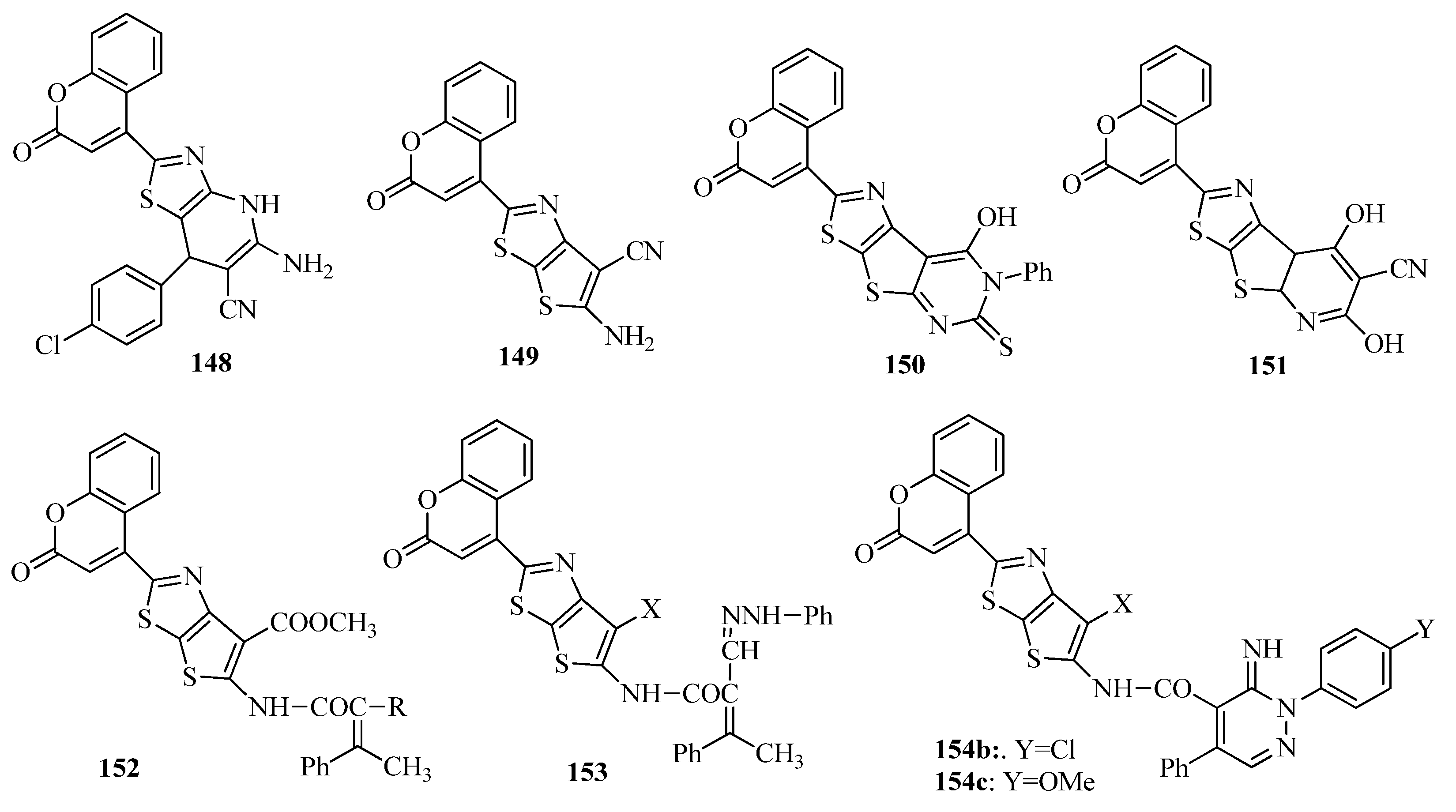{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
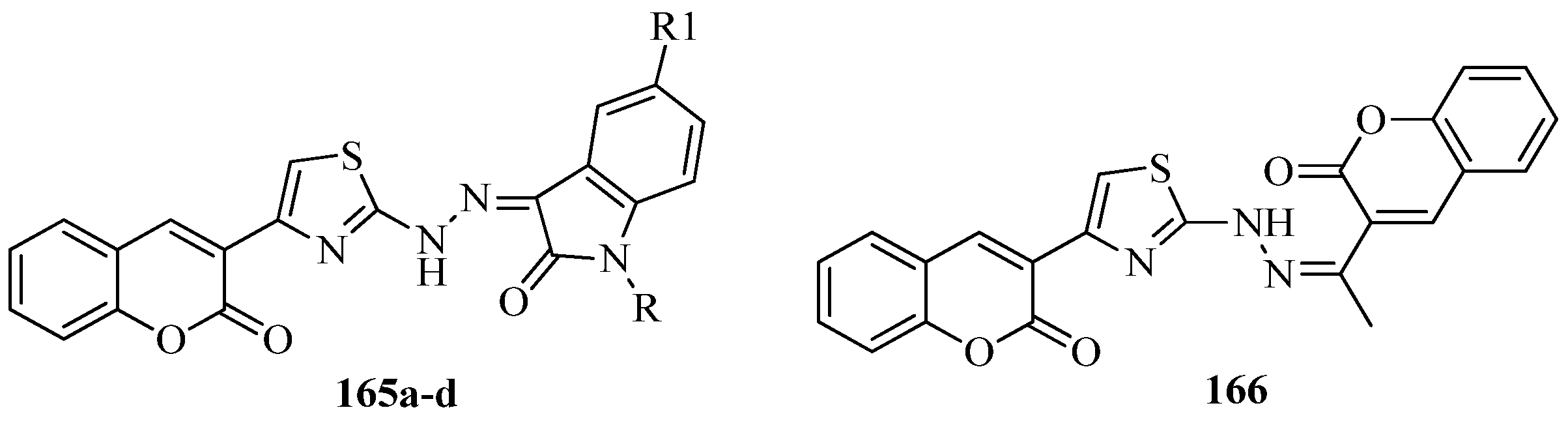{kind=link}
{kind=link}
{kind=link}
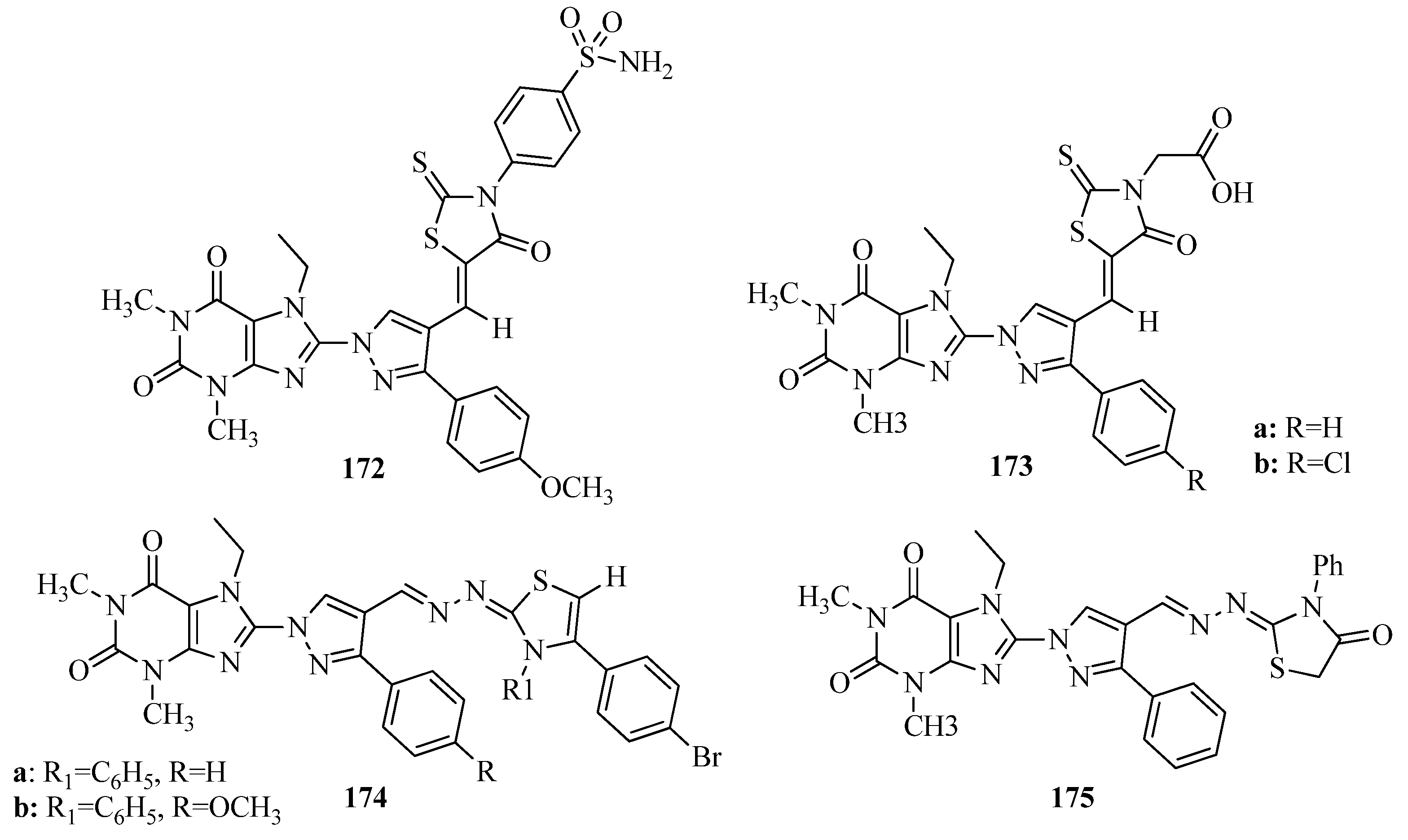{kind=link}
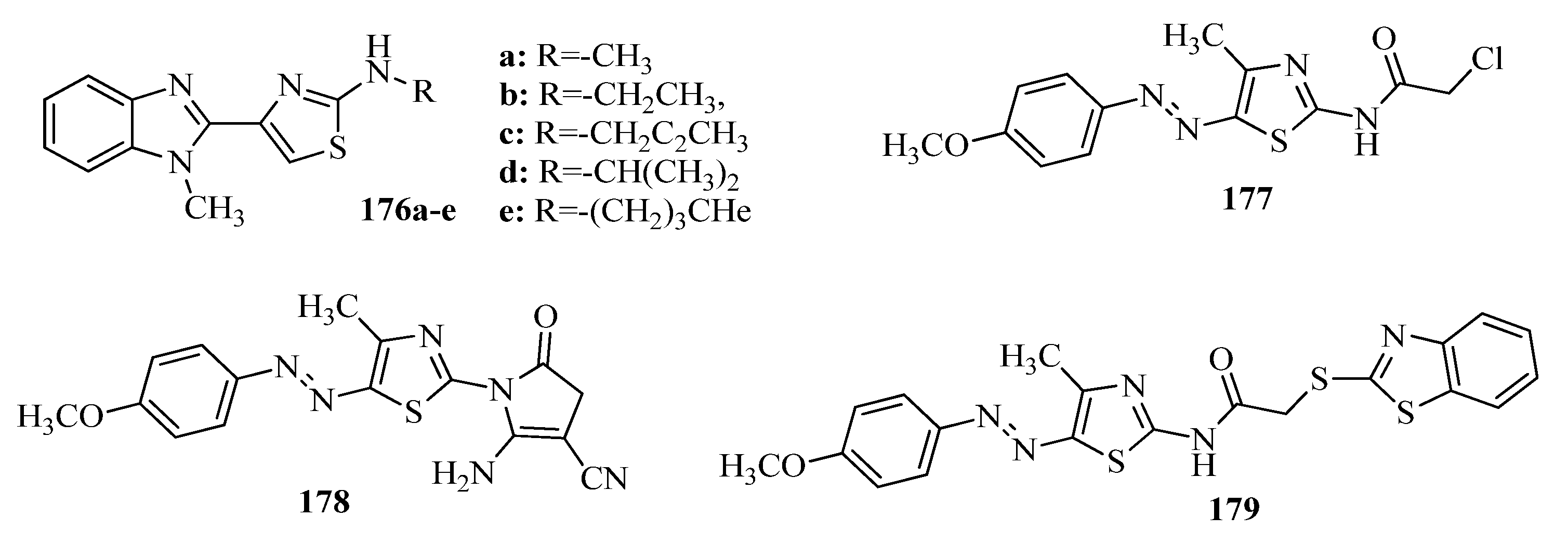{kind=link}
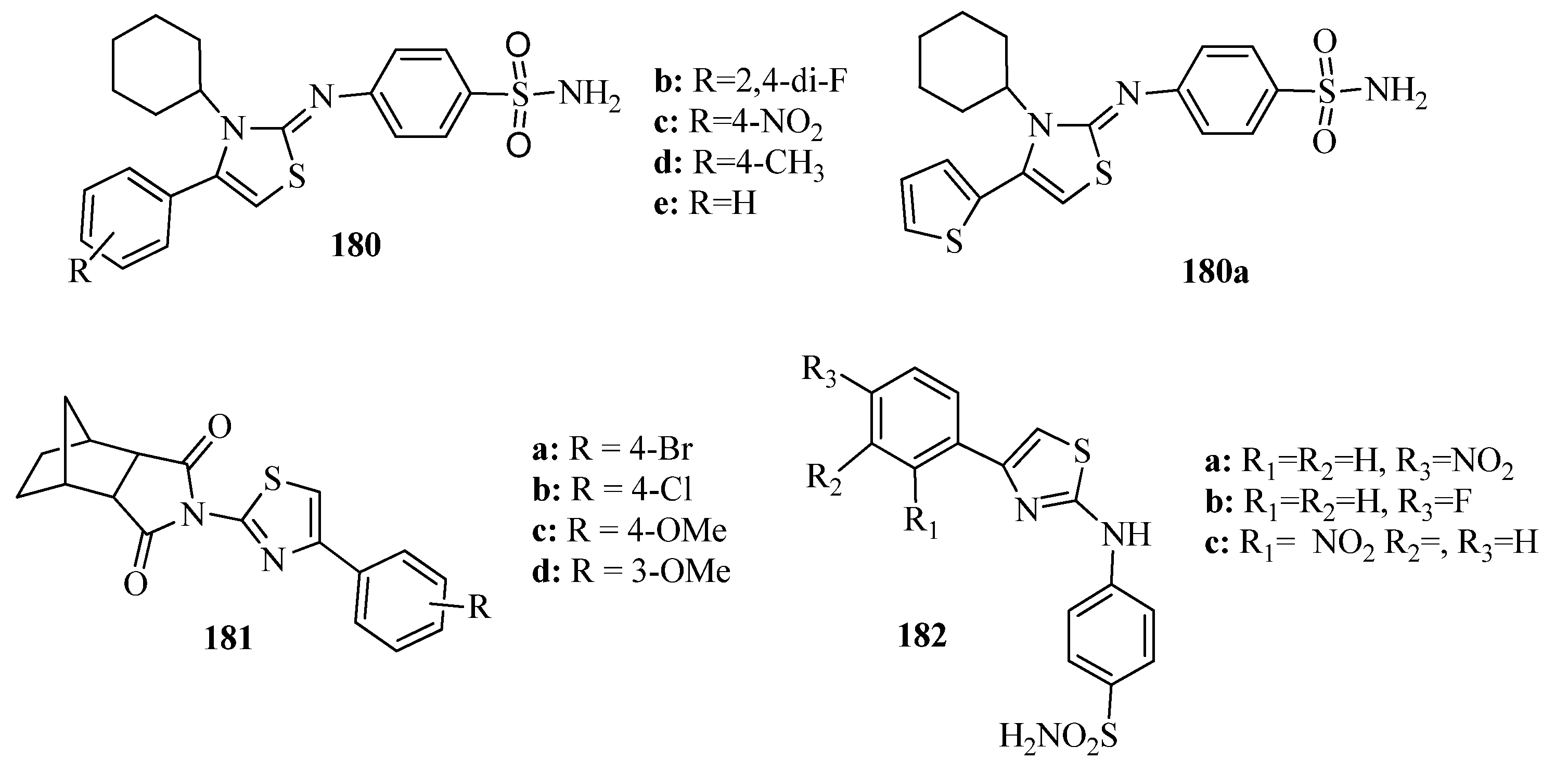{kind=link}
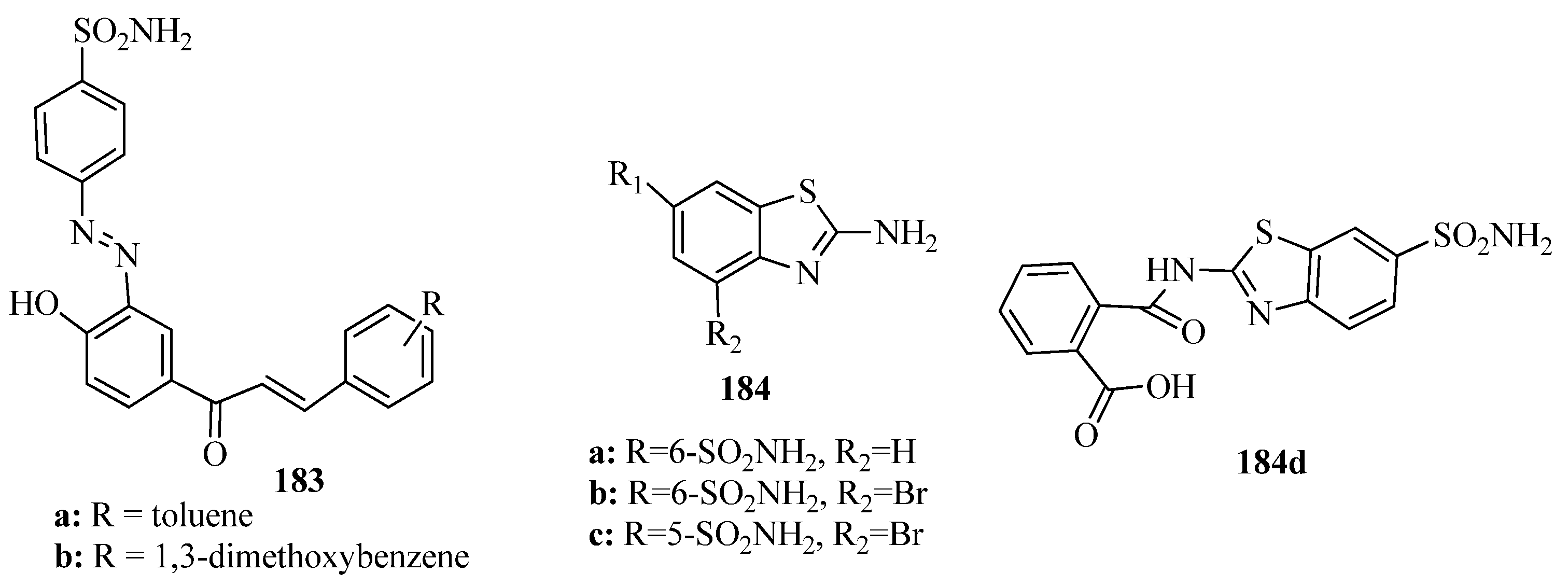{kind=link}
{kind=link}
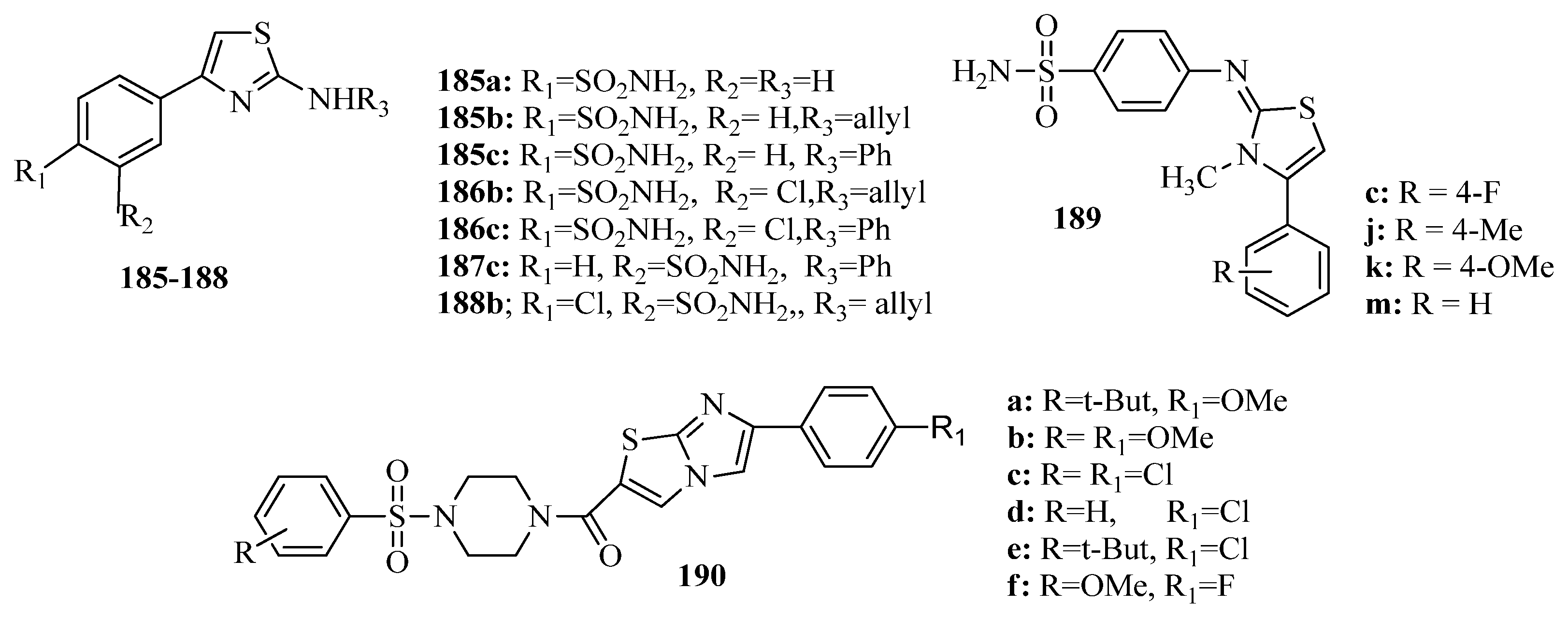
{kind=link}
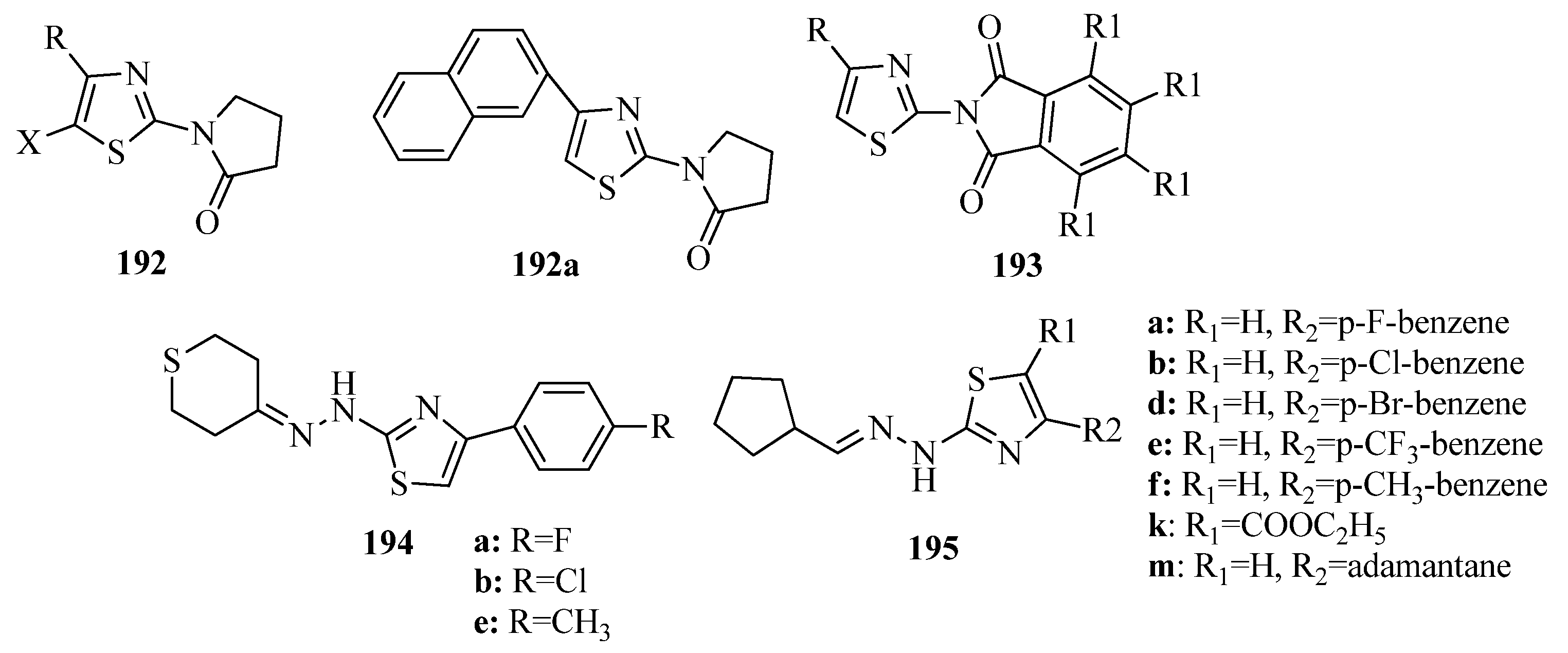{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
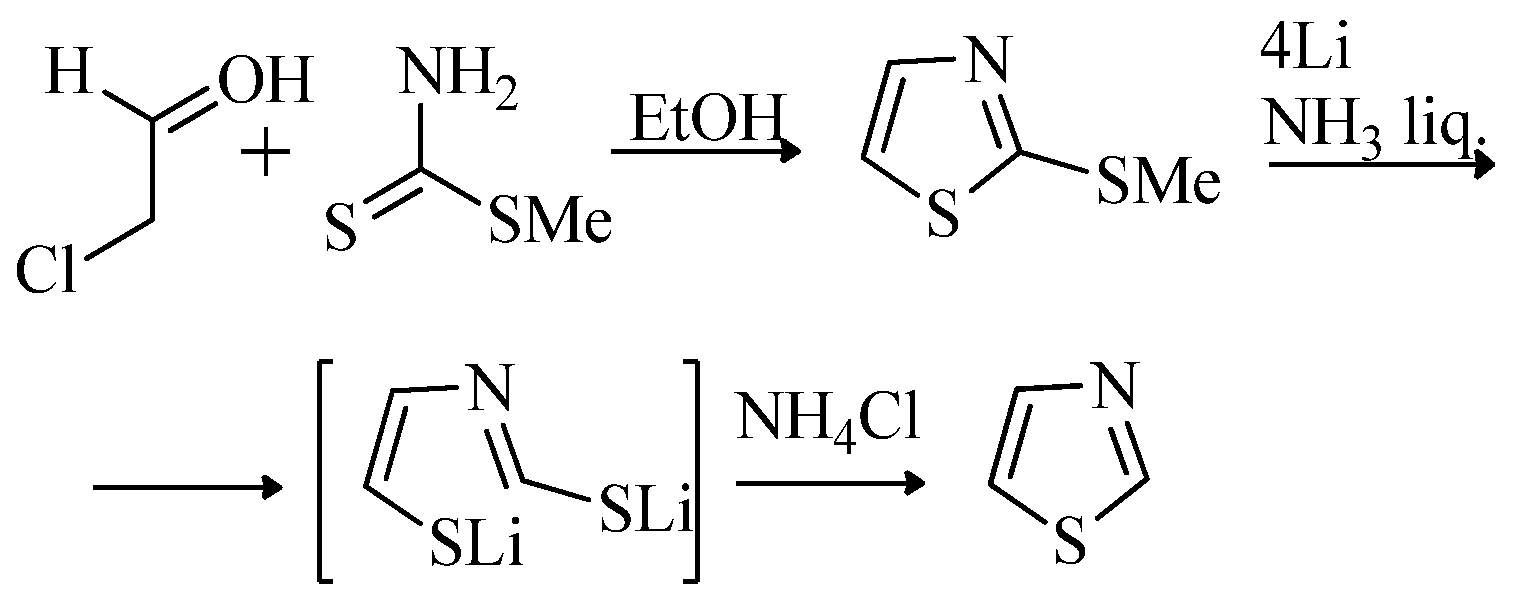{kind=link}
{kind=link}
{kind=link}
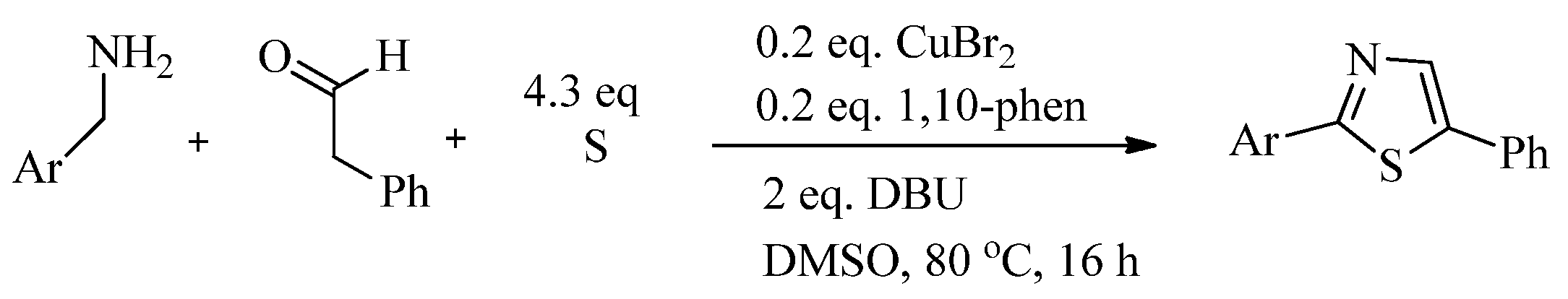{kind=link}
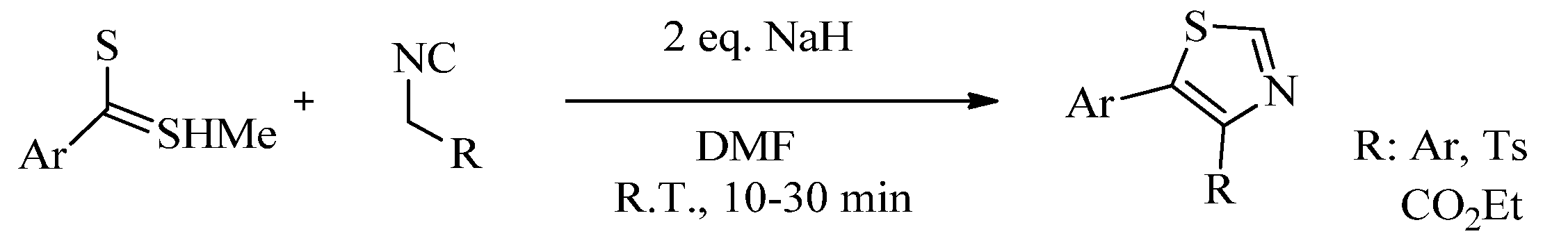{kind=link}
{kind=link}
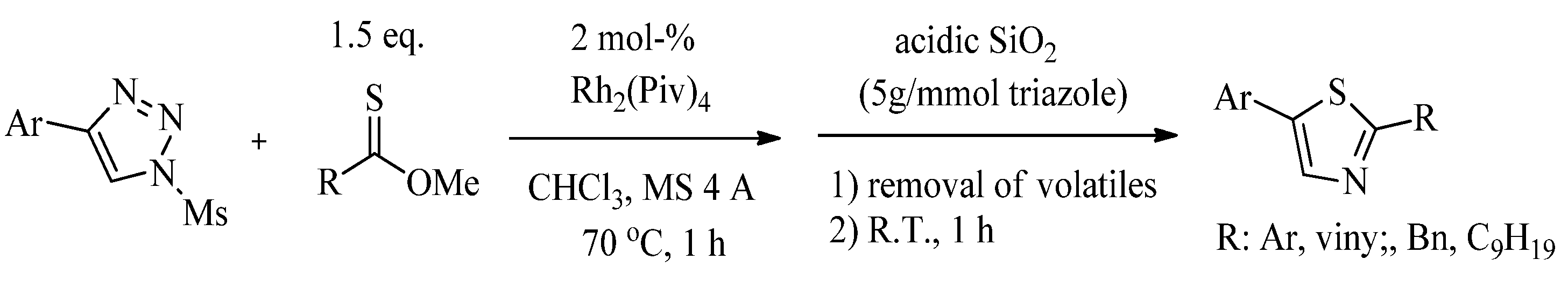{kind=link}
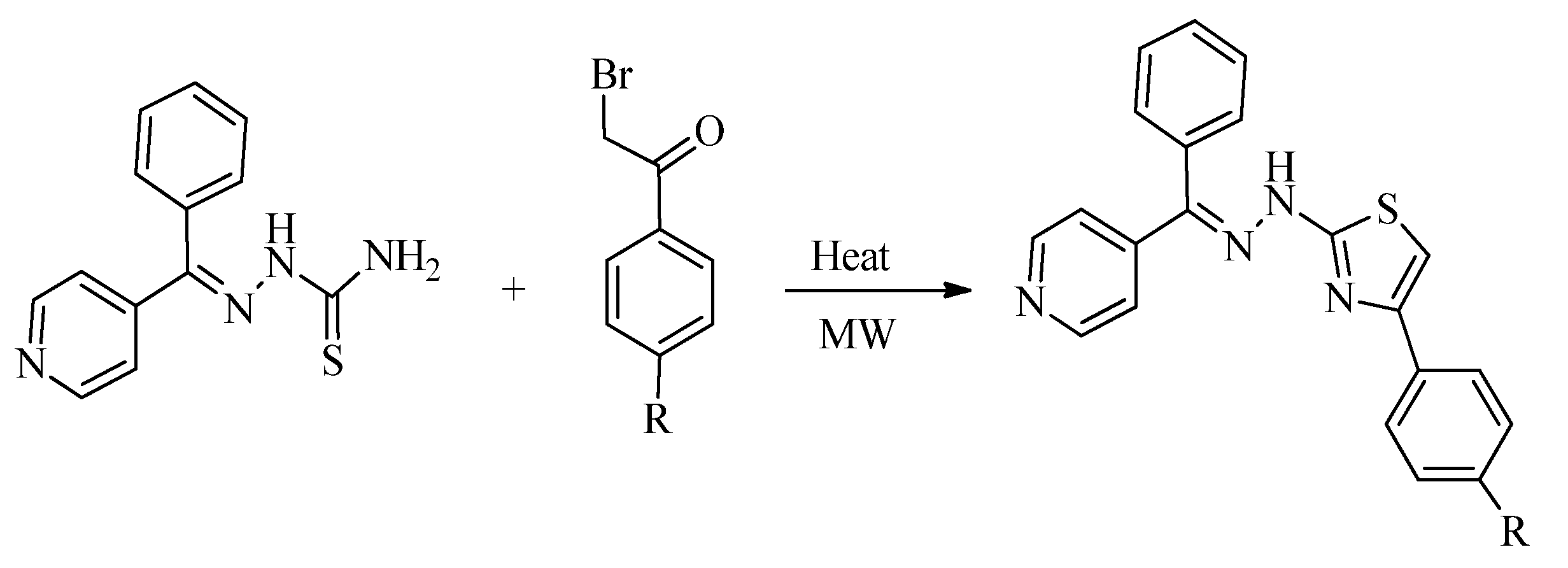{kind=link}
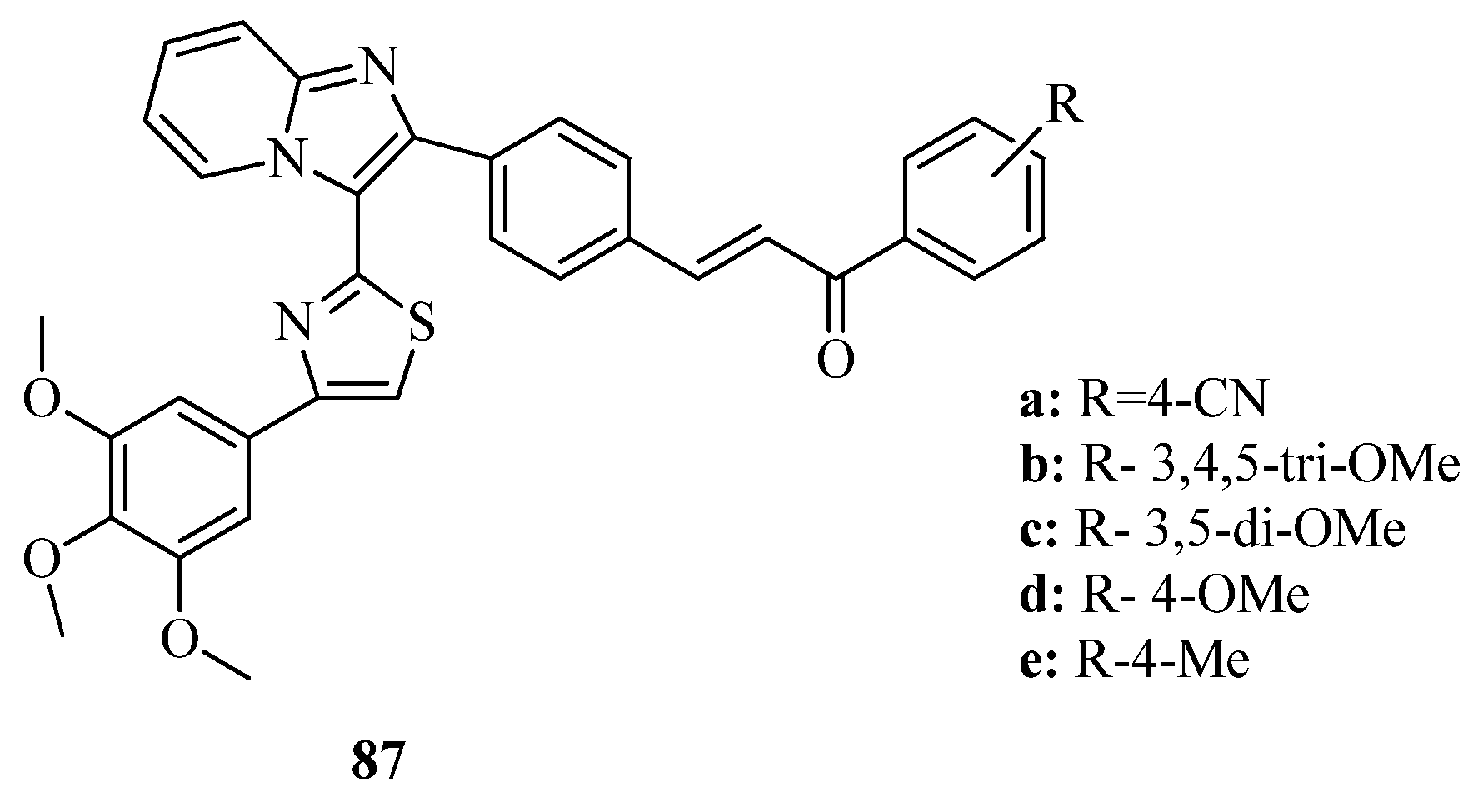
{kind=link}
{kind=link}
{kind=link}
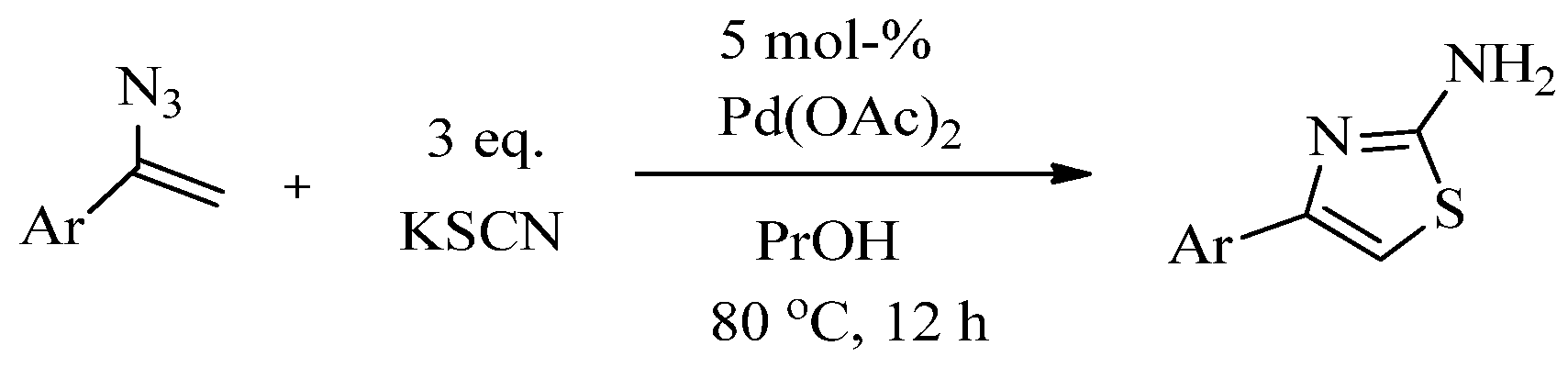{kind=link}
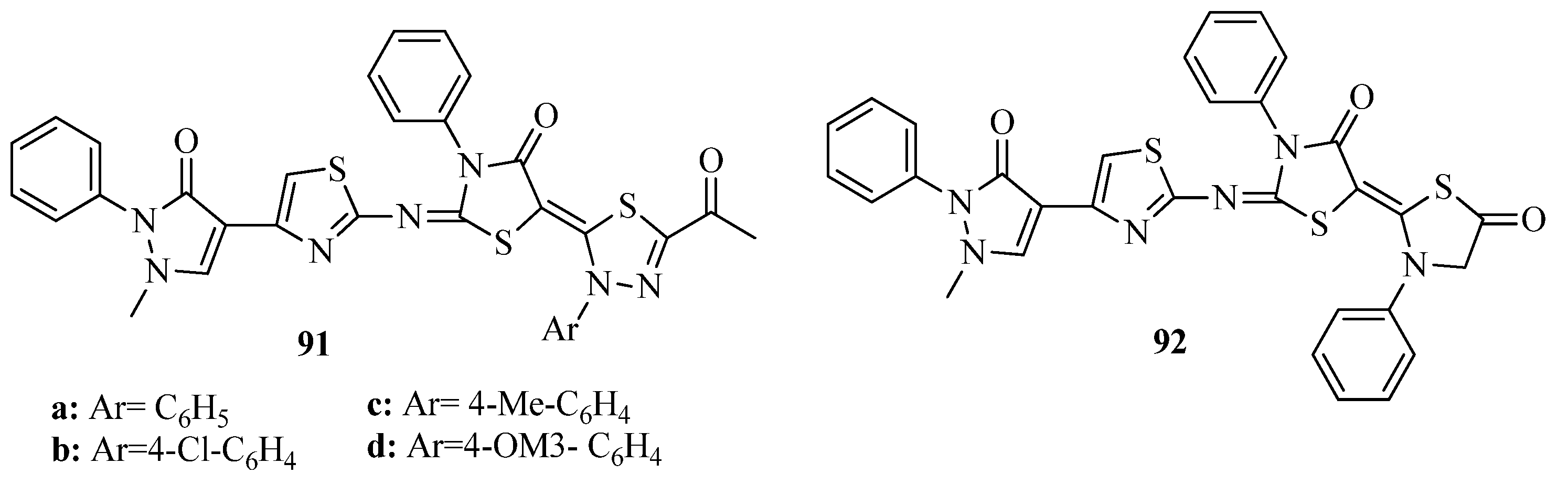
{kind=link}
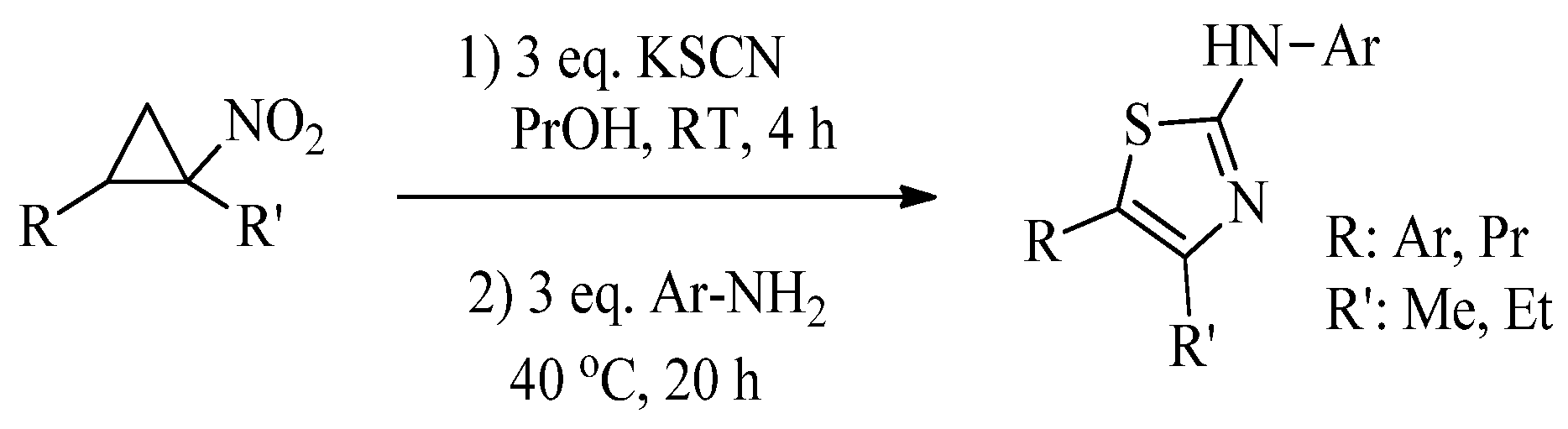{kind=link}
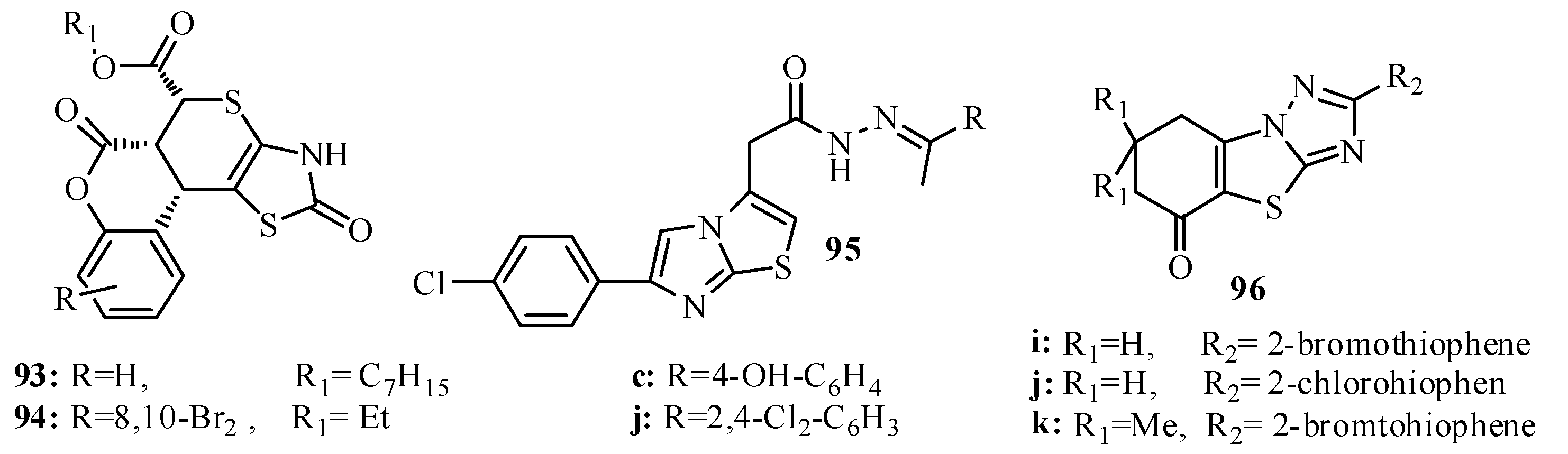
{kind=link}
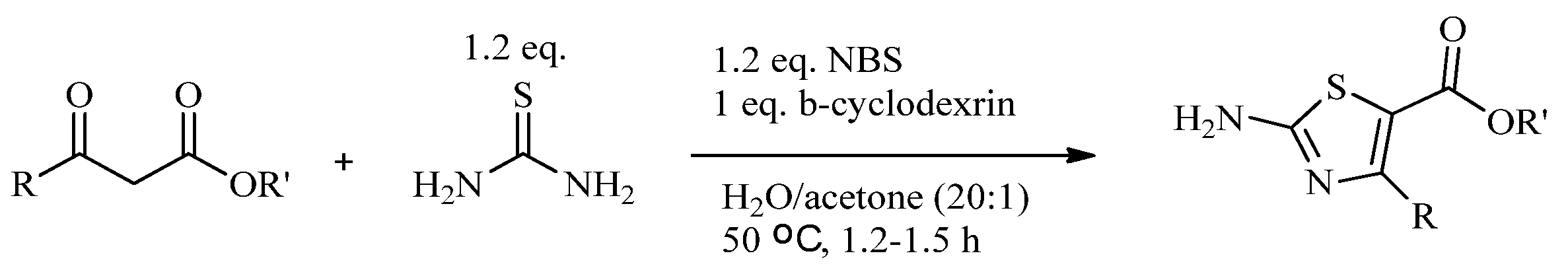{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Thiazole Derivatives
2.1. Chemistry of Thiazole Derivatives
2.2. Synthesis of 2-Aminothiazoles
3. Biological Activities of Thiazoles
3.1. Thiazole Derivatives as Antimicrobial Agents
3.2. Antituberculosis
3.3. Thiazole Derivatives as Anticancer Agents
3.4. Thiazole Derivatives as Antiviral Agents
Thiazole Derivatives as Anti-HIV Agents
3.5. Thiazole Derivatives as Antidiabetic Agents
3.5.1. Protein Tyrosine Phosphatase 1 B (PTP1B) Inhibitors
3.5.2. Dipeptidyl Peptidase-4 (DPP-4) Inhibitors
3.5.3. Aldose Reductase (ALR) Inhibitors
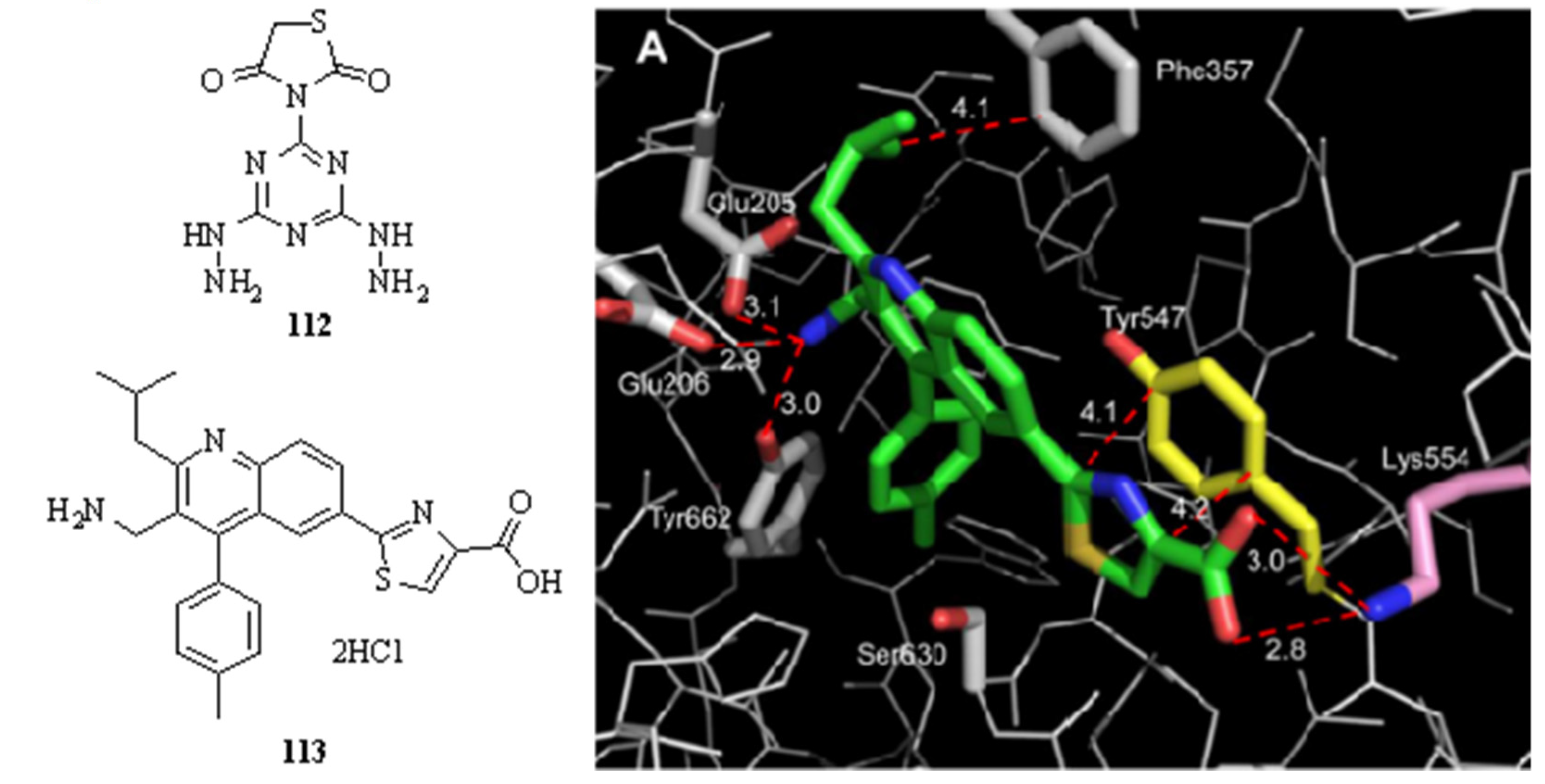
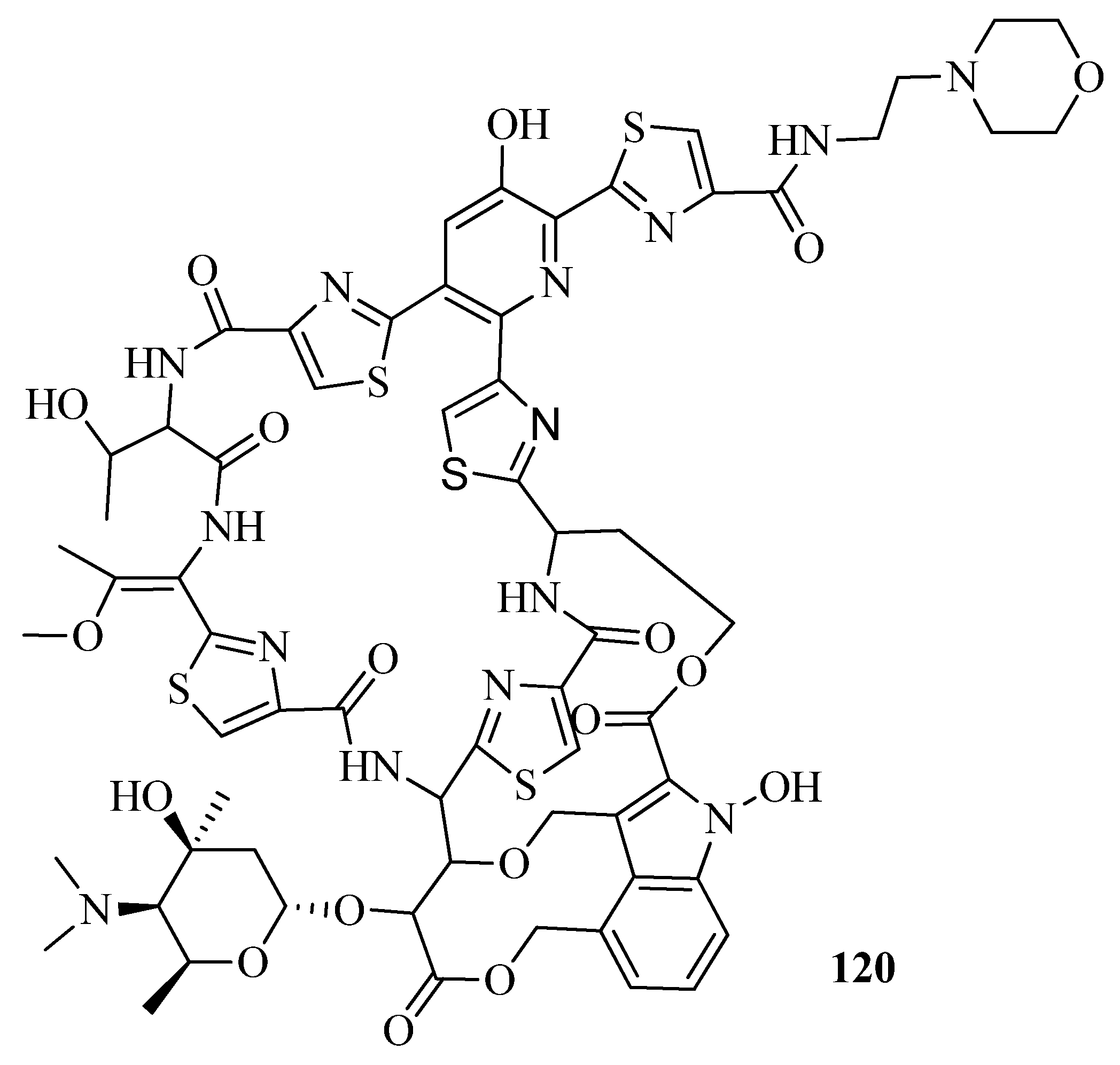
3.6. Neglected Diseases
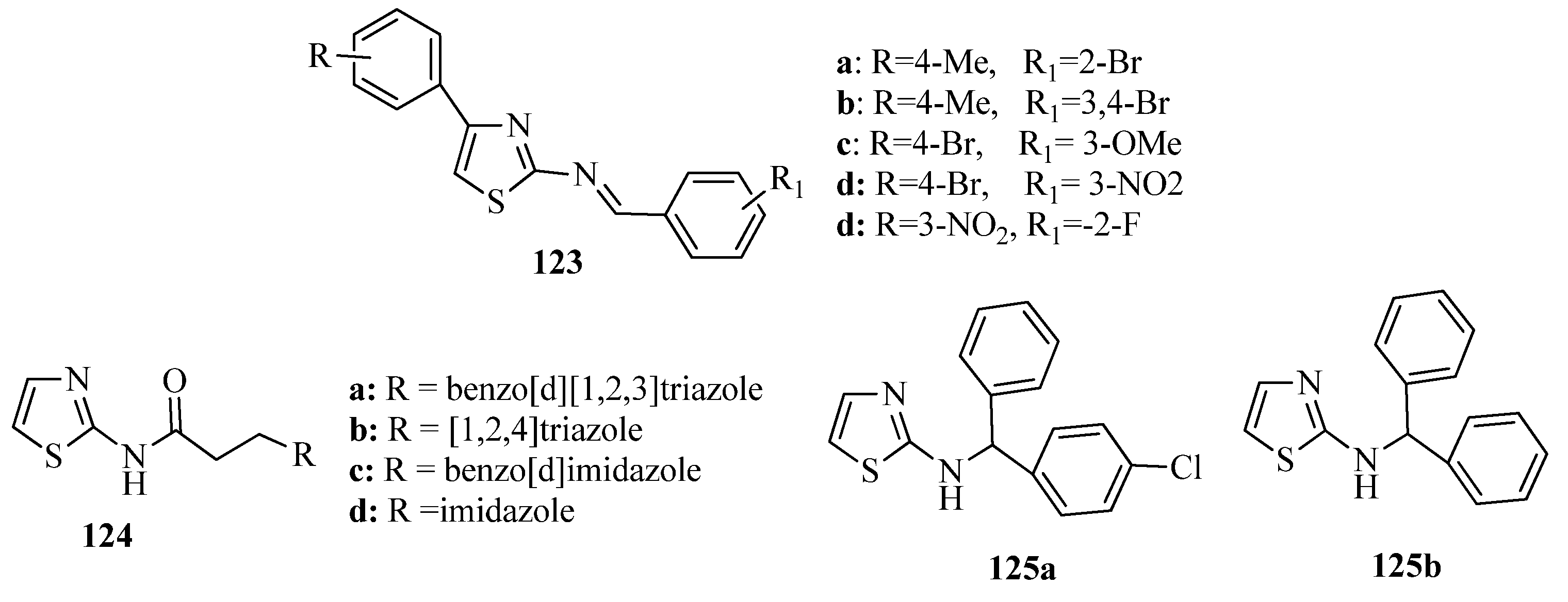
3.7. Thiazoles as Antiflammatory Agents
3.8. Antioxidant
3.9. Thiazole Derivatives as Carbonic Anhydrase Inhibitors
3.10. Anticonvulsant
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Horton, D.A.; Bourne, G.T.; Smythe, M.L. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chem. Rev. 2003, 103, 893–930. [Google Scholar] [CrossRef]
- Frazen, R.G. Recent Advances in the Preparation of Heterocycles on Solid Support: A Review of the Literature. J. Comb. Chem. 2000, 2, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liu, Q.; Wang, C.; Meng, Q.; Sun, H.; Peng, J.; Ma, X.; Liu, K. Benzylpenicillin inhibits the renal excretion of acyclovir by OAT1 andOAT3. Pharmacol. Rep. 2013, 65, 505–512. [Google Scholar] [CrossRef]
- Rahmutulla, B.; Matsushita, K.; Satoh, M.; Seimiya, M.; Tsuchida, S.; Kubo, S.; Shimada, H.; Ohtsuka, M.; Miyazaki, M.; Nomura, F. Alternative splicing of FBP-interacting repressor coordinates c-Myc, P27Kip1/cyclinE and Ku86/XRCC5 expression as a molecular sensor for bleomycin induced DNA damage pathway. Oncotarget 2014, 15, 2404–2417. [Google Scholar] [CrossRef] [PubMed]
- Popsavin, V. Synthesis and antiproliferative activity of two new tiazofurin analogues with 2′-amido functionalities. Bioorg. Med. Chem. Lett. 2006, 16, 2773–2776. [Google Scholar] [CrossRef]
- Wei, L.; Cheng, J.; Meng, Y.; Ren, Y.; Deng, H.; Guo, Y. A novel formulation of thiamine dilaurylsulphate and its preservative effect on apple juice and sterilised milk. Food Chem. 2014, 1, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F.; Poulos, T.L. Dissecting cytochrome P450 3A4-ligand interactions using ritonavir analogues. Biochemistry 2013, 52, 4474–4481. [Google Scholar] [CrossRef]
- Novakova, I.; Subileau, E.A.; Toegel, S.; Gruber, D.; Lachmann, B.; Urban, E.; Chesne, C.; Noe, C.R.; Neuhaus, W. Transport rankings of nonsteroidal antiinflammatory drugs across blood-brain barrier in vitro models. PLoS ONE 2014, 9, e86806. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.J.; McCarthy, M.W. Cefiderocol: A novel siderophore cephalosporin. Expert Opin. Investig. Drugs 2018, 27, 193–197. [Google Scholar] [CrossRef]
- Aoki, T.; Yoshizawa, H.; Yamawaki, K.; Yokoo, K.; Sato, J.; Hisakawa, S.; Hasegawa, Y.; Kusano, H.; Sano, M.; Sugimoto, H.; et al. Cefiderocol (S-649266), A new siderophore cephalosporin exhibiting potent activities against Pseudomonas aeruginosa and other gram-negative pathogens including multi-drug resistant bacteria: Structure activity relationship. Eur. J. Med. Chem. 2018, 155, 847–868. [Google Scholar] [CrossRef]
- Portsmouth, S.; van Veenhuyzen, D.; Echols, R.; Machida, M.; Ferreira, J.C.A.; Ariyasu, M.; Tenke, P.; Nagata, T.D. Cefiderocol versus imipenem-cilastatin for the treatment of complicated urinary tract infections caused by Gram-negative uropathogens: A phase 2, randomised, double-blind, non-inferiority trial. Lancet Infect. Dis. 2018, 18, 1319–1328. [Google Scholar] [CrossRef]
- Ghoncheh, M.; Pournamdar, Z.; Salehiniya, H. Incidence and Mortality and Epidemiology of Breast Cancer in the World. Asian Pac. J. Cancer Prev. 2016, 17, 43–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. Food and Drug Administration (FDA). FDA Approves Lusutrombopag for Thrombocytopenia in Adults with Chronic Liver Disease 2018. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lusutrombopag-thrombocytopenia-adults-chronic-liver-disease (accessed on 20 January 2021).
- Mishra, C.B.; Kumari, S.; Tiwari, M. Thiazole: A promising heterocycle for the development of potent CNS active agents. Eur. J. Med. Chem. 2015, 92, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.I.; Tatchell, A.R.; Furnis, B.S.; Hannaford, A.J.; Smith, P.W.G. Vogel’s Textbook of Practical Organic Chemistry, 5th ed.; Harlow Longman: London, UK, 1989; pp. 1151–1154. [Google Scholar]
- Warburton, W.K. Arylthiazathiolium Salts and o-aminoaryl Thiols—The Herz Reaction. Chem. Rev. 1957, 57, 1011–1020. [Google Scholar] [CrossRef]
- Cook, A.H.; Heilbron, I.; MacDonald, S.F.; Mahadevan, A.P. Synthesis and cytotoxicity evaluation of thiazole derivatives obtained from 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene- 3-carbonitrile. J. Chem. Soc. 1949, 67, 1064–1068. [Google Scholar] [CrossRef]
- Nayak, S.; Gaonkar, S.L. A Review on Recent Synthetic Strategies and Pharmacological Importance of 1,3-Thiazole Derivatives. Mini Rev. Med. Chem. 2019, 19, 215–238. [Google Scholar] [CrossRef]
- Brandsma, L.; De Jong, R.L.P.; Ver Kruijsse, H.D. An Efficient Synthesis of 1, 3-Thiazole. Synthesis 1985, 1985, 948–949. [Google Scholar] [CrossRef]
- Sheldrake, P.W.; Matteucci, M.; McDonald, E. Facile Generation of a Library of 5-Aryl-2-arylsulfonyl-1,3-thiazoles. Synlett 2006, 460–462. [Google Scholar] [CrossRef]
- Tang, X.; Yang, J.; Zhu, Z.; Zheng, M.; Wu, W.; Jiang, H. Access to Thiazole via Copper-Catalyzed [3+1+1]-Type Condensation Reaction under Redox-Neutral Conditions. J. Org. Chem. 2016, 81, 11461–11466. [Google Scholar] [CrossRef]
- Wang, X.; Qiu, X.; Wei, J.; Liu, J.; Song, S.; Wang, W.; Jiao, N. Cu-Catalyzed Aerobic Oxidative Sulfuration/Annulation Approach to Thiazoles via Multiple Csp3-H Bond Cleavage. Org. Lett. 2018, 20, 2632–2636. [Google Scholar] [CrossRef]
- Lingaraju, G.S.; Swaroop, T.R.; Vinayaka, A.C.; Kumar, K.S.S.; Sadashiva, M.P.; Ragappa, K.S. An Easy Access to 4,5-Disubstituted Thiazoles via Base-Induced Click Reaction of Active Methylene Isocyanides with Methyl Dithiocarboxylates. Synthesis 2012, 44, 1373–1379. [Google Scholar] [CrossRef]
- Sanz-Cervera, J.F.; Blasco, R.; Piera, J.; Cynamon, M.; Ibáñez, I.; Murguía, M.; Fustero, S. Solution versus Fluorous versus Solid-Phase Synthesis of 2,5-Disubstituted 1,3-Azoles. Preliminary Antibacterial Activity Studies. J. Org. Chem. 2009, 74, 8988–8996. [Google Scholar] [CrossRef]
- Miura, T.; Funakoshi, Y.; Fujimoto, Y.; Nakahashi, J.; Murakami, M. Facile Synthesis of 2,5-Disubstituted Thiazoles from Terminal Alkynes, Sulfonyl Azides and Thionoesters. Org. Lett. 2015, 17, 2454–2457. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Edrees, M.M.; Faty, R.A.M.; Muhammad, Z.A.; Mabkhot, Y.N. Microwave-assisted one pot three-component synthesis of some novel pyrazole scaffolds as potent anticancer agents. Chem. Cent. J. 2017, 11, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamthulla, S.; Pal, S.; Khan, M.N.; Choudhury, L.H. “On-water” synthesis of novel trisubstituted 1,3-thiazoles via microwaveassisted catalyst-free domino reactions. RSC Adv. 2014, 4, 37889–37899. [Google Scholar] [CrossRef] [Green Version]
- Chinnaraja, D.; Rajalakshmi, R. A facile, solvent and catalyst free, microwave assisted one pot synthesis of hydrazinyl thiazole derivatives. J. Saudi Chem. Soc. 2015, 19, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Kiran, K.R.; Swaroop, T.; Rajeev, N.; Anil, S.; Rangappa, J.; Sadashiva, M. Cyclization of Active Methylene Isocyanides with α-Oxodithioesters Induced by Base: An Expedient Synthesis of 4-Methylthio/Ethoxycarbonyl-5-acylthiazoles. Synthesis 2020, 52, 1103–1112. [Google Scholar] [CrossRef]
- Mamidala, S.; Peddi, S.R.; Aravilli, R.K.; Jilloju, P.C.; Manga, V.; Vedula, R.R. Microwave irradiated one pot, three component synthesis of a new series of hybrid coumarin based thiazoles: Antibacterial evaluation and molecular docking studies. J. Mol. Struct. 2021, 1225, 129114. [Google Scholar] [CrossRef]
- Facchinetti, V.; Avellar, M.M.; Nery, A.S.C.; Gomes, C.R.B.; Vasconcelos, T.R.A.; de Souza, N.V.M. An Eco-friendly, Hantzsch-Based, Solvent-Free Approach to 2-Aminothiazoles and 2-Aminoselenazoles. Synthesis 2016, 48, 437–440. [Google Scholar]
- Chen, B.; Guo, S.; Guo, X.; Zhang, G.; Yu, Y. Selective Access to 4-Substituted 2-Aminothiazoles and 4-Substituted 5-Thiocyano-2-aminothiazoles from Vinyl Azides and Potassium Thiocyanate Switched by Palladium and Iron Catalysts. Org. Lett. 2015, 17, 4698–4701. [Google Scholar] [CrossRef]
- Tang, X.; Zhu, Z.; Qi, C.; Wu, W.; Jiang, H. Copper-Catalyzed Coupling of Oxime Acetates with Isothiocyanates: A Strategy for 2-Aminothiazoles. Org. Lett. 2016, 18, 180–183. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, W.; Zhao, D.; Zhang, G.; Yu, Y. One-Pot Three-Component Strategy for Polysubstituted 2-Aminothiazoles via Ring Opening of α-Nitro Epoxides. Synthesis 2019, 51, 2023–2029. [Google Scholar] [CrossRef]
- Castagnolo, D.; Pagano, M.; Bernardini, M.; Botta, M. Domino Alkylation-Cyclization Reaction of Propargyl Bromides with Thioureas/Thiopyrimidinones: A New Facile Synthesis of 2-Aminothiazoles and 5H-Thiazolo[3,2-a]pyrimidin-5-ones. Synlett 2009, 12, 2093–2096. [Google Scholar]
- Narender, M.; Reddy, M.S.; Kumar, V.P.; Srinivas, B.; Sridhar, R.; Nageswar, Y.; Rao, K.R. Aqueous-Phase One-Pot Synthesis of 2-Aminothiazole- or 2-Aminoselenazole-5-carboxylates from β-Keto Esters, Thiourea or Selenourea, and N-Bromo-succinimide under Supramolecular Catalysis. Synthesis 2007, 4, 3469–3472. [Google Scholar]
- De Andrade, V.S.C.; de Mattos, M.C.S. One-Pot Telescoped Synthesis of Thiazole Derivatives from β-Keto Esters and Thioureas Promoted by Tribromoisocyanuric Acid. Synthesis 2018, 50, 4867–4874. [Google Scholar]
- Fu, R.-G.; Wang, Y.; Xia, F.; Zhang, H.-L.; Sun, Y.; Yang, D.-W.; Wang, Y.-W.; Yin, P. Synthesis of 2-Amino-5-acylthiazoles by a Tertiary Amine-Promoted One-Pot Three-Component Cascade Cyclization Using Elemental Sulfur as a Sulfur Source. J. Org. Chem. 2019, 84, 12237–12245. [Google Scholar] [CrossRef]
- Van Duin, D.; Paterson, D.L. Multidrug-Resistant Bacteria in the Community. Infect. Dis. Clin. N. Am. 2016, 30, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Berkow, E.; Lockhart, S. Fluconazole resistance in Candida species: A current perspective. Infect. Drug Resist. 2017, 10, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, A.H.; Moore, L.S.; Sundsfjord, A.; Steinbakk, M.; Regmi, S.; Karkey, A.; Guerin, P.J.; Piddock, L.J. Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 2016, 387, 176–187. [Google Scholar] [CrossRef]
- Yurttas, L.; Özkay, Y.; Gençer, K.; Acar, U. Synthesis of Some New Thiazole Derivatives and Their Biological Activity Evaluation. Hindawi Publ. Corp. J. Chem. 2015, 464379, 7. [Google Scholar] [CrossRef]
- Arora, P.; Narang, R.; Bhatia, S.; Kumar Nayak, S.; Kumar Singh, S.; Narasimhan, B. Synthesis, molecular docking and QSAR studies of 2, 4- disubstituted thiazoles as antimicrobial agents. J. Appl. Pharm. Sci. 2015, 5, 28–42. [Google Scholar] [CrossRef] [Green Version]
- European Committee for Antimicrobial Susceptibility Testing (EUCAST). Determination of minimum inhibitory concentrations (MICs) of antibacterial agents by broth dilution: EUCAST discussion document E. Dis 5.1. Clin. Microbiol. Infect. 2003, 9, 1–7. [Google Scholar]
- Łączkowski, K.; Jachowicz, K.; Misiura, K.; Biernasiuk, A.; Malm, A. Synthesis and biological evaluation of novel 2-(1H-imidazol-2-ylmethylidene)hydrazinyl-1,3-thiazoles as potential antimicrobial agents. Heterocycl. Commun. 2015, 21, 109–114. [Google Scholar] [CrossRef]
- Mohammad, H.; Reddy, P.V.N.; Monteleone, D.; Mayhoub, A.S.; Mark Cushman, M.; Seleem, M.N. Synthesis and antibacterial evaluation of a novel series of synthetic phenylthiazole compounds against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 2015, 94, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, H.; Mayhoub, A.S.; Ghafoor, A.; Soofi, M.; Alajlouni, R.A.; Mark Cushman, M.; Seleem, M.N. Discovery and Characterization of Potent Thiazoles versus Methicillin- and Vancomycin-Resistant Staphylococcus aureus. J. Med. Chem. 2014, 57, 1609–1615. [Google Scholar] [CrossRef] [Green Version]
- Karale, B.K.; Takate, S.J.; Salve, S.P.; Zaware, B.H.; Jadhav, S.S. Synthesis and antibacterial screening of novel fluorine containing heterocycles. Orient. J. Chem. 2015, 31, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Nagavelli, V.; Narsimha, S.; Swamy Battula, K.; Sudhakar, L.; Thatipamula, K. Synthesis, cytotoxic and antibacterial activities of 6-bromobenzo[d]thiazol-2(3H)-one-[1,2,3] triazole hybrids. Org. Commun. 2016, 9, 32–41. [Google Scholar]
- Mert, S.; Kasimogullari, R.; Ica, T.; Colak, F.; Altun, A.; Ok, S. Synthesis, structure-activity relationships and in vitro anti-bacterial and antifungal activity evaluations of novel pyrazolecarboxylic and dicarboxylic acid derivatives. Eur. J. Med. Chem. 2014, 78, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, B.; Najimi, M. Antibacterial effect of thiazole derivatives on Rhodoccocus equi, Brucella abortus, and Pasteurella multocida. Iran. J. Vet. Med. 2016, 10, 47–52. [Google Scholar]
- Nasr, T.; Bondock, S.; Eid, S. Design, synthesis, antimicrobial evaluation and molecular docking studies of some new 2,3-dihydrothiazoles and 4-thiazolidinones containing sulfisoxazole. J. Enzyme Inhib. Med. Chem. 2016, 31, 236–246. [Google Scholar] [CrossRef]
- Seleem, M.A.; Disouky, A.M.; Mohammad, H.; Abdelghany, T.M.; Mancy, A.S.; Bayoumi, S.A.; Elshafeey, A.; El-Morsy, A.; Seleem, M.N.; Mayhoub, A.S. Second-Generation Phenylthiazole Antibiotics with Enhanced Pharmacokinetic Properties. J. Med. Chem. 2016, 59, 4900–4912. [Google Scholar] [CrossRef]
- Desai, N.C.; Makwana, A.H.; Rajpar, K.M. Synthesis and study of 1,3,5-triazine basedthiazole derivatives as antimicrobial agent. J. Saudi Chem. Soc. 2016, 20, S334–S341. [Google Scholar] [CrossRef] [Green Version]
- Łaczkowski, K.; Motylewska, K.; Baranowska, A.; Biernasiuk, A.; Misiura, K.; Malm, A.; Fernande, B. Synthesis, antimicrobial evaluation and theoretical prediction of NMR chemical shifts of thiazole and selenazole derivatives with high antifungal activity against Candida spp. J. Mol. Struct. 2016, 1108, 427–437. [Google Scholar] [CrossRef]
- Wayne, P. Clinical and Laboratory Standards Institute, Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; M27-S4; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Subhashini, N.J.P.; Bhadraiah, B.; Janak, P. Synthesis and Biological Evaluation of Thiazole-Acetamide Derivatives as Antibacterial Agents. J. Heterocycl. Chem. 2017, 1–19. [Google Scholar] [CrossRef]
- Salem, M. Synthesis of New Thiazole, Bithiazolidinone and Pyrano[2,3-d]thiazole Derivatives as Potential Antimicrobial Agents. Croat. Chem. Acta 2017, 90, 7–15. [Google Scholar] [CrossRef]
- Gençer, H.K. Synthesis of thiazole-phenylacetic acid compounds as dual antibacterial-COX enzymes inhibitors. Cukurova Med. J. 2017, 42. [Google Scholar] [CrossRef] [Green Version]
- Cankılıc, M.Y.; Yurttaş, L. Study on the Antimicrobial Effects of Novel Thiazole Derivatives. Marmara Pharm. J. 2017, 21, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Bikobo, D.; Vodnar, D.; Stana, A.; Tiperciuc, B.; Nastasa, C.; Douchet, M.; Oniga, O. Synthesis of 2-phenylamino-thiazole derivatives as antimicrobial agents J. Saudi Chem. Soc. 2017, 21, 861–868. [Google Scholar] [CrossRef]
- Hagras, M.; Mohammad, H.; Mandour, M.S.; Hegazy, Y.A.; Ghiaty, A.; Seleem, M.N.; Mayhoub, A.S. Investigating the Antibacterial Activity of Biphenylthiazoles against Methicillin-and Vancomy cin-Resistant Staphylococcus aureus (MRSA and VRSA). J. Med. Chem. 2017, 60, 4074–4085. [Google Scholar] [CrossRef]
- Shaikh, F.; Kulkarni, R.; Naik, N.; Madar, J.; Joshi, S.; Sunagar, V.; Shastri, L. Synthesis and Characterization of Chlorophenyl- thiazolocoumarinyl Hydrazides as Promising Antimicrobial and Anti-Inflammatory Agents. IOSR J. Appl. Chem. 2018, 11, Ver. II, 9–39. [Google Scholar]
- Schwable, R.; Steele, L.; Goodwin, A.C. Antimicrobial Susceptibility Testing Protocols; CRC Press: New York, NY, USA, 2007. [Google Scholar]
- Eid, I.; Elsebaei, M.M.; Mohammad, H.; Abouf, M.; Abutaleb, N.S.; Hegazy, Y.A.; Ghiaty, A.; Chen, L.; Zhang, J.; Malwal, S.; et al. Alkynyl containing phenylthiazoles: Systemically active antibacterial agents effective against methicillin-resistant Staphylococcus aureus (MRSA). Eur. J. Med. Chem. 2018, 148, 195–209. [Google Scholar]
- Bondock, S.; Fouda, A.M. Synthesis and evaluation of some new 5-(hetaryl)thiazoles as potential antimicrobial agents. Synth. Commun. 2018, 48, 561–573. [Google Scholar] [CrossRef]
- Lino, C.I.; Gonçalves de Souza, I.; Borelli, B.M.; Silvério Matos, T.T.; Santos Teixeira, I.N.; Ramos, J.P.; Maria de Souza Fagundes, E.; de Oliveira Fernandes, P.; Maltarollo, V.G.; Johann, S.; et al. Synthesis, molecular modeling studies and evaluation of antifungal activity of a novel series of thiazole derivatives. Eur. J. Med. Chem. 2018, 151, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Abu-Melha, S.; Edrees, M.; Salem, H.; Kheder, N.; Gomha, S.; Abdelaziz, M. Synthesis and Biological Evaluation of Some Novel Thiazole-Based Heterocycles as Potential Anticancer and Antimicrobial Agents. Molecules 2019, 24, 539. [Google Scholar] [CrossRef] [Green Version]
- Althagafi, I.; El-Metwaly, N.; Farghaly, T.A. New Series of Thiazole Derivatives: Synthesis, Structural Elucidation, Antimicrobial Activity, Molecular Modeling and MOE Docking. Molecules 2019, 24, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pricopie, A.; Ionut, I.; Marc, J.; Arseniu, A.; Vlase, L.; Grozav, A.; Găină, L.; Vodnar, D.; Pîrnău, A.; Oniga, O. Design and Synthesis of Novel 1,3-Thiazoleand 2-Hydrazinyl-1,3-Thiazole Derivativesas Anti-Candida Agents: In Vitro Antifungal Screening, Molecular Docking Study, and Spectroscopic Investigation of their Binding Interaction with Bovine Serum Albumin. Molecules 2019, 24, 3435. [Google Scholar] [CrossRef] [Green Version]
- Biernasiuk, A.; Kawczyńska, M.; Berecka-Rycerz, A.; Rosada, B.; Gumieniczek, A.; Malm, A.; Dzitko, K.; Łączkowsk, K.Z. Synthesis, antimicrobial activity, and determination of thelipophilicity of (cyclhex-3-enylmethylene)hydrazinyl)thiazolederivatives. Med. Chem. Res. 2019, 28, 2023–2036. [Google Scholar] [CrossRef] [Green Version]
- Pricopie, A.; Focs, M.; Ionut, I.; Marc, G.; Vlase, L.; Găină, L.; Vodnar, D.; Simon, E.; Barta, G.; Pîrnău, A.; et al. Novel 2,4-Disubstituted-1,3-Thiazole Derivatives: Synthesis, Anti Candida Activity Evaluation and Interaction with Bovine Serum Albumine. Molecules 2020, 25, 1079. [Google Scholar] [CrossRef] [Green Version]
- Kamat, V.; Santosh, R.; Poojary, B.; Nayak, S.; Karan Kumar, B.; Sankaranarayanan, M.; Khanapure, S. Pyridine- and Thiazole-Based Hydrazides with Promising Anti-inflammatory and Antimicrobial Activities along with Their in Silico Studies. ACS Omega 2020, 5, 25228–25239. [Google Scholar] [CrossRef]
- Fogel, N. Tuberculosis: A disease without boundaries. Tuberculosis 2015, 95, 527–531. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Tuberculosis Fact Sheet. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 20 January 2021).
- Ran, K.; Gao, C.; Deng, H.; Lei, Q.; You, X.; Wang, N.; Shi, Y.; Liu, Z.; Wei, W.; Peng, C.; et al. Identification of novel 2-aminothiazole conjugated nitrofuran as antitubercular and antibacterial agents. Bioorg. Med. Chem. Lett. 2016, 26, 3669–3674. [Google Scholar] [CrossRef]
- Bekker, B.; Sokolov, D.; Luzina, O.; Komarova, N.; Maslov, D.; Chernousova, L.; Salakhutdinov, N.; Danilenko, V. Synthesis and activity of (+)-usnic acid and (2)-usnic acid derivatives containing 1,3-thiazole cycle against Mycobacterium Tuberculosis. Med. Chem. Res. 2015, 24, 2926–2938. [Google Scholar] [CrossRef]
- Guzeldemirci, N.U.; Gursoy, E. Synthesis and evaluation of new imidazo[2,1-b]thiazoles as antituberculosis agents. Marmara Pharm. J. 2017, 21, 102–109. [Google Scholar]
- Guzeldemirci, N.U.; Karaman, C.; Küçükbasmaci, O. Antibacterial, Antitubercular and Antiviral Activity Evaluations of Some Arylidenehydrazide Derivatives Bearing Imidazo[2,1-b]thiazole Moiety. Turk. J. Pharm. Sci. 2017, 14, 157–163. [Google Scholar] [CrossRef]
- Collins, L.A.; Franzblau, S.G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surineni, G.; Gao, Y.; Hussain, M.; Liu, Z.; Chhotaray, C.; Islam, M.D.M.; Hameed, H.M.A.; Zhang, T. Design, synthesis, and in vitro biological evaluation of novel benzimidazole tethered allylidenehydrazinylmethylthiazole derivatives as potent inhibitors of Mycobacterium tuberculosis. Med. Chem. Commun. 2019, 10, 49–60. [Google Scholar] [CrossRef]
- Tang, J.; Wang, B.; Wu, T.; Wan, J.; Tu, Z.; Njire, N.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Lu, X.; et al. Design, Synthesis, and Biological Evaluation of Pyrazolo[1,5-a]pyridine-3-carboxamides as Novel Antitubercular Agents. ACS Med. Chem. Lett. 2015, 6, 814–818. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Njire, M.M.; Liu, J.; Wu, T.; Wang, B.; Liu, T.; Wan, J.; Tu, Z.; Tan, Y.; Tan, S.; et al. Engineering more stable, selectable marker-free autoluminescent mycobacteria by one step. PLoS ONE 2015, 10, e0119341. [Google Scholar] [CrossRef] [PubMed]
- Shelke, R.N.; Pansare, D.N.; Sarkate, A.P.; Karnik, K.S.; Sarkate, A.P.; Shinde, D.B.; Thopate, S.R. Synthesis of (Z)-5-(substituted benzylidene)-2-((substituted phenyl) amino)thiazol-4(5H)-one analogues with antitubercular activity. J. Taibah Univ. Sci. 2019, 13, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Akhtar, S.; Mishra, A.; Sarkar, D. A novel screening method based on menadione mediated rapid reduction of tetrazolium salt for testing of anti-mycobacterial agents. J. Microbiol. Methods 2011, 84, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Sarkar, S.; Sarkar, D. Bactericidal activity of 2-nitroimidazole against the active replicating stage of Mycobacterium bovis BCG and Mycobacterium tuberculosis with intracellular efficacy in THP-1 macrophages. Int. J. Antimicrob. Agents 2008, 32, 40–45. [Google Scholar] [CrossRef]
- Shinde, V.; Mahulikar, P.; Mhaske, P.C.; Chakraborty, S.; Choudhari, A.; Phalle, S.; Choudhari, P.; Sarkar, D. Synthesis and antimycobacterial evaluation of new 5-(1-benzyl-1H-1,2,3-triazol-4-yl)-4-methyl-2-arylthiazole derivatives. Med. Chem. Res. 2019, 28, 805–819. [Google Scholar] [CrossRef]
- Khan, A.; Sarkar, D. A simple whole cell based high through put screening protocol using Mycobacterium bovis BCG for inhibitors against dormant and active tubercle bacilli. J. Microbiol. Methods 2008, 73, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Nawale, L.; Arkile, M.; Shedbalkar, U.; Wadhwani, S.; Sarkar, D.; Chopade, B. Chemical and biological metal nanoparticlesas antimycobacterialagents: A comparative study. Int. J. Antimicrob. Agents 2015, 46, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Sarkar, D. Potential use of nitrate reductase as a bio-marker for the identification of active and dormant inhibitors of Mycobacterium tuberculosis in a THP1 infection model. J. Biomol. Screen 2012, 17, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Karale, U.B.; Krishna, V.S.; Krishna, E.V.; Choudhari, A.S.; Shukla, M.; Gaikwad, V.R.; Mahizhaveni, B.; Chopra, S.; Misra, S.; Sarkar, D.; et al. Synthesis and biological evaluation of 2,4,5-trisubstituted thiazoles as antituberculosis agents effective against drug-resistant tuberculosis. Eur. J. Med. Chem. 2019, 178, 315–328. [Google Scholar] [CrossRef]
- Cordeiro, R.; Kachroo, M. Synthesis and biological evaluation of anti-tubercular activity of Schiff bases of 2-Amino thiazoles. Bioorg. Med. Chem. Lett. 2020, 30, 127655–127662. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2015, 6, 5–29. [Google Scholar] [CrossRef]
- Kenny, P.A.; Lee, G.Y.; Bissell, M.J. Targeting the tumor microenvironment. Front. Biosci. 2007, 12, 3468–3474. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathwas in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef]
