Abstract
Tiagabine is an antiepileptic drug used for the treatment of partial seizures in humans. Recently, this drug has been found useful in several non-epileptic conditions, including anxiety, chronic pain and sleep disorders. Since tachycardia—an impairment of cardiac rhythm due to cardiac ion channel dysfunction—is one of the most commonly reported non-neurological adverse effects of this drug, in the present paper we have undertaken pharmacological and numerical studies to assess a potential cardiovascular risk associated with the use of tiagabine. A chemical interaction of tiagabine with a model of human voltage-gated ion channels (VGICs) is described using the molecular docking method. The obtained in silico results imply that the adverse effects reported so far in the clinical cardiological of tiagabine could not be directly attributed to its interactions with VGICs. This is also confirmed by the results from the isolated organ studies (i.e., calcium entry blocking properties test) and in vivo (electrocardiogram study) assays of the present research. It was found that tachycardia and other tiagabine-induced cardiac complications are not due to a direct effect of this drug on ventricular depolarization and repolarization.
1. Introduction
Epidemiological studies have consistently shown that people with epilepsy have a higher prevalence of structural cardiac disease than those without it [1]. The functioning of neurons, muscles and cardiac myocytes is based on action potentials (APs) generated by transmutational ion currents mediated mainly by sodium, calcium and potassium [2,3]. According to the guidelines of the Comprehensive in vitro Proarrhythmia Assay (CiPA), a set of six ion channels has been selected for which currents are important for both the repolarization and depolarization of the cardiac action potential (AP) [4]. There is some evidence, based on the effect of clinical drugs on cardiac APs, indicating the classification of cardiac ion channels into two classes [5,6,7]. The first class contains the most important cardiac ion channels, such as KV11.1, NaV1.5 and CaV1.2. The second class comprises KV4.3, KvLQT1/mink and Kir2.1 and is less critical for the assessment of all drugs under CiPA [7,8,9].
A common feature of both neurological disorders (e.g., epilepsy and chronic pain) and cardiac dysrhythmias is cell (neuronal cell and cardiac myocyte, respectively) hyperexcitability [2]. Therefore, drugs affecting cell excitability threshold within the nervous tissue can also interact with cardiac cell APs and vice versa [10,11,12]. It has to be emphasized that this drug-induced effect might sometimes be harmful as it may lead to the occurrence of additional alterations in neuronal or cardiac cell reactivity, thus being a cause for additional drug-induced (iatrogenic) complications. From the safety pharmacology point of view, it is of key importance to recognize these potential risk factors as early as possible. The cardiac VGICs assay is an indispensable step and a high-quality assay must accompany any investigational new drug application. The in silico studies of drug binding to KV11.1, NaV1.5 and CaV1.2 may be valuable assays for drugs and drug candidates at present [13].
Currently, several antiepileptic drugs were reported to have cardiotoxic and metabolic adverse effects [1,14]. Tiagabine (TGB) is an anticonvulsant drug used to treat partial seizures in humans. Recent results of clinical trials and animal studies indicate that it might be also effective in patients suffering from pain, insomnia or mood disorders and these activities are attributed to its inhibitory effect on GABA uptake [15,16]. TGB is a 96% protein-bound molecule [17]. The drug is a potent inhibitor of [3H]GABA uptake into synaptosomal membranes (IC50 = 67 nM) or neurons (IC50 = 446 nM) and glial cells (IC50 = 182 nM) in primary cell cultures. The in vivo tests have shown that TGB neuronal inhibiting is 2.5-fold more potent than glial GABA uptake [18,19,20]. The enhancement of GABA neurotransmission due to GABA transporter subtype 1 (GAT-1) inhibition might also explain most of adverse effects of TGB, including drowsiness, confusion and dizziness [18,21]. However, some other TGB-induced complications do not to be directly related to its influence on GABA concentration in the brain and other tissues.
Several recent reports have demonstrated that tachycardia is observed in about 1.0% of patients treated with TGB [18,22] but the mechanisms underlying this cardiotoxic effect are not known. In the literature there is a limited amount of data regarding the influence of TGB on cardiovascular functions and metabolism [23].
Since tachycardia is most frequently caused by impaired ion channel functions [24,25,26,27], in the in silico part of the present study a detailed analysis of the interactions between the human KV11.1, NaV1.5, CaV1.2 and TGB was performed. In order to compare the strength of TGB’s binding to individual ion channels terfenadine (TEF) [28], batrachotoxin (BTX) [29,30] and nifedipine (NFD) [31] were selected as compounds strongly affecting these molecular targets.
In the course of present study, in vivo tests in rats were performed. The in vivo assay comprised the assessment of TGB’s proarrhythmic potential and its effects on ECG components were studied; i.e., we conducted in vivo evaluation of TGB to assess its influence on PQ, QRS, QT and QTc intervals. Its effect on heart rate was also investigated. The relevant changes in PQ, QRS, QT and QTc intervals were interpreted as the effect of the test drug predominantly on Nav, Kv and Cav channels. In contrast, the changes in heart rate were treated as a measure of the effect of the test compound, particularly, on the Nav1.5 channel or autonomic system function. Calcium blocking properties of TGB were tested in the isolated rat aorta contracted with depolarizing KCl solution.
2. Results
2.1. Pharmacological Part
2.1.1. The Effect on Normal Electrocardiogram
In vivo ECG study showed that TGB marked no significant effect on PQ, QRS, QT and QTc intervals. TGB also did not influence the heart rate significantly. It decreased the heart rhythm maximally at 30 min (by 6.9%) but this result did not reach statistical significance (Table 1). What is most important to note is that TGB at a dose as high as 100 mg/kg i.p did not prolong QT interval, which suggests that it did not prolong cardiac repolarization and probably did not block IKR currents. Similar observations have also been made in the published studies [32,33]. We also did not observe the prolongation of PQ interval and QRS widening, which reflects the slowed conduction and disturbances in ventricular depolarization, usually due to the INa block. This is in line with the results obtained and presented here from molecular docking studies, where we found that TGB did not bind to cardiac voltage-gated ion channels Kv11.1 (hERG) and Nav1.5.

Table 1.
Effects of TGB (100 mg/kg i.p.) on the heart rate and ECG intervals in anesthetized rat (thiopental 75 mg/kg i.p.).
2.1.2. Voltage-Dependent Calcium Channels
During the course of our study, we investigated the calcium entry blocking properties of TGB in vasculature by employing the isolated rat aorta contracted with depolarizing KCl solution. In this experiment, the KCl-induced contraction was caused by an increase in extracellular potassium that leaded to membrane depolarization, which increases calcium influx from extracellular sources involving voltage-dependent calcium channels [34] (Cav1.2). The reference compounds we used were verapamil [35] and NFD [36]. TGB was not able to relax KCl-precontracted aortic rings at the range of concentration 1–30 μM (Figure 1). At a higher concentration, TGB was not tested as it precipitated in Krebs–Henseleit solution. NFD, verapamil and voltage-dependent calcium channel blockers relaxed KCl (60 mmol/L)-precontracted aortic rings in a dose-dependent manner (Figure 1) by 95–97%, with the IC50 values of 4.7 ± 0.2 nM and 32.9 ± 7.4 nM, respectively [35,36]. On the basis of these results, we may state that TGB does not possess voltage-dependent calcium channel blocking properties at the tested range of concentrations.
