
New Acridone- and (Thio)Xanthone-Derived 1,1-Donor–Acceptor-Substituted Alkenes: pH-Dependent Fluorescence and Unusual Photooxygenation Properties



Abstract
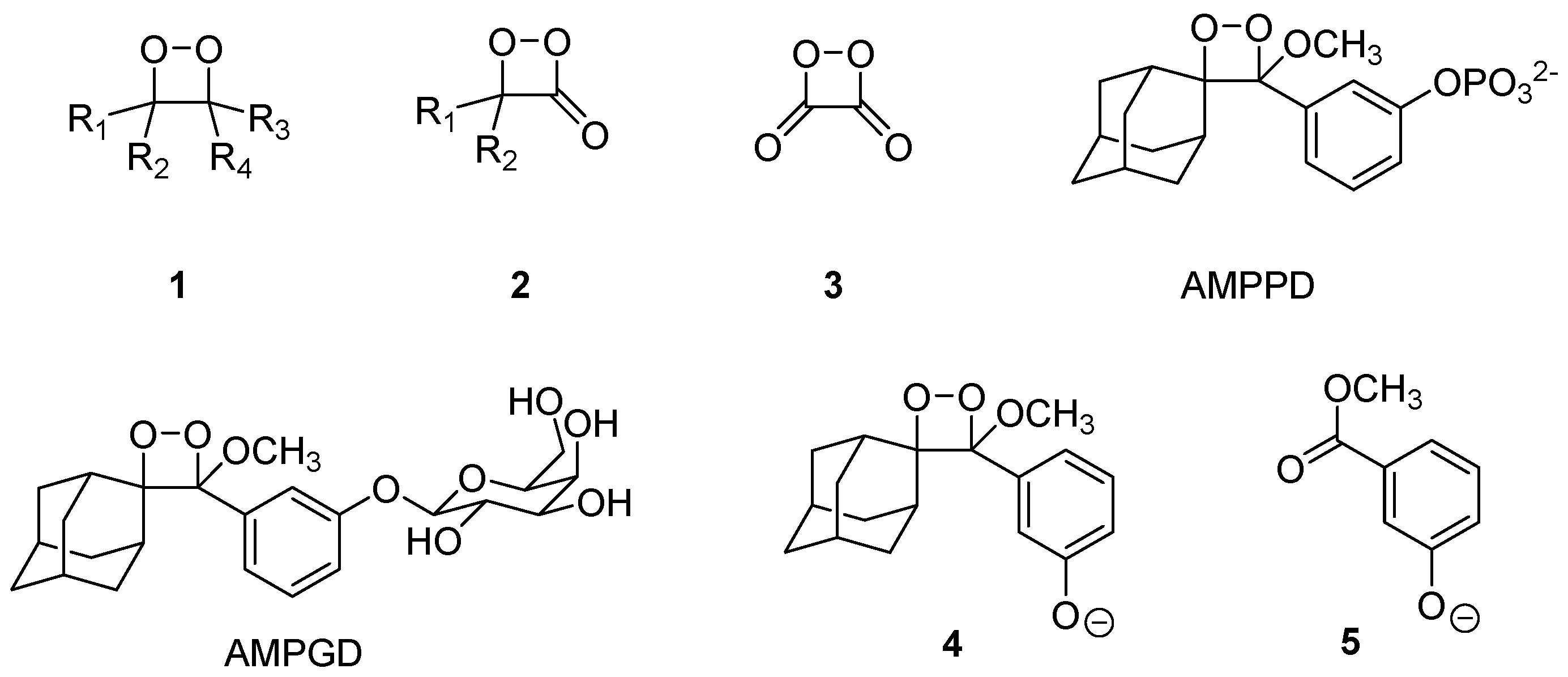
:1. Introduction
2. Results
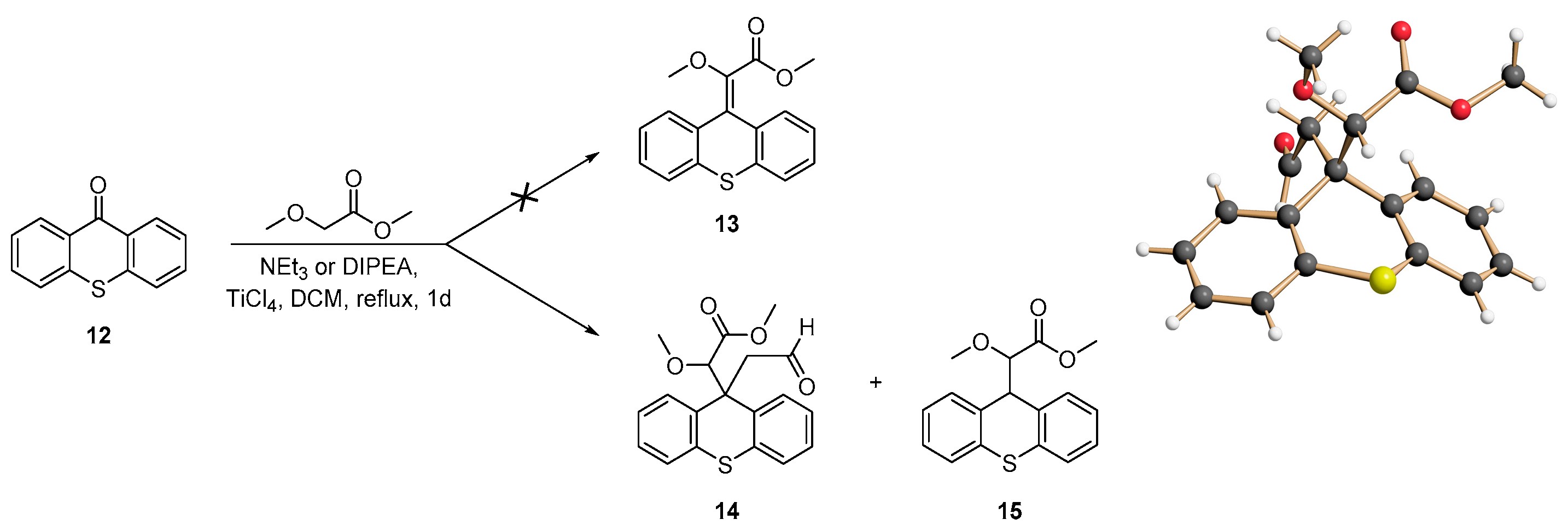
2.1. Synthesis of Donor–Acceptor Alkenes
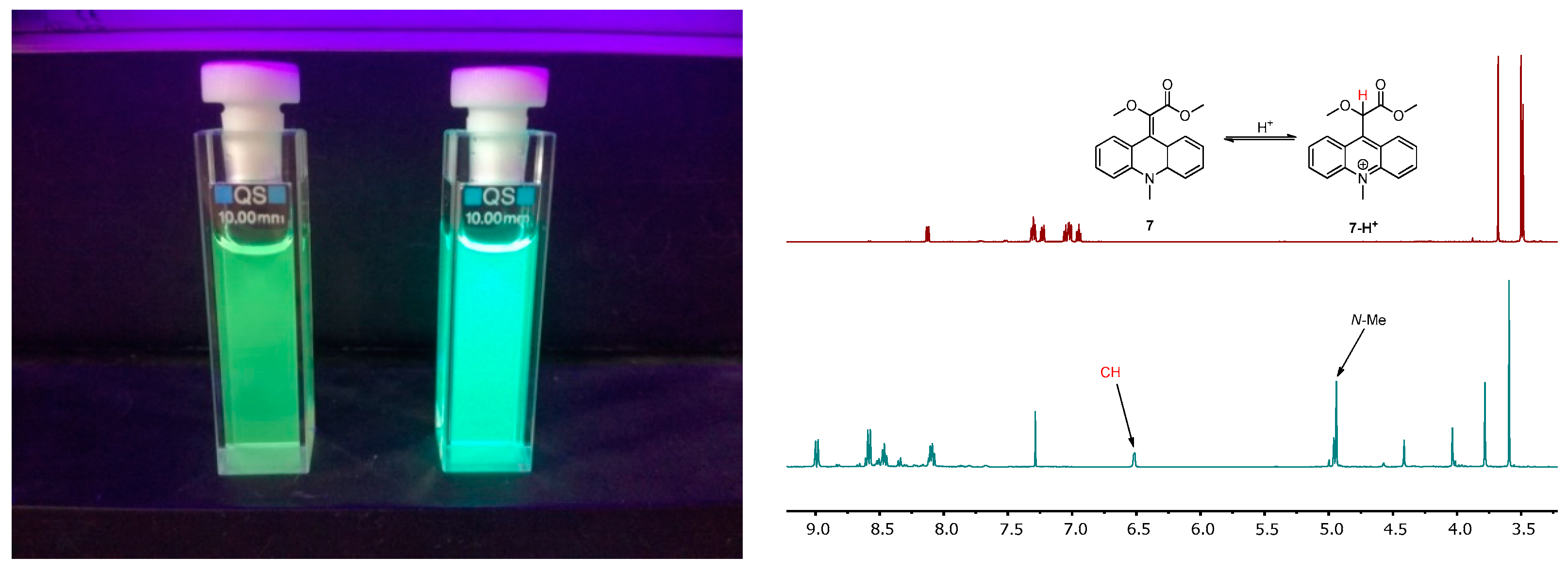
2.2. Fluorescence and UV–VIS Absorption Properties
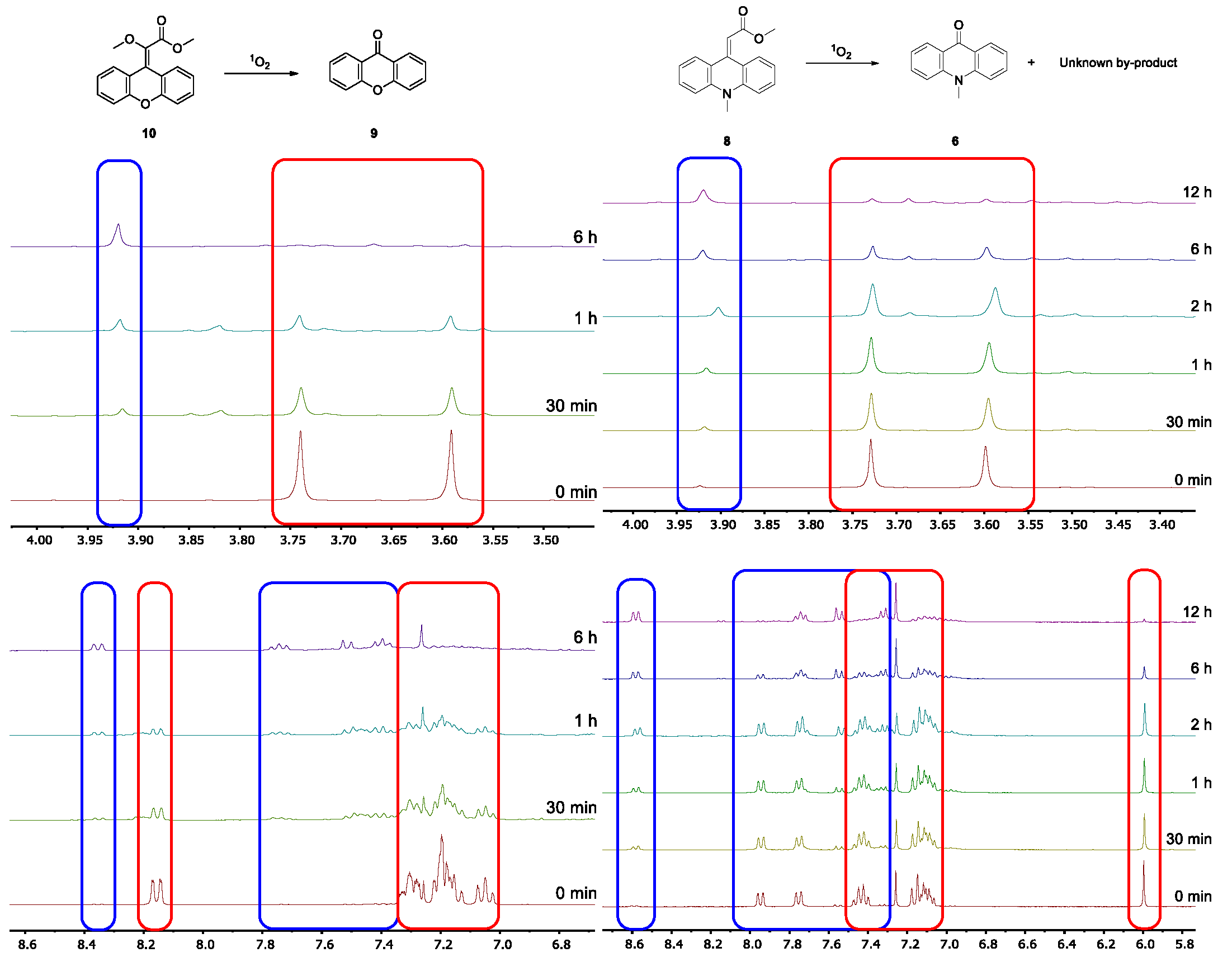
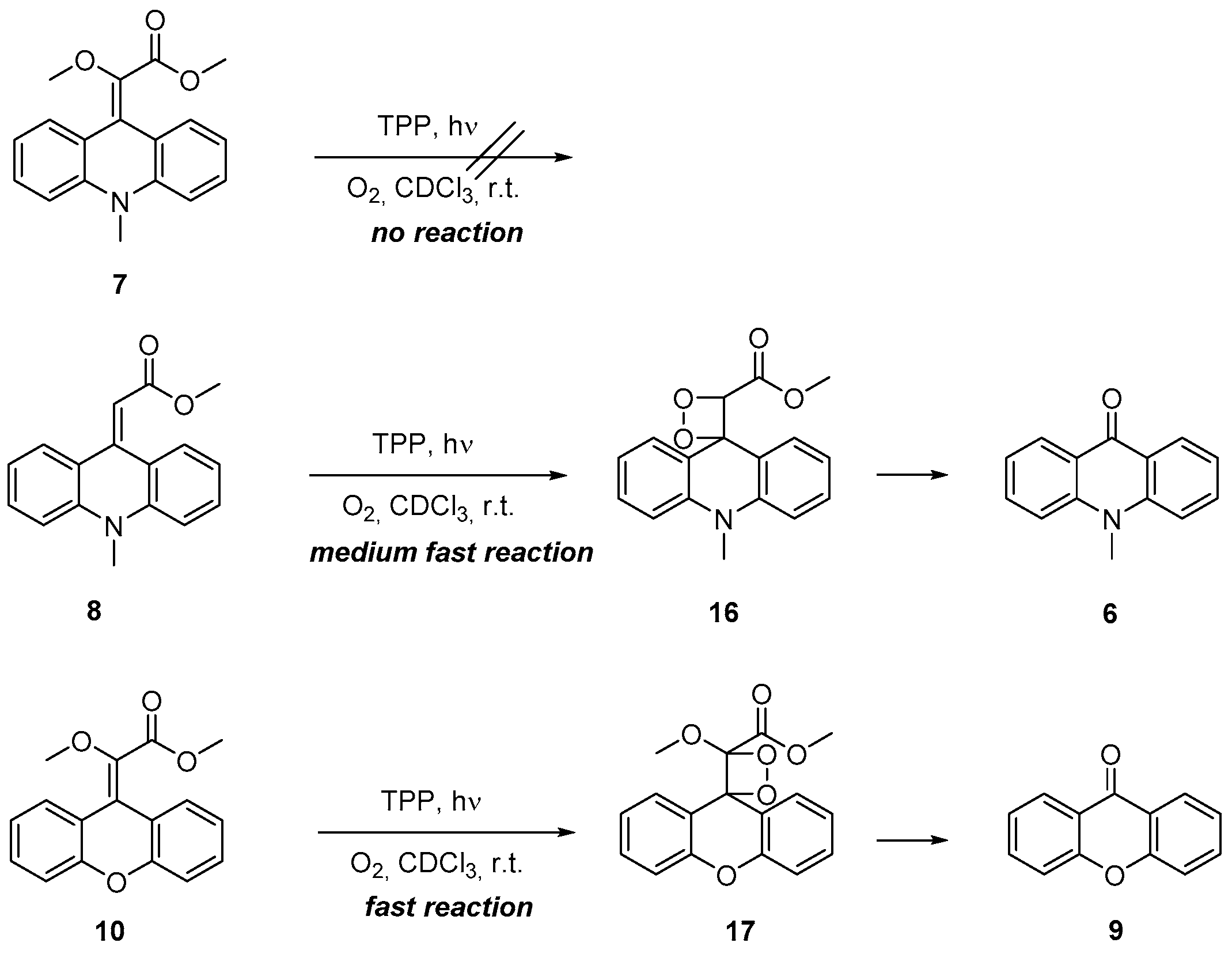
2.3. Photooxygenation of the Geminal Acceptor–Donor Systems
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shimomura, O.; Goto, T.; Johnson, F.H. Source of oxygen in the CO2 produced in the bioluminescent oxidation of firefly luciferin. Proc. Natl. Acad. Sci. USA 1977, 7, 2799–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.-Y.; Schuster, G.B. Chemically initiated electron exchange luminescence. A new chemiluminescent reaction path for organic peroxides. J. Am. Chem. Soc. 1977, 99, 6107–6109. [Google Scholar] [CrossRef]
- Harvey, E.N. A History of Luminescence, from the Earliest Times until 1900; Memoirs of the American Philosophical Society; The American Philosophical Society: Philadelphia, USA, 1957; p. 44. [Google Scholar]
- Albrecht, S.; Brandl, H.; Zimmermann, T. Chemilumineszenz; Hüthig Verlag: Heidelberg, Germany, 1996. [Google Scholar]
- Radziszewski, B. Untersuchungen über Hydrobenzamid, Amarin und Lophin. Ber. Dtsch. Chem. Ges. 1877, 10, 70–75. [Google Scholar] [CrossRef] [Green Version]