- Braga, S.; Fonseca, N.; Ramos, J.; de Souza-Fagundes, E.; de Oliveira, R. Synthesis and cytotoxicity evaluation of thiosemicarbazones and their thiazole derivatives. Braz. J. Pharm. Sci. 2016, 52, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Küster, T.; lense, N.; Barna, F.; HemphilL, A.; Kindermann, M.K.; Heinicke, J.W.; VocK, C.A. A new promising application for highly cytotoxic metal compounds: η6-areneruthenium(II) phosphite complexes for the treatment of alveolar echinococcosis. J. Med. Chem. 2012, 55, 4178–4188. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Emami, S.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Recent applications of 1,3-thiazole core structure in the identification of new lead compounds and drug discovery. Eur. J. Med. Chem. 2015, 97, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Brancale, A.; Ricci, A.; Hamel, E.; Bortolozzi, R.; Basso, G.; Viola, G. Convergent Synthesis and Biological Evaluation of 2-Amino-4-(3′,4′,5′-trimethoxyphenyl)-5-aryl Thiazoles as Microtubule Targeting Agents. J. Med. Chem. 2011, 1, 5144–5153. [Google Scholar] [CrossRef]
- Chang, S.; Zhang, Z.; Zhuang, X.; Luo, J.; Cao, X.; Li, J.; Tu, Z.; Lu, X.; Ren, X.; Ding, K. New thiazole carboxamides as potent inhibitors of Akt kinases. Bioorg. Med. Chem. Lett. 2012, 22, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Hassan, G.S.; El-Messery, S.M.; Al-Omary, F.A.M.; El-Subbagh, H.I. Substituted thiazoles VII. Synthesis and antitumor activity of certain 2-(substituted amino)-4-phenyl-1,3-thiazole analogs. Bioorg. Med. Chem. Lett. 2012, 22, 6318–6323. [Google Scholar] [CrossRef]
- Reddy, P.R.; Seenaiah, D.; Padmaja, A.; Padmavathi, V.; Krishna, N.S. Synthesis, antioxidant, and cytotoxic activities of bis(oxazolyl/thiazolyl/imidazolyl)amidomethanesulfonyl Acetamides. Med. Chem. Res. 2015, 24, 86–98. [Google Scholar] [CrossRef]
- Rouf, A.; Tanyeli, C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. [Google Scholar] [CrossRef]
- Abruzzese, E.; Breccia, M.; Latagliata, R. Second-Generation Tyrosine Kinase Inhibitors in First-Line Treatment of Chronic Myeloid Leukaemia (CML). BioDrugs 2014, 28, 17–26. [Google Scholar] [CrossRef]
- Tromp, V.N.M.F.; Timmers, L.; Koningen, L.; Janssen, J.J.W.M.; Westerweel, P.E.; Geelen, I.G.P.; Jong, J.; Beckeringh, J.J.; Boons, C.C.L.M.; Hugtenburg, J.G. Tyrosine kinase inhibitor treatment discontinuation in chronic myeloid leukemia: Patient views. Leuk. Lymphoma 2021, 62, 649–658. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Kaplancıklı, Z.A.; Alkalin-Çiftçi, G.; Demiamide, R. Moieties: Synthesis and biological evaluation of thiazoline derivatives as new antimicrobial and anticancer agents. Eur. J. Med. Chem 2014, 74, 264–277. [Google Scholar] [CrossRef]
- Altıntop, M.D.; Özdemir, A.; Turan-Zitouni, G.; Ilgın, S.; Atlı, O.; Demirci, F.; Kaplancıklı, Z.A. Synthesis and in Vitro Evaluation of New Nitro-Substituted Thiazolyl Hydrazone Derivatives as Anticandidal and Anticancer Agents. Molecules 2014, 19, 14809–14820. [Google Scholar] [CrossRef] [Green Version]
- Turan-Zitouni, G.; Altıntop, M.D.; Ozdemir, A.; Kaplancıkli, Z.A.; Alkalin-Çiftçi, G.; Temel, H.E. Synthesis and evaluation of bis-thiazole derivatives as new anticancer agents. Eur. J. Med. Chem. 2016, 107, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Abdallah, A.E.M.; Ahmed, E.A. Synthesis and cytotoxicity evaluation of thiazole derivatives obtained from 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile. Acta Pharm. 2017, 67, 495–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finiuk, N.S.; Hreniuh, V.P.; Ostapiuk, V.; Matiychuk, V.; Frolov, D.A.; Obushak, M.D.; Stoika, R.S.; Babsky, A.M. Antineoplastic activity of novel thiazole derivatives. Biopolym. Cell 2017, 33, 135–146. [Google Scholar] [CrossRef] [Green Version]
- Gomha, S.M.; Farghaly, T.A.; Alqurashi, N.T.; Abdou, Y.; Mousa, E.K. Synthesis, molecular docking and anticancer evaluation of new arylazothiazoles. Curr. Org. Synth. 2017, 14, 620–631. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Wu, H.-M.; Deng, S.-N.; Cai, X.-Y.; Yao, Y.; Mwenda, M.C.; Wang, J.-Y.; Cai, D.; Chen, Y. Design, synthesis, and anticancer activities of novel2-amino-4-phenylthiazole scaffold containing amide moieties. Hindawi J. Chem. 2018, 1–8. [Google Scholar] [CrossRef]
- De Santana, T.I.; Barbosa, M.O.; Gomes, P.A.T.M.; da Cruz, A.C.N.; da Silva, T.G.; Leite, A.C.L. Synthesis, anticancer activity and mechanism of action of new thiazole derivatives. Eur. J. Med. Chem. 2018, 144, 874–886. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Wu, Y.; Zhang, J.; Mei, Q.; Zhang, Y.; Zhu, N.; Liu, R.; Zhang, H. Design, synthesis and biological evaluations of novel pyridonethiazole hybrid molecules as antitumor agents. Eur. J. Med. Chem. 2018, 145, 35–40. [Google Scholar] [CrossRef]
- Mohammed, F.Z.; Barakat, L.A.A.; Gad, E.M.; Shetya, N.M. In vivo Biochemical Evaluation of Some synthesized Thiazole Derivatives Containing Coumarin Moiety as Antioxidant and AntitumorAgents. Asian J. Res. Biochem. 2019, 5, 1–15. [Google Scholar]
- Nocentini, A.; Carta, F.; Ceruso, M.; Bartolucci, G.; Supuran, C.T. Click-tailed coumarins with potent and selective inhibitory action against thetumor-associated carbonicanhydrases IX and XII. Bioorg. Med. Chem. 2015, 23, 6955–6966. [Google Scholar] [CrossRef] [PubMed]
- Elhalim, S.A.; Ibrahim, I.T. Radioiodination of 2,3-dimethyl-4H-furo[3,2-c] coumarin and biological evaluation in solid tumor bearing mice. Appl. Radiat. Isot. 2015, 95, 153–158. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Al-Said, M.S. Antitumo ractivity of novel pyridine, thiophene and thiazole derivatives. Arch. Pharm. Res. 2012, 35, 965–973. [Google Scholar] [CrossRef]
- Gabr, M.T.; El-Gohary, N.S.; El-Bendary, E.R.; El-Kerdawy, M.M.; Ni, N. Microwave assisted synthesis and antitumor evaluation of a new series of thiazolylcoumarine derivatives. EXCLI J. 2017, 6, 1114–1131. [Google Scholar]
- Bakare, S.B. Anticancer activity of some new series of 2-(substituted)amino-1,3-thiazole Derivatives. Pol. J. Chem. Technol. 2019, 21, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zaki, I.; Abdelhameid, M.K.; El-Deen, I.M.; Wahab, A.H.; Ashmawy, A.M.; Mohamed, K.O. Design, synthesis and screening of 1, 2, 4-triazinone derivativesas potential antitumor agents with apoptosis inducing activityon MCF-7 breast cancer cell line. Eur. J. Med. Chem. 2018, 156, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Sayed, A.R.; Gomha, S.M.; Abdelrazek, F.M.; Mohamed, S.; Farghaly, M.S.; Hassan, S.A.; Metz, P. Design, efficient synthesis and molecular docking of some novel thiazolyl-pyrazole derivatives as anticancer agents. BMC Chem. 2019, 13, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wu, C.; Zhang, Q.; Shan, Y.; Gu, W.; Wang, S. Design, synthesis and biological evaluation of novel β-pinene-based thiazole derivatives as potential anticancer agents via mitochondrial-mediated apoptosis pathway. Bioorg. Chem. 2019, 84, 468–477. [Google Scholar] [CrossRef]
- Al-Said, M.S.; Bashandy, S.I.; Al-Qasoumi, S.I.; Ghorab, M.M. Anti-breast cancer activity of some novel 1,2-dihydropyridine, thiophene and thiazole derivatives. Eur. J. Med. Chem. 2011, 46, 137–141. [Google Scholar] [CrossRef]
- Bao, M.K.; Xiong, S.S.; Cao, M.Z. Synthesis and Antibacterial, Antitumor Activity of 2,6,6-Thrimethyl- bicyclo[3,1,1]heptan-3-(4-aryl-2-thiazoyl)hydrazones. Chin. J. Org. Chem. 2014, 34, 2146–2251. [Google Scholar] [CrossRef] [Green Version]
- Rui, J.; Yang, J.; Huang, J.; Wang, J.; Xu, X.; Xu, H.; Wang, S. Synthesis, antitumor and anti-inflammatory activities of isolongifolanonyl pyrazole derivatives. Chin. J. Org. Chem. 2016, 36, 2183–2190. [Google Scholar] [CrossRef] [Green Version]
- Yurttas, L.; Ciftci, G.A.; Temel, H.E.; Saglik, B.N.; Demir, B.; Levent, S. Biological Activity Evaluation of Novel 1,2,4-triazine Derivatives Containing Thiazole/benzothiazole Rings. Anticancer Agents Med. Chem. 2017, 17, 1846–1853. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A. Synthesis and in Vitro Antitumor Evaluation of Some Indeno[1,2-c]pyrazol(in)es Substituted with Sulfonamide, sulfonylurea(-Thiourea) Pharmacophores, and Some Derived Thiazole Ring Systems. Bioorg. Med. Chem. 2006, 14, 6475–6485. [Google Scholar] [CrossRef]
- Yurttas, L.; Demir, B.; Ciftci, G.A. Some Thiazole Derivatives Combined with Different Heterocycles: Cytotoxicity Evaluation and Apoptosis Inducing Studies. Anticancer Agents Med. Chem. 2018. [Google Scholar] [CrossRef]
- Evren, A.E.; Yurttas, L.; Ekselli, B.; Akalin-Ciftci, G. Synthesis and biological evaluation of 5-methyl-4-phenyl thiazole derivatives as anticancer agents. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 820–828. [Google Scholar] [CrossRef]
- Nagireddy, P.K.R.; Kommalapati, V.K.; Krishna, V.S.; Sriram, D.; Tangutur, A.D.; Kantevari, S. Imidazo[2,1-b]thiazole-Coupled Natural Noscapine Derivatives as Anticancer Agents. ACS Omega 2019, 4, 19382–19398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Bharathi, E.V.; Sagar, M.V.P.; Pushpavalli, S.N.C.V.; Ray, P.; Pal-Bhadra, M. Design, synthesis and biological evaluation of imidazopyridine/pyr-imidine-chalcone derivatives as potential anticancer agents. Med. Chem. Commun. 2010, 1, 355–360. [Google Scholar] [CrossRef]
- Madhavi, S.; Sreenivasulu, R.; Jyotsna, Y.; Raju, R.R. Synthesis ofchalcone incorporated quinazoline derivatives as anticancer agents. Saudi Pharm. J. 2017, 25, 275–279. [Google Scholar] [CrossRef] [Green Version]
- Go, M.L.; Wu, X.; Liu, X.L. Chalcones: An update on cytotoxic and chemo protective properties. Curr. Med. Chem. 2005, 12, 481–499. [Google Scholar] [CrossRef]
- Haik, A.B.; Prasad, Y.R.; Shahanaaz, S. Design, Facile Synthesis, Characterization and Computational Evaluation of Novel Isobutyl chalcones as Cytotoxic Agents: Part-A. FABAD J. Pharm. Sci. 2015, 40, 1–16. [Google Scholar]
- Suma, V.R.; Sreenivasulu, R.; Rao, M.V.B.R.; Subramanyam, M.; Ahsan, M.J.; Alluri, R.; Rao, K.R.M. Design, synthesis, and biological evaluation of chalcone-linked thiazole-imidazopyridine derivatives as anticancer agents. Med. Chem. Res. 2020, 29, 1643–1654. [Google Scholar] [CrossRef]
- Verma, R.P. Anti-cancer activitiesof1,4-naphthoquinones: A QSAR study. Anticancer Agents Med. Chem. 2006, 6, 489–499. [Google Scholar] [CrossRef]
- Spyroudis, S. Hydroxyquinones: Synthesis and reactivity. Molecules 2000, 5, 1291–1330. [Google Scholar] [CrossRef]
- Liu, H.B.; Cai, B.; Cui, C.B.; Gu, Q.Q.; Zhao, Q.C.; Guan, H.S. Pterocaryquinone, a novel naphthoquinone derivative from Pterocarya tonkinesis. Chin. J. Chem. 2006, 24, 1683–1686. [Google Scholar] [CrossRef]
- Gu, C.; Zhai, J.; Jiang, J.; Liu, H.; Wang, L.; Zhu, D.; Ji, Y. An Efficient One-pot Synthesis of Aryl-substituted 1-(Thiazol-2-yl)-1H-pyrazole-3-carboxylates via a Hantzsch Synthesis-Knorr Reaction Sequence. Chin. J. Chem. 2014, 32, 179–190. [Google Scholar] [CrossRef]
- Aly, A.A.; El-Sheref, E.M.; Brown, A.B.; Bräse, S.; Nieger, M.; Abdelhafez, E.-S.M. New one-pot synthesis of 2-ylidenehydrazono-thiazoles. J. Sulfur Chem. 2019, 40, 641–647. [Google Scholar] [CrossRef]
- Aly, A.A.; Bräse, S.; Hassan, A.A.; Mohamed, N.K.; Abd El-Haleem, L.E.; Nieger, M.; Morsy, N.M.; Alshammari, M.B.; Ibrahim, M.A.A.; Abdelhafez, E.M.N. Design, Synthesis, and Molecular Docking of Paracyclophanyl-Thiazole Hybrids as Novel CDK1 Inhibitors and Apoptosis Inducing Anti-Melanoma Agents. Molecules 2020, 25, 5569. [Google Scholar] [CrossRef] [PubMed]
- Anuradha, S. Antiviral agents and treatment of viral infections. J. Int. Med. Sci. Acad. 2014, 27, 191. [Google Scholar]
- Dawood, K.M.; Eldebss, T.M.A.; El-Zahabi, H.S.A.; Yousef, M.H. Synthesis and antiviral activity of some new bis-1,3-thiazole derivatives. Eur. J. Med. Chem. 2015, 102, 266–276. [Google Scholar] [CrossRef]
- ZelIsko, N.I.; Demchuk, I.L.; Λesyk, R.B. New thiopyrano-[2,3-d][1,3]thiazole derivatives as potential antiviral agents. Ukr. Biochem. J. 2016, 2, 105–112. [Google Scholar] [CrossRef]
- Sidwell, R.W.; Smee, D.F. In vitro and in vivo assay systems for study of influenza virus inhibitors. Antivir. Res. 2000, 48, 1–16. [Google Scholar] [CrossRef]
- Galochkina, A.V.; Bollikanda, R.K.; Zarubaev, V.V.; Tentler, D.G.; Lavrenteva, I.N.; Slita, A.V.; Chirra, N.; Kantevari, S. Synthesis of novel derivatives of 7,8-dihydro-6H-imidazo[2,1-b][1,3]benzothiazol-5-one and their virus-inhibiting activity against influenza A virus. Arch. Pharm. 2018, 352, e1800225. [Google Scholar] [CrossRef]
- Leang, S.K.; Hurt, A.C. Fluorescence-based Neuraminidase Inhibition Assay to Assess the Susceptibility of Influenza Viruses to The Neuraminidase Inhibitor Class of Antivirals. J. Vis. Exp. 2017, 122, 55570. [Google Scholar] [CrossRef]
- Gürsoy, E.; Dincel, E.D.; Naesens, L.; Güzeldemirci, N.U. Design and synthesis of novel Imidazo[2,1-b]thiazole derivatives as potent antiviral and antimycobacterial agents. Bioorg. Chem. 2020, 95, 103496–103505. [Google Scholar] [CrossRef] [PubMed]
- Meleddu, R.; Distinto, S.; Corona, A.; Tramontano, E.; Bianco, G.; Melis, C.; Cottiglia, F.; Maccioni, E. Isatin thiazoline hybrids as dual inhibitors of HIV-1 reverse transcriptase. J. Enzym. Inhib. Med. Chem. 2017, 32, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Kasralikar, H.M.; Jadhavar, S.C.; Goswami, S.V.; Kaminwar, N.S.; Bhusare, S.R. Design, synthesis and molecular docking of pyrazolo [3,4d] thiazole hybrids as potential anti-HIV-1 NNRT inhibitors. Bioorg. Chem. 2019, 86, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Petrou, A.; Eleftheriou, P.; Geronikaki, A.; Akrivou, M.G.; Vizirianakis, I. Novel Thiazolidin-4-ones as Potential Non-nucleoside Inhibitors of HIV-1 Reverse Transcriptase. Molecules 2019, 24, 3821. [Google Scholar] [CrossRef] [Green Version]
- Meleddu, R.; Corona, A.; Distinto, S.; Cottiglia, F.; Deplano, S.; Sequeira, L.; Secci, D.; Onali, A.; Sanna, E.; Esposito, F.; et al. Exploring New Scaffolds for the Dual Inhibition of HIV-1 Rt Polymerase and Ribonuclease Associated Functions. Preprints 2021, 2021020525. [Google Scholar] [CrossRef]
- Ottana, R.; Paoli, P.; Alexandra, N.; Lori, G.; Cardile, V.; Adornato, I.; Maccari, R. Discovery of 4-[(5-arylidene-4- oxothiazolidin-3-yl)methyl]benzoic acid derivatives active as novel potent allosteric inhibitors of protein tyrosine phosphatase 1B: In silico studies and in vitro evaluation as insulinomimetic and anti-inflammatory agents. Eur. J. Med. Chem. 2017, 127, 840–858. [Google Scholar] [CrossRef] [PubMed]
- Ottana, R.; Maccari, R.; Amuso, S.; Camici, G.; Rotondo, A.; Wolber, G.; Paoli, P. Synthesis, biological activity and structure-activity relationships of new benzoic acid-based protein tyrosine phosphatase inhibitors endowed with insulinomimetic effects in mouse C2C12 skeletal muscle cells. Eur. J. Med. Chem. 2014, 71, 112–127. [Google Scholar] [CrossRef]
- Ottana, R.; Maccari, R.; Amuso, S.; Wolber, G.; Schuster, D.; Herdlinger, S.; Manao, G.; Camici, G.; Paoli, P. New 4-[(5-Arylidene-2-arylimino-4-oxo-3-thiazolidinyl)methyl]benzoic acids active as protein tyrosine phosphataseinhibitors endowed with insulinomimetic effect on mouse C2C12 skeletalmuscle cells. Eur. J. Med. Chem. 2012, 50, 332–343. [Google Scholar] [CrossRef]
- Ottanà, R.; Ciurleo, R.; Paoli, P.; Manao, G.; Camici, G.; Laggner, C.; Langer, T. Structure based optimization of benzoic acids as protein tyrosine phosphatase 1B and low molecular weight protein tyrosinephosphatase. ChemMedChem 2009, 4, 957–962. [Google Scholar]
- Verma, S.; Thareja, S. Molecular docking assisted 3D-QSAR study of benzylidene-2, 4- thiazolidinedione derivatives as PTP-1B inhibitors for the the management of Type-2 diabetes mellitus. RSC Adv. 2016, 6, 33857–33867. [Google Scholar] [CrossRef]
- Meng, G.; Zheng, M.; Wang, M.; Tong, J.; Ge, W.; Zhang, J.; Zheng, A.; Li, J.; Gao, L.; Li, J. Design and Synthesis of New Potent PTP1B Inhibitors with the Skeleton of 2-Substituted Imino-3-Substituted-5-Heteroarylidene-1,3-Thiazolidine-4-One: Part I. Eur. J. Med. Chem. 2016, 122, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Ganou, C.A.; Eleftheriou, P.T.; Theodosis-Nobelos, P.; Fesatidou, M.; Geronikaki, A.A.; Lialiaris, T. Docking analysis targeted to the whole enzyme: An application to the prediction of inhibition of PTP1B by thiomorpholine and thiazolyl derivatives. SAR QSAR Environ. Res. 2018, 29, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Figueroa, S.; Estrada-Soto, S.; Ramirez-Espinosa, J.J.; Paoli, P.; Lori, G.; Leon-Rivera, I. Synthesis and evaluation of thiazolidine-2,4-dione/benzazole derivatives as inhibitors of protein tyrosine phosphatase 1B (PTP-1B): Antihyperglycemic activity with molecular docking study. Biomed. Pharmacother. 2018, 107, 1302–1310. [Google Scholar] [CrossRef]
- Liu, W.-S.; Wang, R.-R.; Wang, S.-Q.; Dong, W.-L.; Wang, R.-L.; Yue, H. Design, synthesis, biological evaluation and molecular dynamics studies of 4-thiazolinone derivatives as protein tyrosine phosphatase 1B (PTP1B) inhibitors. J. Biomol. Struct. Dyn. 2019, 38, 1–11. [Google Scholar] [CrossRef]
- Wu, J.; Ma, Y.; Zhou, H.; Zhou, L.; Du, S.; Sun, Y.; Li, W.; Dong, W.; Wang, R. Identification of protein tyrosine phosphatase 1B (PTP1B) inhibitors through De Novo Evoluton, synthesis, biological evaluation and molecular dynamics simulation. Biochem. Biophys. Res. Commun. 2020, 526, 273–280. [Google Scholar] [CrossRef]
- Patel, A.D.; Pasha, T.Y.; Lunagariya, P.; Shah, U.; Bhambharoliya, T.; Tripathi, R.K.P. A library of thiazolidin-4-one derivatives as protein tyrosine phosphatase 1B (PTP1B) inhibitors: An attempt to discover novel antidiabetic agents. Chem. Med. Chem. 2020, 15, 1229–1242. [Google Scholar] [CrossRef]
- Srivastava, J.K.; Dubey, P.; Singh, S.; Bhat, H.; Kumawatb, M.K.; Singh, U. Discovery of novel 1,3,5-triazine-thiazolidine-2,4-diones as dipeptidyl peptidase-4 inhibitors with antibacterial activity targeting the S1 pocket for the treatment of type 2 diabetes. RSC Adv. 2015, 5, 14095–14102. [Google Scholar] [CrossRef]
- Maezaki, H.; Tawada, M.; Yamashita, T.; Banno, Y.; Miyamoto, Y.; Yamamoto, Y.; Ikedo, K.; Kosaka, T.; Tsubotani, S.; Tani, A.; et al. Design of potent dipeptidyl peptidase IV (DPP-4) inhibitors by employing a strategy to form a salt bridge with Lys554. Bioorg. Med. Chem. Lett. 2017, 27, 3565–3571. [Google Scholar] [CrossRef]
- Nishimura, C.Y. Aldose reductase in glucose toxicity a potential target for the prevention of diabetic complications. Pharmacol. Rev. 1988, 50, 21–33. [Google Scholar]
- Ibrar, A.; Iqbal, J. Coumarin-thiazole and -oxadiazole derivatives: Synthesis, bioactivity and docking studies for aldose/aldehyde reductase inhibitors. Bioorg. Chem. 2016, 68, 177–186. [Google Scholar] [CrossRef]
- Andleeb, H.; Tehseen, Y.; Shah, S.; Khan, I.; Iqbal, J.; Hameed, S. Identification of novel pyrazole–rhodanine hybrid scaffolds as potent inhibitors of aldose reductase: Design, synthesis, biological evaluation and molecular docking analysis. RSC Adv. 2016, 6, 77688–77700. [Google Scholar] [CrossRef]
- Demir, Y.; Taslimi, P.; Koçyiğit, Ü.M.; Akkuş, M.; Özaslan, M.S.; Duran, H.E.; Budak, Y.; Tüzün, B.; Gürdere, M.B.; Ceylan, M.; et al. Determination of the inhibition profiles of pyrazolyl-thiazole derivatives against aldose reductase and α-glycosidase and molecular docking studies. Arch. Pharm. 2020, 353, e2000118. [Google Scholar] [CrossRef]
- Celestina, S.K.; Sundaram, K.; Ravi, S. In vitro studies of potent aldose reductase inhibitors: Synthesis, characterization, biological evaluation and docking analysis of rhodanine-3-hippuric acid derivatives. Bioorg. Chem. 2020, 97, 103640. [Google Scholar] [CrossRef] [PubMed]
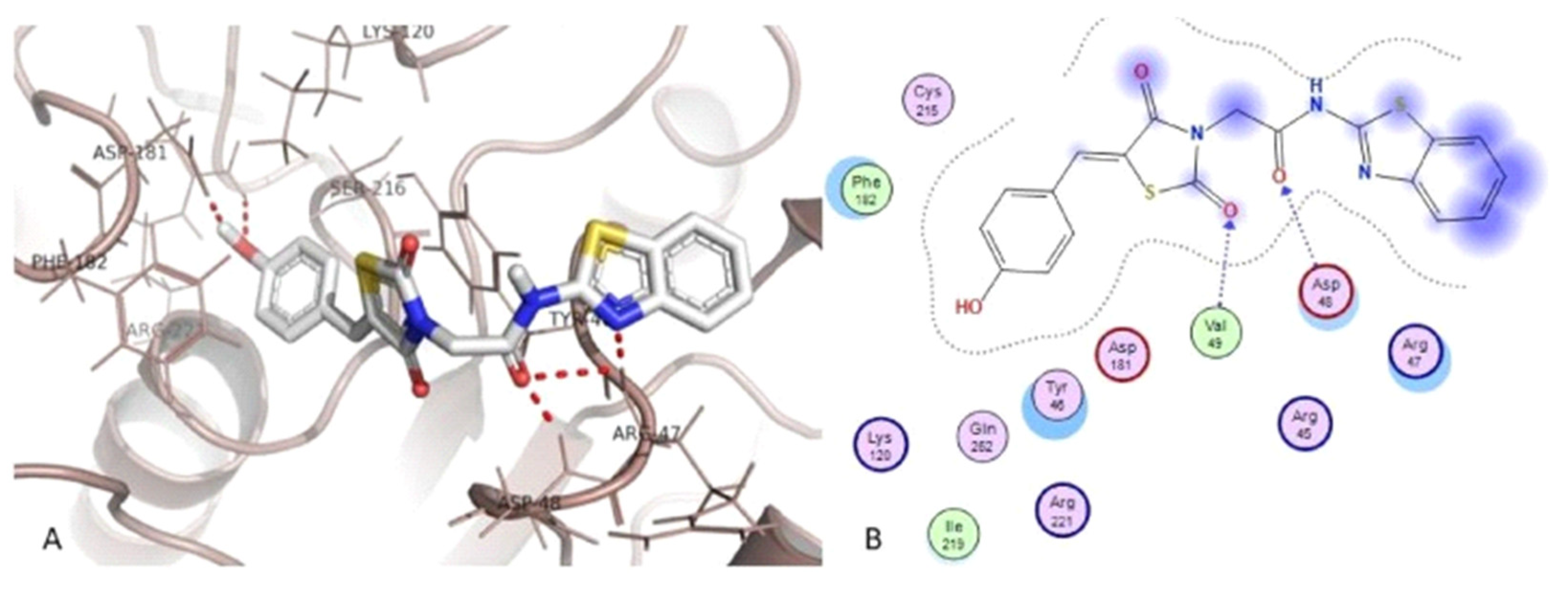
- Sever, B.; Altıntop, M.D.; Demir, Y.; Akalın Çiftçi, G.; Beydemir, Ş.; Özdemir, A. Design, synthesis, in vitro and in silico investigation of aldose reductase inhibitory effects of new thiazole-based compounds. Bioorg. Chem. 2020, 102, 104110. [Google Scholar] [CrossRef] [PubMed]
- Sever, B.; Altıntop, M.; Demir, Y.; Pekdoğan, M.; Akalın Çiftçi, G.; Beydemir, Ş.; Özdemir, A. An extensive research on aldose reductase inhibitory effects of new 4H-1,2,4-triazole derivatives. J. Mol. Struct. 2021, 1224, 129446. [Google Scholar] [CrossRef]
- Carter, R.; Mendis, K.N. Evolutionary and Historical Aspects of the Burden of Malaria. Clin. Microbiol. Rev. 2002, 15, 564–594. [Google Scholar] [CrossRef] [Green Version]
- Desjeux, P. Leishmaniasis: Current situation and new perspectives. Comp. Immunol. Microbiol. Infect. Dis. 2004, 27, 305–318. [Google Scholar] [CrossRef]
- Sarkari, B.; Ahmadpour, N.B.; Motazedian, M.H.; Mirjalali, H.; Akhoundi, M.; Mohebali, M.; Hajjaran, H. Inter-and intraspecific variations of Leishmania strains isolated from patients with cutaneous and visceral leishmaniases in Fars Province, South of Iran. Iran. J. Med. Sci. 2016, 41, 209–216. [Google Scholar] [PubMed]
- Sarkari, B.; Hatam, G.; Ghatee, M. Epidemiological features of visceral leishmaniasis in fars province, southern iran. Iran. J. Public Health 2012, 41, 94–99. [Google Scholar]
- Sarkari, B.; Naraki, T.; Ghatee, M.A.; Abdolahi Khabisi, S.; Davami, M.H. Visceral leishmaniasis in Southwestern Iran: A retrospectiveclinico-hematological analysis of 380 consecutive hospitalizedcases (1999–2014). PLoS ONE 2016, 11, e0150406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.S.A.; Silveira, G.O.; Amaral, M.S.; Sinara, M.V.; Almeida, S.M.V.; Oliveira, J.F.; Lima, M.C.A.; Sergio Verjovski-Almeida, S.I.D. In vitro activity of aryl-thiazole derivatives against Schistosoma mansoni schistosomula and adult worms. PLoS ONE 2019, 14, e0225425. [Google Scholar] [CrossRef] [PubMed]
- Sharma, I.; Sullivan, M.; Thomas, F.; McCutchan, T.F. In Vitro Antimalarial Activity of Novel Semisynthetic Nocathiacin I Antibiotics. Antimicrob. Agents Chemother. 2015, 59, 3174–3179. [Google Scholar] [CrossRef] [Green Version]
- Pucci, M.J.; Bronson, J.J.; Barrett, J.F.; DenBleyker, K.L.; Discotto, L.F.; Fung-Tomc, J.C.; Ueda, Y. Antimicrobial evaluation of nocathiacins, athiazole peptide class of antibiotics. Antimicrob. Agents Chemother. 2004, 48, 3697–3701. [Google Scholar] [CrossRef] [Green Version]
- Naidu, B.N.; Sorenson, M.E.; Zhang, Y.; Kim, O.K.; Matiskella, J.D.; Wichtowski, J.A.; Connolly, T.P.; Li, W.; Lam, K.S.; Bronson, J.J.; et al. Nocathiacin I analogues: Synthesis, in vitro and in vivo biological activity of novel semi-synthetic thiazolyl peptide antibiotics. Bioorg. Med. Chem. Lett. 2004, 14, 5573–5577. [Google Scholar] [CrossRef] [PubMed]
- Bueno, J.M.; Carda, M.; Crespo, B.; Cuñat, A.C.; de Cozaa, C.; León, M.L.; Marco, J.A.; Roda, N.; Sanz-Cervera, J.F. Design, synthesis and antimalarial evaluation of novel thiazole derivative. Bioorg. Med. Chem. Lett. 2016, 26, 3938–3944. [Google Scholar] [CrossRef]
- Sahu, S.; Ghosha, S.K.; Gahtori, P.; Singh, U.P.; Bhattacharyya, D.R.; Raj Bhat, H.R. In silico ADMET study, docking, synthesis and antimalarial evaluation of thiazole-1,3,5-triazine derivatives as Pf-DHFR inhibitor. Pharmacol. Rep. 2019, 71, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.C.B.; da Silva, H.V.C.; Brondani, D.J.; de Faria, A.R.; Ximenes, R.M.; da Silva, I.M.; de Albuquerque, J.F.C.; Castilho, M.S. Synthesis and biological evaluation of thiazole derivatives as LbSOD inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 34, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.A.; Freire, P.; da Costa, M.O.L.; Pavani, T.F.A.; Patrícia Xander, P.; Geraldo, M.M.; Ana Mengarda, A.; de Moraes, J.; Rando, D.G.G. 4-Phenyl-1,3-thiazole-2-amines as scaffolds for new antileishmanial agents. J. Venom. Anim. Toxins Incl. Trop. Dis. 2018, 24, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alianc, A.S.S.; Oliveira, A.R.; Feitosa, A.P.S.; Ribeiro, K.R.C.; de Castro, M.C.A.B.; Leite, A.C.L.; Alves, L.C.; Brayner, F.A. In vitro evaluationof cytotoxicity and leishmanicidal activity of phthalimido-thiazole derivatives. Eur. J. Pharm. Sci. 2017, 105, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.J.; Skripchenko, A.; Salata, J.; O’Sullivan, A.M.; Cardo, L.J. Inactivation of Leishmania donovani infantum and Trypanosoma cruzi in red cell suspensions with thiazole orange. Transfusion 2008, 48, 1363–1367. [Google Scholar] [CrossRef]
- Prajapati, A.K.; Modi, V.P. Synthesis and biological activity of n-{5-(4-methylphenyl) diazenyl-4-phenyl-1, 3-thiazol-2-yl} ben-zamide derivatives. Química Nova 2011, 34, 771–774. [Google Scholar]
- Amnerkar, N.D.; Bhongade, B.A.; Bhusari, K.P. Synthesis and biological evaluation of some 4-(6-substituted-1, 3-benzothiazol-2-yl) amino-1, 3-thiazole-2-amines and their Schiff bases. Arab. J. Chem. 2015, 8, 545–552. [Google Scholar] [CrossRef]
- Rezaei, Z.; Khabnadideh, S.; Pakshir, K.; Hossaini, Z.; Amiri, F.; Assadpour, E. Design, synthesis, and antifungal activity of triazole and benzotriazole derivatives. Eur. J. Med. Chem. 2009, 44, 3064–3067. [Google Scholar] [CrossRef] [PubMed]
- Rezaei, Z.; Sarkari, B.; Khabnadideh, S.; Farjami, M.; Mehrjou, M.; Atefeh Yazdi, A.; Elham Riazimontazer, E.; Fararouei, M. Synthesis and Biological Activity of Some Aminothiazole Derivatives as Antileishmanial Agent. Anti-Infect. Agents 2020, 18, 178–189. [Google Scholar] [CrossRef]
- Emami, S.; Tavangar, P.; Keighobadi, M. An overview of azoles targeting sterol 14α-demethylase for antileishmanial therapy. Eur. J. Med. Chem. 2017, 135, 241–259. [Google Scholar] [CrossRef]
- De Oliviera Cardoso, M.; de Siqueira, L.R.P.; da Silva, E.B.; Costa, L.B.; Hernandes, M.Z.; Rabello, M.M.; Ferreira, R.S.; da Cruz, L.F.; Moreira, D.R.M.; Pereira, V.R.A.; et al. 2-Pyridyl thiazoles as novel anti-Trypanosoma cruzi agents: Structural design, synthesis and pharmacological evaluation. Eur. J. Med. Chem. 2014, 86, 48–59. [Google Scholar] [CrossRef]