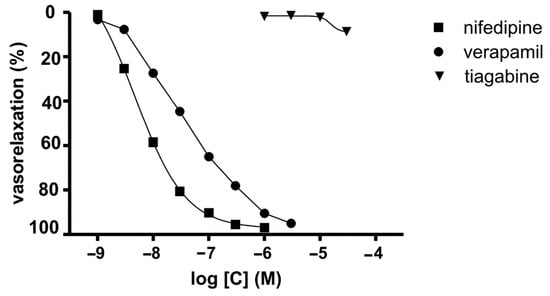
Figure 1.
Inhibitory potencies of TGB and reference drugs (NFD and verapamil) on sustained contraction of aortic rings induced by KCl (60 mM).
2.2. Molecular Docking Studies
2.2.1. Terfenadine
TEF is (RS)-1-(4-tert-butylphenyl)-4-{4-[hydroxy(diphenyl)methyl]piperidin-1-yl}-butan-1-ol and, from a chemical point of view, belongs to piperidine derivatives (Figure 2).
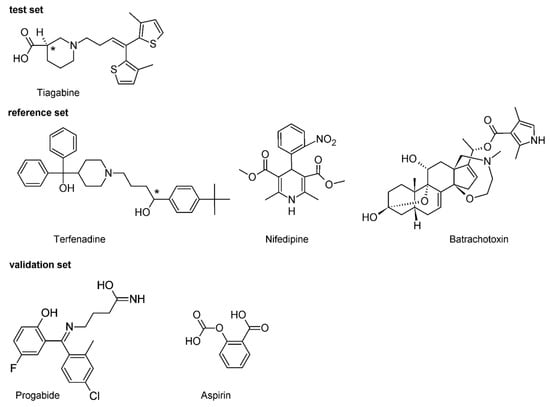
Figure 2.
Chemical structure of the investigated compounds. Carbons at a tetrahedral stereogenic center are distinguished by *.
The obtained results from molecular docking confirmed the strongest blocking effects of TEF on the hKv11.1 channel. R-TEF-hKv11.1 is the most stable complex in the studied set. Comparison of the data (such as EB and pKi, Table 2) obtained for R-TEF and S-TEF complexes leads to the conclusion that there are significant stereoselective differences in the potential interaction for all studied channels. Docking experimentation predicted more effective potential interactions of all studied channels and S-TEF than its counterpart R-TEF (Table 2). Nevertheless, all calculated h-bonds formed are weak interactions. Moreover, obtained data suggests that R/S-TEF displays non inhibitory effects for hCav1.2. According to data in Table 2, pKi < 3.5, while by convention pKi ≤ 4 indicates the lack of a biological effect. Based on the CiPA studies, including the examination of the effect of 30 clinical drugs on the 7-ion channel [7], it can be concluded that the risk level of torsade de pointes (TdP) is correlated with the blocking effects of hKv11.1, hNav1.5 and hCav1.2. Crumb et al. [7,37] in their research proposed a classification of drugs into three categories of TdP entry risk (high, medium and low). According to their studies, drugs belonging to the high and medium risk stand out with block hERG to a much greater extent than any other tested currents. The drugs belonging to the low risk category is distinct with the non-specific blocking of hKv11.1, hNav1.5 and hCav1.2 channels (Figure S1). These results clearly indicate the need for testing drug candidates on the ion channel panel. In our study it was found, on the molecular level, that the high risk of TdP is a result of TEF’s strong blocking effects on hERG. R-TEF-hKv11.1 complex has one normal h-bond in which the hydroxyl group of the (4-tert-butylphenyl)methanol moieties of R-TEF donates energetically weak (−3.62 kcal/mol), short (2.12 Å) and almost linear interactions (158°) to Tyr652 (Figure S2). Regarding the interaction of R-TEF and S-TEF with the hNav1.5 channel, it should be noted that both enantiomers of TEF practically strongly interact with this protein (pKi > 5, Table 2), which also indicates their arrhythmogenic effects through the hNav1.5. This observation is also confirmed by previously presented pharmacological studies [38].
Table 2.
The summary of hNav1.5, hCav1.2 and hKv11.1 (hERG) channel docking experiment results.
2.2.2. Nifedipine
NFD is a 3,5-dimethyl 2.6-dimethyl-4-(2-nitrophenyl)-1.4-dihydropyridine-3,5-dicarboxylate (Figure 2). It is classified as a dihydropyridine subclass compound. It is a highly apolar photosensitive compound. The NFD docking experiment revealed that this molecule can interact with an active site of all the studied proteins (Table 2). It can be set to the following descending order of binding energies EB(NFD-hKv11.1) ≈ −4.42, EB(NFD-hNav1.5) ≈ −7.00 and EB(NFD-hCav1.2) ≈ −10.71 kcal/mol, respectively. The binding energies obtained in the docking experiment show that NFD forms a more stable complex in the case of the hCav1.2 channel. The in silico data obtained for blocking of hCav1.2 channel (pKi = 7.85) are in line with data in the literature (pIC50 = 7.48) [35] and (pKi = 7.66) [39]. Based on the predicted binding affinity, the highest affinity was observed with the hCav1.2 and NFD in all cases of studied channels. Additionally, the blocking effect of NFD with hNav1.5 was also proven (Table 2). It has to be highlighted that in NFD-hNav1.5 and NFD-hKv11.1 complexes, NFD interacts via one normal h-bond. In contrast, the complex NFD-hCav1.2 shows two h-bonds, both of which are short (≈1.68 Å) and strong (≈−3.8 kcal/mol) (Table 2.). The assessment of the data obtained for the hydrogen bonds clearly leads to the conclusion that they all have incomparable energies and bond lengths. Interestingly, NFD forms the strongest h-bond with hKv11.1 and the weakest one with hNav1.5 channel (Table 2. Figure S2). The h-bond with hCav1.2 is characterized by an indirect force of influence. However, taking into account the data from Table 2, there is no correlation between the binding affinities of NFD and h-bond energy. The obtained distribution of estimated pKi measure shows that in case of the hKv11.1 channel, NFD has no inhibitory effect for this channel (pKi < 4. Table 2). These data prove that NFD has potency only in inhibiting the hCav1.2 and hNav1.5 channel, which is in agreement with the pharmacological data previously presented in pharmacological literature [40].
2.2.3. Batrachotoxin
BTX as a steroidal alkaloid belongs to class A channel opening toxins [41]. Its molecule contains an oxazepane ring with tertiary amine and an aromatic pyrrole ring connected to the rigid polycyclic steroidal core via the ester group (Figure 2). Its 3D structure adopts a horseshoe conformation [42,43]. The outer surface of the horseshoe is hydrophobic, while the inner one is rather hydrophilic and forms the oxygen triad (at C3, C9 and C11) [44,45]. The outer surface of the horseshoe is hydrophobic, while the inner one is rather hydrophilic and forms oxygen triad (at C3, C9 and C11) [44]. It was suggested in the literature that this oxygen triad forms a hydrophilic arc, which can be regarded as a chelating site attracting some cations [46]. In the preliminary analysis of the docking, it was observed that BTX interacts with the active site of all studied proteins (Figure S2). The achieved data demonstrated that Nav1.5 forms one h-bond, while hCav1.2 and hKv11.1 form two h-bonds. The energies obtained in the present in silico experiment show that hCav1.2 and hKv11.1 form more and form stronger hydrogen bonds than the remaining one (hNav1.5). The predicted binding affinity can be arranged in the following increasing order: hKv11.1 < hCav1.2 < hNav1.5 (Table 2). Obtained results revealed that BTX-hNav1.5 is an energetically more stable complex with a binding energy of −9.01 kcal/mol. The stability of the complex with the hCav1.2 and hKv11.1 is slightly lower than that with hNav1.5 (i.e., EB BTX-hCav1.2 = −7.17 and EB BTX-hKv11.1 = −6.75 kcal/mol, respectively). The calculated BTX affinity value of pKi = 6.68 is consistent with the relevant inhibitory potential data known from the specialized literature (pIC50 = 6.71) [47]. The BTX-hNav1.5 complex is formed via a single key interaction with residues of hNav1.5. The hydroxyl group at position C-11 of the steroid skeleton donates one h-bond to the sulfur atom of Ser1458. This interaction is non-linear (157°) and with weak energy, i.e., −3.43 kcal/mol and a small bond length equal to 2.18 Å. This is in line with the literature data according to which the atom included in oxygen triad is responsible for the toxic interaction of BTX with its molecular target [44]. We can treat these data as a strong argument proving that BTX acts on the cytoplasmic side of the channel just as other Class A neurotoxic compounds do, as it is shown in Figure S2.