- Gleu, K.; Petsch, W. Die Chemiluminescenz der Dimethyldiacridyliumsalze. Angew. Chem. 1935, 48, 57–72. [Google Scholar] [CrossRef]
- Albrecht, H.O. Über die Chemiluminescenz des Aminophthalsäurehydrazids. Z. Phys. Chem. 1928, 136, 321–330. [Google Scholar] [CrossRef]
- Mayer, M.; Takegami, S.; Neumeier, M.; Rink, S.; Jacobi von Wangelin, A.; Schulte, S.; Vollmer, M.; Griesbeck, A.G.; Duerkop, A.; Bäumner, J. Electrochemiluminescence Bioassays with a Water-Soluble Luminol Derivative Can Outperform Fluorescence Assays. Angew. Chem. Int. Ed. 2018, 57, 408–411. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Diaz-Miara, Y.; Jacobi von Wangelin, A.; Pérez-Ruiz, R.; Sampredo, D. Steric Enhancement of the Chemiluminescence of Luminols. Chem. Eur. J. 2015, 21, 9975–9979. [Google Scholar] [CrossRef] [PubMed]
- Chandross, E.A. A new chemiluminescent system. Tetrahedron Lett. 1963, 12, 761–765. [Google Scholar]
- Ciscato, L.F.M.L.; Bartoloni, F.H.; Bastos, E.L.; Baader, W.J. Direct Kinetic Observation of the Chemiexcitation Step in Peroxyoxalate Chemiluminescence. J. Org. Chem. 2009, 74, 8974–8979. [Google Scholar] [CrossRef] [PubMed]
- Ciscato, L.F.M.L.; Augusto, F.A.; Weiss, D.; Bartoloni, F.H.; Albrecht, S.; Brandl, H.; Zimmermann, T.; Baader, W.J. The chemiluminescent peroxyoxalate system: State of the art almost 50 years from its discovery. ARKIVOC 2012, 2012, 391–430. [Google Scholar] [CrossRef] [Green Version]