- Nava-Zuazo, C.; Chávez-Silva, F.; Moo-Puc, R.; Chan-Bacab, M.J.; Ortega-Morales, B.O.; Moreno-Díaz, H.; Díaz-Coutiño, D.; Hernández-Núñez, E.; Navarrete-Vázquez, G. 2-Acylamino-5-nitro-1,3-thiazoles: Preparation and in vitro bioevaluation against four neglected protozoan parasites. Bioorg. Med. Chem. 2014, 22, 1626–1633. [Google Scholar] [CrossRef]
- De Oliveira, V.V.G.; de Souza, M.A.A.; Cavalcanti, R.R.M.; De Oliviera Cardoso, M.V.; Leiete, A.C.L.; Silva Junior, V.A.; de Figueiredo, R.C.B.Q. Study of in vitro biological activity of thiazoles on Leishmania (Leishmania) infantum. J. Glob. Antimicrob. Resist. 2020, 22, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.A.; Wenzler, T.; Yang, S.; Weiser, P.T.; Wang, M.Z.; Tidwell, R.R. Synthesis of novel amide and urea derivatives of thiazol-2-ethylamines and their activity against derivatives 27–55. Bioorg. Med. Chem. 2016, 24, 2451–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadich, E.; Kryshchyshyn-Dylevych, A.; Holota, S.; Polishchuk, P.; Džubak, P.; Gurska, S.; Hajduch, M.; Lesyk, R. Assessing different thiazolidine and thiazole-based compounds as antileishmanial scaffolds. Bioorg. Med. Chem. Lett. 2020, 30, 127616–127620. [Google Scholar] [CrossRef]
- Karade, H.; Acharya, B.N.; Sathe, M.; Kaushik, M.P. Design, synthesis, and antimalarial evaluation of thiazole-derived amino acids Med. Chem. Res. 2008, 17, 19–29. [Google Scholar]
- Kryshchyshyn, A.; Kaminskyy, D.; Karpenko, O.; Gzella, A.; Grellier, P.; Lesyk, R. Thiazolidinone/thiazole based hybrids—New class of antitrypanosomal agents. Eur. J. Med. Chem. 2019, 174, 292–308. [Google Scholar] [CrossRef]
- Holota, S.; Kryshchyshyn, A.; Derkach, H.; Trufin, Y.; Demchuk, Ι.; Gzella, A.; Grellier, P.; Lesyk, R. Synthesis of 5-enamine-4-thiazolidinone derivatives with trypanocidal and anticancer activity. Bioorg. Chem. 2019, 86, 126–136. [Google Scholar] [CrossRef]
- Georgiadis, Μ.-O.; Kourbeli, V.; Papanastasiou, I.P.; Tsotinis, A.; Taylor, M.C.; Kelly, J.M. Synthesis and evaluation of novel 2,4-disubstituted arylthiazoles against T. brucei. RSC Med. Chem. 2020, 11, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Kryshchyshyn, A.; Kaminskyy, D.; Nektegayev, I.; Grellier, P.; Lesyk, R. Isothiochromenothiazoles—A Class of Fused Thiazolidinone Derivatives with Established Anticancer Activity That Inhibits Growth of Trypanosoma brucei brucei. Sci. Pharm. 2018, 86, 47. [Google Scholar] [CrossRef] [Green Version]
- Kryshchyshyn, A.P.; Atamanyuk, D.V.; Kaminskyy, D.V.; Grellier, P.; Lesyk, R.B. Investigation of anticancer and anti-parasitic activity of thiopyrano[2,3-d]thiazoles bearing norbornane moiety. Biopolym. Cell 2017, 33, 183–205. [Google Scholar] [CrossRef] [Green Version]
- Gomes, P.A.T.M.; Barbosa, M.O.; Santiago, E.F.; Cardoso, M.V.O.; Costa, N.T.C.; Hernandes, M.Z.; Moreira, D.R.M.; da Silva, A.C.; dos Santos, T.A.R.; Pereira, V.R.A.; et al. New 1,3-thiazole derivatives and their biological and ultrastructural effects on Trypanosoma cruzi. Eur. J. Med. Chem. 2016, 121, 387–398. [Google Scholar] [CrossRef]
- Du, X.; Guo, C.; Hansell, E.; Doyle, P.S.; Caffrey, C.R.; Holler, T.P.; McKerrow, J.H.; Cohen, F.E. Synthesis and structure-activity relationship study of potent trypanocidal thiosemicarbazone inhibitors of the trypanosomal cysteine protease cruzain. J. Med. Chem. 2002, 1, 2695–2707. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira Filho, G.B.; de Oliveira Cardoso, M.V.; Espíndola, J.W.P.; Silva, D.A.O.; Rafaela Salgado Ferreira, R.S.; Coelho, P.L.; dos Anjos, P.S.; Santos, E.S.; Meira, C.S.; Diogo Rodrigo Magalhaes Moreira, D.R.M.; et al. Structural design, synthesis and pharmacological evaluation of thiazoles against Trypanosoma cruzi. Eur. J. Med. Chem. 2017, 141, 346–361. [Google Scholar] [CrossRef]
- Da Silva, E.B.; Oliveira e Silva, D.A.; Oliveira, A.R.; Mendes, C.H.S.; dos Santos, T.A.R.; da Silva, A.C.; de Castro, M.C.A.; Ferreira, R.S.; Moreira, D.R.M.; Cardoso, M.V.O.; et al. Design and synthesis of potent anti-Trypanosoma cruzi agents’ new thiazoles derivatives which induce apoptotic parasite death. Eur. J. Med. Chem. 2017, 130, 39–50. [Google Scholar] [CrossRef] [PubMed]
- De Siqueira, L.R.P.; de Oliveira Barbosa, M.; Oliveira, A.R.; de Moraes Gomes, P.A.T.; de Oliveira Filho, G.B.; de Oliveira Cardoso, M.V.; Pereira, V.R.A.; da Silva Santos, A.C.; Júnior, P.A.S.; Romanha, A.J.; et al. Synthesis and anti-Trypanosoma cruzi profile of the novel 4-thiazolidinone and 1,3-thiazole derivatives. Front. Drug Chem. Clin. Res. 2019, 2, 1–12. [Google Scholar]
- Rostom, A.; Dube, C.; Wells, G.; Tugwell, P.; Welch, V.; Jolicoeur, E.; McGowan, J. Prevention of NSAID-induced gastroduodenal ulcers. Cochrane Database Syst. Rev. 2002, 4, CD002296. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Cryer, B. Gastrointestinal injury associated with NSAID use: A case study and review of risk factors and preventative strategies. Drug Healthc. Patient Saf. 2015, 7, 31–41. [Google Scholar] [CrossRef] [Green Version]
- Bally, M.; Dendukuri, N.; Rich, B.; Nadeau, L.; Helin-Salmivaara, A.; Garbe, E.; Brophy, J.M. Risk of acute myocardial infarction with NSAIDs in real world use: Bayesian meta-analysis of individual patient data. BMJ 2017, 357, j1909, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Cooper, M.G.; Craig, J.C.; Knight, J.F.; Keneally, J.P. Effects of nonsteroidal anti-inflammatory drugs on postoperative renal function in adults with normal renal function. Cochrane Database Syst. Rev. 2007, 2, CD002765. [Google Scholar] [CrossRef]
- Lucas, G.N.C.; Leitão, A.C.C.; Alencar, R.L.; Xavier, R.M.F.; Daher, E.D.F.; da Silva Junior, G.B. Pathophysiological aspects of nephropathy caused by non-steroidal anti-inflammatory drugs. Braz. J. Nephrol. 2019, 41, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.L.; Tian, Z.G.; Wang, F.; Li, W.G.; Cheng, D.Y.; Yang, Y.F.; Gao, H.M. Characteristics and clinical outcome of nonsteroidal anti-inflammatory drug-induced acute hepato-nephrotoxicity among Chinese patients. World J. Gastroenterol. 2014, 20, 13956–13965. [Google Scholar] [CrossRef]
- Telliez, A.; Furman, C.; Pommery, N.; Hénichart, J.P. Mechanisms leading to COX-2 expression and COX-2 induced tumorigenesis: Topical therapeutic strategies targeting COX-2 expression and activity. Anticancer Agents Med. Chem. 2006, 6, 187–208. [Google Scholar] [CrossRef] [PubMed]
- Khloya, P.; Kumar, S.; Kaushik, P.; Surain, P.; Kaushik, O.; Sharma, K. Synthesis and biological evaluation of pyrazolylthiazole carboxylic acids as potent anti-inflammatory–antimicrobial agents. Bioorg. Med. Chem. Lett. 2015, 25, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Kamble, R.; Meshram, R.; Hese, S.; More, R.; Kamble, S.; Gacche, R.; Dawan, B. Synthesis and In Silico Investigation of Thiazoles Bearing Pyrazoles Derivatives as Anti-Inflammatory Agents. Comput. Biol. Chem. 2016, 61, 86–96. [Google Scholar] [CrossRef]
- Mohareb, R.; Al-Omran, F.; Abdelaziz, M.; Ibrahim, R. Anti-inflammatory and Anti-ulcer Activities of New Fused Thiazole Derivatives Derived from 2-(2-Oxo-2H-chromen-3-yl)thiazol-4(5H)-one. Acta Chim. Slov. 2017, 64, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Ali, Y.; Alam, M.S.; Hamid, H.; Husain, A.; Dhulap, A.; Banoa, S.; Kharband, C. Novel 2,4-dichlorophenoxy acetic acid substituted thiazolidin-4-ones asanti-inflammatory agents: Design, synthesis and biological screening. Bioorg. Med. Chem. Lett. 2017, 22, 1017–1025. [Google Scholar] [CrossRef]
- Abdelazeem, A.; El-Saadia, M.; Safi El-Din, A.; Omar, H.; El-Moghazy, S. Design, synthesis and analgesic/anti-inflammatory evaluation of novel diarylthiazole and diarylimidazole derivatives towards selective COX-1inhibitors with better gastric profile. Bioorg. Med. Chem. 2017, 25, 665–676. [Google Scholar] [CrossRef]
- Kakuta, H.; Zheng, X.; Oda, H.; Harada, S.; Sugimoto, Y.; Sasaki, K.; Tai, A. Cyclooxygenase-1-selective inhibitors are attractive candidates for analgesics that do not cause gastric damage. design and in vitro/in vivo evaluation of a benzamide-type cyclooxygenase-1 selective inhibitor. J. Med. Chem. 2008, 51, 2400–2411. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, R.; Morham, S.G.; Tiano, H.F.; Loftin, C.D.; Ghanayem, B.I.; Chulada, P.C.; Mahler, J.F.; Lee, C.A.; Goulding, E.H.; Kluckman, K.D.; et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 1995, 83, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Oda, H.; Takamatsu, K. Analgesic agents without gastric damage: Design and synthesis of structurally simple benzenesulfonanilide-type cyclooxygenase-1-selective inhibitors. Bioorg. Med. Chem. 2007, 15, 1014. [Google Scholar] [CrossRef]
- Koster, R.; Anderson, M.; Ej, D.B. Acetic acid-induced analgesic screening. Fed. Proc. 1959, 18, 412. [Google Scholar]
- Abdelazeem, A.H.; Salama, S.A.; Maghrabi, I.A. Synthesis and evaluation of noveldiphenylthiazole derivatives as potentialanti-inflammatory agents. Arch. Pharm. 2015, 348, 518. [Google Scholar] [CrossRef] [PubMed]
- Borne, R.; Mark, L.; Wilson, N. Nonsteroidal anti-inflammatory drugs. In Foye’s Principles of Medicinal Chemistry; Thomas, L.L., David, A.W., Victoria, F.R., Zito, W., Eds.; Wolters Kluwer Health Inc.: Alphen aan den Rijn, The Netherlands; Lippincott Williams & Wilkins: Baltimore, MD, USA, 2013; p. 1021. ISBN 9781609133450. [Google Scholar]
- Oniga, S.; Pacureanu, L.; Stoica, C.; Palage, M.; Crăciun, A.; Araniciu, C. COX Inhibition Profile and Molecular Docking Studies of Some 2-(Trimethoxyphenyl)-Thiazoles. Molecules 2017, 22, 1507. [Google Scholar] [CrossRef] [Green Version]
- Araniciu, C.; Pârvu, A.E.; Tiperciuc, B.; Palage, M.; Oniga, S.; Verité, P.; Oniga, O. Synthesis andevaluation of the anti-inflammatory activity of some 2-(Trimethoxyphenyl)-4-R1-5-R2-Thiazoles. Dig. J. Nanomater. Biostruct. 2013, 8, 699–709. [Google Scholar]
- Araniciu, C.; Palage, M.; Oniga, S.; Pirnau, A.; Verité, P.; Oniga, O. Synthesis and characterization of some novel 5,2 -and 4,2-bisthiazoles derivatives. Revista Chimie 2013, 64, 1067–1071. [Google Scholar]
- Araniciu, C.; Pârvu, A.E.; Palage, M.D.; Oniga, S.D.; Benedec, D.; Oniga, I.; Oniga, O. The effect of some 4,2and 5,2 bisthiazole derivatives on nitro-oxidative stress and phagocytosis in acute experimental inflammation. Molecules 2014, 19, 9240–9925. [Google Scholar] [CrossRef] [Green Version]
- Sirsat, D.; Kate, P.; Bachute, M. synthesis and biological evaluation of novel thiazole-pyrazole integrated chalcones as antioxidant and anti-inflammatory agents. Asian J. Pharm. Clin. Res. 2019, 12, 311–315. [Google Scholar] [CrossRef]
- El-Kerdawy, M.; Ghaly, A.; Darwish, S.; Abdel-Aziz, A.; Elsheakh, A.; Abdelrahman, R.; Hassan, G. New benzimidazothiazole derivatives as anti-inflammatory, antitumor active agents: Synthesis, in-vitro and in-vivo screening and molecular modeling studies. Bioorg. Chem. 2019, 83, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Son, M.; Ju, H.K.; Lin, C.X.; Moon, T.C.; Choi, H.G.; Chang, H.W. Dual inhibition of cyclooxygenases-2 and 5-libooxgynase by deoxypodophyllotoxin (anthricin) in mouse bone marrow-derived mast cells. Biol. Pharm. Bull. 2004, 27, 786–788. [Google Scholar] [CrossRef] [Green Version]
- Manivannan, E.; Chaturvedi, C.S. Analogue-based design, synthesis and molecular docking analysis of 2,3-diarylquinazolinones as non-ulcerogenic anti-inflammatory agents. Bioorg. Med. Chem. 2011, 19, 4520–4528. [Google Scholar] [CrossRef]
- Shahrasbi, M.; Movahed, M.; Dadras, O.; Daraei, B.; Zarghi, A. Design, Synthesis and Biological Evaluation of New Imidazo[2,1-b]Thiazole Derivatives as Selective COX-2 Inhibitors. Iran. J. Pharm. Sci. 2018, 17, 1288–1296. [Google Scholar]
- Mahsa Azami, M.; Bahram, D.; Zarghi, A. Synthesis and biological evaluation of new imidazo[1,2-a]pyridine derivatives as selective cox-2 inhibitors. Lett. Drug Des. Discov. 2016, 13, 1–7. [Google Scholar]
- Jacob, J.P.; Manju, S.L. Novel approach of multi-targeted thiazoles and thiazolidenes toward anti-inflammatory and anticancer therapy—Dual inhibitionof COX-2 and 5-LOX enzymes. Med. Chem. Res. 2021, 30, 236–257. [Google Scholar]
- Abdelall, E.K. Synthesis and biological evaluations of novel iso-xazoles and furoxan derivative as anti-inflammatory agents. Bioorg. Chem. 2020, 94, 103441. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, M.H.; Youssif, B.G.; Abdelazeem, A.H.; Ibrahim, H.M.; Abd, E.L.; Ghany, A.M.; Treamblu, L.; Bukhari, S.N.A. Synthesis, Bio-logical evaluation, docking study and ulcerogenicity profiling ofsome novel quinoline-2-carboxamides as dual COXs/LOX inhibitors endowed with anti-inflammatory activity. Eur. J. Med. Chem. 2017, 127, 972–985. [Google Scholar] [CrossRef]
- Jacob, J.P.; Manju, S.L. Identification and development of thiazole lead as COX-2/5-LOX inhibitors through in-vitro and in-vivo biological evaluation for anti-inflammatory activity. Bioorg. Chem. 2020, 100, 103882–103898. [Google Scholar] [CrossRef] [PubMed]
- Ziakas, G.; Rekka, E.; Gavalas, A.; Eleftheriou, P.; Kourounakis, P. New analogues of butylated hydroxytoluene as anti-inflammatory and antioxidant agents. Bioorg. Med. Chem. 2006, 14, 5616–5624. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.; Gawande, S.; Bodade, R.; Totre, J.; Khobragade, C. Synthesis and biological evaluation of simple methoxylated chalcones as anticancer, anti-inflammatory and antioxidant agents. Bioorg. Med. Chem. 2010, 18, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Jimenez, M.T.B.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.F.; Wang, G. Environmental Agents, Oxidative Stress and Autoimmunity. Curr. Opin. Toxicol. 2018, 7, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Thota, S.; Nadipelly, K.; Shenkesi, A.; Yerra, R. Design, synthesis, characterization, antioxidant and in vitro cytotoxic activities of novel coumarin thiazole derivatives. Med. Chem. Res. 2015, 24, 1162–1169. [Google Scholar] [CrossRef]
- Nastasă, C.; Tiperciuc, B.; Duma, M.; Benedec, D.; Oniga, O. New Hydrazones Bearing Thiazole Scaffold: Synthesis, Characterization, Antimicrobial, and Antioxidant Investigation. Molecules 2015, 20, 17325–17338. [Google Scholar] [CrossRef] [Green Version]
- Lozynskyi, A.; Zasidko, V.; Atamanyuk, D.; Kaminskyy, D.; Derkach, H.; Karpenko, O.; Ogurtsov, V.; Kutsyk, R.; Lesyk, R. Synthesis, antioxidant and antimicrobial activities of novel thiopyrano[2,3-d]thiazoles based on aroylacrylic acids. Mol. Divers. 2017, 21, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Grozav, A.; Porumb, I.; Gaina, L.; Filip, L.; Hanganu, D. Cytotoxicity and Antioxidant Potential of Novel 2-(2-((1H-indol-5yl)methylene)-hydrazinyl)-thiazole Derivatives. Molecules 2017, 22, 260. [Google Scholar] [CrossRef] [Green Version]
- Djukic, M.; Fesatidou, M.; Xenikakis, I.; Geronikaki, A.; Angelova, V.; Savic, V.; Pasic, M.; Krilovic, B.; Djukic, D.; Gobeljic, B.; et al. In vitro antioxidant activity of thiazolidinone derivatives of 1,3-thiazole and 1,3,4-thiadiazole. Chem. Biol. Interact. 2018, 286, 119–131. [Google Scholar] [CrossRef]
- Afifi, O.; Shaaban, O.; Abd El Razik, H.; El-Dine, S.; Ashour, F.; El-Tombary, A.; Abu-Serie, M. Synthesis and biological evaluation of purine-pyrazole hybrids incorporating thiazole, thiazolidinone or rhodanine moiety as 15-LOX inhibitors endowed with anticancer and antioxidant potential. Bioorg. Chem. 2019, 87, 821–837. [Google Scholar] [CrossRef]
- Chithra, V.; Abbs Ven Reji, T. Synthesis and antioxidant activity of novel benzimidazolyl thiazole derivatives complemented by DFT studies. Indian J. Chem. 2019, 58B, 1279–1283. [Google Scholar]
- Hossan, A. Synthesis, modelling and molecular docking of new 5-arylazo-2-chloroacetamido thiazole derivatives as antioxidant agent. J. Mol. Struct. 2020, 1206, 127712. [Google Scholar] [CrossRef]
- Erzengin, M.; Bilen, G.; Ergun, A.; Gencer, N. Antipsychotic agents screened as human carbonic anhydrase I and II inhibitors. Arch. Physiol. Biochem. 2014, 120, 29. [Google Scholar] [CrossRef] [PubMed]
- Pinard, M.A.; Lotlikar, S.R.; Boone, C.D.; Vullo, D.; Supuran, C.T.; Patrauchan, M.A.; McKenna, R. Structure and inhibition studies of a type II beta-carbonic anhydrase psCA3 from Pseudomonas aeruginosa. Bioorg. Med. Chem. 2015, 23, 54831–54838. [Google Scholar] [CrossRef] [PubMed]
- Ceruso, M.; Antel, S.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Inhibition studies of new ureido-substituted sulfonamides incorporating a GABA moiety against human carbonic anhydrase isoforms I–XIV. Bioorg. Med. Chem. 2014, 22, 6768–6775. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, D.; Çavdar, H.; Durdagi, S.O.; Şentürk, M. Structure–activity relationships for the interaction of 5,10-dihydroindeno[1,2-b]indole derivatives with human and bovine carbonic anhydrase isoforms I, II, III, IV and VI. Eur. J. Med. Chem. 2012, 49, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Yaseen, R.; Ekinci, D.; Senturk, M.; Hammed, A.D.; Ovais, S.; Rathore, P.; Samim, M.; Javed, K.; Supuran, C.T. Pyridazinone substituted benzenesulfonamides as potent carbonic anhydrase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1337–1341. [Google Scholar] [CrossRef]
- Sarikaya, S.B.O.; Kaya, A.A.; Kaya, E.C.; Senturk, M. Synthesis and Determination of Some Biological Activities of Novel 2,4-Dinitrophenyl Derivatives. Arch. Pharm. Chem. Life Sci. 2015, 348, 214–220. [Google Scholar] [CrossRef]
- Balaydına, H.T.; Sentürkc, M.; Göksua, S.; Menzek, A. Synthesis and carbonic anhydrase inhibitory properties of novel bromophenolsand their derivatives including natural products: Vidalol B. Eur. J. Med. Chem. 2012, 54, 1423–1428. [Google Scholar]
- Korkmaz, N.; Obaidi, O.A.; Senturk, M.; Astley, D.; Ekinci, D.; Supuran, C.T. Synthesis and biological activity of novel thioureaderivatives as carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2015, 30, 75–80. [Google Scholar] [CrossRef]
- Karatas, M.O.; Alici, B.; Cakir, U.; Cetinkaya, E.; Demir, D.; Ergün, A.; Gençer, N.; Oktay Arslan, O. Synthesis and carbonic anhydrase inhibitoryproperties of novel coumarin. J. Enzym. Inhib. Med. Chem. 2013, 28, 299–304. [Google Scholar] [CrossRef] [Green Version]
- Fidan, I.; Salmas, R.E.; Arslan, M.; Şentürk, M.; Durdagi, S.; Ekinci, D.; Şentürk, E.; Coşgun, S.; Supuran, C.T. Carbonic anhydrase inhibitors: Design, synthesis, kinetic, docking and molecular dynamics analysis of novel glycine and phenylalanine sulfonamide derivatives. Bioorg. Med. Chem. 2015, 23, 7353–7358. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Ferraroni, M.; Scozzafava, A.; Supuran, C.T. Fluorescent sulfonamide carbonic anhydrase inhibitors incorporating 1,2,3-triazole moieties: Kinetic and X-ray crystallographic studies. Bioorg. Med. Chem. 2016, 24, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Chandak, N.; Ceruso, M.; Supuran, C.T.; Sharma, P.K. Novel sulfonamide bearing coumarin scaffolds as selective inhibitors of tumor associated carbonic anhydrase isoforms IX and XII. Bioorg. Med. Chem. 2016, 24, 2882–2886. [Google Scholar] [CrossRef] [PubMed]
- Pala, N.; Micheletto, L.; Sechi, M.; Aggarwal, M.; Carta, F.; McKenna, R.; Supuran, C.T. Carbonic Anhydrase Inhibition with Benzenesulfonamides and Tetrafluorobenzenesulfonamides Obtained via Click Chemistry. ACS Med. Chem. Lett. 2014, 5, 927–930. [Google Scholar] [CrossRef]
- Meleddu, R.; Maccioni, E.; Distinto, S.; Bianco, G.; Melis, C.; Alcaro, S.; Cottiglia, F.; Ceruso, M.; Supuran, C.T. New 4-[(3-cyclohexyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino]benzene-1-sulfonamides, synthesis and inhibitory activity toward carbonic anhydrase I., II, IX, XII. Bioorg. Med. Chem. Lett. 2015, 25, 3281–3284. [Google Scholar] [CrossRef]
- Kocyigit, U.M.; Aslan, O.N.; Gulcin, I.; Temel, Y.; Ceylan, M. Synthesis and Carbonic Anhydrase Inhibition of Novel 2-(4-(Aryl)-thiazole-2-yl)-3a,4,7,7a-tetrahydro-1H-4,7-methanoisoindole-1,3(2H)-dione Derivatives. Arch. Pharm. Chem. Life Sci. 2016, 349, 955–963. [Google Scholar] [CrossRef]
- Kılıcaslan, S.; Arslan, M.; Ruya, Z.; Bilen, C.; Ergün, A.; Gençer, N.; Arslan, O. Synthesis and evaluation of sulfonamide-bearing thiazole as carbonic anhydrase isoforms hCA I andhCA II. J. Enzym. Inhib. Med. Chem. 2016, 31, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Arslan, T.; Türkoğlu, E.A.; Murat Sentürk, M.; Claudiu, T.; Supuran, C.T. Synthesis and carbonic anhydrase inhibitory properties of novel chalcone substituted benzenesulfonamides. Bioorg. Med. Chem. Lett. 2016, 26, 5867–5870. [Google Scholar] [CrossRef]
- Abdoli, M.; Angeli, A.; Bozdag, M.; Carta, F.; Kakanejadifard, A.; Hamid Saeidian, H.; Supuran, C.T. Synthesis and carbonic anhydrase I, II, VII, and IX inhibition studies with a series of [benzo[d]thiazole-5- and 6-sulfonamides. J. Enzym. Inhib. Med. Chem. 2017, 32, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase: I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Capkauskaite, Ε.; Zubriene, A.; Paketuryte, V.; Timm, D.D.; Tumkevicius, S.; Matulis, D. Thiazole-substituted benzenesulfonamides as inhibitors of 12 humancarbonic anhydrases. Bioorg. Chem. 2018, 77, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Melis, D.; Meleddu, R.; Angeli, A.; Distinto, S.; Bianco, G.; Capasso, C.; Cottiglia, F.; Angius, R.; Supuran, C.T.; Maccioni, E. Isatin: A privileged scaf-fold for the design of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 68–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, G.; Meleddu, R.; Distinto, S.; Cottiglia, F.; Gaspari, M.; Melis, C.; Maccioni, E. N-Acylbenzenesulphonamide dihydro-1,3,4-oxadiazole hybrids:seeking selectivity toward carbonic anhydrase isoforms. ACS Med. Chem. Lett. 2017, 8, 792–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meleddu, R.; Distinto, S.; Cottiglia, F. Tuning the dualinhibition of carbonic anhydrase and cyclooxygenase bydihydrothiazole benzensulphonamides. ACS Med. Chem. Lett. 2018, 9, 1045. [Google Scholar] [CrossRef]
- Melis, C.; Distinto, S.; Bianco, G. Targeting tumor associ-ated carbonic anhydrases IX and XII: Highly isozyme select-ive coumarin and psoralen inhibitors. ACS Med. Chem. Lett. 2018, 9, 725–729. [Google Scholar] [CrossRef]
- Distinto, S.; Meleddu, R.; Ortuso, F.; Cottiglia, F.; Deplano, S.; Sequeira, L.; Melis, C.; Fois, B.; Angeli, A.; Capasso, C.; et al. Exploring new structural features of the 4-[(3-methyl-4-aryl-2,3-dihydro-1,3-thiazol-2-ylidene)amino] benzene sulphon amide scaffold for the inhibition of human carbonic anhydrases. J. Enzym. Inhib. Med. Chem. 2019, 34, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Manasa, K.L.; Pujitha, S.; Sethi, A.; Arifuddin, M.; Alvala, M.; Angeli, A.; Supuran, C.T. Synthesis and Biological Evaluation ofImidazo[2,1-b]Thiazole based Sulfonyl Piperazines as Novel Carbonic Anhydrase II Inhibitor. Metabolites 2020, 10, 136. [Google Scholar] [CrossRef] [Green Version]
- Al-Jaidi, B.A.; Deb, P.K.; Telfah, S.T.; Dakkah, A.N.; Bataineh, Y.A.; Ahmed Al Khames Aga, Q.A.; Al-dhoun, M.A.; Ala’Ali Ahmad Al-Subeihi, A.A.; Odetallah, H.M.; Bardaweel, S.K.; et al. Synthesis and evaluation of 2,4,5-trisubstituted thiazoles as carbonic anhydrase-III inhibitors. J. Enzym. Inhib. Med. Chem. 2020, 35, 1483–1490. [Google Scholar] [CrossRef]
- Lzweiri, M.; Al-Balas, Q.; Al-Hiari, Y. Chromatographic evaluation and qsar optimization for benzoic acid analogues against carbonic anhydrase iii. J. Enzym. Inhib. Med. Chem. 2015, 30, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Jarrar, N.; Alzweiri, M.; Al-Hiari, Y.; Farah, S.; Khanfar, M.A. Modified hummel-dreyer method and molecular modeling studies identified nicotinic acid analogues as carbonic anhydrase iii ligands. Lett. Drug Design Discov. 2016, 13, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Alzweiri, M.; Al-Hiari, Y. Evaluation of vanillic acid as inhibitor of carbonic anhydrase isozyme iii by using a modified Hummel-Dreyer method: Approach for drug discovery. Biomed. Chromatogr. 2013, 27, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Ghabbour, H.A.; Kadi, A.; ElTahir, K.; Angawi, R.; El-Subbagh, H. Synthesis, biological evaluation and molecular docking studies of thiazole-based pyrrolidinones and isoindolinediones as anticonvulsant agents. Med. Chem. Res. 2015, 24, 3194–3211. [Google Scholar] [CrossRef]
- Łączkowski, K.; Biernasiuk, A.; Baranowska-Łączkowska, A.; Zielińska, S.; Sałat, K.; Furgała, A.; Konrad Misiura, A.; Malm, A. Synthesis, antimicrobial and anticonvulsant screening of small library of tetrahydro-2H-thiopyran-4-yl based thiazoles and selenazoles. J. Enzym. Inhib. Med. Chem. 2016, 31, 24–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łączkowski, K.; Sałat, K.; Misiura, K.; Podkowa, A.; Malikowska, N. Synthesis and anticonvulsant activities of novel 2-(cyclopentylmethylene)hydrazinyl-1,3-thiazoles in mouse models of seizures. J. Enzym. Inhib. Med. Chem. 2016, 31, 1576–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łączkowski, K.; Konklewska, N.; Biernasiuk, A.; Malm, A.; Sałat, K.; Furgała, A.; Dzitko, K.; Bekier, A.; Baranowska-Łączkowska, A.; Paneth, A. Thiazoles with cyclopropyl fragment as antifungal, anticonvulsant, and anti-Toxoplasma gondii agents: Synthesis, toxicity evaluation, and molecular docking study. Med. Chem. Res. 2018, 27, 2125–2140. [Google Scholar] [CrossRef] [Green Version]
- Mishchenko, M.; Shtrygol, S.; Kaminskyy, D.; Lesyk, R. Thiazole-Bearing 4-Thiazolidinones as New Anticonvulsant Agents. Sci. Pharm. 2020, 88, 16. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, A.; Partap, S.; Khisal, S.; Shahar Yar, M.; Mishra, R. Synthesis, anti-convulsant activity and molecular docking study of novel thiazole pyridazinone hybrid analogues. Bioorg. Chem. 2020, 99, 103584. [Google Scholar] [CrossRef]