For hCav1.2 and hKv11.1 channels, BTX has pKi ≈ 5.2, which suggests comparable blocking effects. The BTX-hCav1.2 complex is a more energetically stable form than the BTX-hNav1.5 complex (Table 2). This complex has one normal h-bond. In the bifurcated h-bond, the major component is Ser1132 and the minor component is Gln1060; the associated energies are weak (EHB = −0.37 kcal/mol and −3.23 kcal/mol, respectively). The distances of the hydrogen bonds lay in the range from 1.69 to 2.22 Å and, due to this, the three-centered hydrogen bond is highly not symmetric. In the case of BTX-hCav1.2 complex, it is formed via one normal h-bond interaction (Table 2).
2.2.4. Tiagabine
As it can be seen from the data collected in Table 2, TGB complexes between hNav1.5, hCav1.2 and hKv11.1 have a calculated pKi ≤ 4, which is commonly used as a threshold and this drug can thus be considered as an inactive ligand for those proteins. These data refer to both R/S enantiomers of TGB (Figure 3). Docking TGB into the hNav1.5 channel showed the highest pKi value in Table 2, however, the value is still below the activity threshold level of pKi > 4. The lowest pKi was observed for hKv11.1. We can treat these data as a partial explanation of the reported adverse TGB interactions in the cardiovascular system. It also seems that a small percentage of the observed cardiac disorders can be attributed to the fact that TGB does not interact with hKv11.1, for which inhibition is responsible for QTc prolongation. In all analyzed cases, R-TGB shows slightly higher binding affinities than S-TGB. This observation is in line with many previous pharmacological studies indicating the greater biological activity of R enantiomers compared to the S ones [10,11,12]. It is also worth emphasizing that all h-bonds formed between hCav1.2 channels and R/S-TGB possess very favorable key interaction energy values (EHB ≈ −7.32 for hCav1.2-R-TGB and −5.54 kcal/mol for hCav1.2-S-TGB, Table 2) and geometrically nonlinear systems.
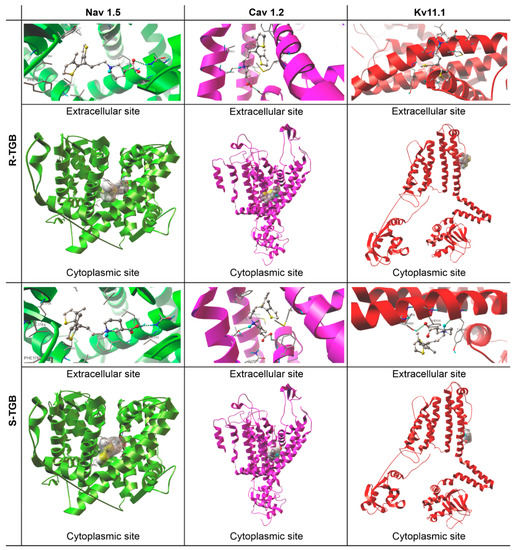
Figure 3.
Pocket locations and binding modes of (R/S)-Tiagabine (R/S-TGB) and the investigated channels: hNav1.5, hCav1.2 and hKv11.1. Ligands (ball and stick model) and calculated hydrogen bonds (dashed green lines).
To sum up, the docking experiment revealed that R/S-TGB has lower intermolecular forces with all studied ion channels (EB ≈ −5.30 kcal/mol, Table 2). The combination of the above-mentioned docking data with pharmacological as well as literature data regarding the risk for tachycardia due to TGB administration suggests that this adverse effect observed in humans is not likely to result from TGB interaction with molecular anti-targets (i.e., ion channels tested in this study) used for the cardiac risk assessment. The data obtained in this study allowed the supplementation of information on the impact of TGB on other than GAT-1 molecular targets. As is currently known, TGB has no significant affinity to other uptake systems, such as those for dopamine, noradrenaline, acetylcholine, adenosine, serotonin, histamine, opiate, glycine, glutamate or GABA [48,49]. It only has a weak affinity towards benzodiazepine receptors and does not affect K+ and Ca2+, while it slightly affects Na+ and cardiovascular channel function [19].
2.3. Validation Experiment
The validation was carried out by the docking of molecules with no affinity to hNav1.5, hCav1.2 and hKv11.1 channels, such as progabide (PRG) [50,51] and acetylsalicylic acid (ASA) [52]. This choice of PRG and ASA was made based on their chemical and biological similarity to TGB (ATC code: N03AG06 [53]). PRG (ATC code: N03AG05 [53]) is a first-generation antiepileptic drug without analgesic properties [12,54]. ASA (ATC code: N02BA01 [52]) is a classical and peripherally-acting nonsteroidal anti-inflammatory drug that is inactive at GAT and recommended for the prevention of several cardiovascular diseases due to its antiplatelet activity [52].
The results of the validation experiments, such as the complex binding energies, the specific hydrogen bond components and detailed data of the hydrogen bond features (energies, lengths and angles) are gathered in Table 3. The binding modes between hNav1.5, hCav1.2 and hKv11.1; and the control compound are illustrated in Figure S3 in the supplementary file. As it can be observed from the data, all control complexes have a calculated pKi < 4, which is commonly used as the threshold and therefore the test compounds can be considered to be inactive ligands for those proteins. In addition, all control complexes have higher binding energy values and lower hydrogen bond energy than the TGB-hVGICs complex.
Table 3.
The summary of validation experiment results.
3. Discussion
Taking into consideration the new action profile of drugs already on the market, pharmacological safety is a particularly important issue. The main reason for this is the fact that for drugs with an extended therapeutic range (i.e., repurposed medications), the number of patients for whom a given drug is recommended will increase significantly. TGB discussed in this study belongs to this group of drugs. TGB is an anticonvulsant medication. It is also used in the treatment of anxiety-related disorders, as are a few other anticonvulsants [21]. In the case of this drug, the above mentioned point is particularly important because TGB was originally prescribed to a relatively small group of patients due to its narrow range of indicators (adjunctive treatment of partial seizures in adults and children 12 years of age and older) [56]. However, when we consider its analgesic and anxiolytic effects in our considerations, we include therapeutic indications for a significantly larger group of patients. Reports presented in the scientific literature indicate an approximate 70% increase in the number of patients on TGB therapy [57]. This is all the more important as the safety and efficacy of TGB have not been systematically evaluated for indications other than epilepsy.