- Vacher, M.; Galván, I.F.; Ding, B.-W.; Schramm, S.; Berraud-Pache, R.; Naumov, P.; Ferré, N.; Liu, Y.-J.; Navizet, I.; Roca-Sanjuán, D.; et al. Chemi- and Bioluminescence of Cyclic Peroxides. Chem. Rev. 2018, 118, 6927–6974. [Google Scholar] [CrossRef] [PubMed]
- Farahani, P.; Roca-Sanjuán, D.; Zapata, F.; Lindh, R. Revisiting the Nonadiabatic Process in 1,2-Dioxetane. J. Chem. Theory Comput. 2013, 9, 5404–5411. [Google Scholar] [CrossRef]
- Tanaka, C.; Tanaka, J. Ab Initio Molecular Orbital Studies on the Chemiluminescence of 1,2-Dioxetanes. J. Phys. Chem. A 2000, 104, 2078–2090. [Google Scholar] [CrossRef]
- Rodríguez, E.; Reguero, M. The DDCI Method Applied to Reactivity: Chemiluminescent Decomposition of Dioxetane. J. Phys. Chem. A 2002, 106, 504–509. [Google Scholar] [CrossRef]
- De Vico, L.; Liu, Y.-J.; Krogh, J.W.; Lindh, R. Chemiluminescence of 1,2-Dioxetane. Reaction Mechanism Uncovered. J. Phys. Chem. A 2007, 111, 8013–8019. [Google Scholar] [CrossRef] [PubMed]
- Baader, W.J.; Stevani, C.V.; Bastos, E.L. The Chemistry of Peroxides; Rappoport, Z., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2006; pp. 1211–1278. [Google Scholar]
- Adam, W. Chemical and Biological Generation of Excited States; Adam, W., Cilento, G., Eds.; Academic Press: Cambridge, MA, USA, 1982; pp. 1115–1152. [Google Scholar]
- El-Sayed, M.A. Spin—Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. [Google Scholar] [CrossRef]
- El-Sayed, M.A. Triplet state. Its radiative and nonradiative properties. Acc. Chem. Res. 1968, 1, 8–16. [Google Scholar] [CrossRef]