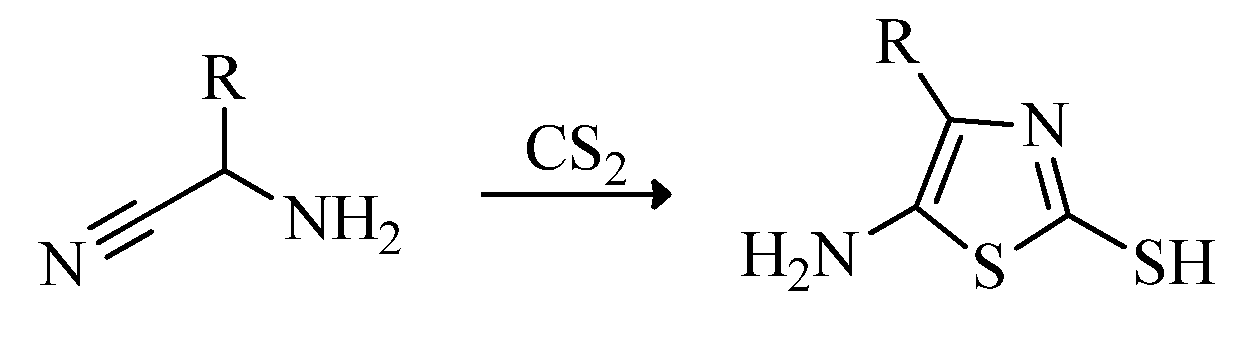

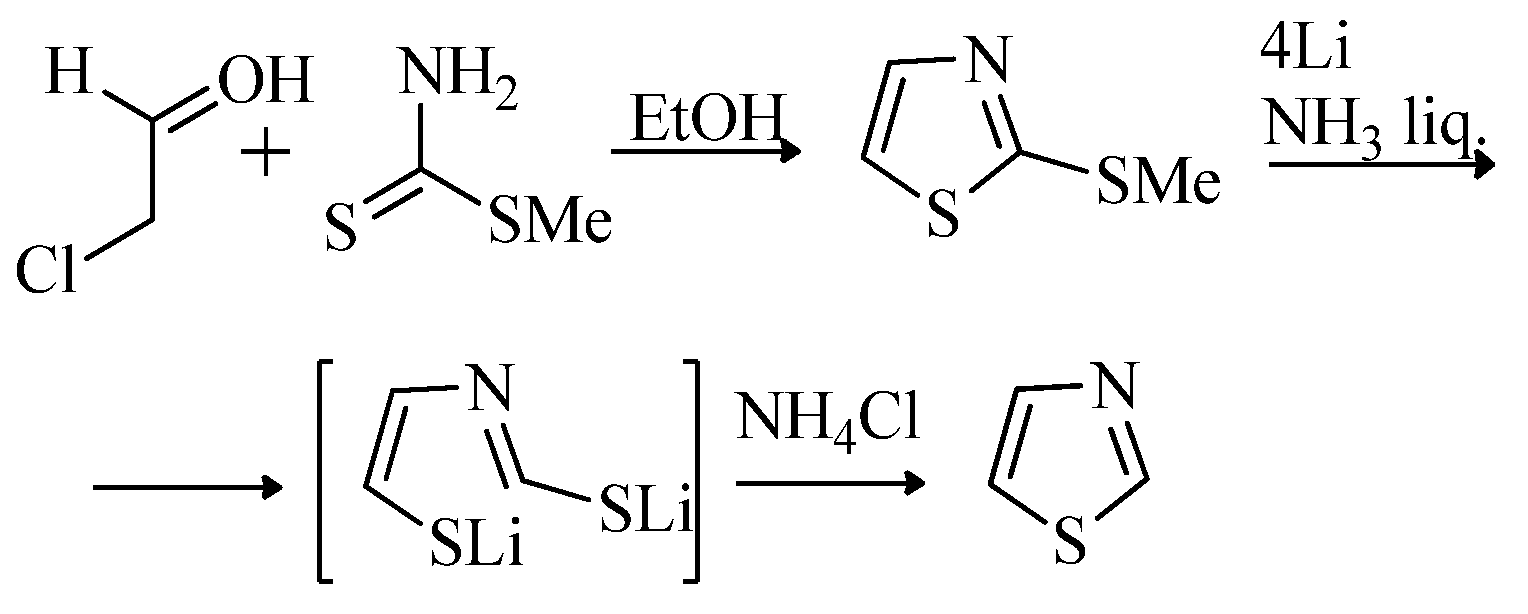
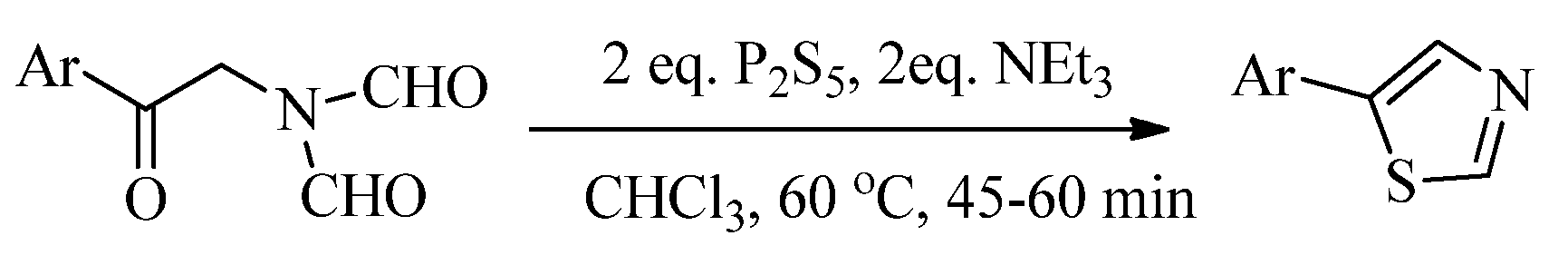