Modern technology provides us with many different possibilities, the use of which should create the conditions to learn about safety pharmacology. Undoubtedly, in silico research is one of many possibilities for seeking answers in this regard. It seems to us that the research presented above makes some important contributions to this issue. The study tried to answer the question about the cardiovascular safety assessment of TGB. This is all the more important in the light of the recent expert discussions focused on extending the pharmacological profile of TBG. Many modern studies indicate that, in addition to the therapeutic use of TGB in epilepsy, we should strongly consider its utilization for non-epileptic indications. On the other hand, it is known that drugs that show this type of biological activity might have a strong effect on a heart. This, in turn, undoubtedly forces us to increase the effort focused on assessing cardiac safety. Considering the assessment of the effect of the compound on so-called anti-targets adopted from CiPA in the cardiac risk assessment, the TGB interaction was investigated with the following channels: hKv11.1, hNav1.5 and hCav1.2. Drugs strongly affecting individual channels (such as TEF, BTX and NFD) were selected as reference compounds for this study. TEF is a prodrug metabolized by intestinal CYP3A4 to fexofenadine, the active form being a selective histamine H1-receptor antagonist with antihistaminic and non-sedative effects. As it is well known, antihistamines may increase the rate of heart beat [58,59]. TEF causes prolonged repolarization, as is reflected in the broadening of the electrocardiographic QT interval, with the potential for serious ventricular arrhythmia and death [60,61,62]. Due to this, in the U.S. TEF was superseded by its active metabolite fexofenadine in the 1990s [59]. Numerous studies have proven that the ability of TEF to extend the QT interval depends on its binding to the Kv11.1 (Figure S1) protein encoded by hERG [63,64]. Nevertheless, TEF does not readily cross the blood–brain barrier and due to this its CNS, depression is minimal. NFD is a calcium channel blocker, a specific antagonist of Cav1.2 channels [65]. It is used to treat hypertension and chronic stable angina. NFD binds directly to inactive calcium channels and stabilizes their inactive conformation. By inhibiting the influx of calcium in smooth muscle cells, NFD prevents calcium-dependent myocyte contraction and vasoconstriction [31,40]. BTX was chosen as a reference compound in our docking experiment study due to its extremely potent cardiotoxic and neurotoxic characteristics [29,42,46,47,66]. In animals, BTX inactivates sodium channels in nerve cells and muscle cells, thereby interfering with the electrical signals sent throughout the body and causing fibrillation, arrhythmias, cardiac failure and death [29]. It is worth emphasizing that the obtained data from molecular modeling confirmed the high selectivity of the reference compounds for the appropriate ion channels (Table 2). The response obtained from molecular studies also indicates that the mechanisms underlying tachycardia in patients treated with TGB appear to be unrelated to its effect on the hNav1.5, hCav1.2 and hKv11.1 heart ion channels. Moreover, it is known that one of the most common mechanisms of drug-induced ventricular tachycardia is the blocking of hERG channels [67,68]. For this reason, additional evidence supporting the conclusions of the in silico study appears to provide epidemiological data (pharmacological reports) in which tachycardia is noted in approximately 1.0% of patients treated with TGB. In light of these facts, if TGB-induced tachycardia contacts the hKv11.1, hNav1.5 and hCav1.2. blocking mechanism, one would expect a higher rate of these side effects. In addition to the above-mentioned in vivo tests, as a part of safety pharmacology experiments, the TGB effects on PQ, QRS and QT intervals and the effects of TGB on heart rate were assessed. Many years of research have demonstrated that proarrhythmic effect, QT prolongation and hERG blocking cannot be treated as the only determinants of the occurrence of TdP. For instance, verapamil and ranolazines are examples of drugs that are strong inhibitors of the hERG channel and are simultaneously devoid of the risk of inducing arrhythmias and, vice versa, devoid of serious disorders of cardiomyocyte electrophysiology caused by drugs that are weak hERG inhibitors (e.g., sotalol and alfuzosin) [24,69]. Thus, this proves the insufficient specificity of the tests based only on the assessment of the hERG channel blocking potential. The risk of drug-induced TdP is rather balanced by multiple internal cardiac ionic currents that define ventricular repolarization. Therefore, studies that utilize the whole tissue seem to be a good option for the reflection of pharmacodynamics and potential adverse effects of a drug in a living organism. The in vivo results obtained from studies in rats showed that TGB did not prolong the QT interval or alter the QRS complex which suggests that it did not affect ventricular depolarization and repolarization. Taking into account that as in many other animal models of human diseases and also in this particular rat model, there might be basic translational problems. Some fundamental differences in the cardiac electrophysiology and myocyte calcium/potassium handling between rodents and humans have been suggested [70], but, nonetheless, the ECG in rats is still a widely applied experimental method in basic cardiovascular research. The technique of ECG recordings is simple; however, the interpretation of electrocardiographic parameters might be challenging. This is because the analysis may be biased by experimental settings, such as the type of anesthesia and the strain or age of animals. Furthermore, differences and similarities between rat and human ECG are frequently discussed in the context of translational cardiovascular research. Despite this, rat electrocardiography is an important investigational tool in experimental cardiology, even if the interpretation of electrocardiographic parameters is problematic [71]. In addition to this, a number of studies have shown that cardiotoxic drugs prolong QT interval in rodents and ECG recordings in rats have been used as a screening tool to assess the cardiotoxicity of various drugs. However, it needs to be stressed that the translation of the results of those studies to human application also possesses limitations. This is because rats’ hearts do not express hERG, whereas the cardiotoxicity of drugs is strongly associated with the blockade of hERG-related potassium channels. However, rat hearts express a variant of Ether-à-go-go-Related Gene (rat ERG, also known as Kcnh2) [72,73], which may also play a key role in the assessment of drug-induced cardiotoxicity. Taken together, we are aware that extrapolating the results from our rat model to humans should be performed extremely cautiously and this, of course, should be regarded as the main limitation of our study. Therefore, one can assume that its direct interaction with heart sodium and potassium channels can be neglected. TGB also marks no significant effect on the PQ interval, which suggests that it does not influence the atrio-ventricular conduction time. Consequently, we can postulate that TGB has low pro-arrhythmic potential, at least, after a single administration. Given that tachycardia may result due to a number of different mechanisms and not all of them directly affect ion channels, it is also necessary to evaluate the effects of TGB on several neurotransmitters/neuromodulators and the activity of the autonomic nervous system. However, these effects were not investigated in the present research. Further studies are necessary to assess the effect of TGB on the cardiovascular system, especially after chronic administration. On the basis of our research, we can state that TGB did not bind to voltage-gated ion channels and did not affect them directly. Furthermore, the observed accidents of tachycardia are probably not due to the direct effect of TGB on voltage-gated ion channels.
4. Methods
4.1. Pharmacological Studies General Information
The experiments were carried out using male Wistar rats (Krf:(WI) (WU), 200–250 g). The animals were housed in constant temperature facilities exposed to 12:12 h light/dark cycles and were maintained on a standard pellet diet with tap water given ad libitum. All procedures were conducted according to guidelines of ICLAS (International Council on Laboratory Animal Science) and approved by the Second Local Ethics Committee in Krakow, Poland (resolution No. 106/2016, 14 June 2016).
4.1.1. Voltage-Dependent Calcium Channels—Functional Assays
In order to investigate the calcium entry blocking properties of TGB, it was tested on isolated rat aorta precontracted with KCl. Rats were anaesthetized with thiopental sodium (75 mg/kg, i.p., Rotexmedica, Germany) and the thoracic aorta was dissected, cleaned, denuded of endothelium, cut and mounted as described earlier [35]. Briefly, aorta rings were incubated in 30 mL chambers filled with a Krebs–Henseleit solution (NaCl 118 mM, KCl4.7 mM, CaCl2 2.25 mM, MgSO4 1.64 mM, KH2PO4 1.18 mM, NaHCO3 24.88 mM, glucose 10 mM, C3H3O3Na 2.2 mM and EDTA 0.05 mM) at 37 °C and pH 7.4 with constant oxygenation (O2/CO2, 19:1) and connected to an isometric FDT10-A force displacement transducer (BIOPAC Systems, Inc., COMMAT Ltd., Ankara, Turkey). The aortic rings were stretched and maintained at an optimal tension of 2 g and permitted to equilibrate for 2 h. The aortic rings were contracted to submaximal tension with KCl (60 mmol/L). Once the plateau was attained, concentration-relaxation curves were obtained by the addition of cumulative doses of tiagabine to the precontracted preparations.