- Kopecky, K.R.; van de Sande, J.H.; Mumford, C. Preparation and base-catalyzed reactions of some β-halohydroperoxides. Can. J. Chem. 1968, 46, 25–34. [Google Scholar] [CrossRef]
- Kopecky, K.R.; Mumford, C. Luminescence in the thermal decomposition of 3,3,4-trimethyl-1,2-dioxetane. Can. J. Chem. 1969, 47, 709–711. [Google Scholar] [CrossRef]
- Wieringa, J.H.; Strating, J.; Wynberg, H.; Adam, W. Adamantylideneadamantane peroxide, a stable 1,2-dioxetane. Tetrahedron Lett. 1972, 2, 169–172. [Google Scholar] [CrossRef]
- Bronstein, I.; McGrath, P. Chemiluminescence lights up. Nature 1989, 338, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, I.; Edwards, B.; Voyta, J.C. 1,2-Dioxetanes: Novel chemiluminescent enzyme substrates. Applications to immunoassays. J. Biolumin. Chemilumin. 1989, 4, 99–111. [Google Scholar] [CrossRef]
- Schaap, A.P.; Akhavan, H.; Romano, L. Chemiluminescent substrates for alkaline phosphatase: Application to ultrasensitive enzyme-linked immunoassays and DNA probes. Clin. Chem. 1989, 35, 1863–1864. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.A.; Schmidt, S.P.; Schuster, G.B. Bioluminescence of the firefly: Key steps in the formation of the electronically excited state for model systems. Proc. Natl. Acad. Sci. USA 1978, 75, 30–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, J.-Y.; Schuster, G.B. Chemiluminescence of diphenoyl peroxide. Chemically initiated electron exchange luminescence. A new general mechanism for chemical production of electronically excited states. J. Am. Chem. Soc. 1978, 100, 4496–4503. [Google Scholar] [CrossRef]