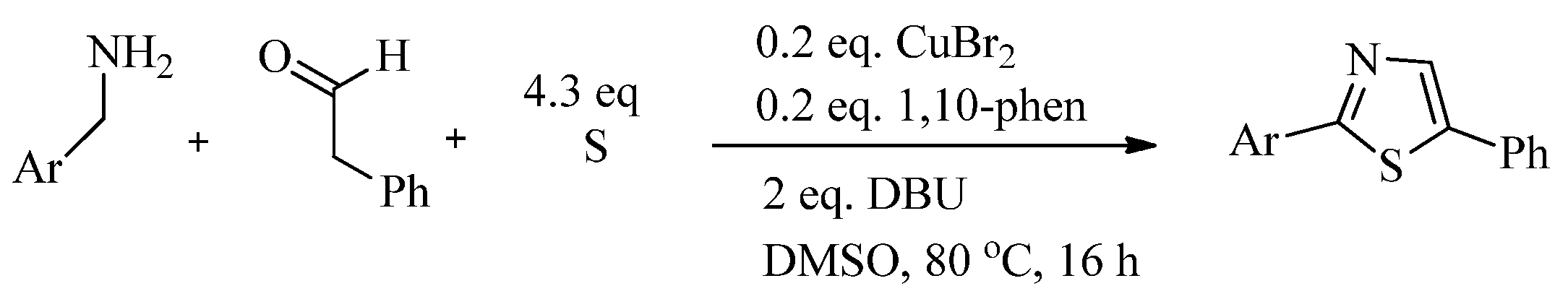
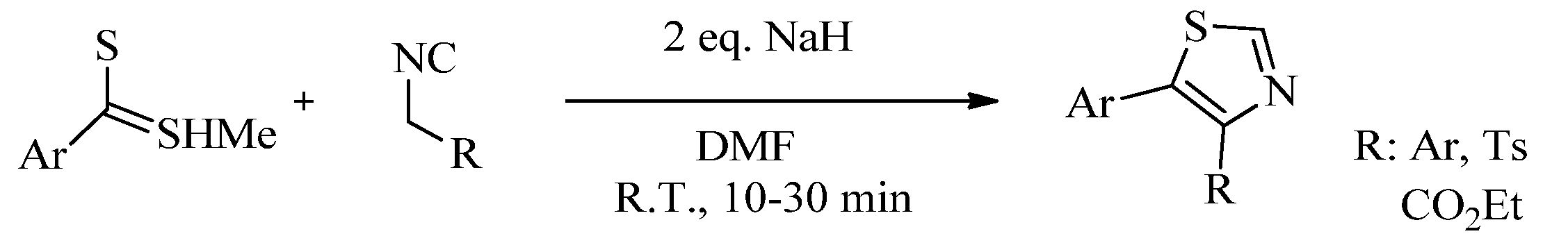
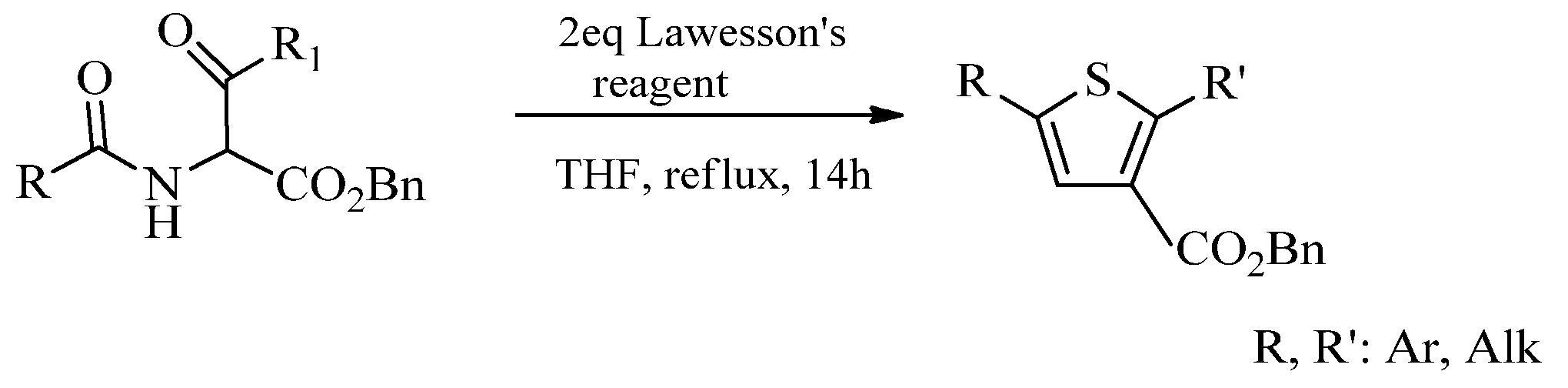
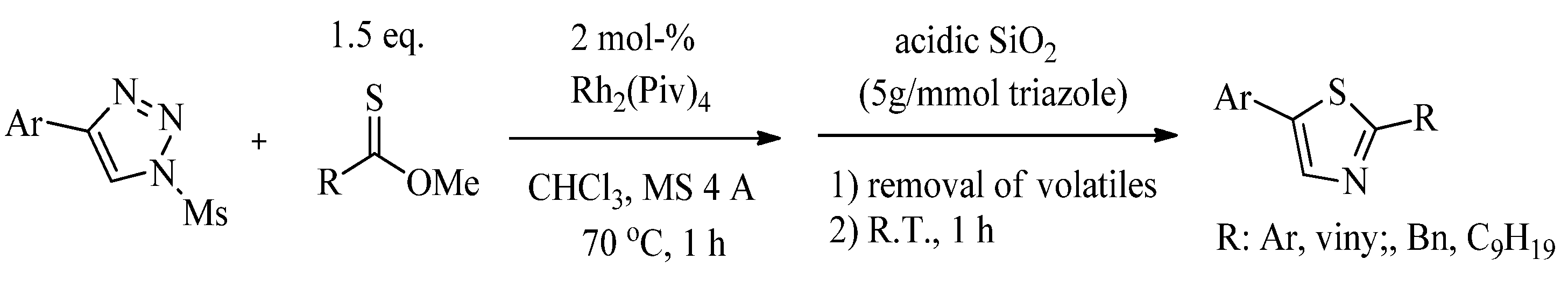
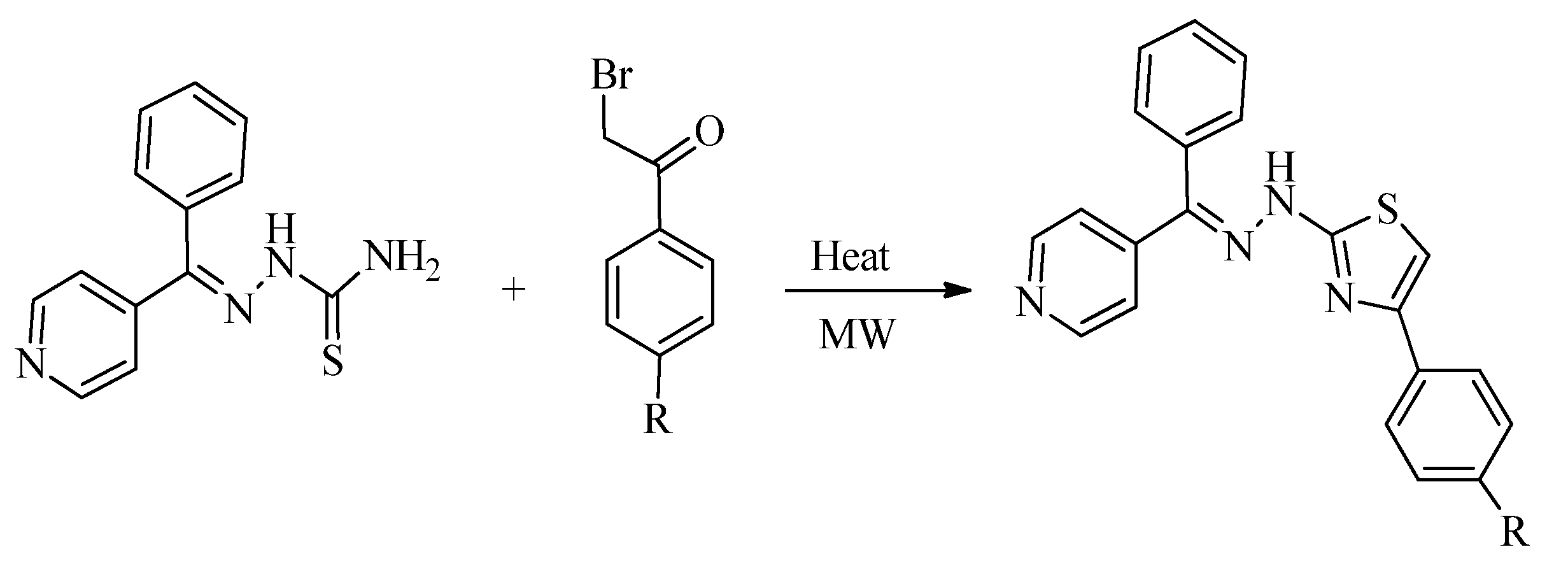
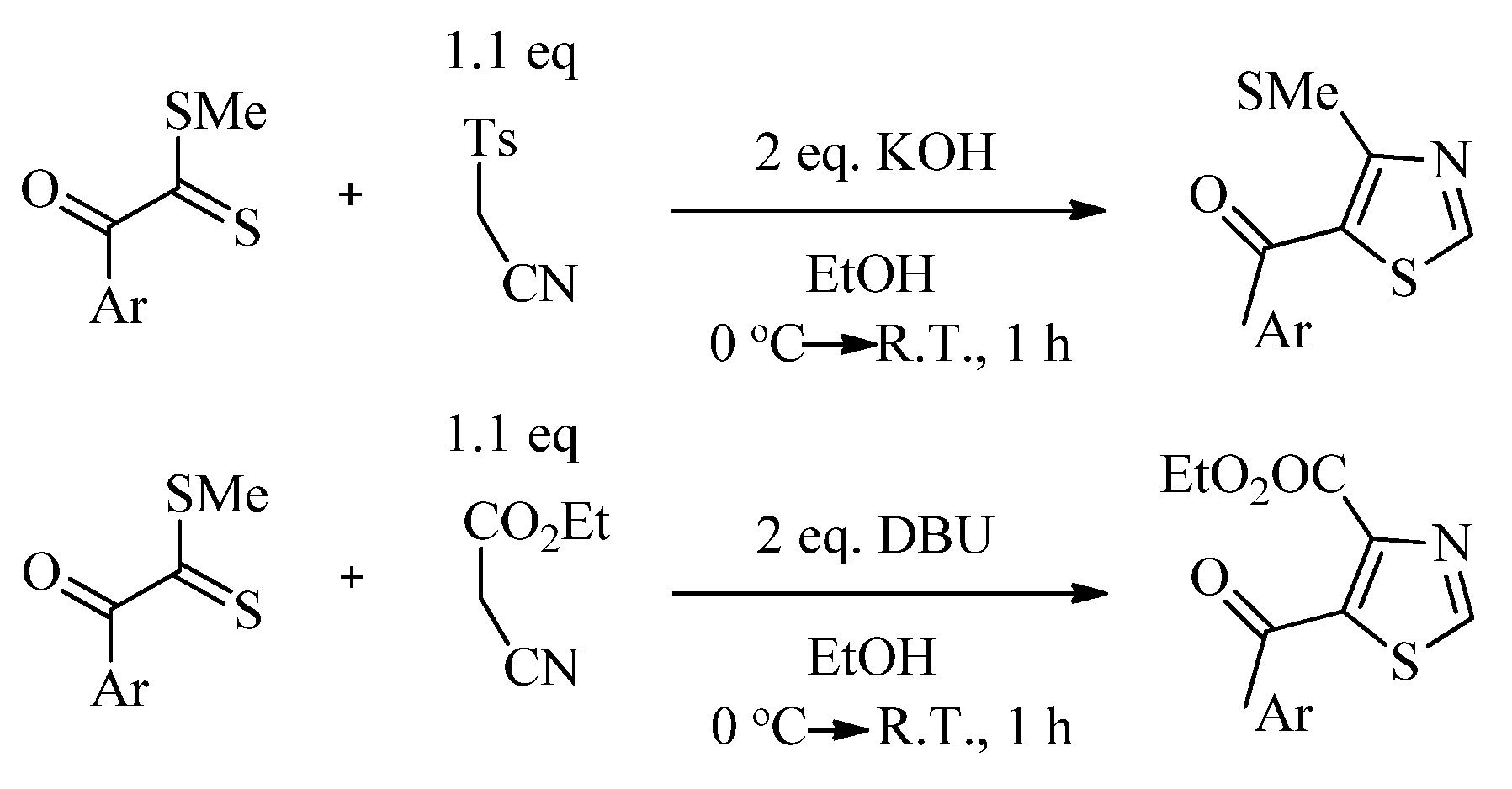
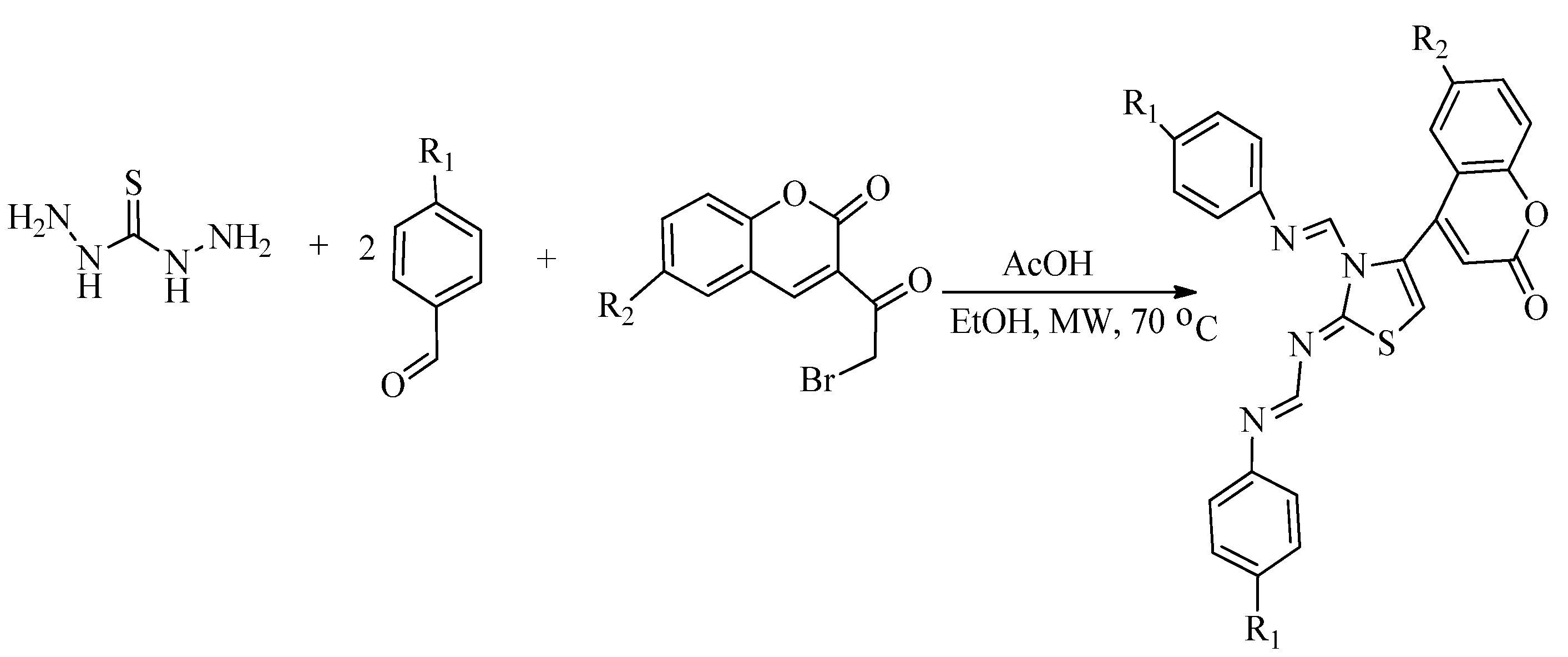
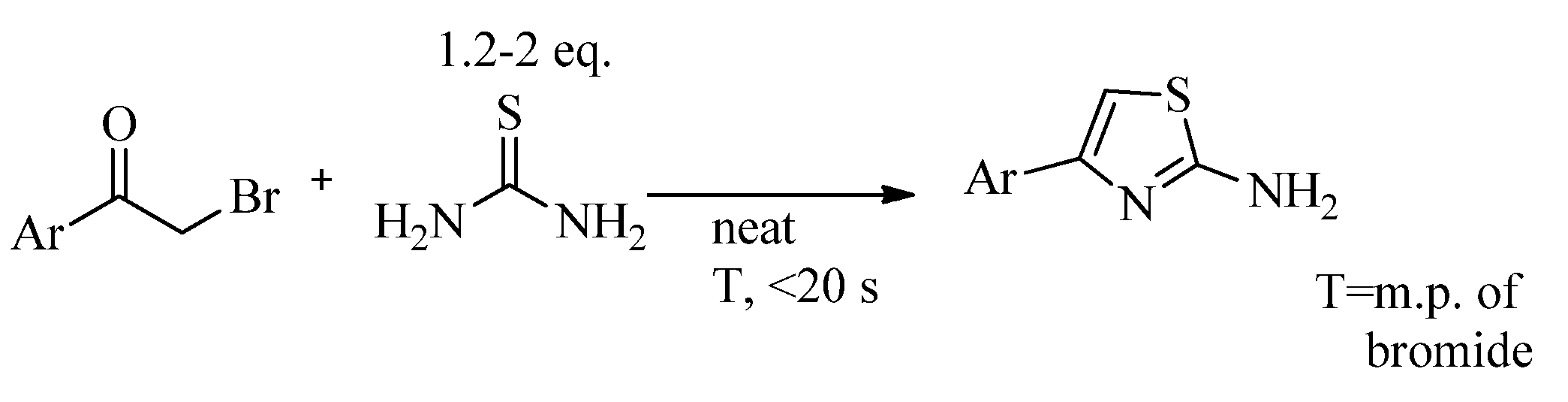
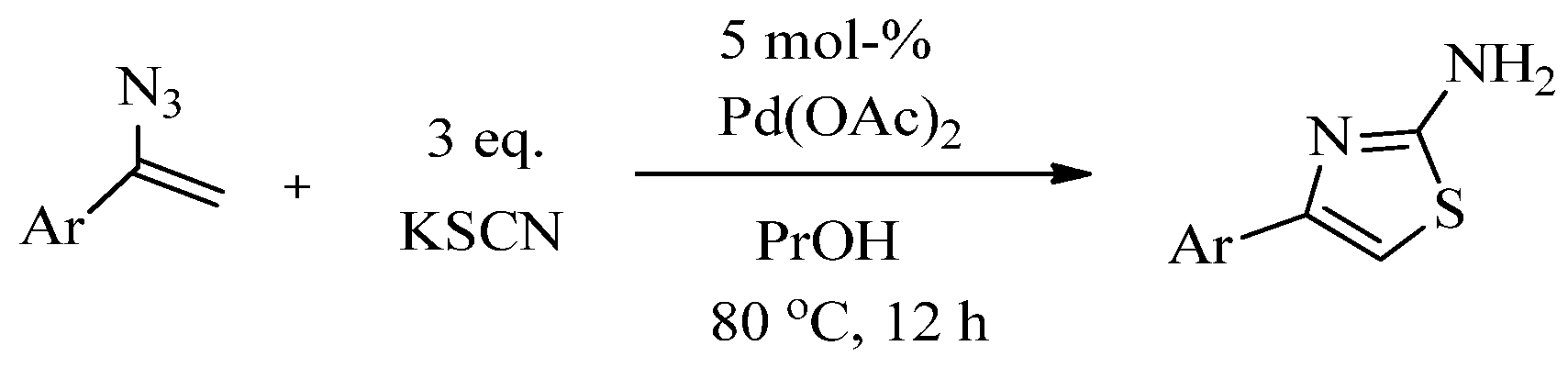

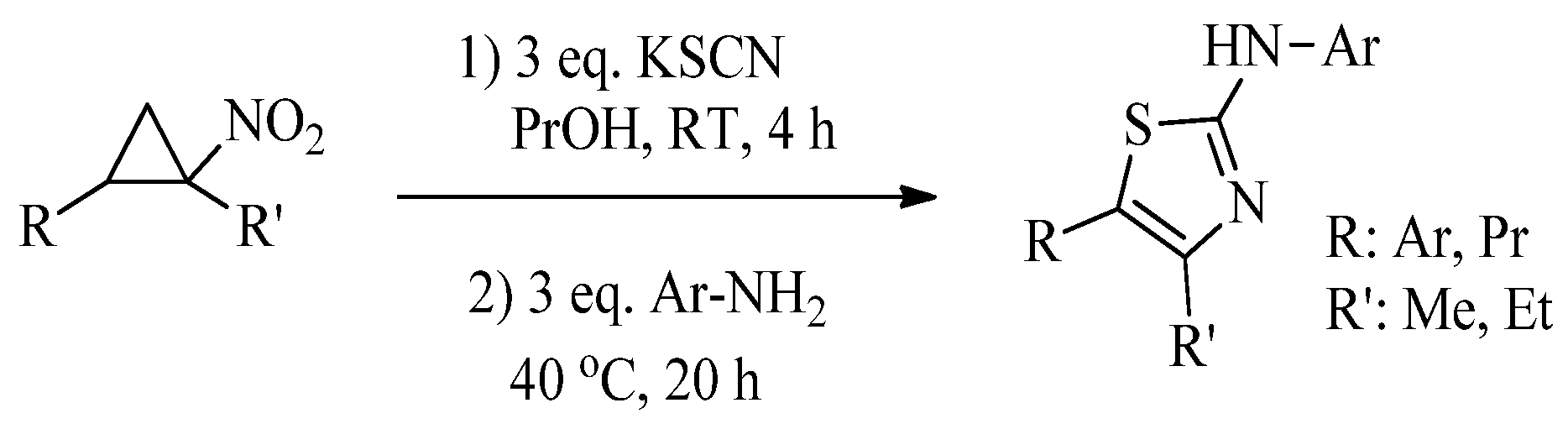
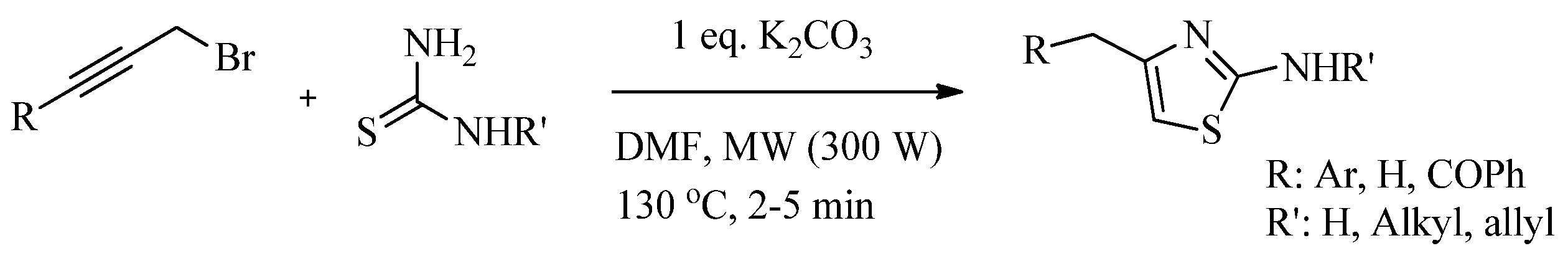
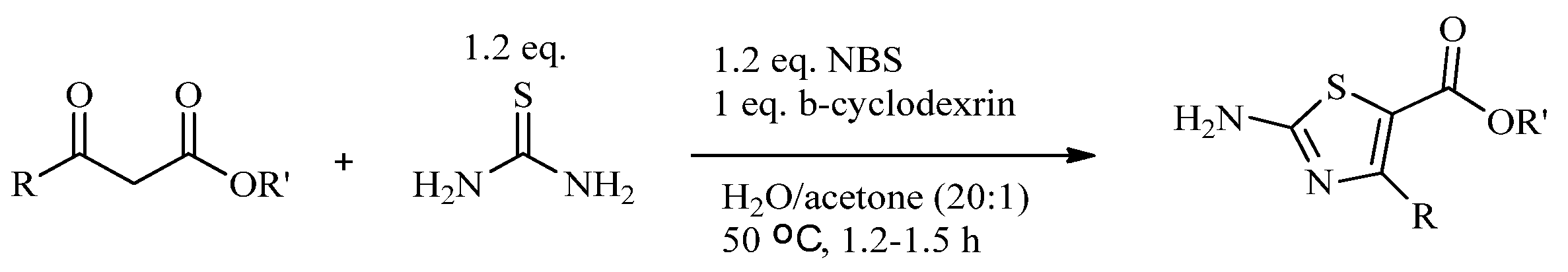
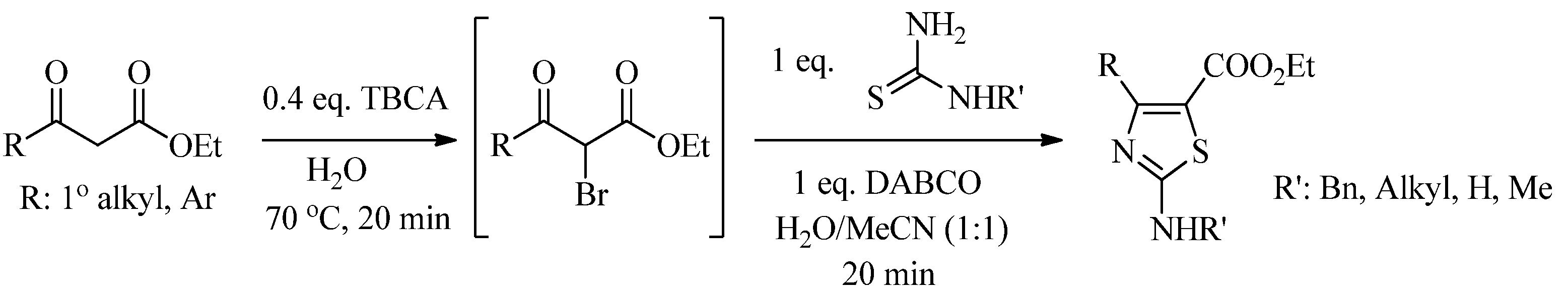
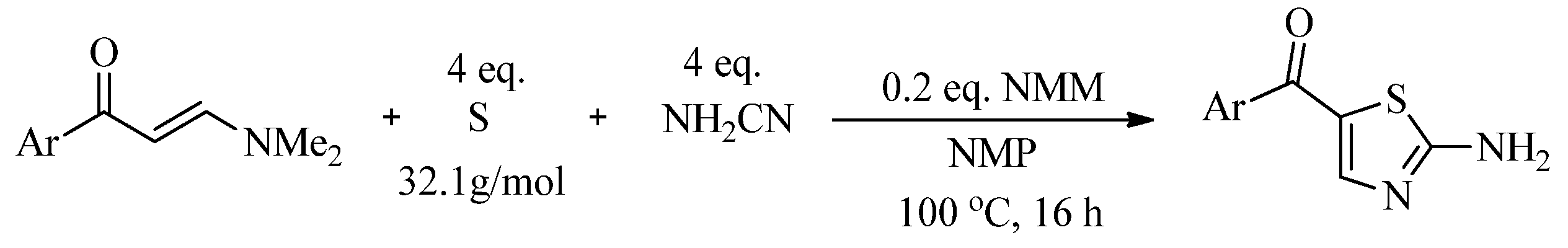

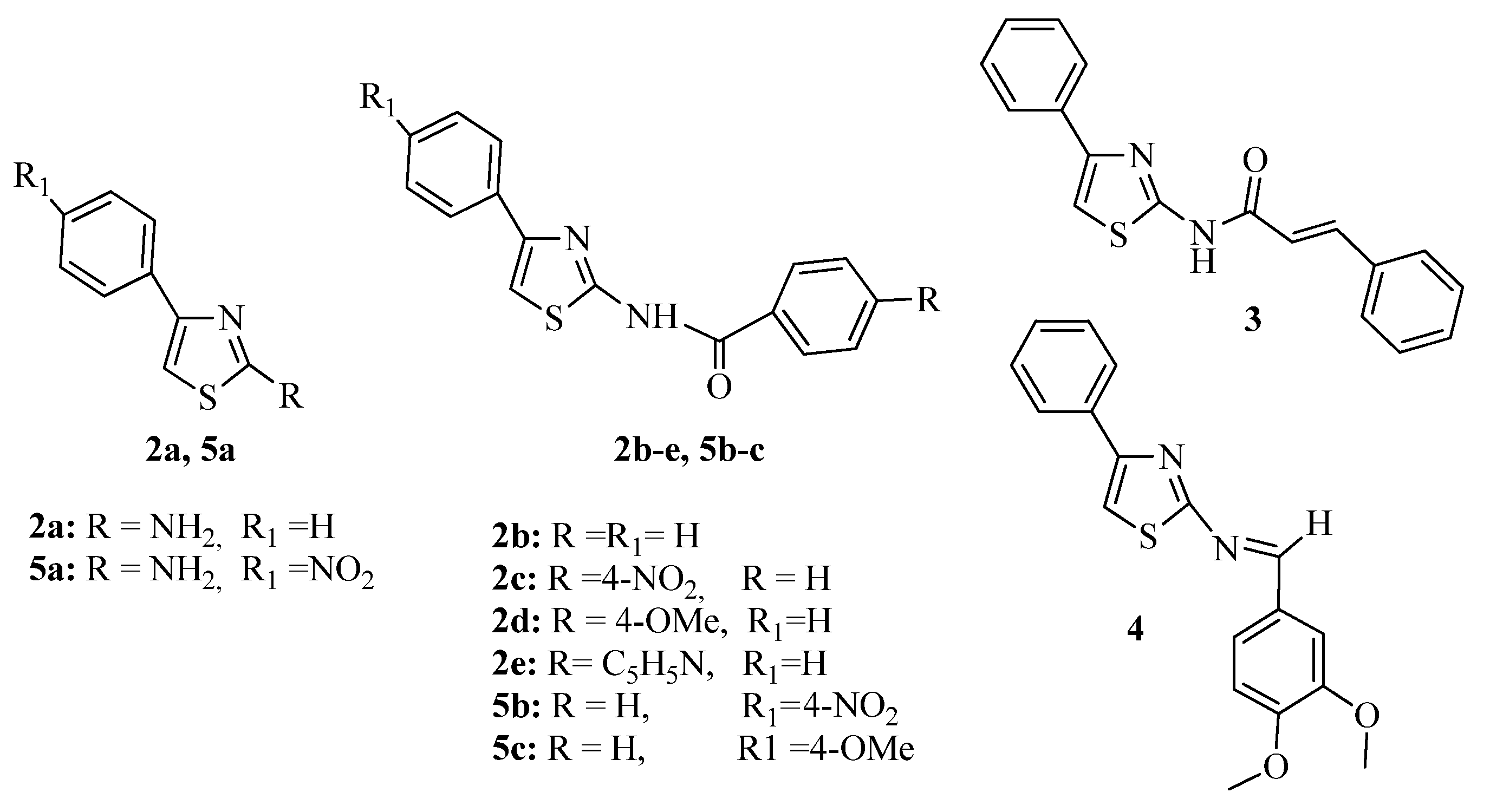
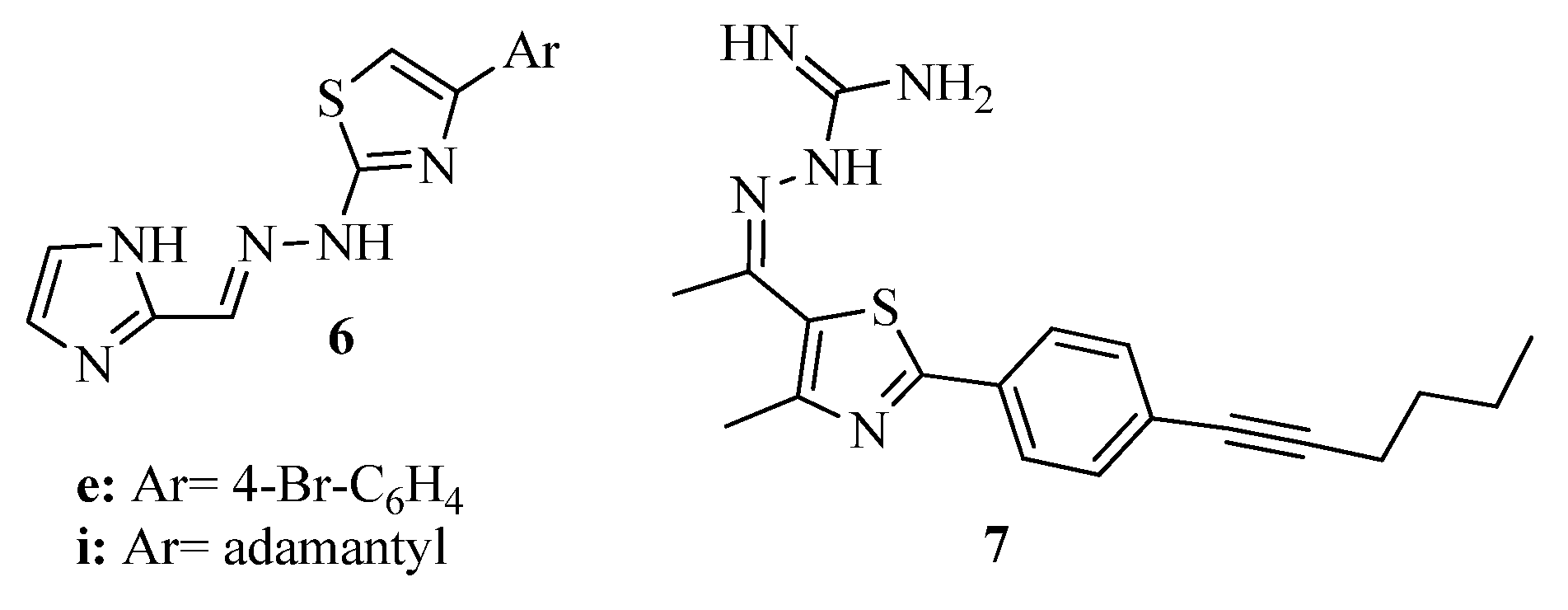
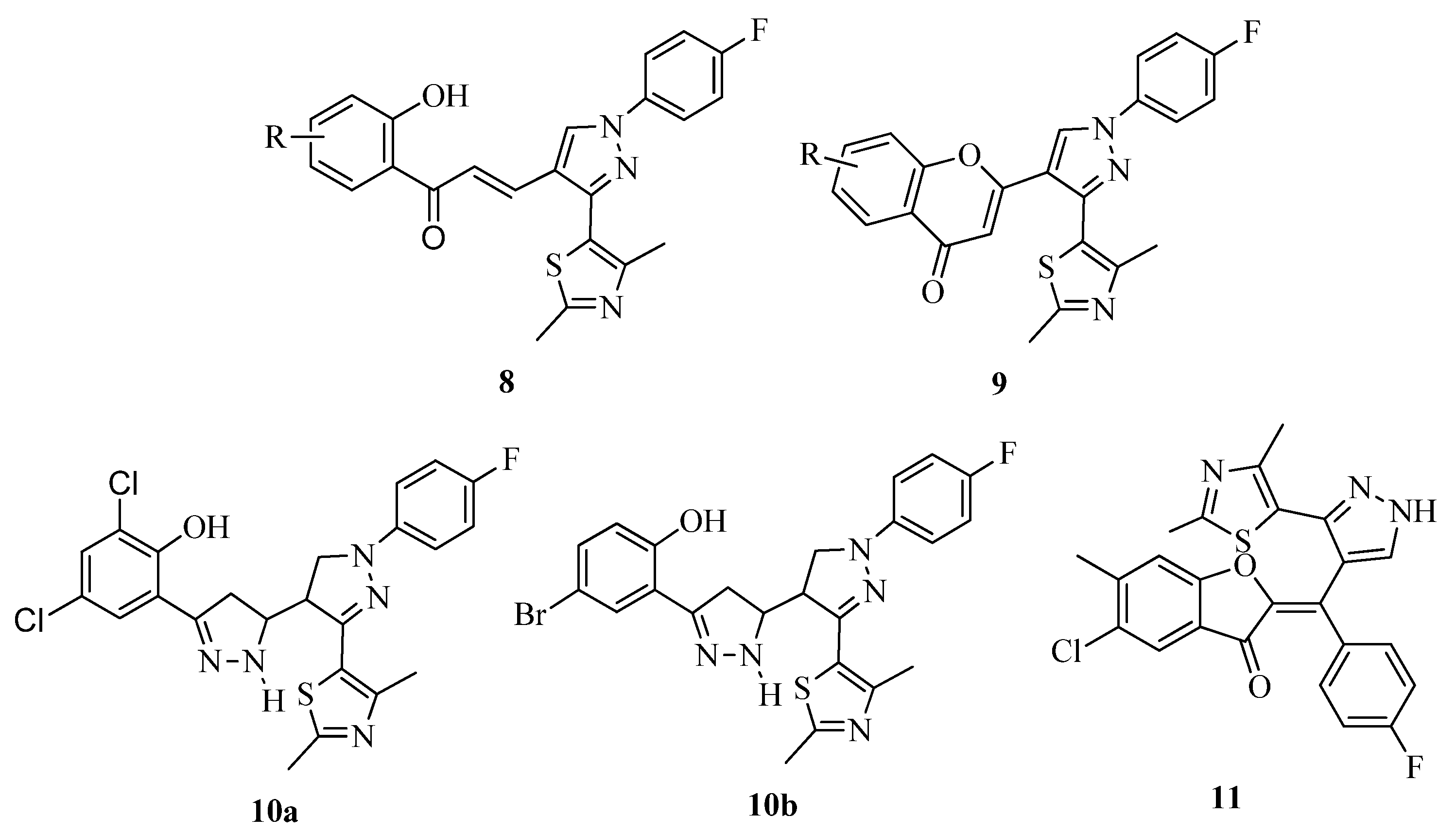
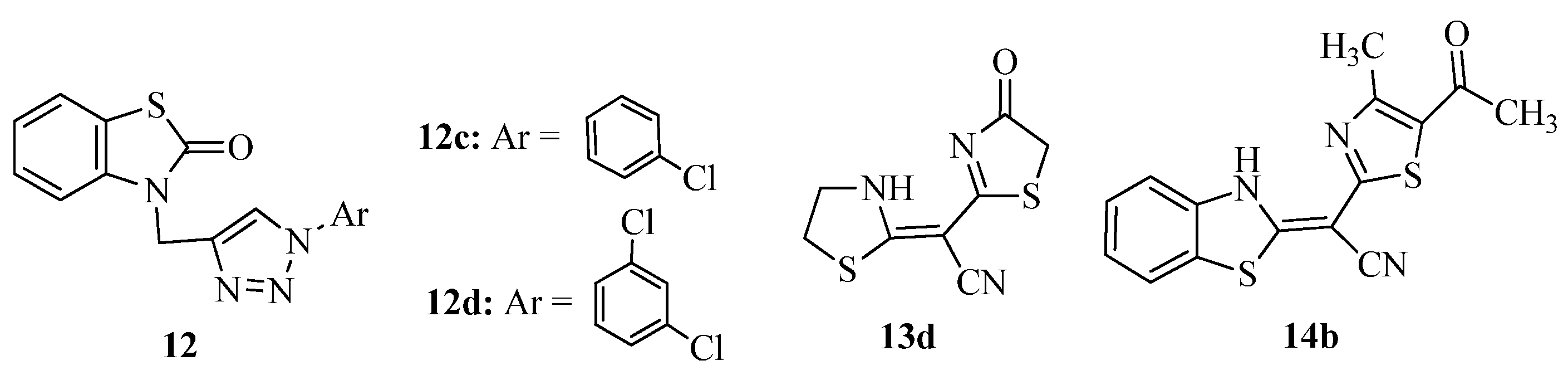