Concentration-response curves were analyzed using GraphPad Prism 5.0 software (GraphPad Software Inc., San Diego, CA, USA). Relaxations are expressed as a percentage of inhibition of the maximal tension obtained with the contractile agent (Emax = 100%). Data are the means ± SEM of at least 4 separate experiments.
4.1.2. The Effect on Normal Electrocardiogram
In vivo electrocardiographic investigations were carried out using an ASPEL ASCARD B5 apparatus (Aspel, Poland), standard lead II and paper speed of 50 mm/s. The ECG was recorded just prior to and also at 1, 5, 10, 20, 30, 40, 50 and 60 min following the i.p. administration of TGB at a dose of 100 mg/kg. The QTc was calculated according to the formula of Bazzett: QTc = QT/√RR [74]
4.2. In Silico Studies
The calculation procedures applied in the study are typical for the processing of docking studies.
4.2.1. Ligand Preparation
For the 3D molecular structure calculations, the Gaussian 09 (version D.01. for Unix/Linux) package was used [75]. The initial acceptable 3D structures of 6 studied compounds (Figure 2) were downloaded (as mol2 file) from ZINC [76]. Later, the GaussView [75,77] was applied for preparation of Gaussian input files. All the molecules were geometry-optimized in water as described by the PCM (polarizable continuum model). DFT/B3LYP level of theory 6311 + G(d, p) basis set was used. After geometrical optimization, (the root-mean-square gradient value smaller than 10−6 a.u.) compounds were saved as mol2 files using the GaussView. Subsequently, torsionals and the number of active torsions for ligands were defined and the Gasteiger charges were assigned to each compound via AutoDockTools (ADT) [78]. Finally, ligands prepared for docking were saved as pdbqt files.
4.2.2. Voltage-Gated Ion Channels Preparation
- hNav1.5 preparation:
The lack of crystal structure of hNaV1.5 pore domain causes the need for preparing homology 3D models for this protein. For our research, the sequence for the hNav1.5 protein was gained from the Swiss Model Repository (SMR). SMR is a database which currently holds over 400,000 high quality 3D protein structure models generated by the automated SWISS-MODEL homology modeling pipeline [79]. The pdb file was downloaded from SWISS-MODEL SERVER (accession number Q14524) [80]. For this alignment, X-ray structure of human Nav1.2-β2-KIIIA ternary complex (PDB entry 6J8E) was employed. Sequence identity between template and the monomer of sodium channel protein type 5 subunit α is 66.70%. Subsequently, the pdb file was opened in ADT [78]. ADT read coordinates, added charges, merged non-polar hydrogens and assigned appropriate atom types. Before formatting a molecule for AutoDock, we removed 9Z9 ((3β,14β,17β,25R)-3-[4-methoxy-3-(methoxymethyl)-butoxyl]-spirost-5-en), which is irrelevant molecule in this experiment. Finally, the prepared protein was saved as a pdbqt file. In the computational part of the study, we pondered the interaction between the investigated ligands and the intracellular pore gate formed from the proper residues of chain A.
- hCav1.2 preparation
The dearth of a 3D structure of hCav1.2 proper region also causes need for preparing homology models of this protein. The sequence for the hCav1.2 was gained from the SMR as well (accession number Q13936). For this alignment, X-ray structures of nifedipine complex with rabbit Cav1.1 (PDB entry 6JP5) were employed. The sequence identity between template and isoform 4 of CAC1C_HUMAN Voltage-dependent L-type calcium channel subunit alpha-1C is 70.31% and, according to the best of our knowledge, this is one of the highest identities currently available. Subsequently, similar to the hNav1.5 protein, the pdb file was opened in ADT [58]. The next steps were also analogous. Before the docking experiment, we removed C8U (methyl (4~{S})-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-1,4-dihydropyridine-3-carboxylate)), which is a pointless ligand in this case. The pore forming and dihydropiridyne binding residues (from ARG1109 to LYS1198) were considered as the ligand binding site [81].
- hKv11.1 preparation:
The sequence for the potassium voltage-gated channel subfamily H member 2 protein was downloaded from the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB entry 5va2) as the crystal structure [82]. As in previous proteins, the pdb file was opened in ADT, read coordinates, added charges, merged non-polar hydrogens and assigned the appropriate atom types. As usual, we also removed crystallographic waters from 5va2. The binding pocket of the studied molecule were composed of the pore forming segment H5 and transmembrane helical fragment-Segment S6 [83].
4.2.3. Molecular Docking
Molecular docking was performed using the AutoDockTools 4.2 suite of the program [55]. A grid box with a dimension of 60 × 60 × 60 Å3 and grid spacing of 0.375 Å, which is large enough for a free rotation of a ligands, was built in the middle of the binding pockets of the studied Voltage Gated Ion Channels (VGICs) channels, which are composed using the appropriate residues (Table 4).
Table 4.
The composition of the binding pocket of the analyzed channel models: hNav1.5, hCav1.2 and hKv11.1.
Torsionals in the residuals of the binding pocket were not rotatable. The rigid docking was carried out using the Lamarckian genetic algorithm 4. The optimized docking parameters were set as default values, with the exception of the number of genetic algorithms run which was 100. Torsionals in the ligands were rotatable-6 active torsions in each ligand (except for terfenadine, where it was 11). A cluster analysis was performed using RMS tolerance of 2 Å. In each case, the best docking result was considered as the complex with the lowest binding energy. Interactions between ligands and the related channel models were analyzed using the AutoDockTools program (ADT. Version 1.5.4) [78].
Supplementary Materials
The following are available online, Figure S1: Model transport proteins selected for the study: hNav1.5, hCav1.2, Kv11.1. Figure S2: Figure S3: Binding modes between hNav1.5, hCav1.2 and Kv11.1 and the four tested compounds () and two validated compounds
Author Contributions
A.N. and M.K. (Magdalena Kowalska) conceived and directed the project. K.S., Ł.F., M.K. (Magdalena Kowalska) and A.N. designed the study. K.S., Ł.F., M.K. (Magdalena Kowalska), M.K collected the data and carried out the experiments. K.S., Ł.F., M.K. (Magdalena Kowalska), M.K. and A.N. analyzed the data. A.N., M.K. (Magdalena Kowalska), Ł.F., M.K., G.G., J.N. and K.S. interpreted the results and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by a research grant from the National Science Centre UMO-2015/17/B/NZ7/02937.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board the Second Local Ethics Committee in Krakow, Poland (resolution No. 106/2016, 14 June 2016).
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
None of the authors declare any conflicts of interest with respect to this study.
Sample Availability
Samples of the compounds are not available from the authors.