- Wöhrle, D.; Tausch, M.W.; Stohrer, W.-D. Photochemie: Konzepte, Methoden, Experimente; Wiley-VCH: Weinheim, Germany, 1998; pp. 1262–1269. [Google Scholar]
- Gnaim, S.; Green, O.; Shabat, D. The emergence of aqueous chemiluminescence: New promising class of phenoxy 1,2-dioxetane luminophores. Chem. Commun. 2018, 54, 2073–2085. [Google Scholar] [CrossRef]
- Yue, L.; Liu, Y.-J. Mechanism of AMPPD Chemiluminescence in a Different Voice. J. Chem. Theory Comput. 2013, 9, 2300–2312. [Google Scholar] [CrossRef]
- Catalani, L.H.; Wilson, T. Electron transfer and chemiluminescence. Two inefficient systems: 1,4-dimethoxy-9,10-diphenylanthracene peroxide and diphenoyl peroxide. J. Am. Chem. Soc. 1989, 111, 2633–2639. [Google Scholar] [CrossRef]
- Takano, Y.; Tsunesada, T.; Isobe, H.; Yoshioka, Y.; Yamaguchi, K.; Saito, I. Theoretical Studies of Decomposition Reactions of Dioxetane, Dioxetanone, and Related Species. CT Induced Luminescence Mechanism Revisited. Bull. Chem. Soc. Jpn. 1999, 72, 213–225. [Google Scholar] [CrossRef]
- Isobe, H.; Takano, Y.; Okumura, M.; Kuramitsu, S.; Yamaguchi, K. Mechanistic Insights in Charge-Transfer-Induced Luminescence of 1,2-Dioxetanones with a Substituent of Low Oxidation Potential. J. Am. Chem. Soc. 2005, 127, 8667–8679. [Google Scholar] [CrossRef]
- Kleczka, M.; But, D.; Dylong, D.; Fendinger, C.; Marmann, V.; Wartke, C.; Griesbeck, A.G. Two Useful Directing Modes in Singlet Oxygen Reactivity: Electrostatic Effects in the Ene Reaction with Allylic Alcoholates and a Chemoselectivity Change with α-Alkoxy Michael Esters. ChemPhotoChem 2018, 2, 964–975. [Google Scholar] [CrossRef]