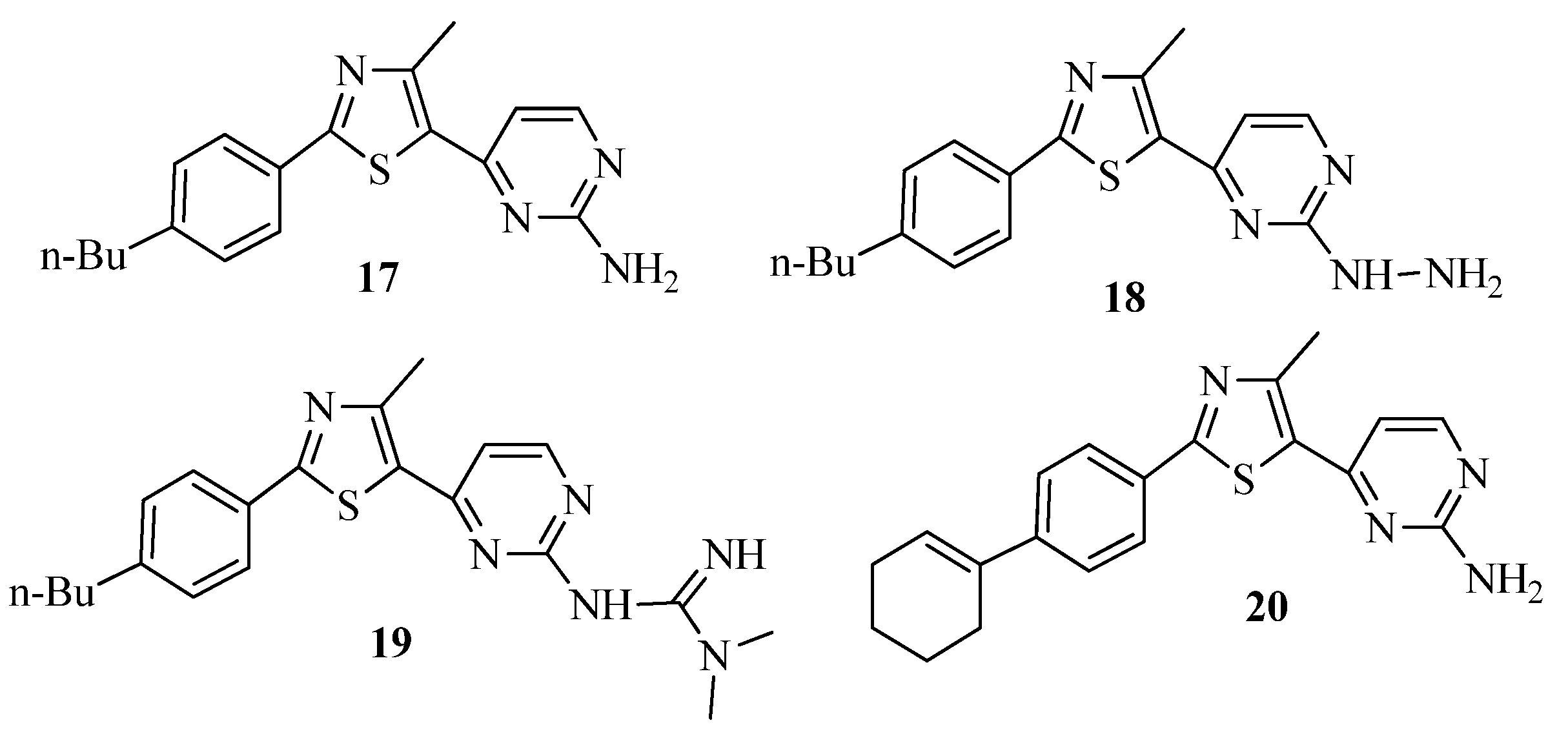
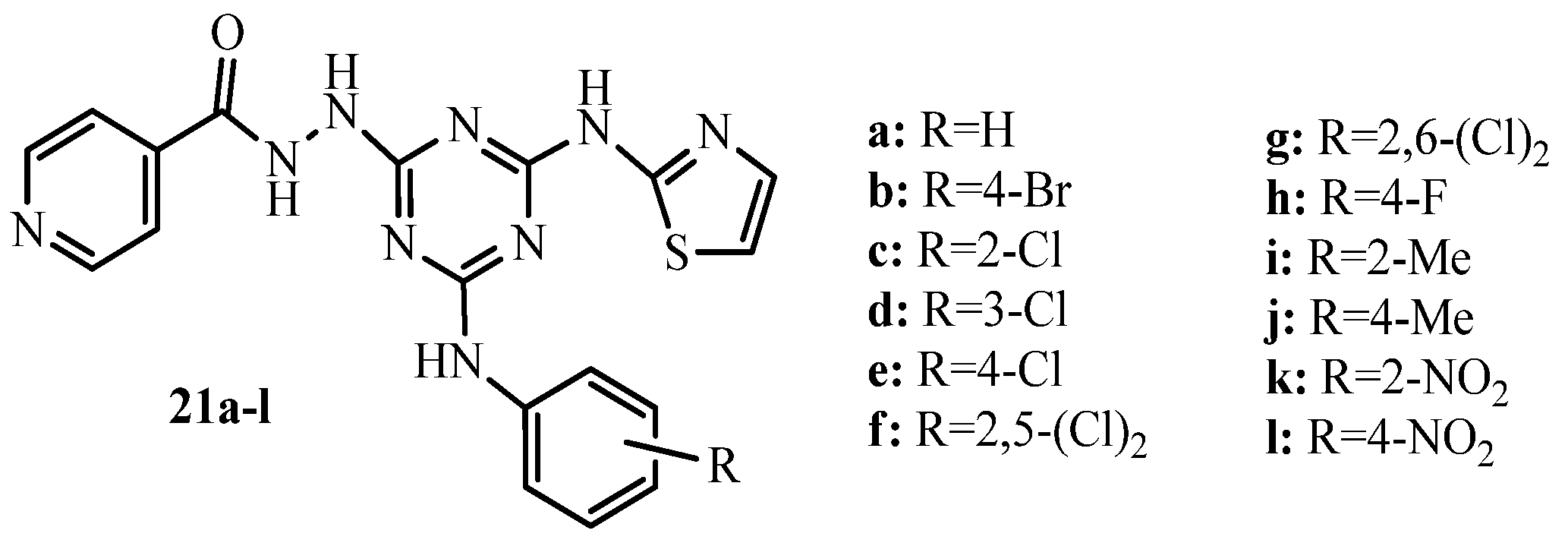
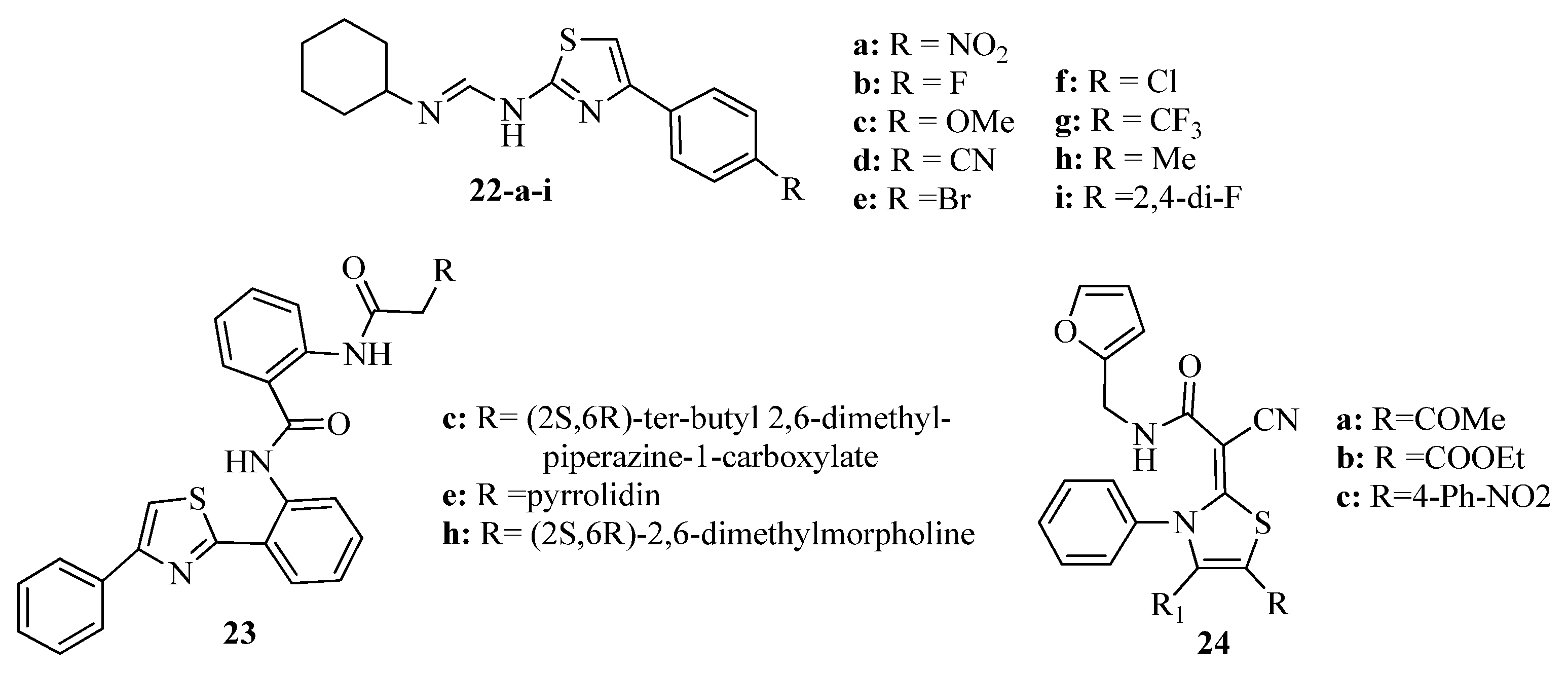
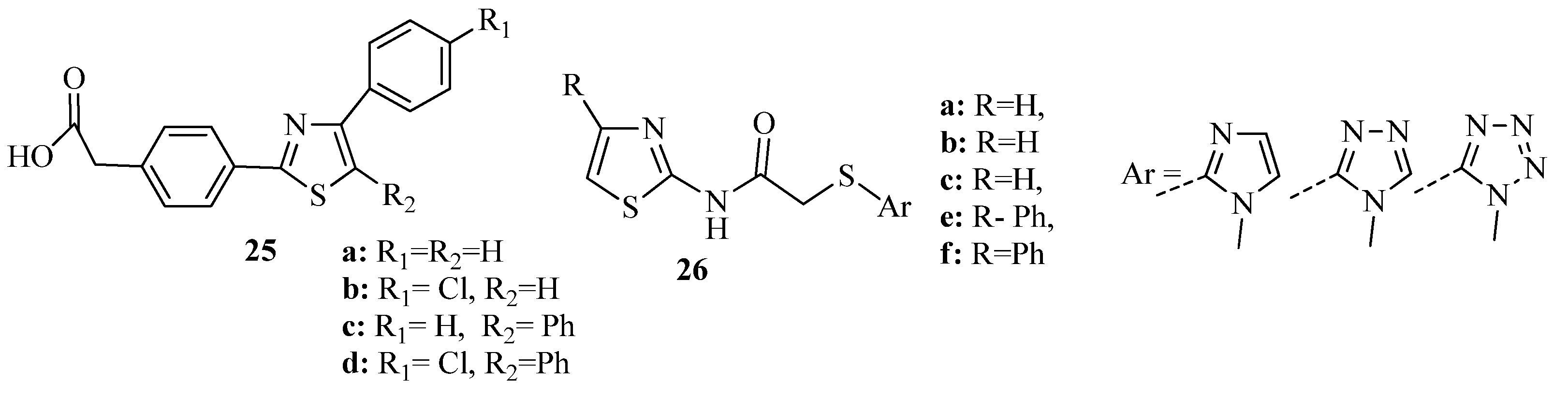
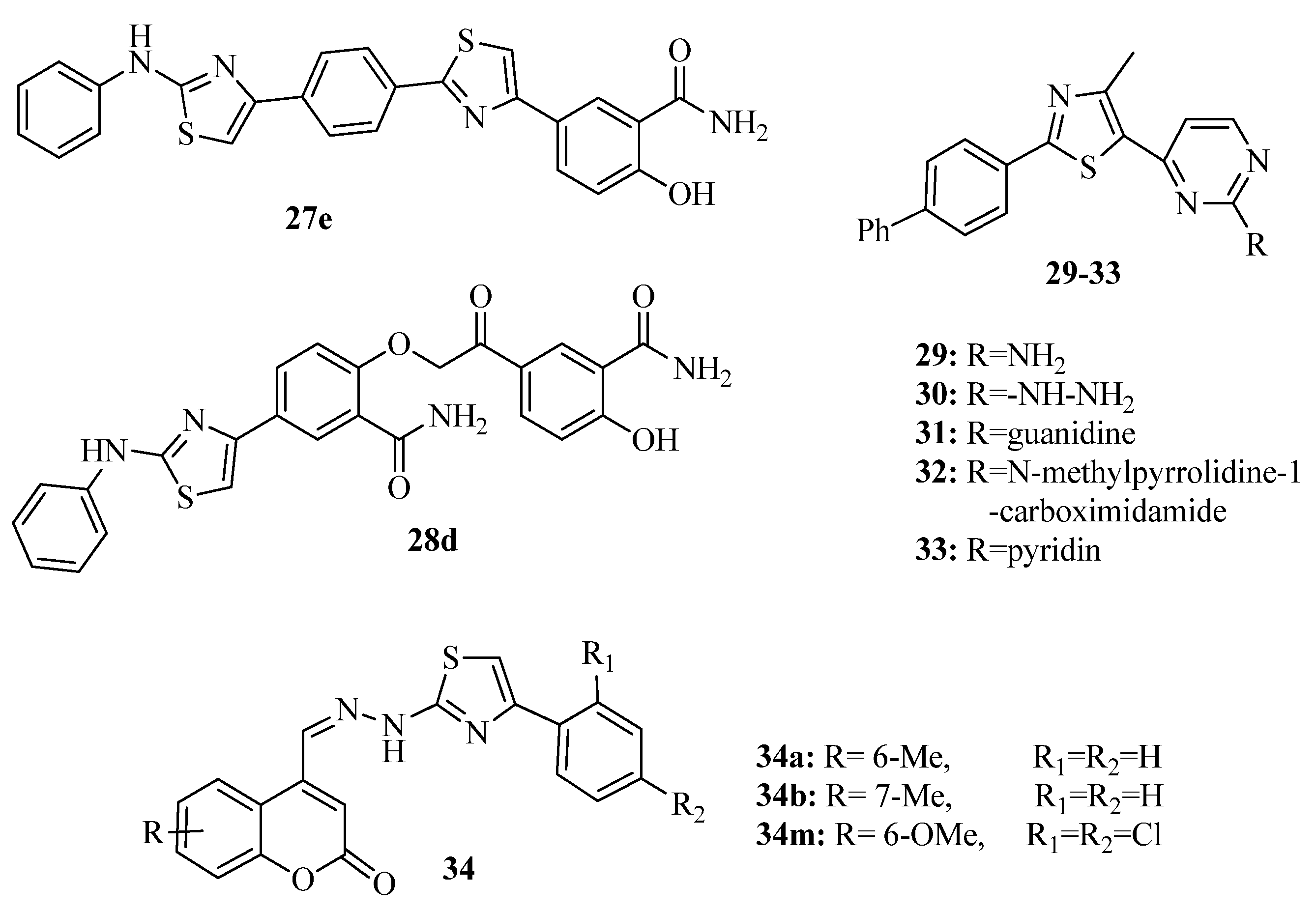

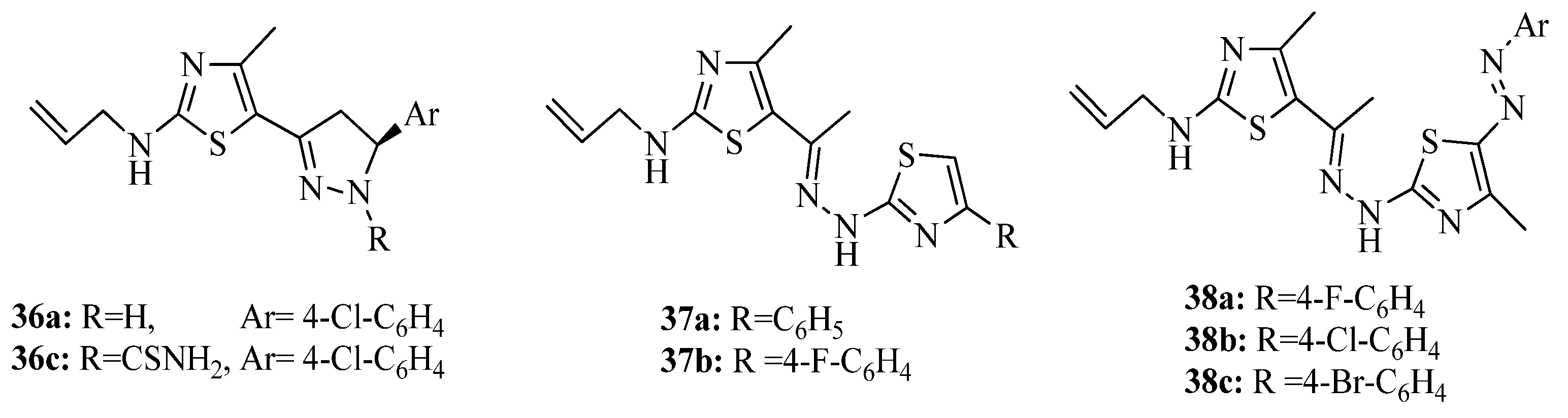
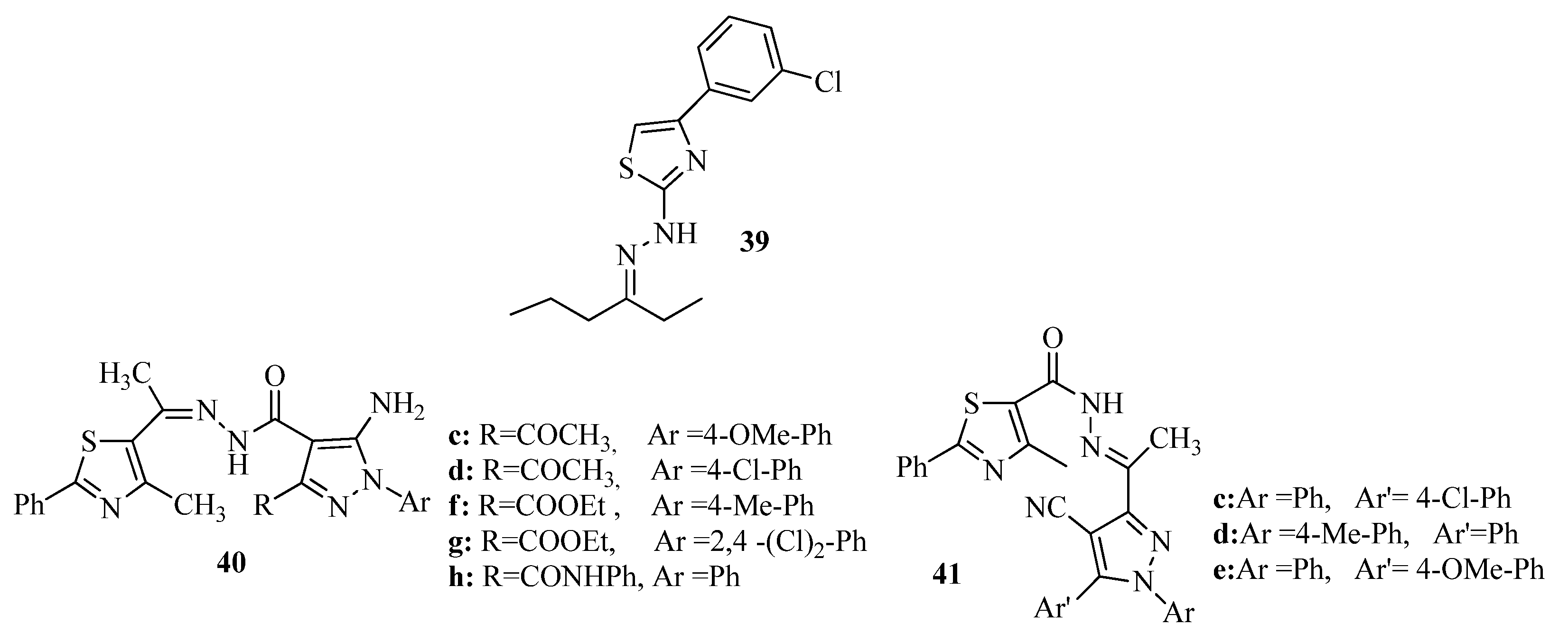
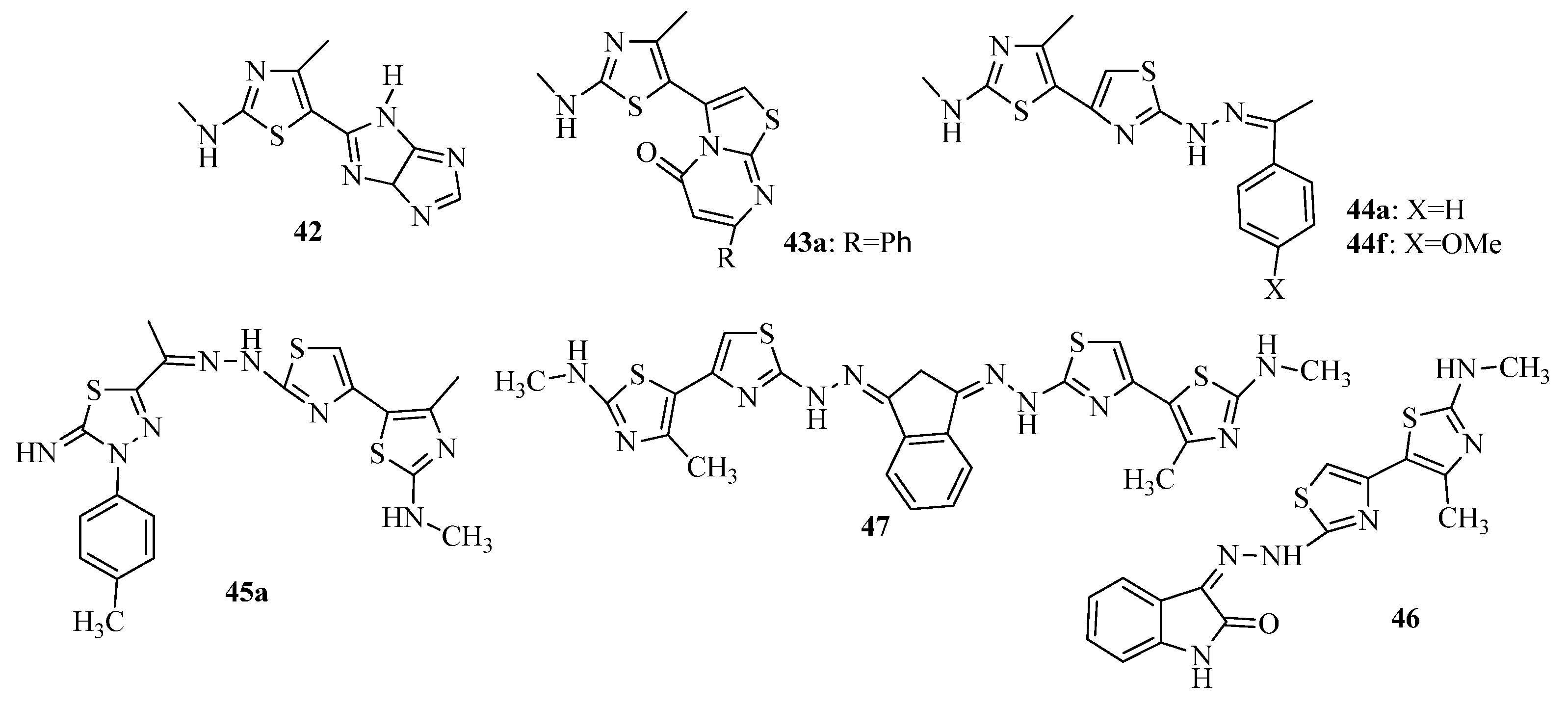
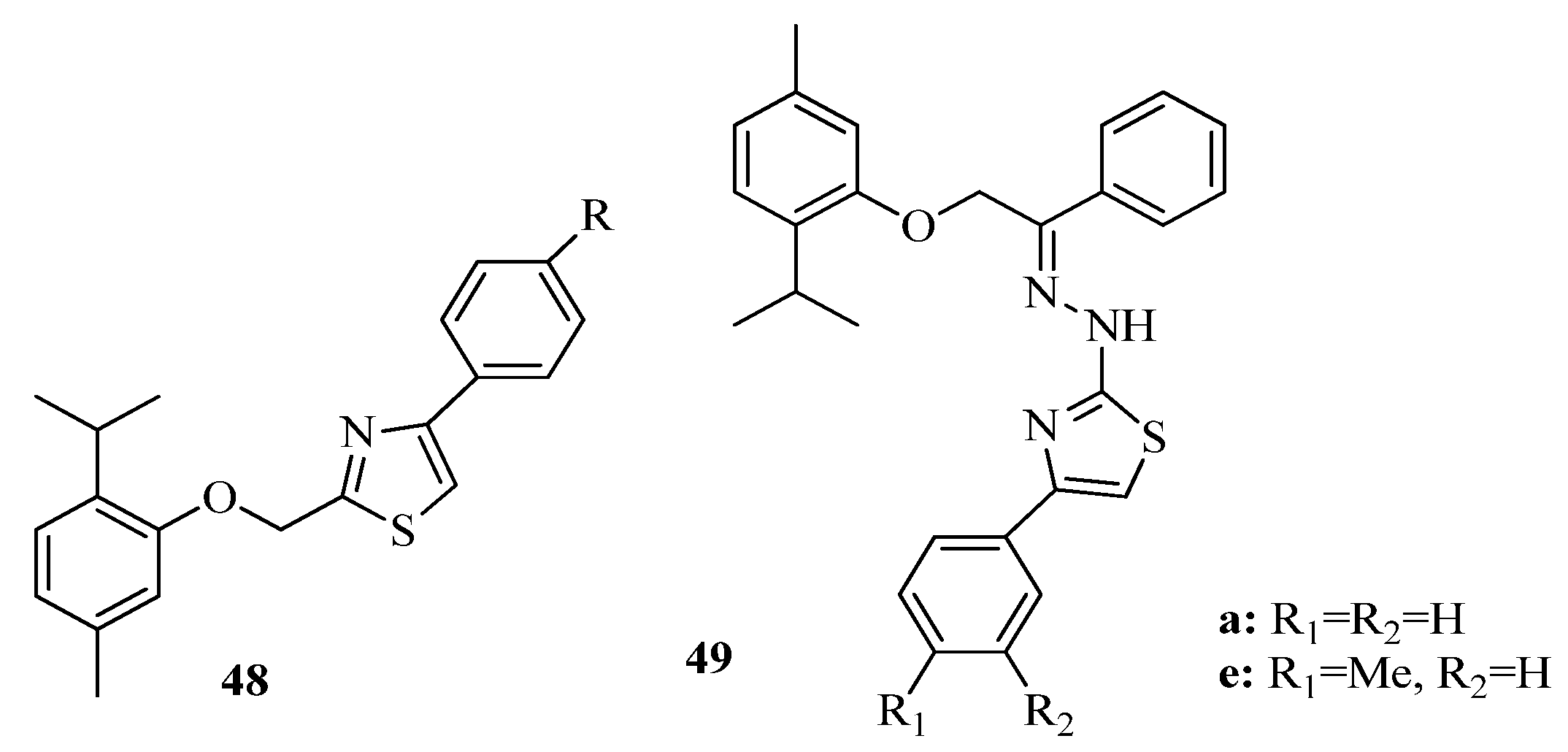


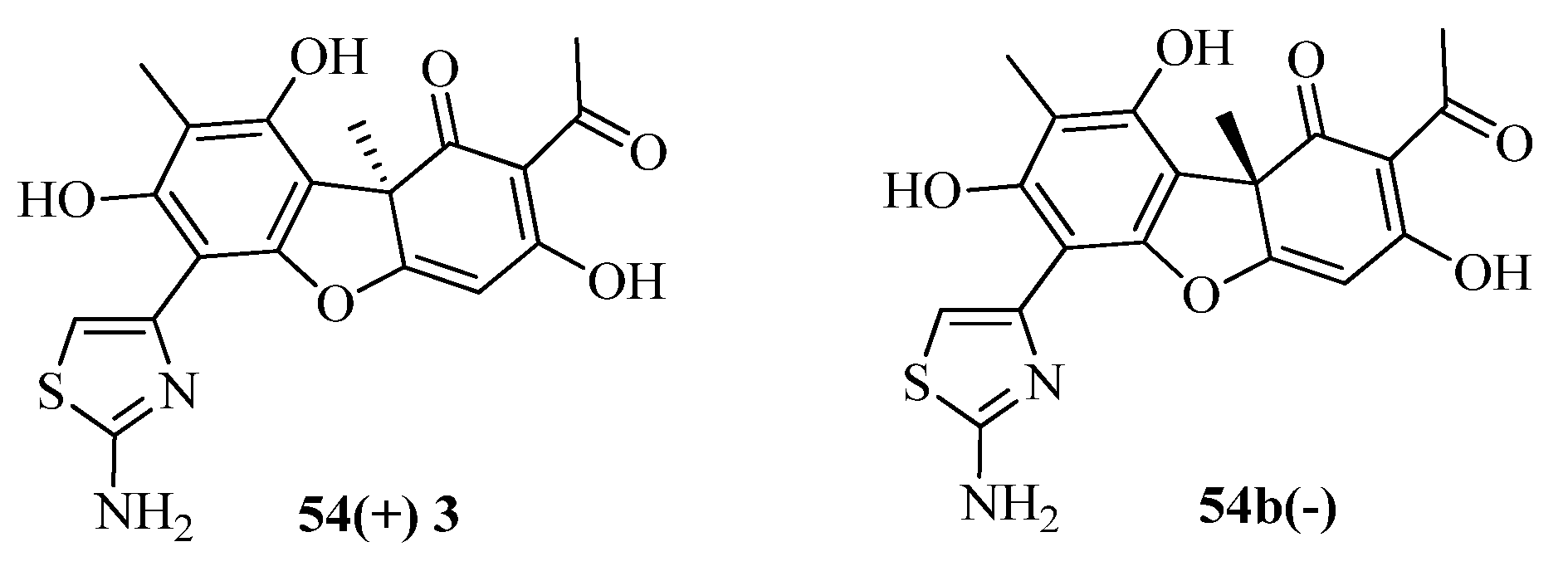

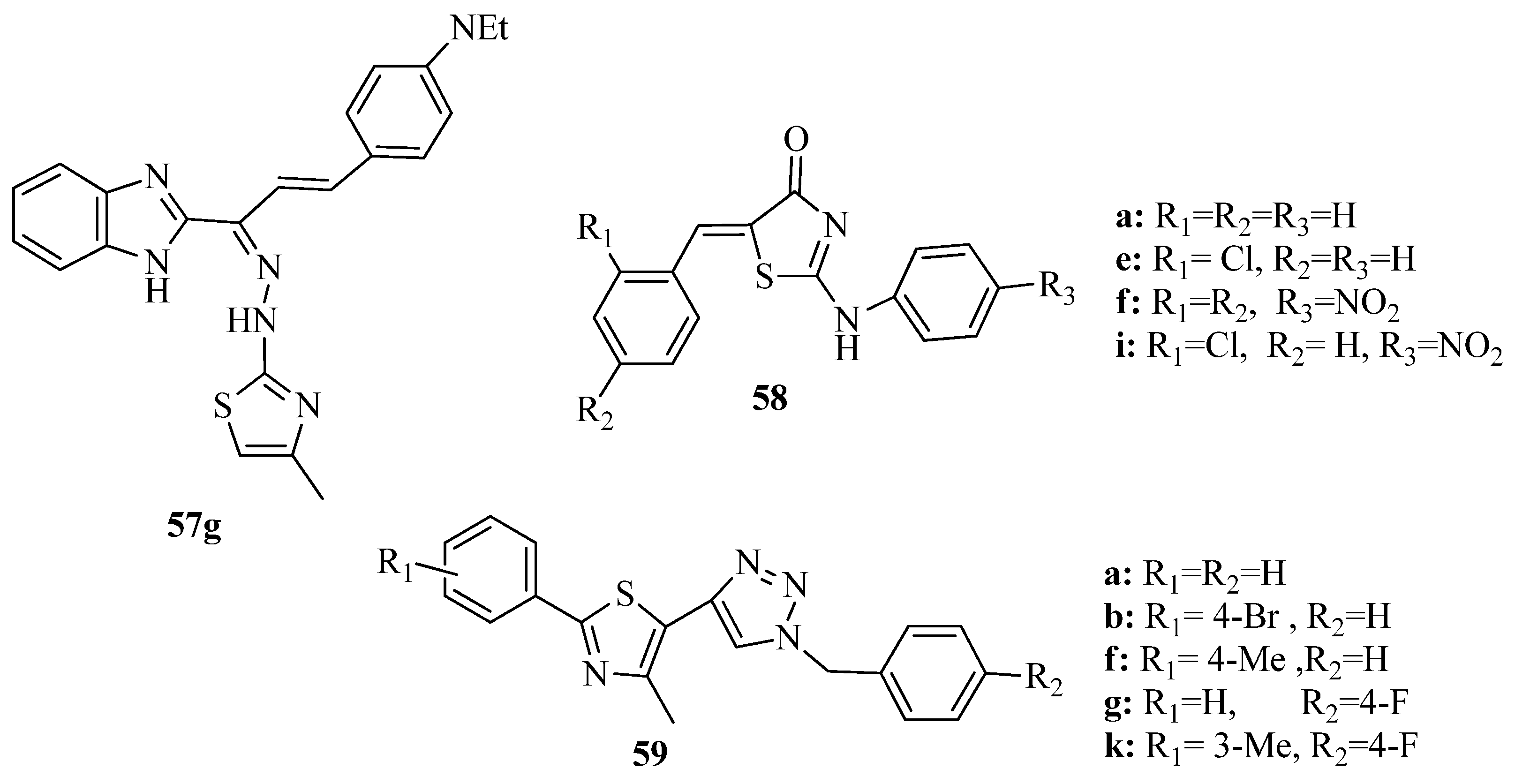
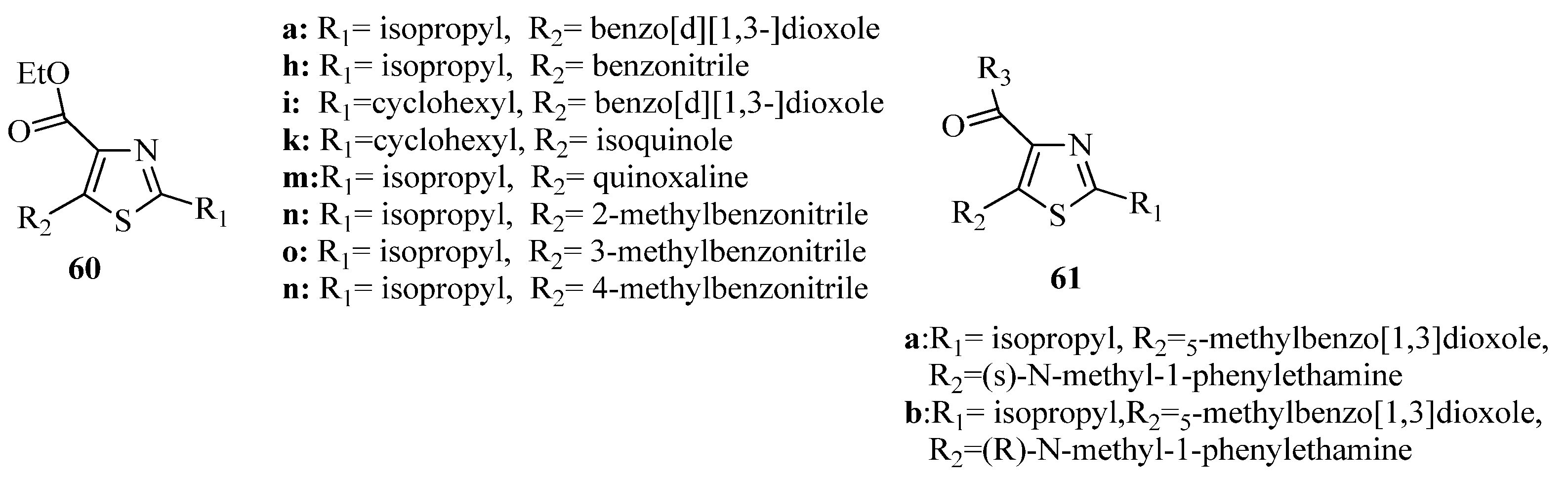
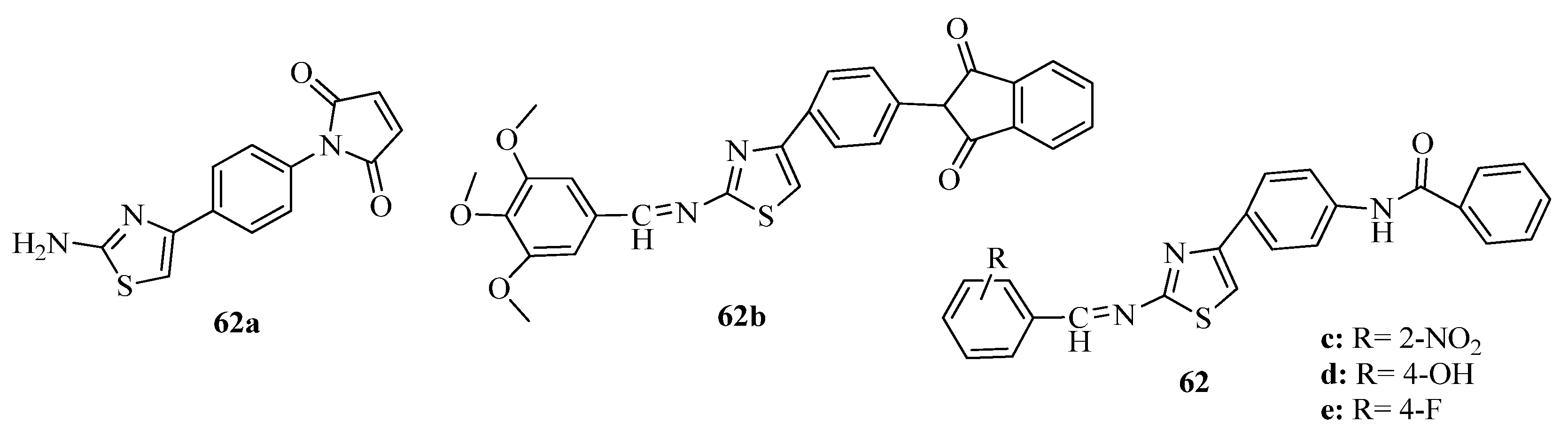


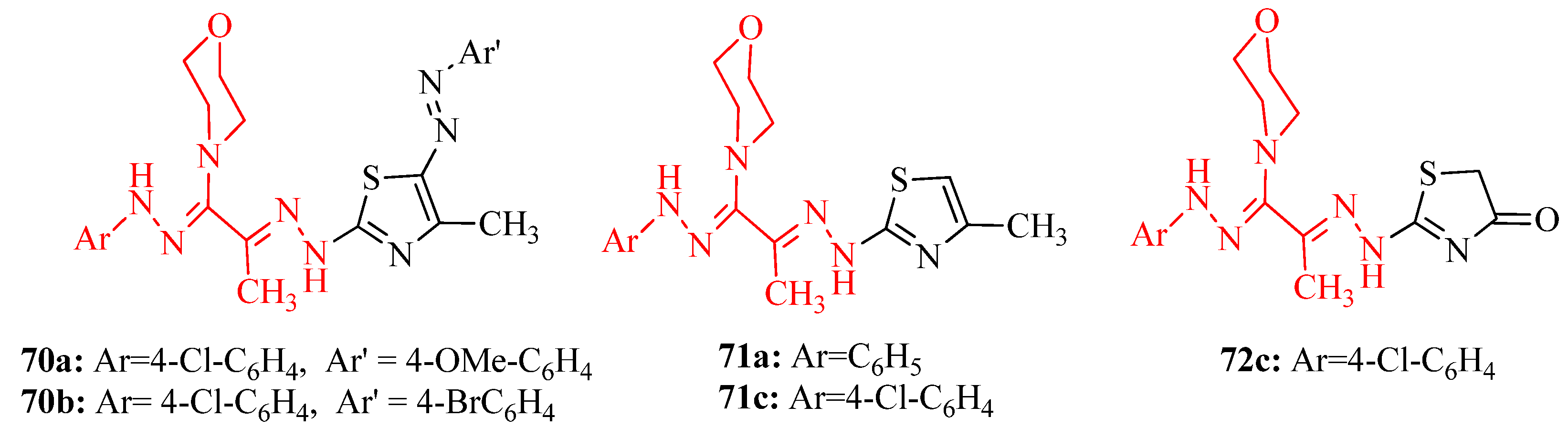
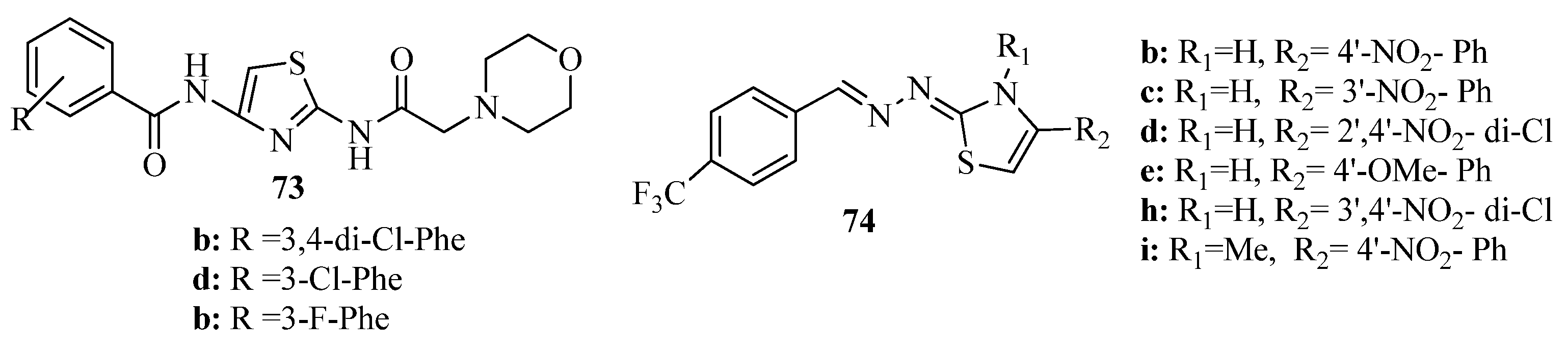
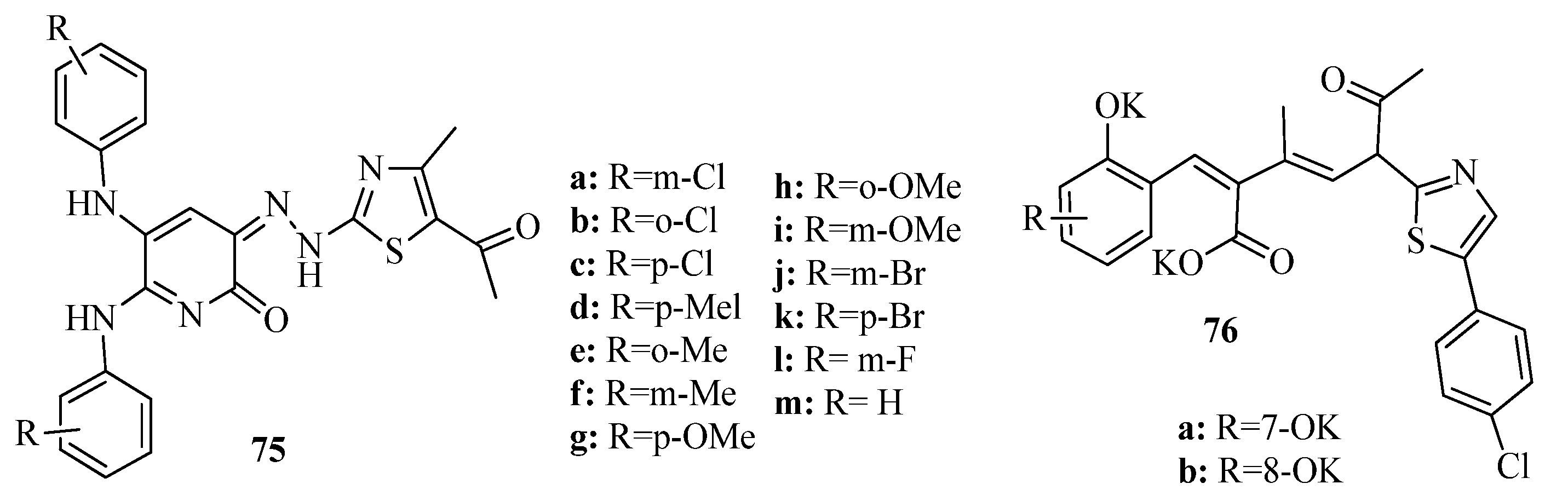
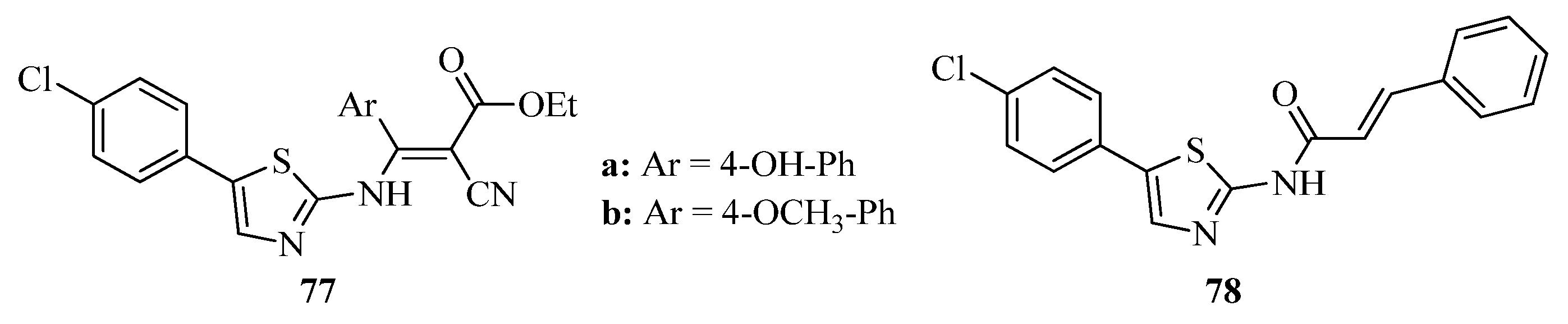
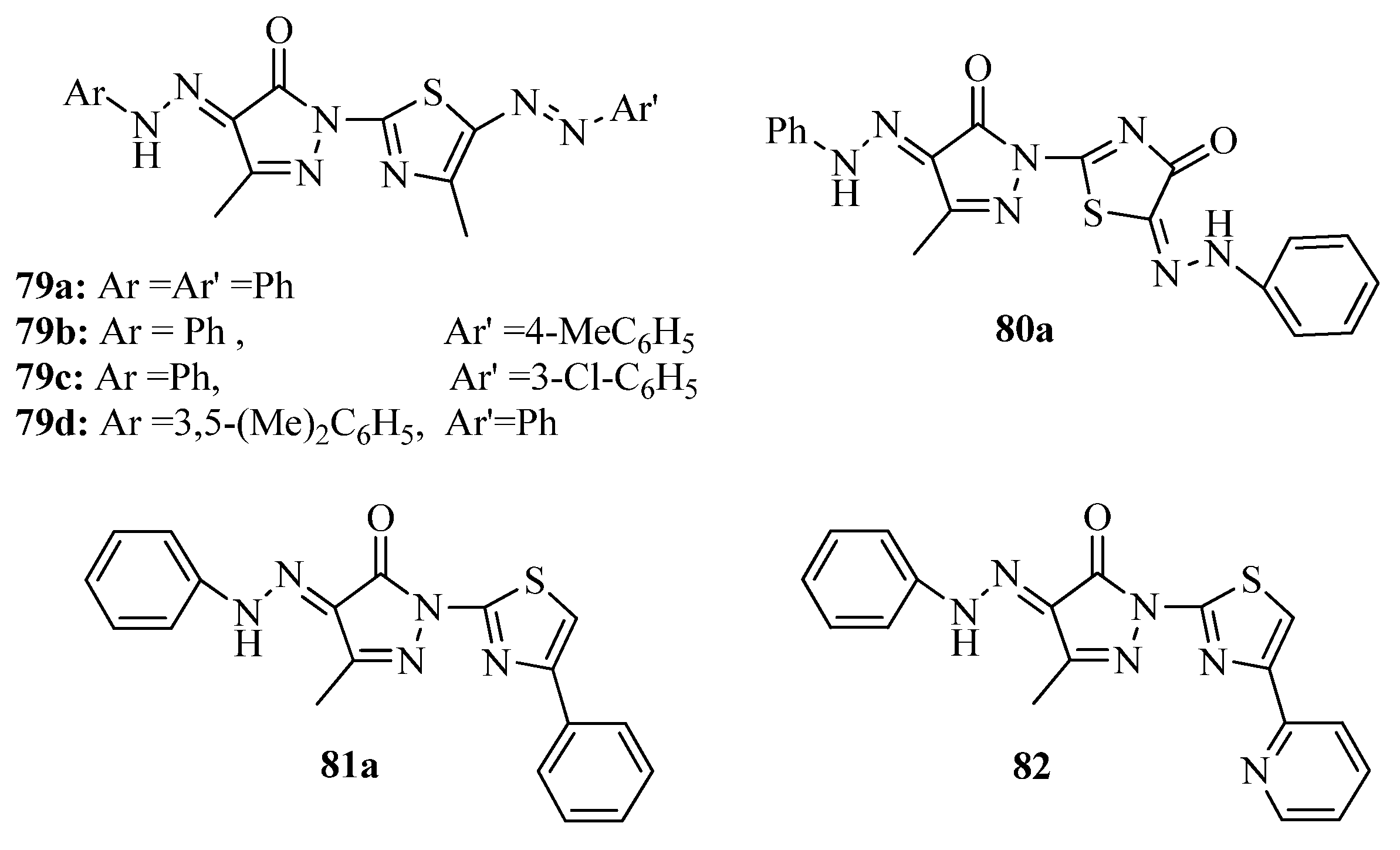