Abbreviations
| AP | Cardiac action potential |
| (R/S)-TGB | (R/S)-Tiagabine |
| (R/S)-TEF | ((R/S)-Terfenadine |
| NFD | Nifedipine |
| BTX | Batrachotoxin |
| # | Hydrogen bond components: from the protein |
| % | Hydrogen bond components: from the ligand |
| Acc | Hydrogen bond acceptor |
| EB | Complex energy binding |
| EHB | Hydrogen bond energy |
| θ | Hydrogen bond angle |
| VGICs | Voltage-Gated Ion Channels |
| VGKCs | Voltage-Gated Potassium. Channels |
| VGNaCs | Voltage-Gated Sodium Channels |
| VGCaCs | Voltage-Gated Calcium Channels |
| KV11.1 (hERG) | Cardiac Voltage-Gated Potassium Channels |
| NaV1.5 | Cardiac Voltage-Gated Sodium Channels |
| CaV1.2 | Cardiac Voltage-Gated Calcium Channels |
References
- Shmuely, S.; Van der Lende, M.; Lamberts, R.J.; Sander, J.W.; Thijs, R.D. The heart of epilepsy: Current views and future concepts. Seizure 2017, 44, 176–183. [Google Scholar] [CrossRef]
- Shah, R.R. Cardiac Effects of Antiepileptic Drugs. In Atlas of Epilepsies; Springer: London, UK, 2010; pp. 1479–1486. [Google Scholar]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2015. [Google Scholar]
- Fermini, B.; Hancox, J.C.; Abi-Gerges, N.; Bridgland-Taylor, M.; Chaudhary, K.W.; Colatsky, T.; Correll, K.; Crumb, W.; Damiano, B.; Erdemli, G. A new perspective in the field of cardiac safety testing through the comprehensive in vitro proarrhythmia assay paradigm. J. Biomol. Screen. 2016, 21, 1–11. [Google Scholar] [CrossRef]
- Cheung, S.; Parkinson, J.; Wählby-Hamrén, U.; Dota, C.; Kragh, Å.; Bergenholm, L.; Vik, T.; Collins, T.; Arfvidsson, C.; Pollard, C. A tutorial on model informed approaches to cardiovascular safety with focus on cardiac repolarisation. J. Pharmacokinet. Pharmacodyn. 2018, 45, 365–381. [Google Scholar] [CrossRef]
- Obejero-Paz, C.A.; Bruening-Wright, A.; Kramer, J.; Hawryluk, P.; Tatalovic, M.; Dittrich, H.C.; Brown, A.M. Quantitative profiling of the effects of vanoxerine on human cardiac ion channels and its application to cardiac risk. Sci. Rep. 2015, 5, 1–15. [Google Scholar] [CrossRef]
- Crumb, W.J., Jr.; Vicente, J.; Johannesen, L.; Strauss, D.G. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods 2016, 81, 251–262. [Google Scholar] [CrossRef]
- Li, Z.; Dutta, S.; Sheng, J.; Tran, P.N.; Wu, W.; Chang, K.; Mdluli, T.; Strauss, D.G.; Colatsky, T. Improving the in silico assessment of proarrhythmia risk by combining hERG (human ether-à-go-go-related gene) channel–drug binding kinetics and multichannel pharmacology. Circ. Arrhythmia Electrophysiol. 2017, 10, e004628. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.; Obejero-Paz, C.A.; Myatt, G.; Kuryshev, Y.A.; Bruening-Wright, A.; Verducci, J.S.; Brown, A.M. MICE models: Superior to the HERG model in predicting Torsade de Pointes. Sci. Rep. 2013, 3, 2100. [Google Scholar] [CrossRef]
- Nowaczyk, A.; Fijałkowski, Ł.; Zaręba, P.; Sałat, K. Selective neuronal and astrocytic inhibition of human GABA transporter isoform 1 (hGAT1) inhibitors in the mechanism of epilepsy and pain-molecular docking and pharmacodynamics studies, part I. JMGM 2018, 85, 171–181. [Google Scholar]
- Nowaczyk, A.; Fijałkowski, Ł.; Kowalska, M.; Podkowa, A.; Sałat, K. Studies on the activity of selected highly lipophilic compounds toward hGAT1 inhibition: Part II. ACS Chem. Neurosci. 2019, 10, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Fijałkowski, Ł.; Sałat, K.; Podkowa, A.; Zaręba, P.; Nowaczyk, A. Potential role of selected antiepileptics used in neuropathic pain as human GABA transporter isoform 1 (GAT1) inhibitors—Molecular docking and pharmacodynamic studies. Eur. J. Pharm. Sci. 2017, 96, 362–372. [Google Scholar] [CrossRef]
- Kirsch, G.E.; Kramer, J.; Bruening-Wright, A.; Obejero-Paz, C.; Brown, A.M. The Comprehensive In Vitro Proarrhythmia Assay (CiPA) Guide: A New Approach to Cardiac Risk Assessment; Charles River Laboratories International: Wilmington, MA, USA, 2016. [Google Scholar]
- Raj, S.R.; Stein, C.M.; Saavedra, P.J.; Roden, D.M. Cardiovascular effects of noncardiovascular drugs. Circulation 2009, 120, 1123–1132. [Google Scholar] [CrossRef]
- Schwartz, T.L.; Nihalani, N. Tiagabine in anxiety disorders. Expert Opin. Pharmacother. 2006, 7, 1977–1987. [Google Scholar] [CrossRef]
- Sałat, K.; Podkowa, A.; Kowalczyk, P.; Kulig, K.; Dziubina, A.; Filipek, B.; Librowski, T. Anticonvulsant active inhibitor of GABA transporter subtype 1, tiagabine, with activity in mouse models of anxiety, pain and depression. Pharmacol. Rep. 2015, 67, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Brodie, M.J. Tiagabine pharmacology in profile. Epilepsia 1995, 36, S7–S9. [Google Scholar] [CrossRef] [PubMed]
- Spiller, H.; Wiles, D.; Russell, J.; Casavant, M. Review of toxicity and trends in the use of tiagabine as reported to US poison centers from 2000 to 2012. Hum. Exp. Toxicol. 2016, 35, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Schachter, S.C. Tiagabine. In Antiepileptic Drugs Pharmacology and Therapeutics; Springer: New York, NY, USA, 1999; pp. 447–463. [Google Scholar]
- Braestrup, C.; Nielsen, E.B.; Sonnewald, U.; Knutsen, L.J.; Andersen, K.E.; Jansen, J.A.; Frederiksen, K.; Andersen, P.H.; Mortensen, A.; Suzdak, P.D. (R)-N-[4, 4-bis (3-methyl-2-thienyl) but-3-en-1-yl] nipecotic acid binds with high affinity to the brain γ-aminobutyric acid uptake carrier. J. Neurochem. 1990, 54, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Khouzam, H.R. A Review of Anticonvulsants use in Psychiatric Conditions. EC Neurol. 2019, 11, 579–591. [Google Scholar]
- Spiller, H.A.; Winter, M.L.; Ryan, M.; Krenzelok, E.P.; Anderson, D.L.; Thompson, M.; Kumar, S. Retrospective evaluation of tiagabine overdose. Clin. Toxicol. 2005, 43, 855–859. [Google Scholar] [CrossRef]
- Jankovic, S.M.; Dostic, M. Choice of antiepileptic drugs for the elderly: Possible drug interactions and adverse effects. Expert Opin. Drug Metab. Toxicol. 2012, 8, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, M.; Nowaczyk, J.; Nowaczyk, A. KV11.1, NaV1.5 and CaV1.2 transporter proteins as antitarget for drug cardiotoxicity. Int. J. Mol. Sci. 2020, 21, 8099. [Google Scholar] [CrossRef]