- Krick, M.; Holstein, J.J.; Wuttke, A.; Mata, R.A.; Clever, G.H. Temperature-Dependent Dynamics of Push–Pull Rotor Systems Based on Acridinylidene Cyanoacetic Esters. Eur. J. Org. Chem. 2017, 2017, 5141–5146. [Google Scholar] [CrossRef]
- Lehnert, W. Verbesserte Variante der Knoevenagel-Kondensation mit TiCl4/THF/Pyridin (I). Alkyliden-und Arylidenmalonester bei 0–25 °C. Tetrahedron Lett. 1970, 54, 4723–4724. [Google Scholar] [CrossRef]
- Lehnert, W. Knoevenagelkondensationen mit Titantetrachlorid/Base—II: Alkyliden- und Arylidenacet- bzw. -Nitroessigester bei 0–22°. Tetrahedron 1972, 28, 663–666. [Google Scholar] [CrossRef]
- Lehnert, W. Knoevenagelkondensationen mit TiCl4/Base—III: Umsetzungen von Ketonen und α-Halogenketonen mit Malonester. Tetrahedron 1973, 29, 635–638. [Google Scholar] [CrossRef]
- Lehnert, W. Knoevenagelkondensationen mit TiCl4/Base-IV: Umsetzungen von Aldehyden und Ketonen mit Phosphonoessigester und Methylendiphosphonsäureestern. Tetrahedron 1974, 30, 301–305. [Google Scholar] [CrossRef]
- Würth, C.; Grabolle, M.; Pauli, J.; Spieles, M.; Resch-Genger, U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 2013, 8, 1535–1550. [Google Scholar] [CrossRef]