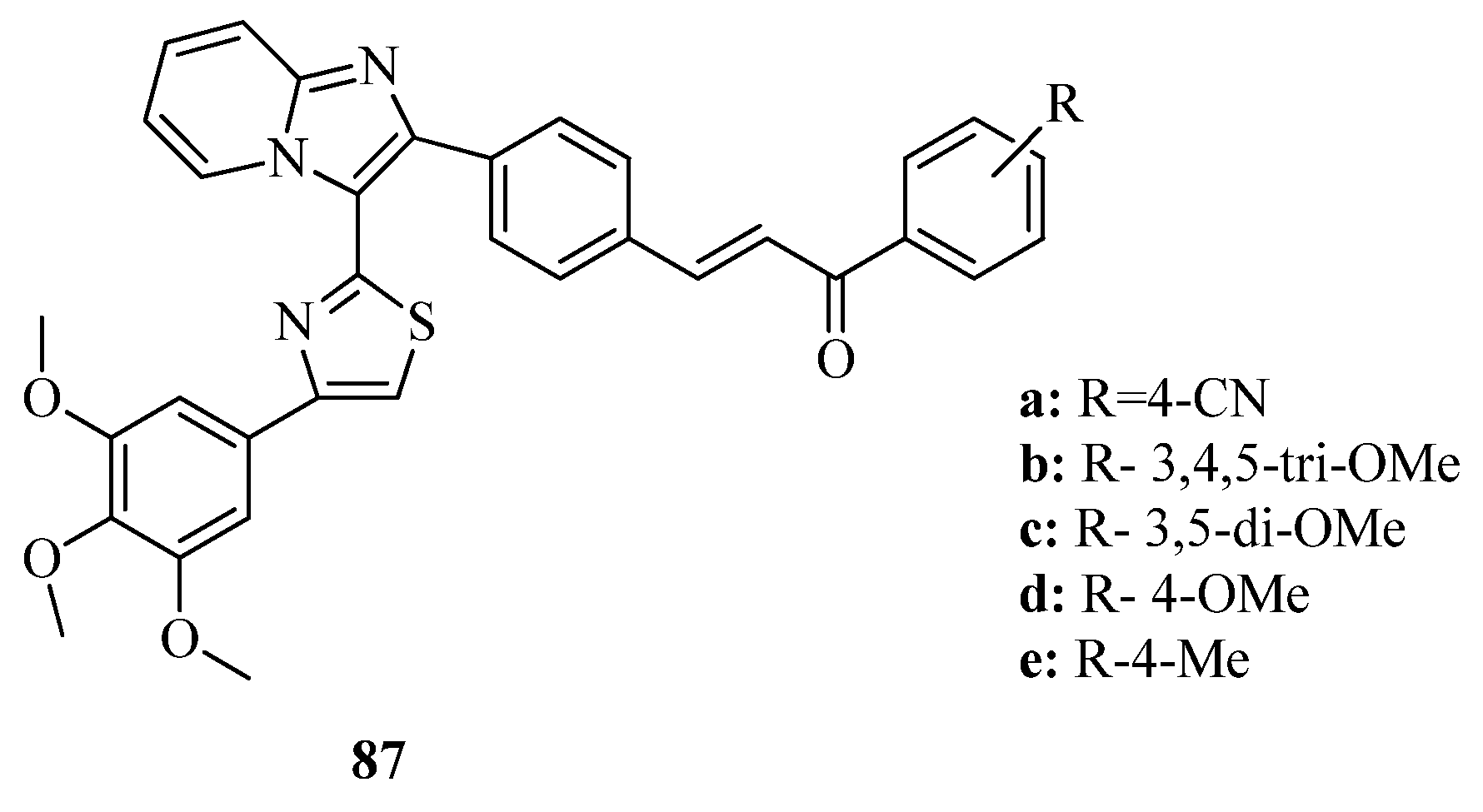
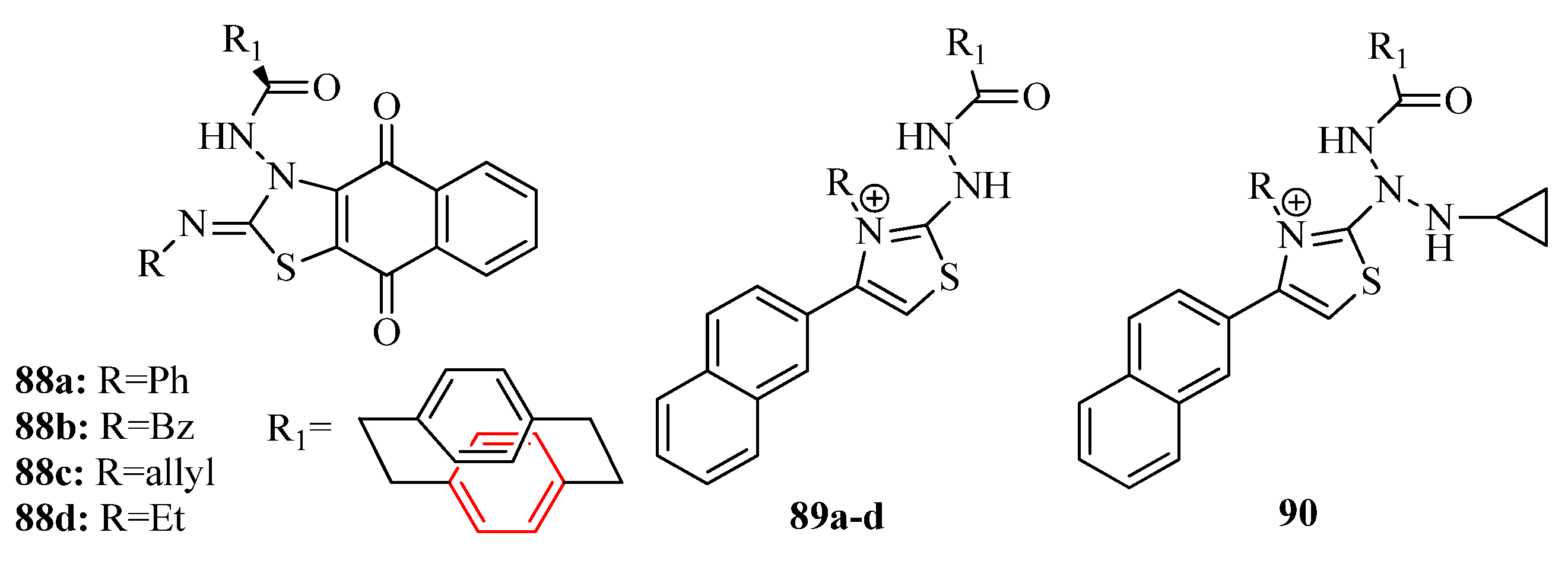
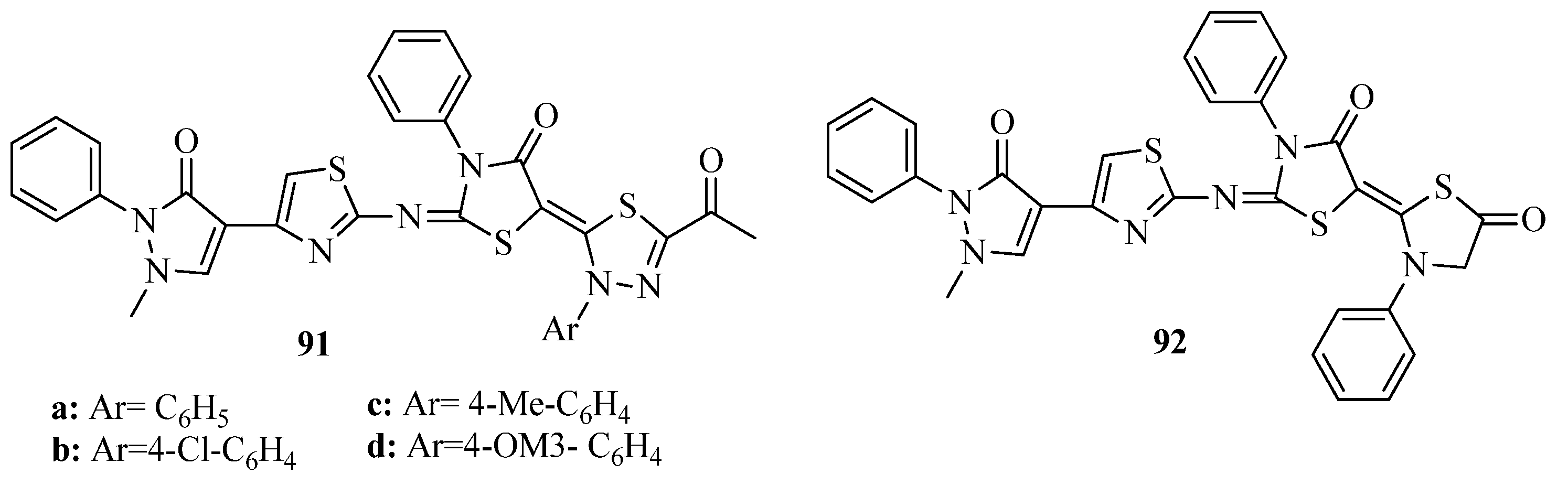
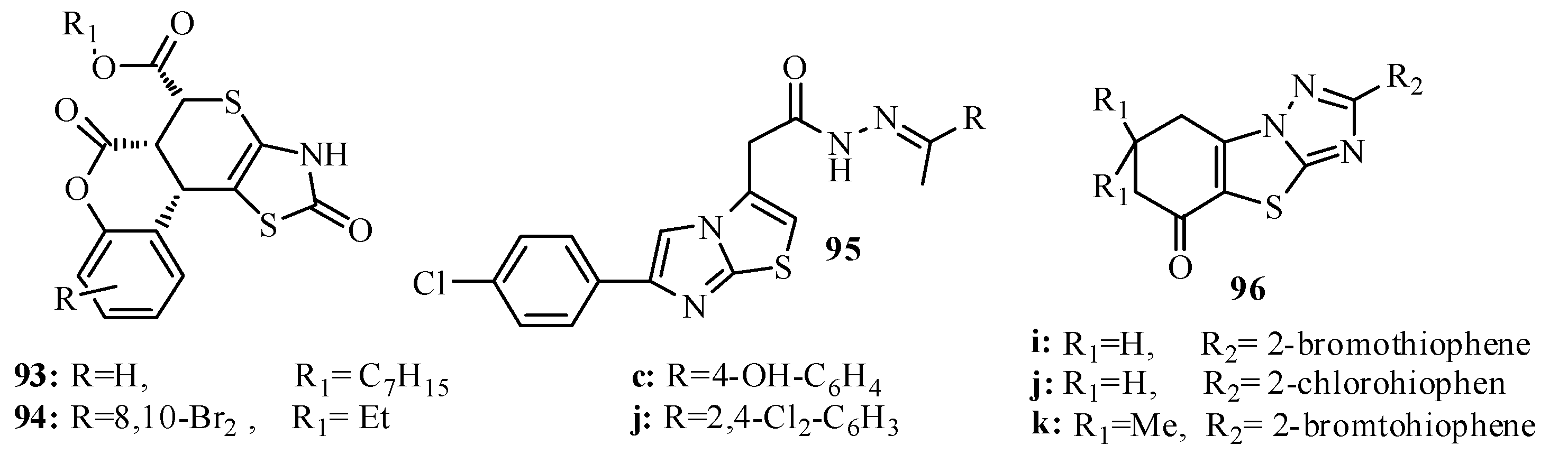
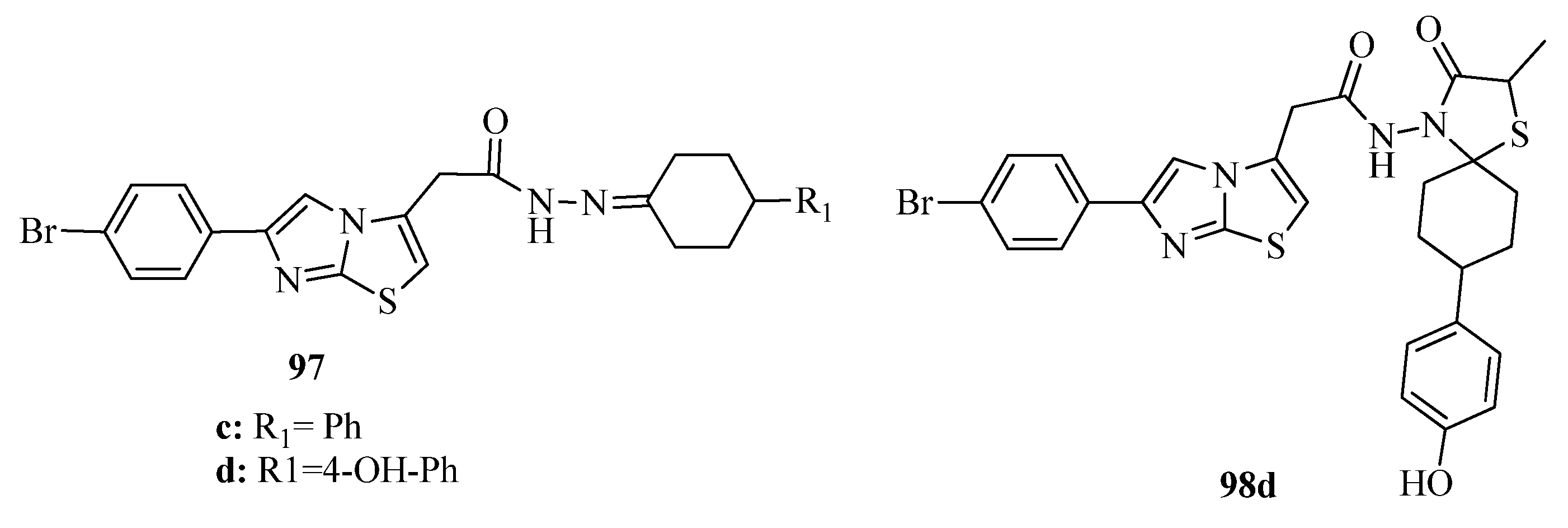
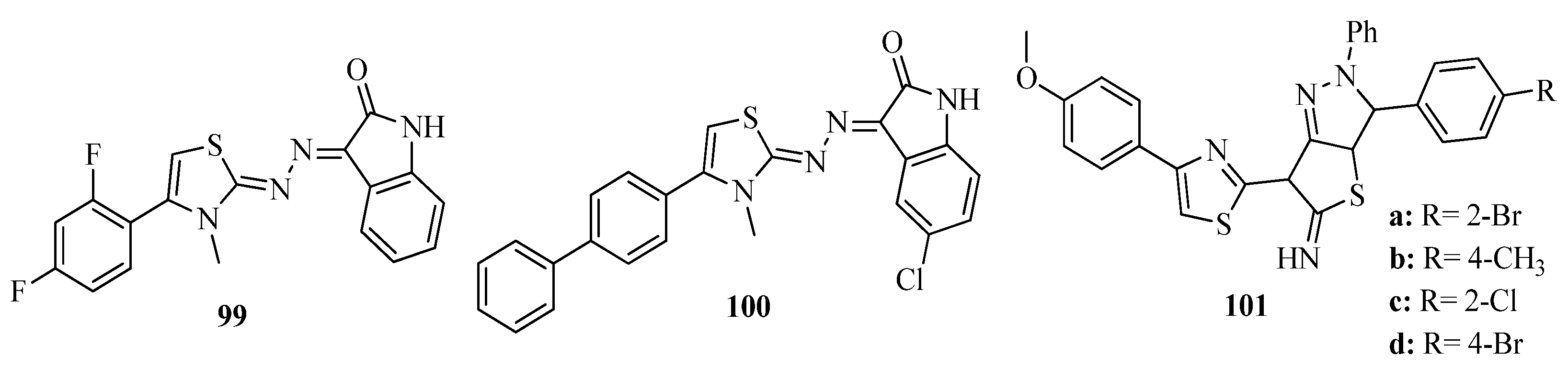
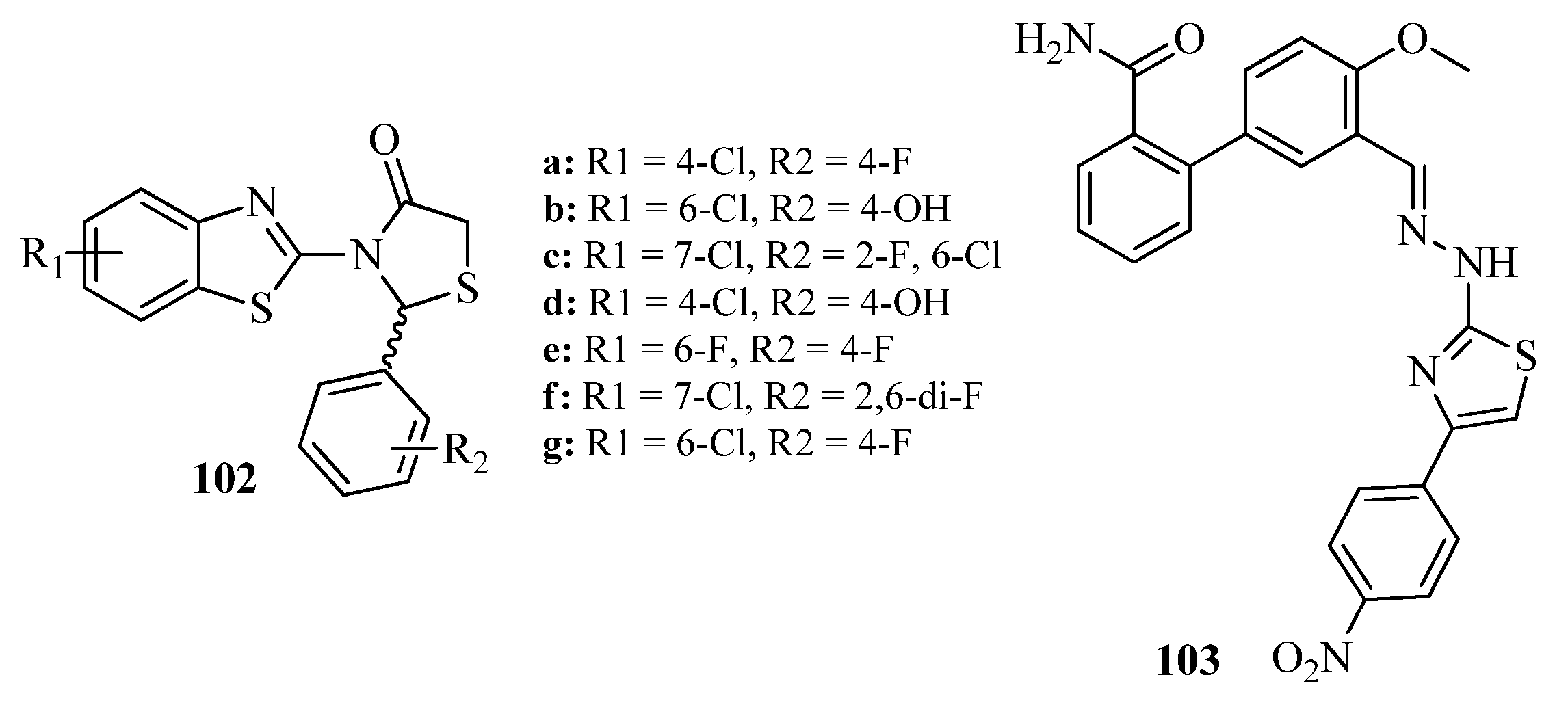



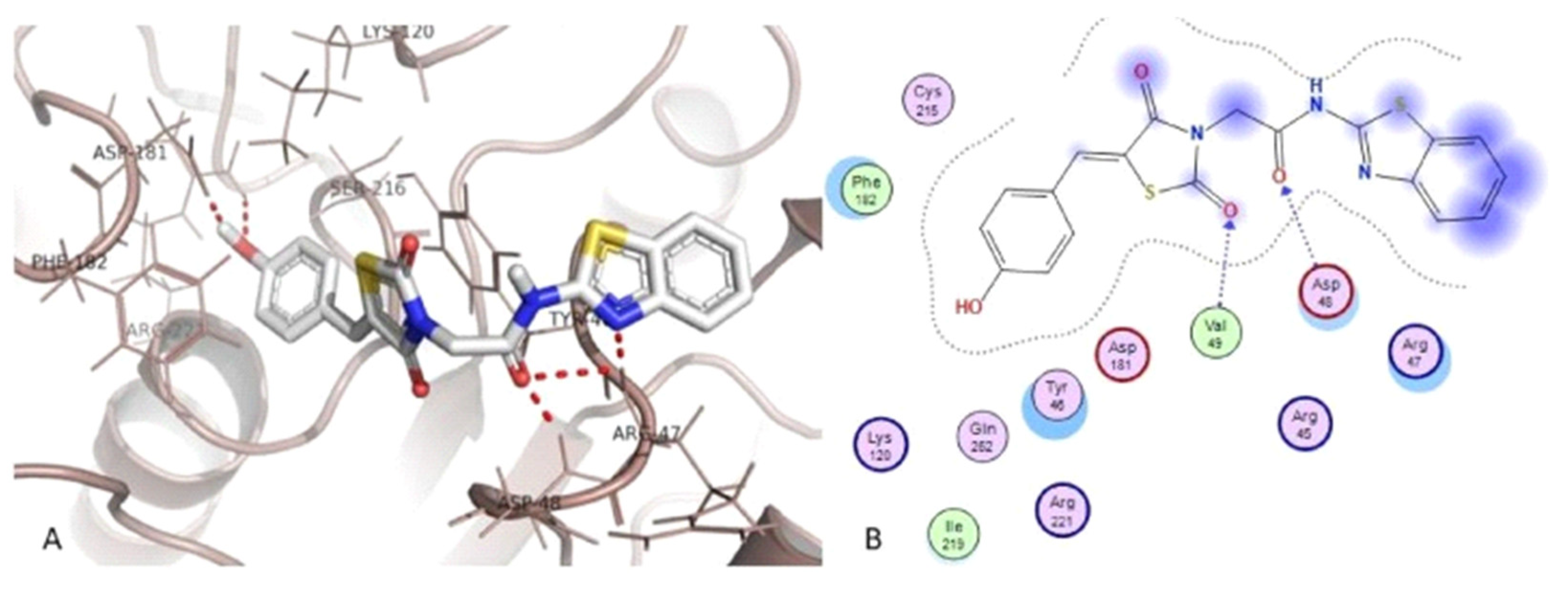
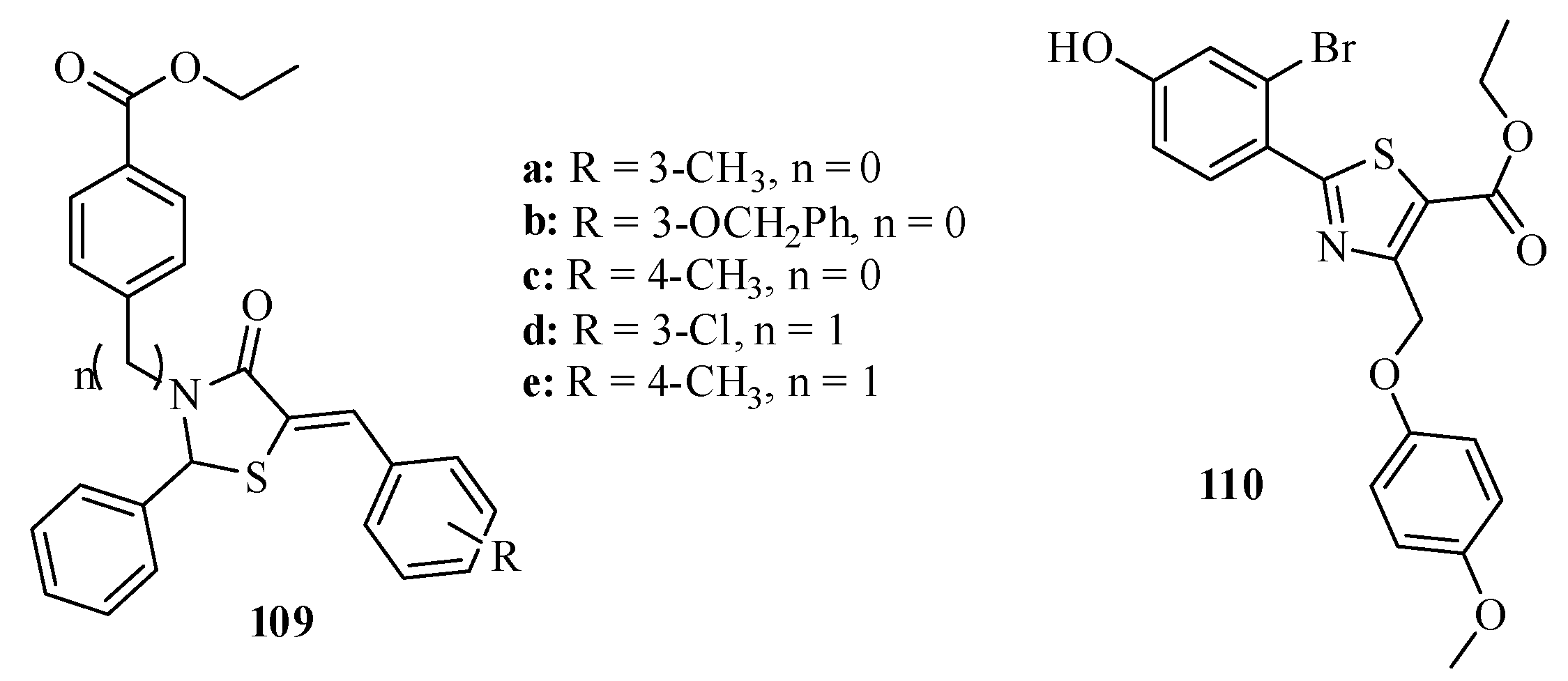
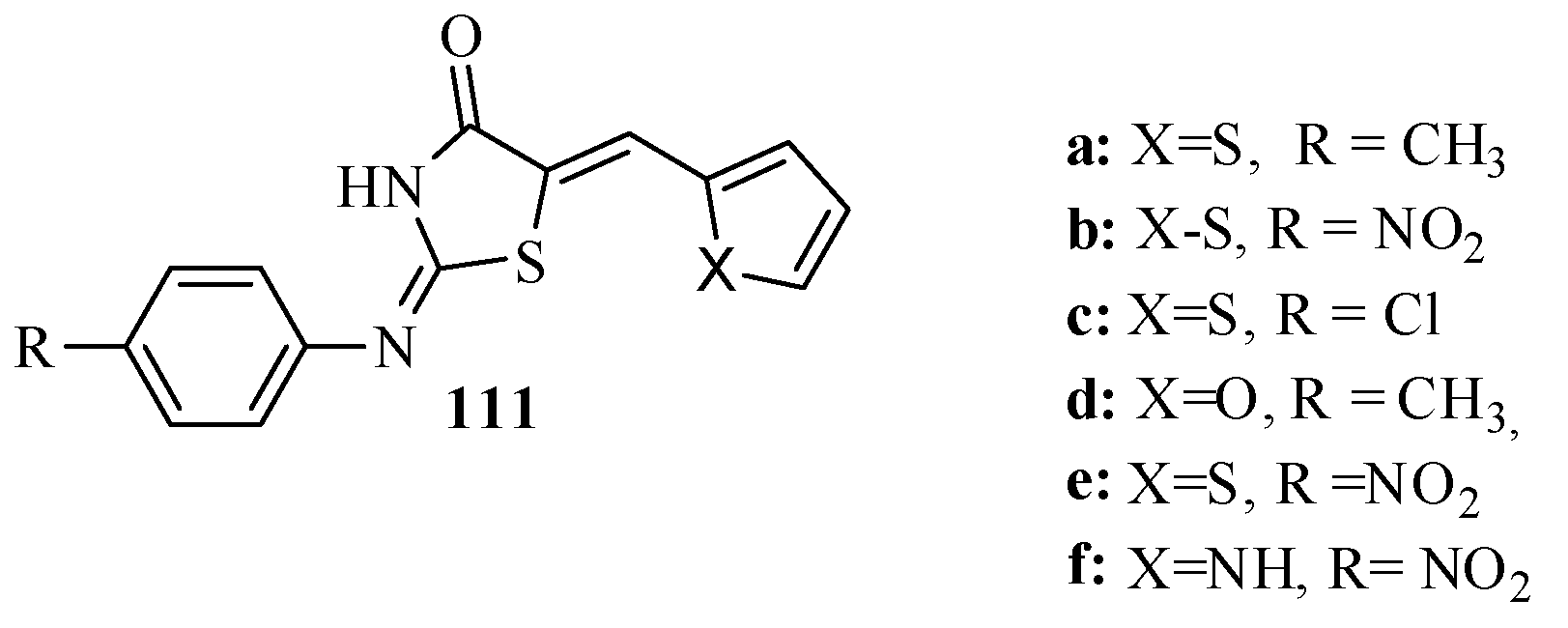
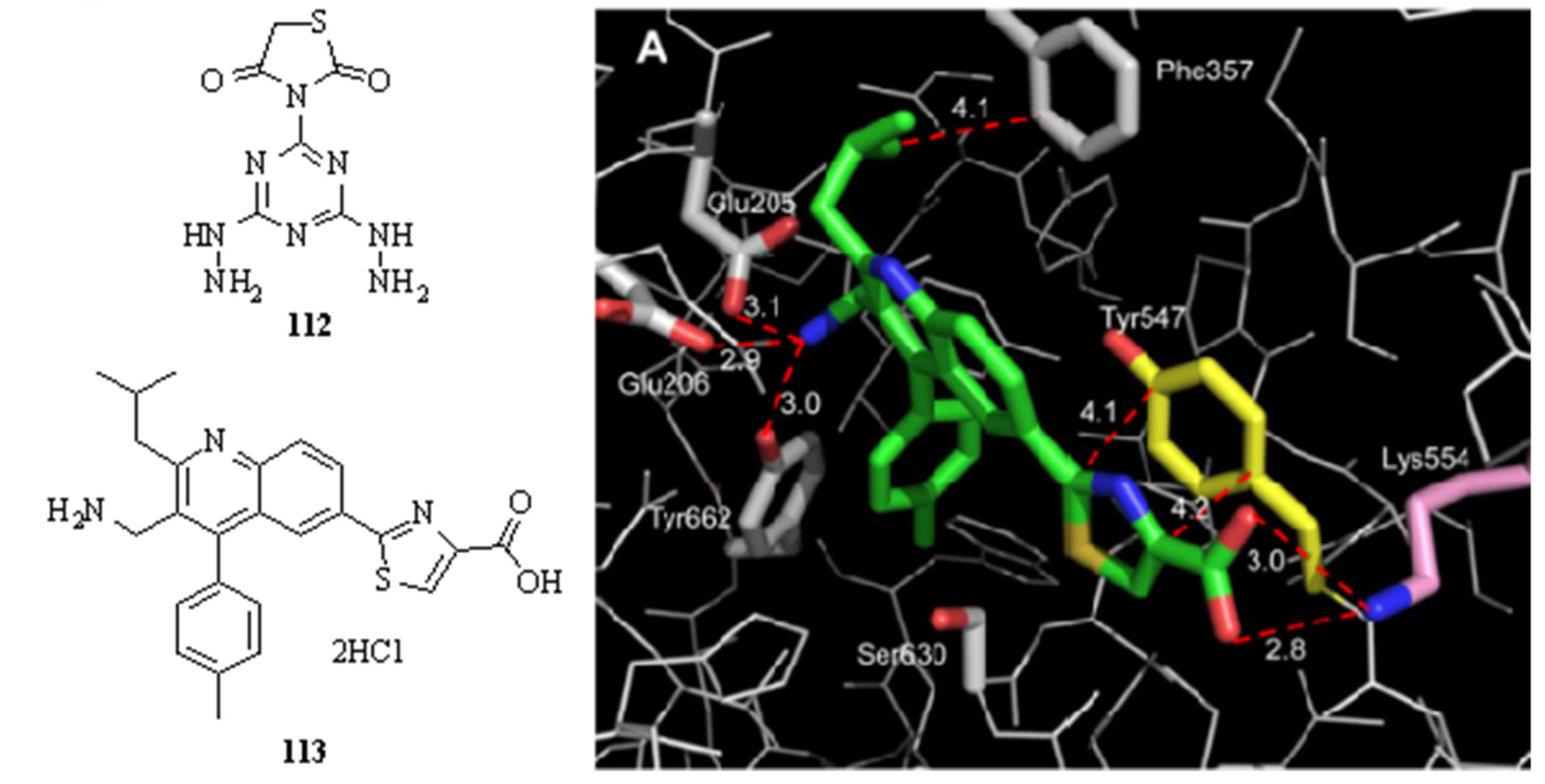