- Passman, R.; Kadish, A. Polymorphic ventricular tachycardia, long QT syndrome and torsades de pointes. Med. Clin. N. Am. 2001, 85, 321–341. [Google Scholar] [CrossRef]
- Rosso, R.; Hochstadt, A.; Viskin, D.; Chorin, E.; Schwartz, A.L.; Tovia-Brodie, O.; Laish-Farkash, A.; Havakuk, O.; Gepstein, L.; Banai, S. Polymorphic ventricular tachycardia, ischaemic ventricular fibrillation, and torsade de pointes: Importance of the QT and the coupling interval in the differential diagnosis. Eur. Heart J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Rahm, A.-K.; Lugenbiel, P.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Role of ion channels in heart failure and channelopathies. Biophys. Rev. 2018, 10, 1097–1106. [Google Scholar] [CrossRef]
- Triggle, D.; Janis, R. Calcium channel ligands. Annu. Rev. Pharmacol. Toxicol. 1987, 27, 347–369. [Google Scholar] [CrossRef]
- Albuquerque, E.; Daly, J.; Witkop, B. Batrachotoxin: Chemistry and pharmacology. Science 1971, 172, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, J.; Olivera, B.M.; Bosmans, F. Animal toxins influence voltage-gated sodium channel function. Volt. Gated Sodium Channels 2014, 221, 203–229. [Google Scholar]
- Sorkin, E.; Clissold, S.; Brogden, R. Nifedipine. Drugs 1985, 30, 182–274. [Google Scholar] [CrossRef]
- Pensel, M.C.; Nass, R.D.; Taubøll, E.; Aurlien, D.; Surges, R. Prevention of sudden unexpected death in epilepsy: Current status and future perspectives. Expert Rev. Neurother. 2020, 20, 497–508. [Google Scholar] [CrossRef]
- Goodnick, P.J.; Parra, F.; Jerry, J. Psychotropic drugs and the ECG: Focus on the QTc interval. Expert Opin. Pharmacother. 2002, 3, 479–498. [Google Scholar] [CrossRef]
- Godfraind, T.; Miller, R.; Wibo, M. Calcium antagonism and calcium entry blockade. Pharmacol. Rev. 1986, 38, 321–416. [Google Scholar]
- Kubacka, M.; Szkaradek, N.; Mogilski, S.; Pańczyk, K.; Siwek, A.; Gryboś, A.; Filipek, B.; Żmudzki, P.; Marona, H.; Waszkielewicz, A.M. Design, synthesis and cardiovascular evaluation of some aminoisopropanoloxy derivatives of xanthone. Bioorganic Med. Chem. 2018, 26, 3773–3784. [Google Scholar] [CrossRef]
- Kubacka, M.; Mogilski, S.; Filipek, B.; Marona, H. The hypotensive activity and alpha1-adrenoceptor antagonistic properties of some aroxyalkyl derivatives of 2-methoxyphenylpiperazine. Eur. J. Pharmacol. 2013, 698, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Strauss, D.G.; Gintant, G.; Li, Z.; Wu, W.; Blinova, K.; Vicente, J.; Turner, J.R.; Sager, P.T. Comprehensive in vitro proarrhythmia assay (CiPA) update from a cardiac safety research consortium/health and environmental sciences institute/FDA Meeting. Ther. Innov. Regul. Sci. 2019, 53, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Orvos, P.; Kohajda, Z.; Szlovák, J.; Gazdag, P.; Árpádffy-Lovas, T.; Tóth, D.; Geramipour, A.; Tálosi, L.; Jost, N.; Varró, A. Evaluation of Possible Proarrhythmic Potency: Comparison of the Effect of Dofetilide, Cisapride, Sotalol, Terfenadine, and Verapamil on hERG and Native I Kr Currents and on Cardiac Action Potential. Toxicol. Sci. 2019, 168, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, B.; Imredy, J.P.; Kim, D.; Penniman, J.; Lagrutta, A.; Salata, J.J. Optimization of Cav1. 2 screening with an automated planar patch clamp platform. J. Pharmacol. Toxicol. Methods 2009, 59, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.M.; Schaefer, T.J. Nifedipine. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK537052/ (accessed on 22 April 2019).
- Kosower, E.M. A hypothesis for the mechanism of sodium channel opening by batrachotoxin and related toxins. FEBS Lett. 1983, 163, 161–164. [Google Scholar] [CrossRef]
- Du, Y.; Garden, D.P.; Wang, L.; Zhorov, B.S.; Dong, K. Identification of New Batrachotoxin-sensing Residues in Segment IIIS6 of the Sodium Channel. J. Biol. Chem. 2011, 286, 13151–13160. [Google Scholar] [CrossRef]
- Tokuyama, T.; Daly, J.; Witkop, B.; Karle, I.L.; Karle, J. The structure of batrachotoxinin A, a novel steroidal alkaloid from the Columbian arrow poison frog, Phyllobates aurotaenia. J. Am. Chem. Soc. 1968, 90, 1917–1918. [Google Scholar] [CrossRef]
- Khodorov, B.I.; Yelin, E.A.; Zaborovskaya, L.D.; Maksudov, M.Z.; Tikhomirova, O.B.; Leonov, V.N. Comparative analysis of the effects of synthetic derivatives of batrachotoxin on sodium currents in frog node of Ranvier. Cell. Mol. Neurobiol. 1992, 12, 59–81. [Google Scholar] [CrossRef]
- Tikhonov, D.B.; Zhorov, B.S. Sodium channel activators: Model of binding inside the pore and a possible mechanism of action. FEBS Lett. 2005, 579, 4207–4212. [Google Scholar] [CrossRef]
- Wang, S.-Y.; Tikhonov, D.B.; Mitchell, J.; Zhorov, B.; Wang, G.K. Irreversible block of cardiac mutant Na+ channels by batrachotoxin. Channels 2007, 1, 179–188. [Google Scholar] [CrossRef]
- Toma, T.; Logan, M.M.; Menard, F.; Devlin, A.S.; Du Bois, J. Inhibition of sodium ion channel function with truncated forms of batrachotoxin. ACS Chem. Neurosci. 2016, 7, 1463–1468. [Google Scholar] [CrossRef] [PubMed]
- Vossler, D.G.; Morris, G.L.; Harden, C.L.; Montouris, G.; Faught, E.; Kanner, A.M.; Fix, A.; French, J.A. The Postmarketing Antiepileptic Drug Survey (PADS) group study investigators. Tiagabine in clinical practice: Effects on seizure control and behavior. Epilepsy Behav. 2013, 28, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Masuda, N.; Peng, Q.; Li, Q.; Jiang, M.; Liang, Y.; Wang, X.; Zhao, M.; Wang, W.; Ross, C.A.; Duan, W. Tiagabine is neuroprotective in the N171-82Q and R6/2 mouse models of Huntington’s disease. Neurobiol. Dis. 2008, 30, 293–302. [Google Scholar] [CrossRef]
- Davydov, O. Antiepileptic Drugs Beyond Epilepsy (use of anticonvulsants in the treatment of pain syndromes). Neurosci. Behav. Physiol. 2014, 44, 772–778. [Google Scholar] [CrossRef]
- Pack, A.M. Epilepsy Overview and Revised Classification of Seizures and Epilepsies. Contin. Lifelong Learn. Neurol. 2016, 25, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Desborough, M.J.; Keeling, D.M. The aspirin story–from willow to wonder drug. Br. J. Haematol. 2017, 177, 674–683. [Google Scholar] [CrossRef]
- ATC Anatomical Therapeutic Chemical Classification System. Available online: http://www.atccode.com/ (accessed on 22 April 2019).