- Yuasa, J.; Yamada, S.; Fukuzumi, S. Detection of a Radical Cation of an NADH Analogue in Two-Electron Reduction of a Protonated p-Quinone Derivative by an NADH Analogue. Angew. Chem. Int. Ed. 2008, 47, 1068–1071. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, Y.-M.; Kotani, H.; Nam, W.; Fukuzumi, S. High-valent manganese(v)–oxo porphyrin complexes in hydride transfer reactions. Chem. Commun. 2009, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Yao, Y.; Xue, M. A novel fluorescent probe for detecting paraquat and cyanide in water based on pillar[5]arene/10-methylacridinium iodide molecular recognition. Chem. Commun. 2014, 50, 5064–5067. [Google Scholar] [CrossRef]
- Albrecht, S.; Brandl, H.; Ciscato, L.F.; Weiss, D.; Augustin, D.; Zimmermann, T. The cold light of the olefins. CHIUZ 2011, 45, 24–30. [Google Scholar]
- Schiessl, K.; Roller, A.; Hammerschmidt, F. Determination of absolute configuration of the phosphonic acid moiety of fosfazinomycins. Org. Biomol. Chem. 2013, 11, 7420–7426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brachais, C.H.; Huguet, J.; Bunel, C. Synthesis, characterization and stabilization of poly(methyl glyoxylate). Polymer 1997, 38, 4959–4964. [Google Scholar] [CrossRef]
- Beutner, S.; Bloedorn, B.; Hoffmann, T.; Martin, H.D. Synthetic singlet oxygen quenchers. Methods Enzym. 2000, 319, 226–241. [Google Scholar]
- Data from Crystal Structure Analyses Are Deposited at the Cambridge Crystallographic Data Centre (CCDC) with Deposition Numbers CCDC 2080452 (Compound 14) and CCDC 2080453 (Compound 15). Available online: https://www.ccdc.cam.ac.uk/ (accessed on 31 May 2021).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | TFA | λabsmax (nm) | Absorption Coefficient ελ (L/mol·cm) | λex (nm) | λemmax (nm) | Quantum Yield ΦF |