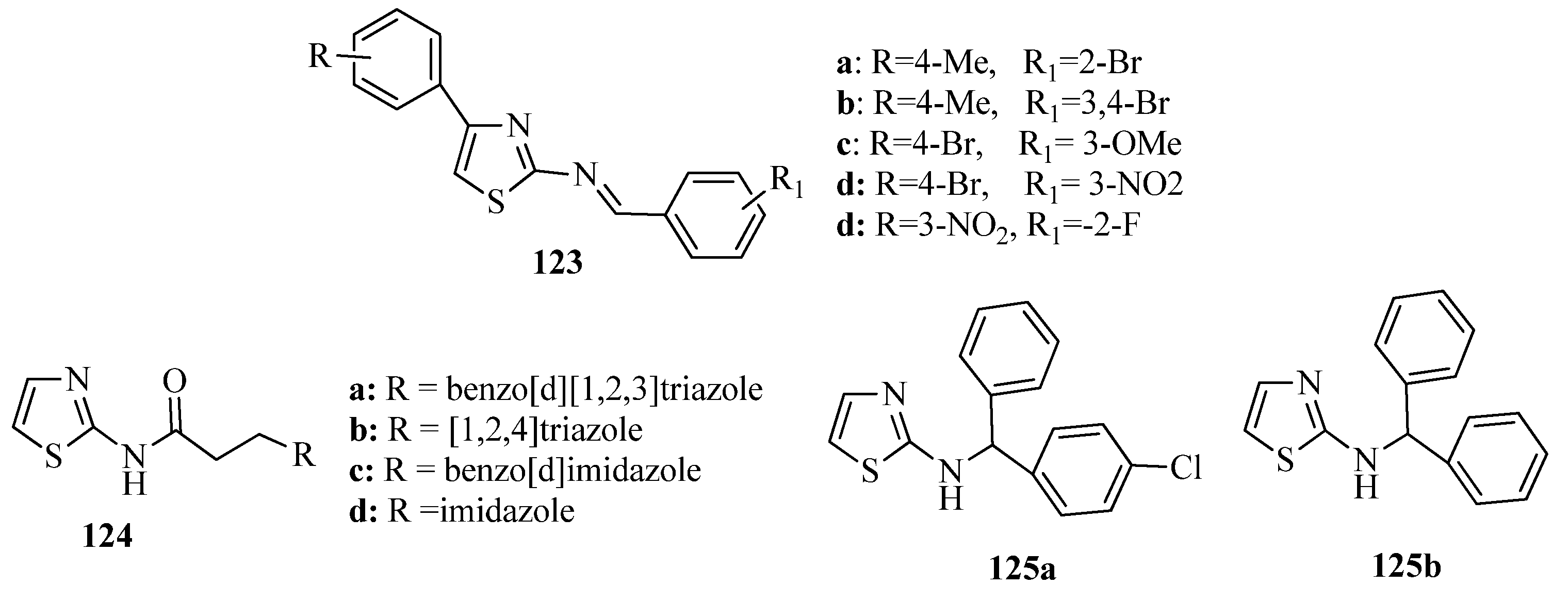
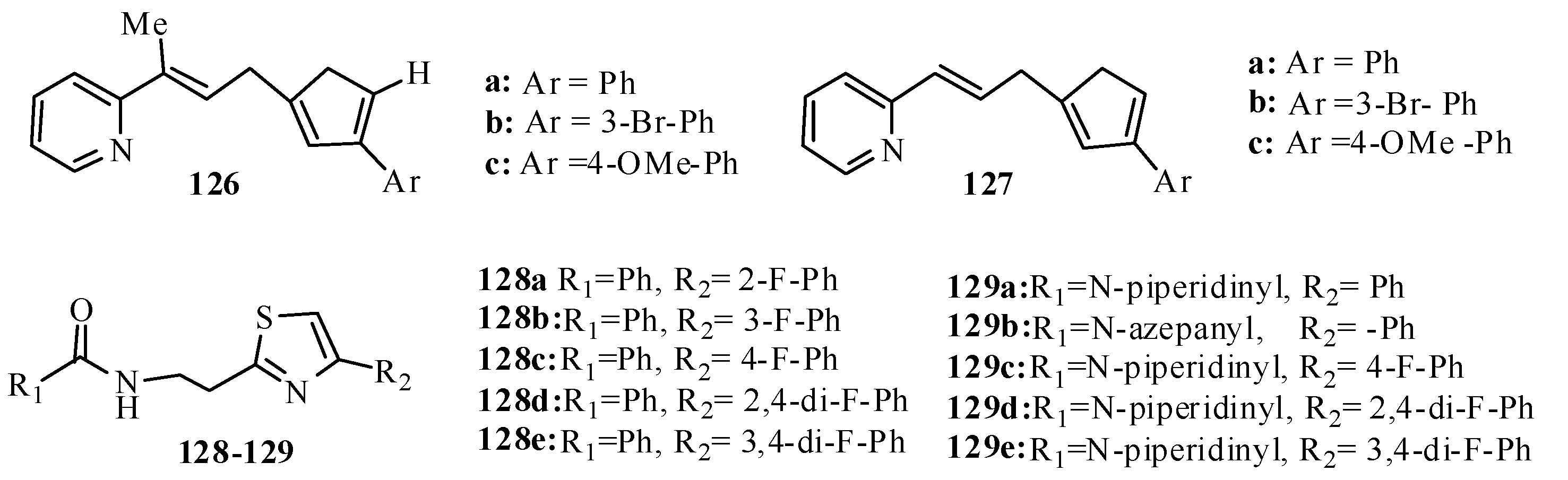
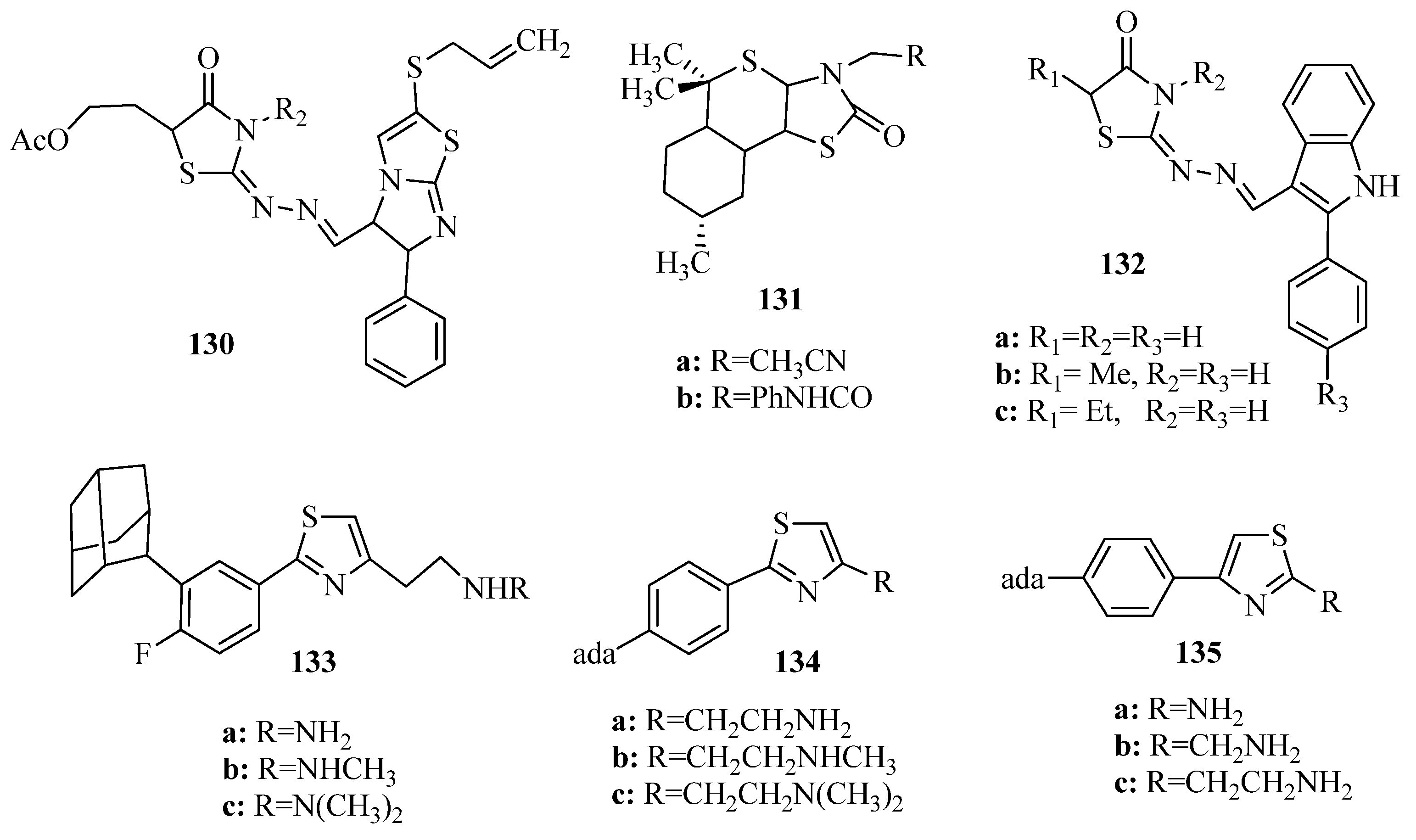
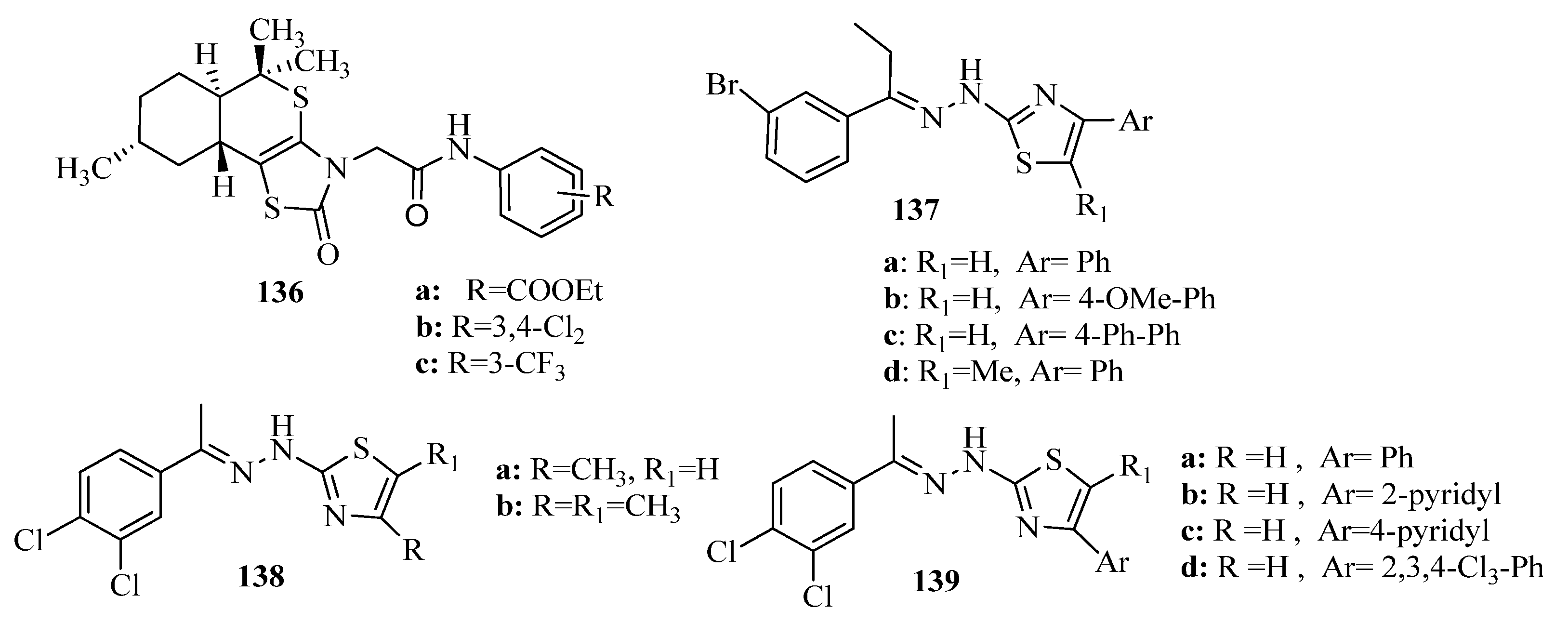
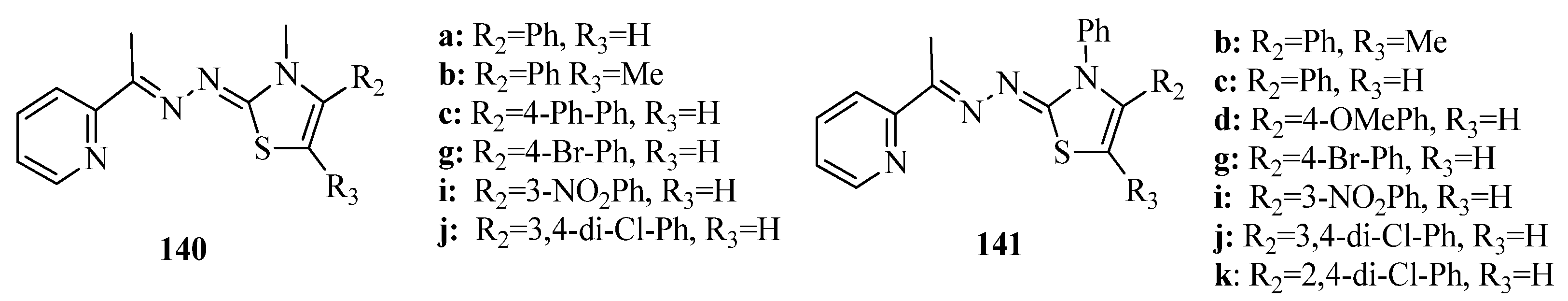

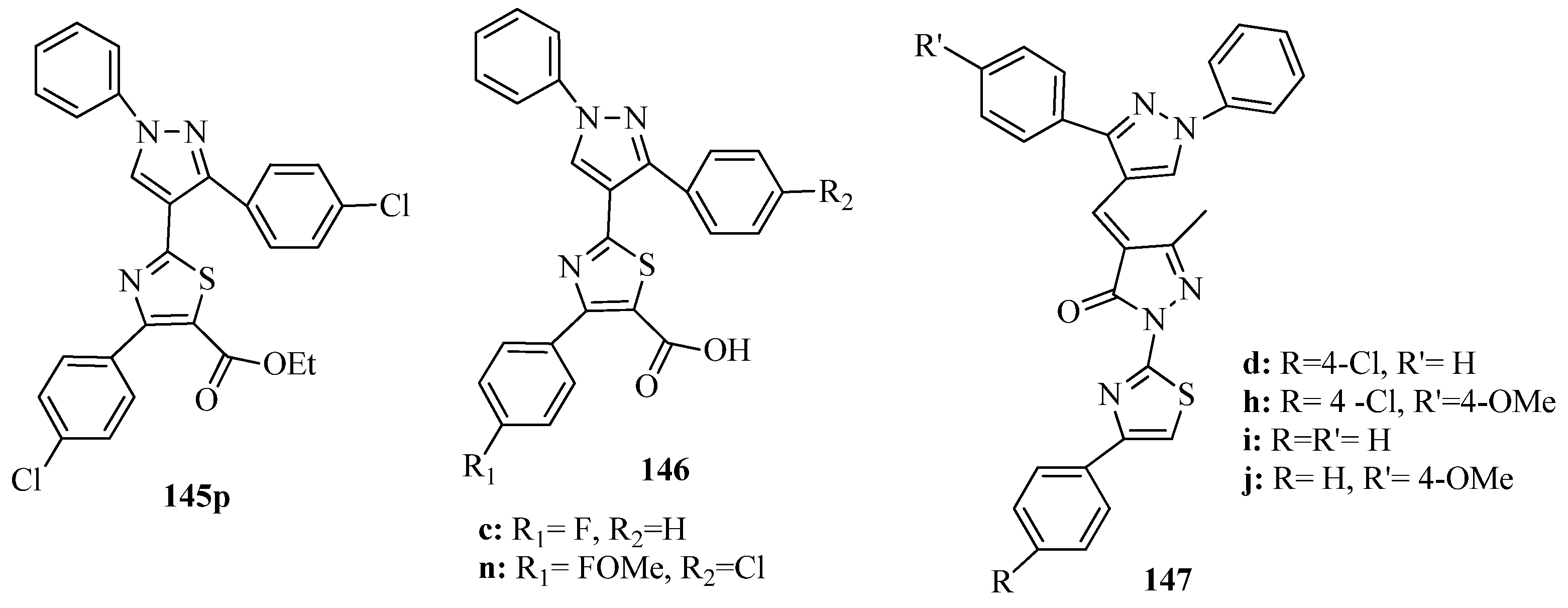

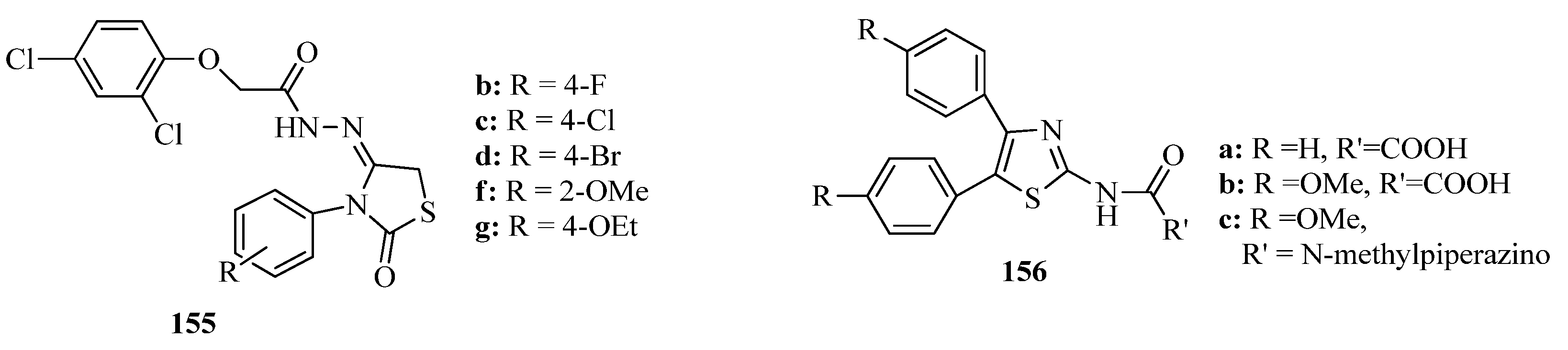
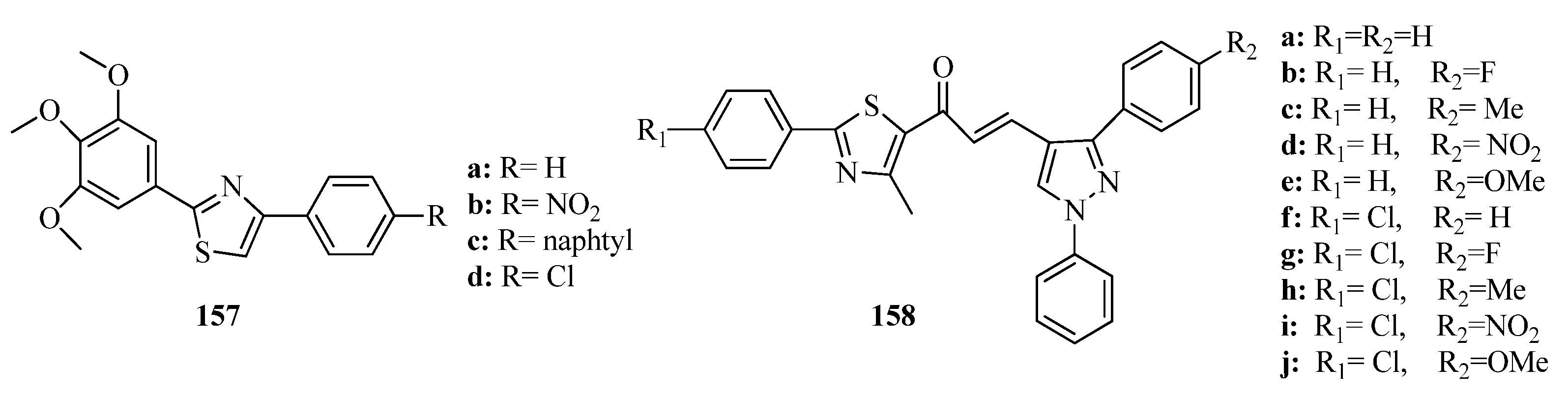
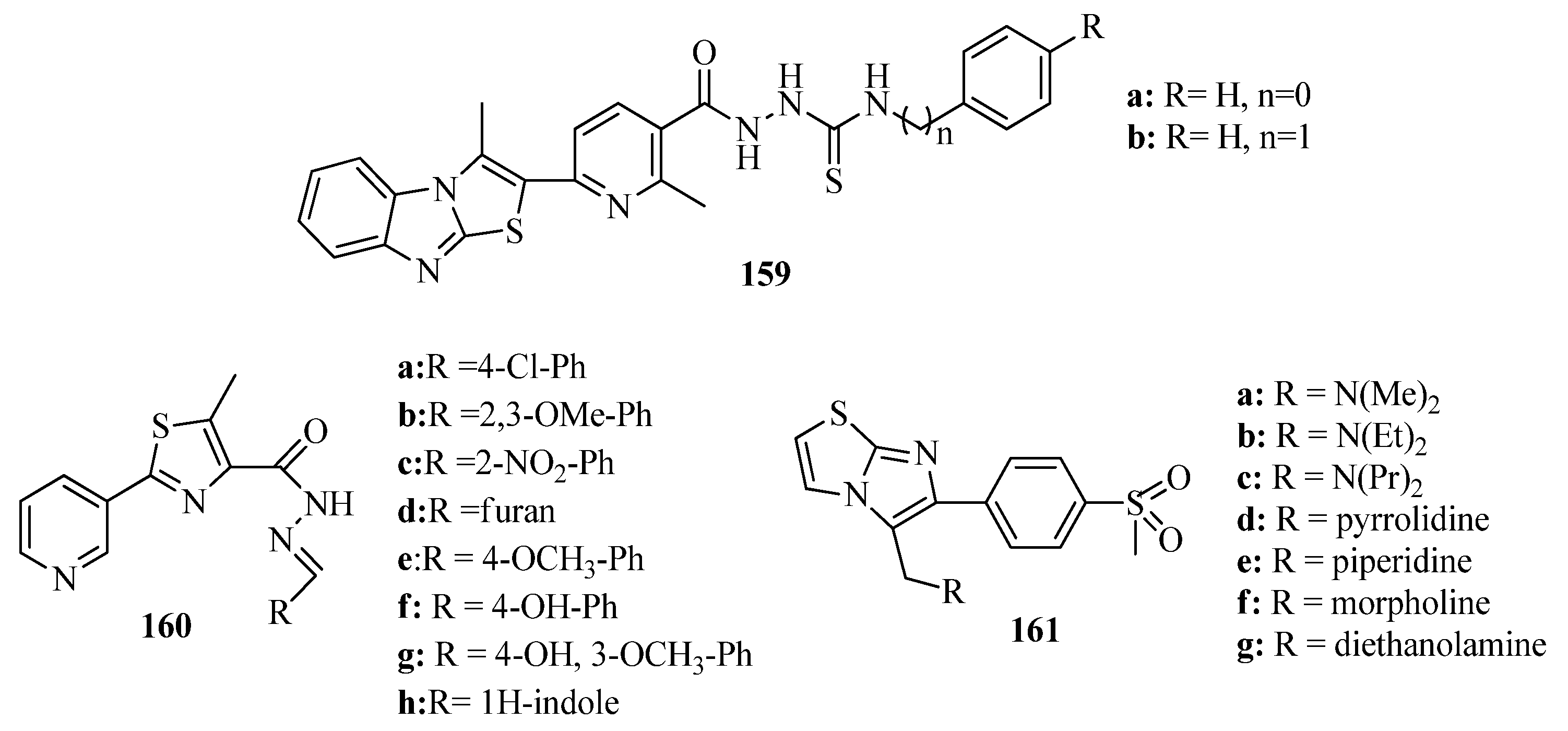

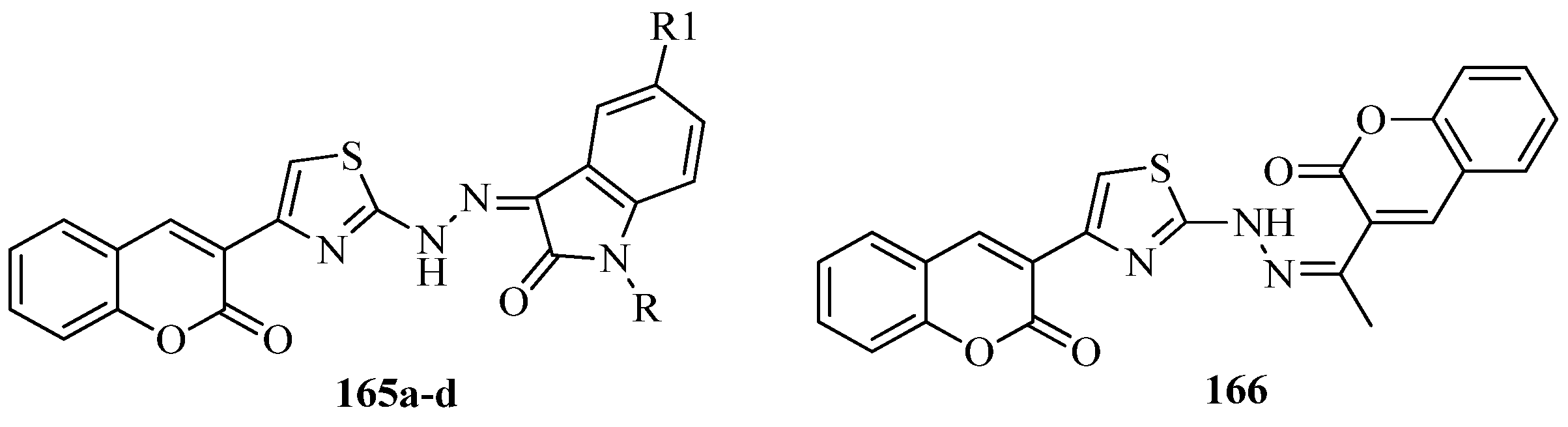
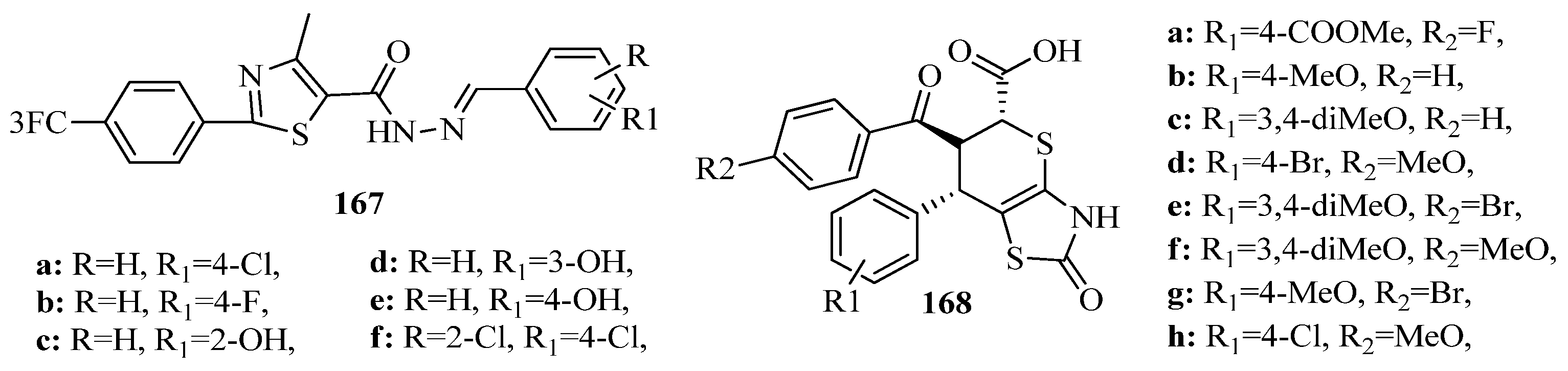
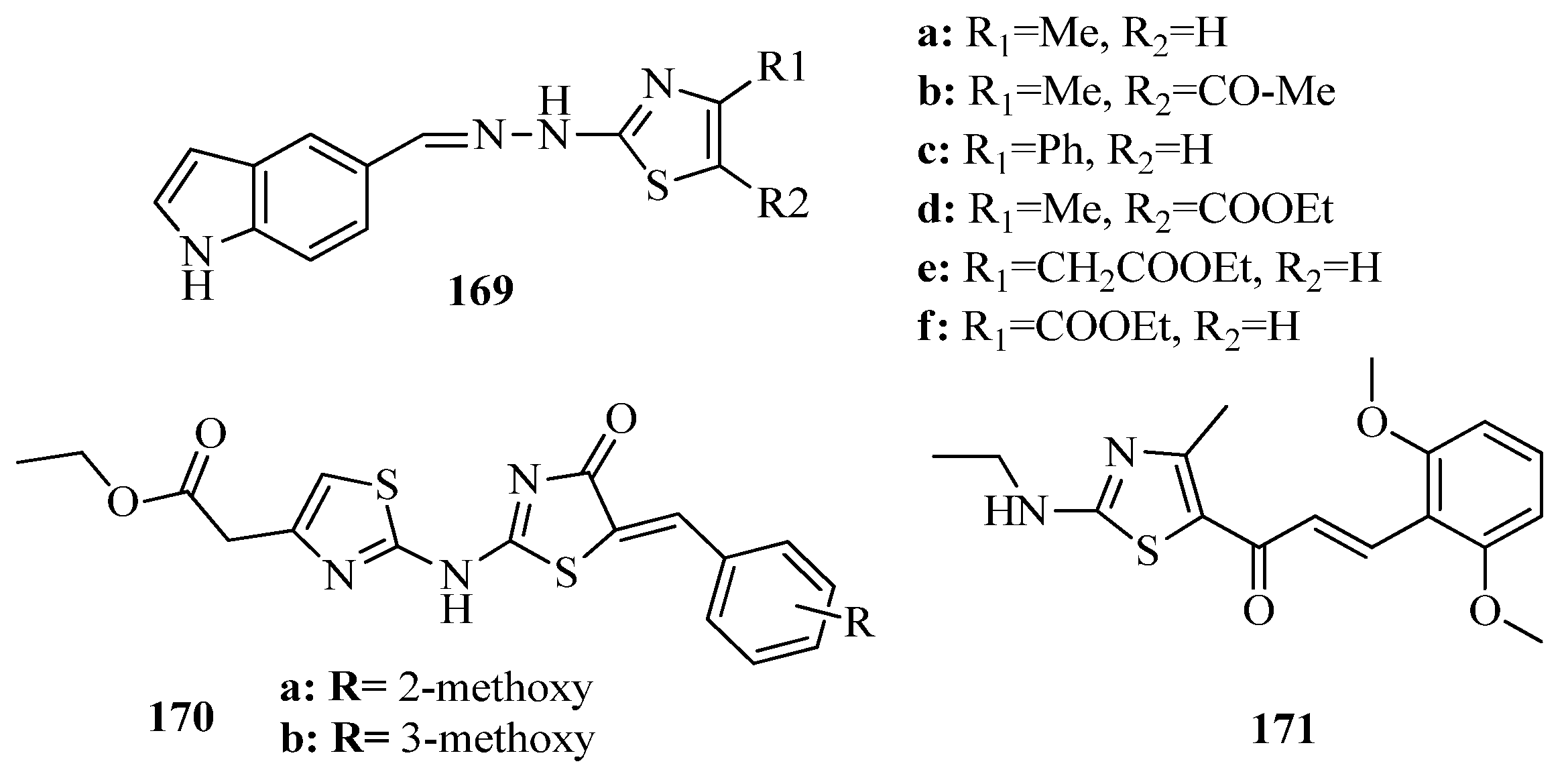


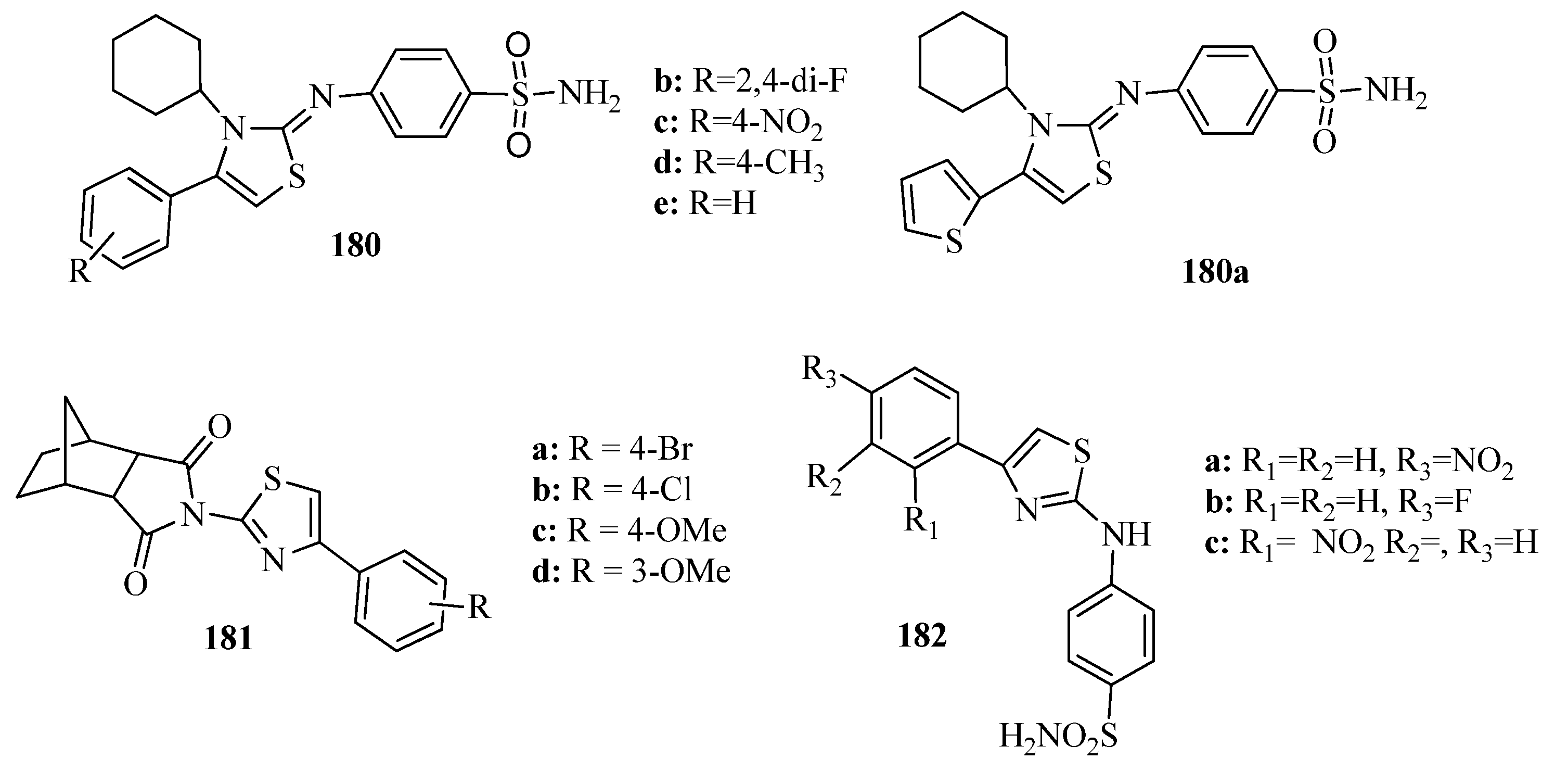
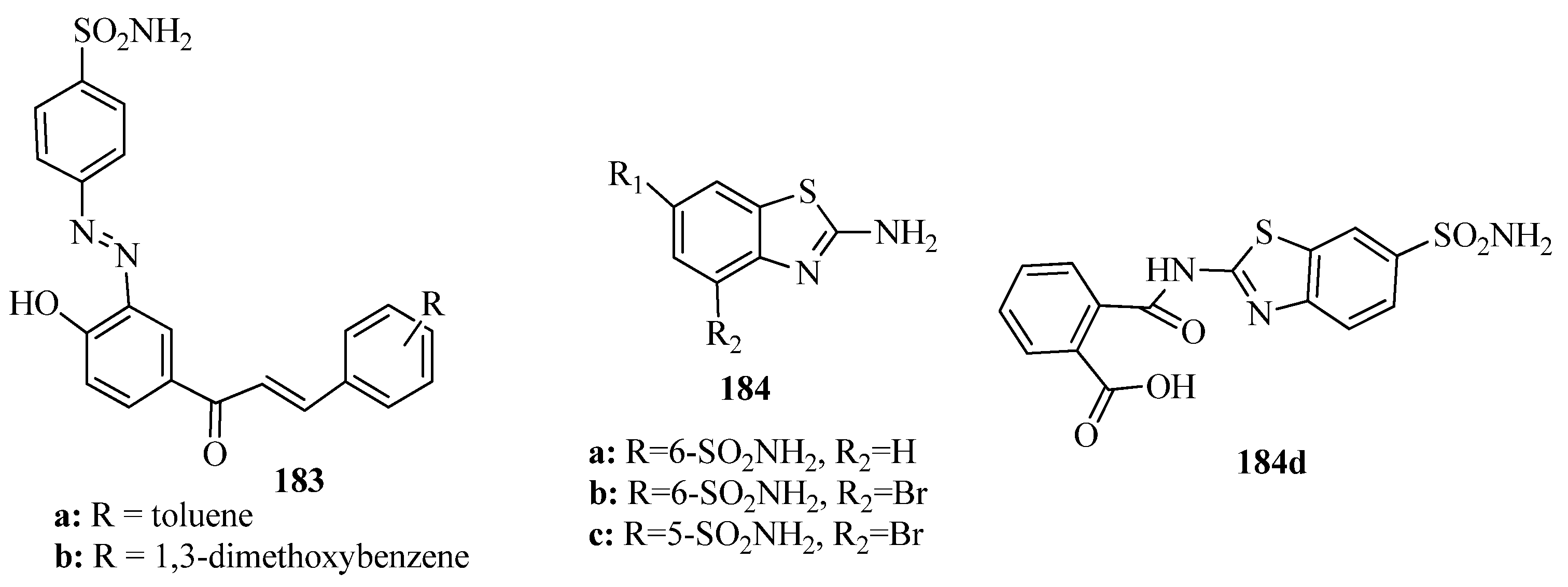
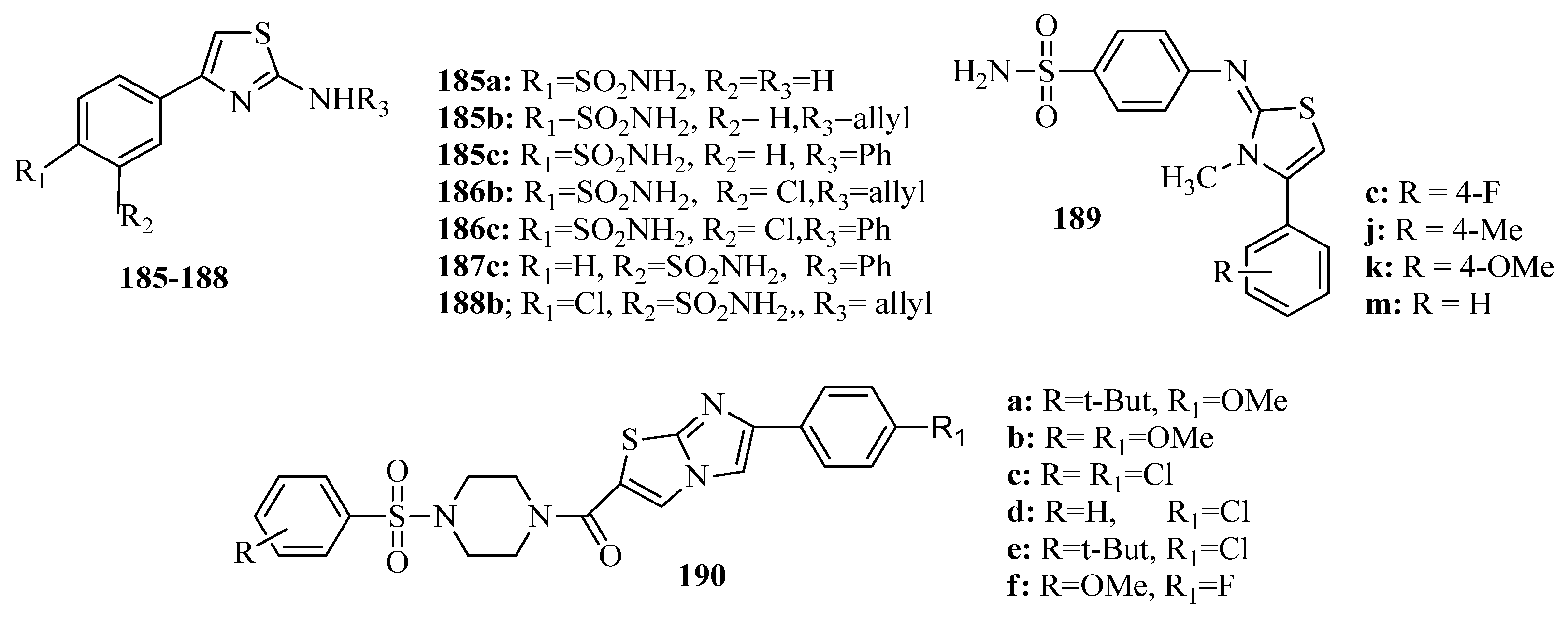
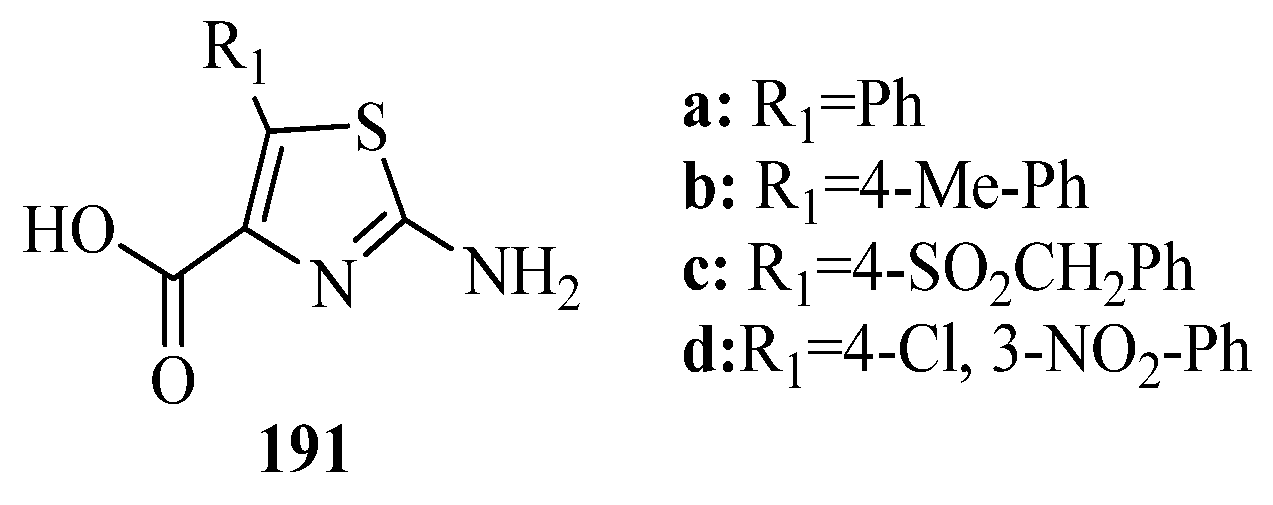


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrou, A.; Fesatidou, M.; Geronikaki, A. Thiazole Ring—A Biologically Active Scaffold. Molecules 2021, 26, 3166. https://doi.org/10.3390/molecules26113166
Petrou A, Fesatidou M, Geronikaki A. Thiazole Ring—A Biologically Active Scaffold. Molecules. 2021; 26(11):3166. https://doi.org/10.3390/molecules26113166
Chicago/Turabian StylePetrou, Anthi, Maria Fesatidou, and Athina Geronikaki. 2021. "Thiazole Ring—A Biologically Active Scaffold" Molecules 26, no. 11: 3166. https://doi.org/10.3390/molecules26113166
APA StylePetrou, A., Fesatidou, M., & Geronikaki, A. (2021). Thiazole Ring—A Biologically Active Scaffold. Molecules, 26(11), 3166. https://doi.org/10.3390/molecules26113166