- Asif, M. A review on antiepileptic drug and their uses, mechanism of actions, adverse effects and drug interaction. Curr. Sci. Perspect. 2016, 2, 19–38. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Meldrum, B.S.; Chapman, A.G. Basic Mechanisms of Gabitril (Tiagabine) and Future Potential Developments. Epilepsia 1999, 40 (Suppl. 9), S2–S6. [Google Scholar] [CrossRef]
- Flowers, C.M.; Racoosin, J.A.; Kortepeter, C. Seizure activity and off-label use of tiagabine. N. Engl. J. Med. 2006, 354, 773–774. [Google Scholar] [CrossRef]
- Yarborough, M.; Johnson, J.G. Histamine Modulators. In Essentials of Pharmacology for Anesthesia, Pain Medicine, and Critical Care; Springer: New York, NY, USA, 2015; pp. 365–379. [Google Scholar]
- Li, M.; Ramos, L.G. Drug-induced QT prolongation and torsades de pointes. Pharm. Ther. 2017, 42, 473–477. [Google Scholar]
- Woosley, R.L.; Chen, Y.; Freiman, J.P.; Gillis, R.A. Mechanism of the cardiotoxic actions of terfenadine. JAMA 1993, 269, 1532–1536. [Google Scholar] [CrossRef]
- Sorkin, E.; Heel, R. Terfenadine A review of its pharmacodynamic properties and terapeutic efficiecy. Drugs 1985, 29, 34–56. [Google Scholar] [CrossRef]
- Ajayi, F.O.; Sun, H.; Perry, J. Adverse drug reactions: A review of relevant factors. J. Clin. Pharmacol. 2000, 40, 1093–1101. [Google Scholar]
- Koch, E.; Plassmann, S. Critical Aspects of Integrated Nonclinical Drug Development: Concepts, Strategies, and Potential Pitfalls. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development; Elsevier: Amsterdam, The Netherlands, 2017; pp. 7–38. [Google Scholar]
- Roy, M.-L.; Dumaine, R.; Brown, A.M. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation 1996, 94, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Pratt, P.F.; Bonnet, S.; Ludwig, L.M.; Bonnet, P.; Rusch, N.J. Upregulation of L-type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension 2002, 40, 214–219. [Google Scholar] [CrossRef]
- Worley, J.; French, R.J.; Krueger, B.K. Trimethyloxonium modification of single batrachotoxin-activated sodium channels in planar bilayers. Changes in unit conductance and in block by saxitoxin and calcium. J. Gen. Physiol. 1986, 87, 327–349. [Google Scholar] [CrossRef]
- Yap, Y.G.; Camm, A.J. Drug induced QT prolongation and torsades de pointes. Heart 2003, 89, 1363–1372. [Google Scholar] [CrossRef]
- Huang, H.; Pugsley, M.K.; Fermini, B.; Curtis, M.J.; Koerner, J.; Accardi, M.; Authier, S. Cardiac voltage-gated ion channels in safety pharmacology: Review of the landscape leading to the CiPA initiative. J. Pharmacol. Toxicol. Methods 2017, 87, 11–23. [Google Scholar] [CrossRef]
- Mirams, G.R.; Cui, Y.; Sher, A.; Fink, M.; Cooper, J.; Heath, B.M.; McMahon, N.C.; Gavaghan, D.J.; Noble, D. Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc. Res. 2011, 91, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ai, X.; Nakayama, H.; Chen, B.; Harris, D.M.; Tang, M.; Xie, Y.; Szeto, C.; Li, Y.; Li, Y. Persistent increases in Ca2+ influx through Cav1. 2 shortens action potential and causes Ca2+ overload-induced afterdepolarizations and arrhythmias. Basic Res. Cardiol. 2016, 111, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Konopelski, P.; Ufnal, M. Electrocardiography in rats: A comparison to human. Physiol. Res. 2016, 65, 717–725. [Google Scholar] [CrossRef]
- Chun, K.; Koenen, M.; Katus, H.A.; Zehelein, J. Expression of the IKr components KCNH2 (rERG) and KCNE2 (rMiRP1) during late rat heart development. Exp. Mol. Med. 2004, 36, 367–371. [Google Scholar] [CrossRef]
- Matus, M.; Kucerova, D.; Kruzliak, P.; Adameova, A.; Doka, G.; Turcekova, K.; Kmecova, J.; Kyselovic, J.; Krenek, P.; Kirchhefer, U. Upregulation of SERCA2a following short-term ACE inhibition (by enalaprilat) alters contractile performance and arrhythmogenicity of healthy myocardium in rat. Mol. Cell. Biochem. 2015, 403, 199–208. [Google Scholar] [CrossRef]
- De Clerck, F.; Van de Water, A.; D’Aubioul, J.; Lu, H.R.; Van Rossem, K.; Hermans, A.; Van Ammel, K. In vivo measurement of QT prolongation, dispersion and arrhythmogenesis: Application to the preclinical cardiovascular safety pharmacology of a new chemical entity. Fundam. Clin. Pharmacol. 2002, 16, 125–140. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.; Stratmann, R.E.; Burant, S.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The SWISS-MODEL Repository—New features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef]
- Schultz, D.; Mikala, G.; Yatani, A.; Engle, D.B.; Iles, D.E.; Segers, B.; Sinke, R.J.; Weghuis, D.O.; Klockner, U.; Wakamori, M.; et al. Cloning, chromosomal localization, and functional expression of the alpha 1 subunit of the L-type voltage-dependent calcium channel from normal human heart. Proc. Natl. Acad. Sci. USA 1993, 90, 6228–6232. [Google Scholar] [CrossRef] [PubMed]
- Cabral, J.H.M.; Lee, A.; Cohen, S.L.; Chait, B.T.; Li, M.; Mackinnon, R. Crystal Structure and Functional Analysis of the HERG Potassium Channel N Terminus: A Eukaryotic PAS Domain. Cell 1998, 95, 649–655. [Google Scholar] [CrossRef]
- Morais-Cabral, J.H.; Robertson, G.A. The enigmatic cytoplasmic regions of KCNH channels. J. Mol. Biol. 2015, 427, 67–76. [Google Scholar] [CrossRef]
- Rayevsky, A.; Samofalova, D.O.; Maximyuk, O.; Platonov, M.; Hurmach, V.; Ryabukhin, S.; Volochnyuk, D. Modelling of an autonomous Nav1. 5 channel system as a part of in silico pharmacology study. J. Mol. Model. 2021, 27, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Hasani, H.J.; Ganesan, A.; Houghton, M.; Barakat, K. Modeling the human Nav1. 5 sodium channel: Structural and mechanistic insights of ion permeation and drug blockade. Drug Des. Dev. Ther. 2017, 11, 2301. [Google Scholar] [CrossRef] [PubMed]
- Hering, S.; Zangerl-Plessl, E.-M.; Beyl, S.; Hohaus, A.; Andranovits, S.; Timin, E. Calcium channel gating. Pflügers Arch. Eur. J. Physiol. 2018, 470, 1291–1309. [Google Scholar] [CrossRef]
- Findeisen, F.; Minor, J.; Daniel, L. Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels 2010, 4, 459–474. [Google Scholar] [CrossRef]
- Al-Owais, M.; Bracey, K.; Wray, D. Role of intracellular domains in the function of the herg potassium channel. Eur. Biophys. J. 2009, 38, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Stansfeld, P.J.; Gedeck, P.; Gosling, M.; Cox, B.; Mitcheson, J.S.; Sutcliffe, M.J. Drug block of the hERG potassium channel: Insight from modeling. Proteins Struct. Funct. Bioinform. 2007, 68, 568–580. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).