|---|---|---|---|---|---|---|
| 7 | − | 378 | 9370 | 360 | 398 | 0.008 |
| + | 366 | 11,580 | 366 | 511 | 0.310 | |
| 8 | − | 417 | 12,540 | 360 | 410/432 | 0.035 |
| + | 362 | 22,890 | 362 | 476/495 | 0.886 | |
| 10 | − | 329 | 13,520 | 329 | − | − |
| + | 329 | 12,040 | 329 | − | − |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lippold, T.; Neudörfl, J.M.; Griesbeck, A. New Acridone- and (Thio)Xanthone-Derived 1,1-Donor–Acceptor-Substituted Alkenes: pH-Dependent Fluorescence and Unusual Photooxygenation Properties. Molecules 2021, 26, 3305. https://doi.org/10.3390/molecules26113305
Lippold T, Neudörfl JM, Griesbeck A. New Acridone- and (Thio)Xanthone-Derived 1,1-Donor–Acceptor-Substituted Alkenes: pH-Dependent Fluorescence and Unusual Photooxygenation Properties. Molecules. 2021; 26(11):3305. https://doi.org/10.3390/molecules26113305
Chicago/Turabian StyleLippold, Tim, Jörg M. Neudörfl, and Axel Griesbeck. 2021. "New Acridone- and (Thio)Xanthone-Derived 1,1-Donor–Acceptor-Substituted Alkenes: pH-Dependent Fluorescence and Unusual Photooxygenation Properties" Molecules 26, no. 11: 3305. https://doi.org/10.3390/molecules26113305
APA StyleLippold, T., Neudörfl, J. M., & Griesbeck, A. (2021). New Acridone- and (Thio)Xanthone-Derived 1,1-Donor–Acceptor-Substituted Alkenes: pH-Dependent Fluorescence and Unusual Photooxygenation Properties. Molecules, 26(11), 3305. https://doi.org/10.3390/molecules26113305