
Structure and IR Spectra of 3(5)-Aminopyrazoles and UV-Induced Tautomerization in Argon Matrix




Abstract
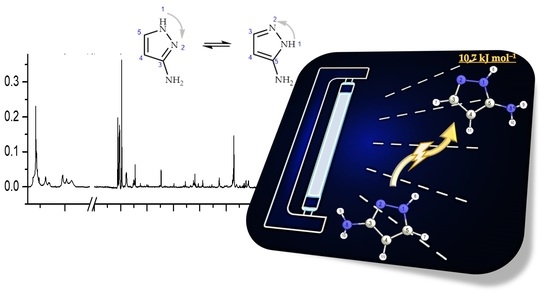
:
1. Introduction
2. Results and Discussion
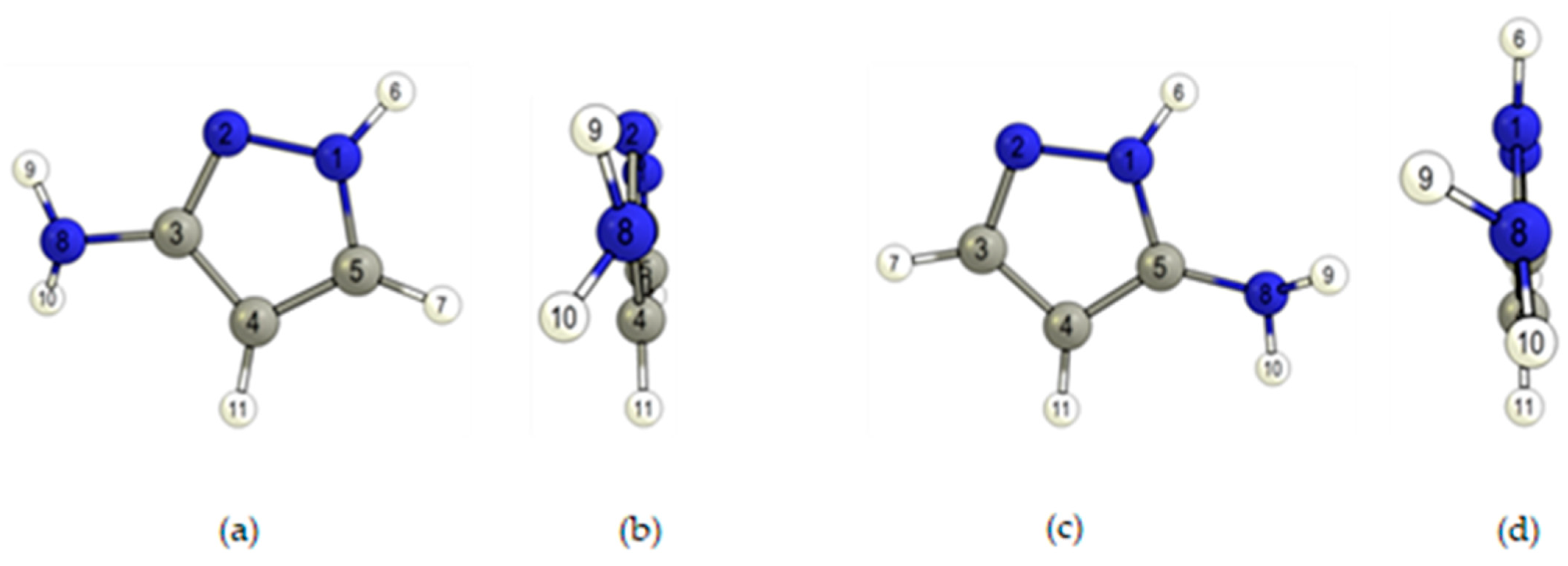
2.1. Geometries and Energies
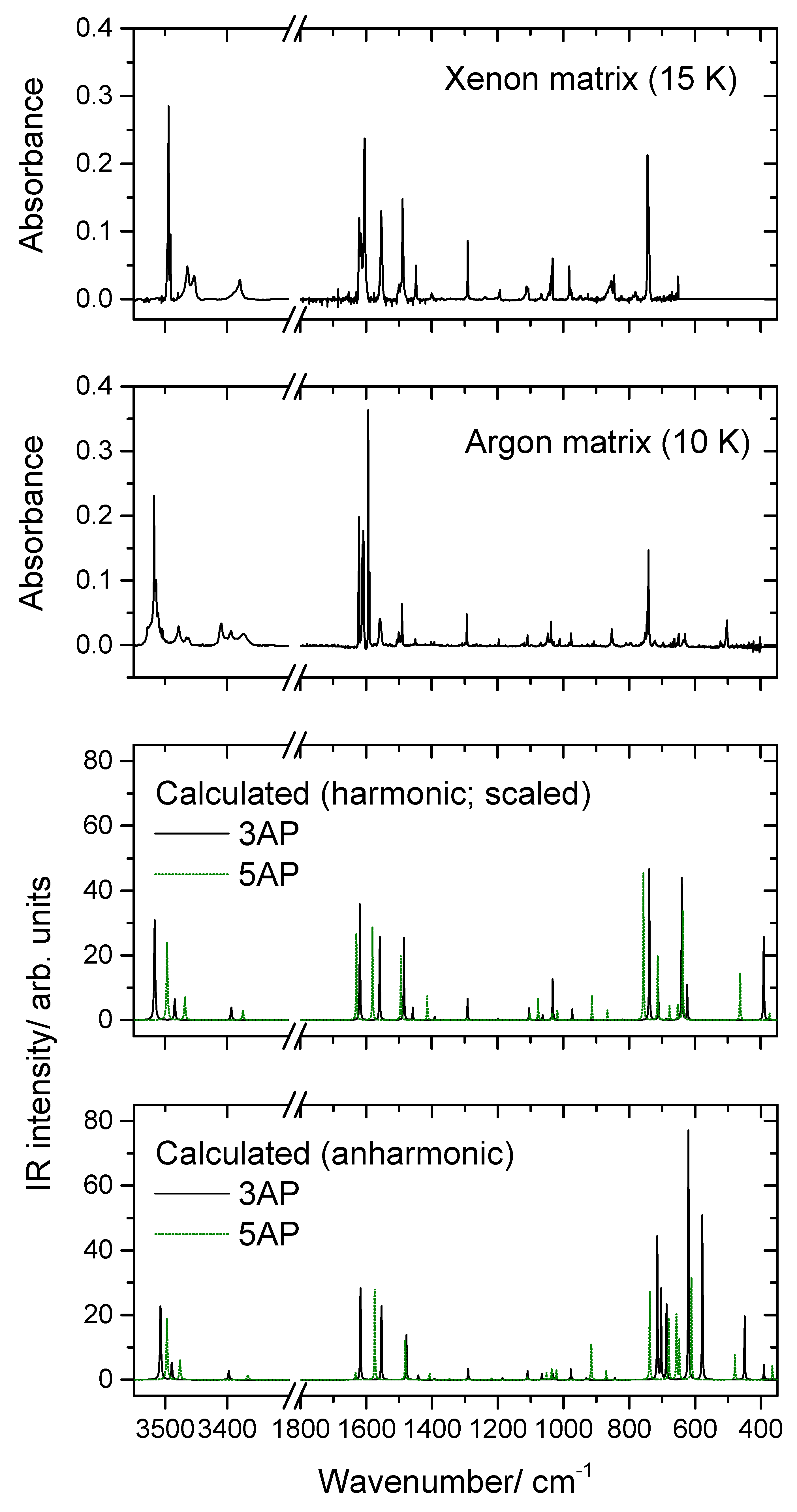
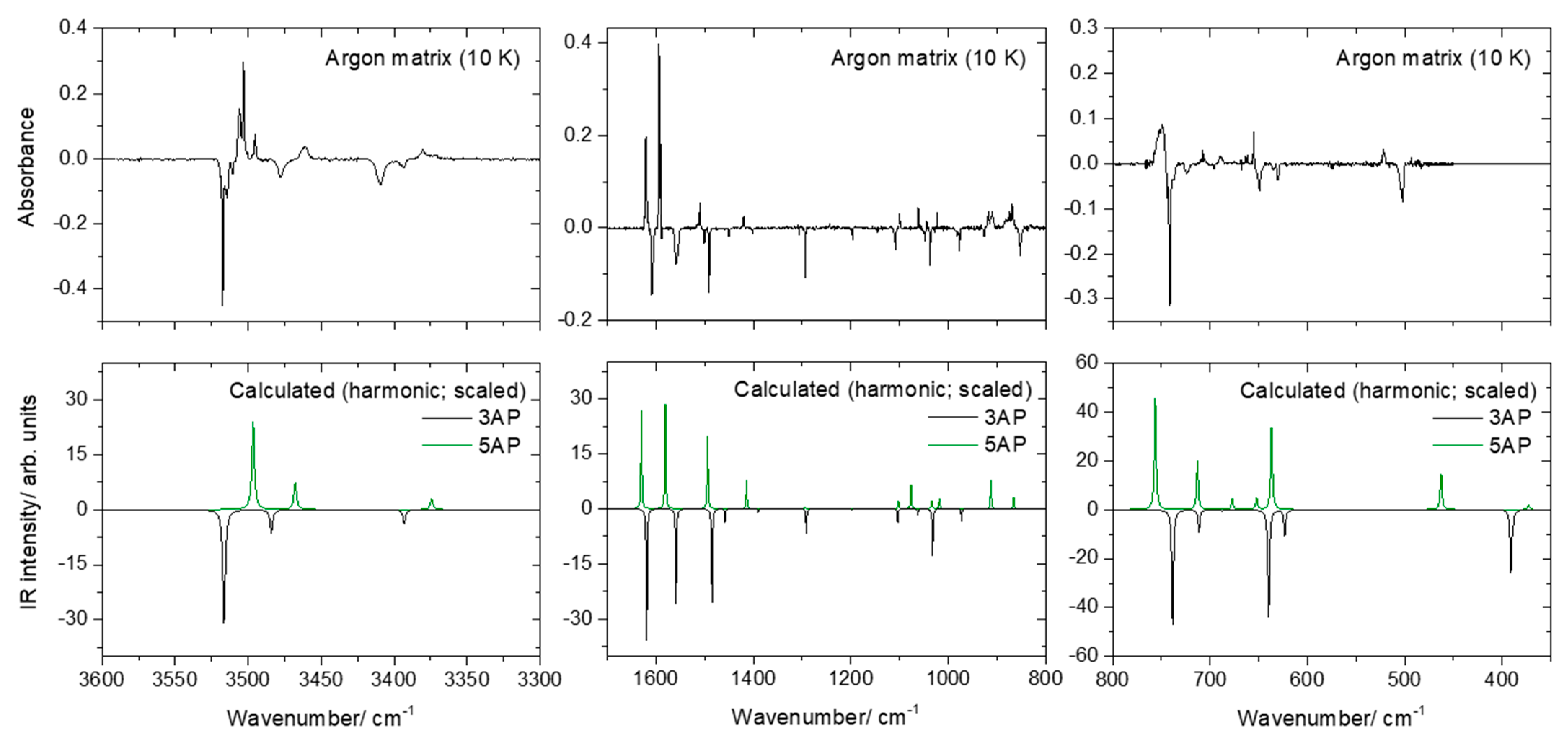
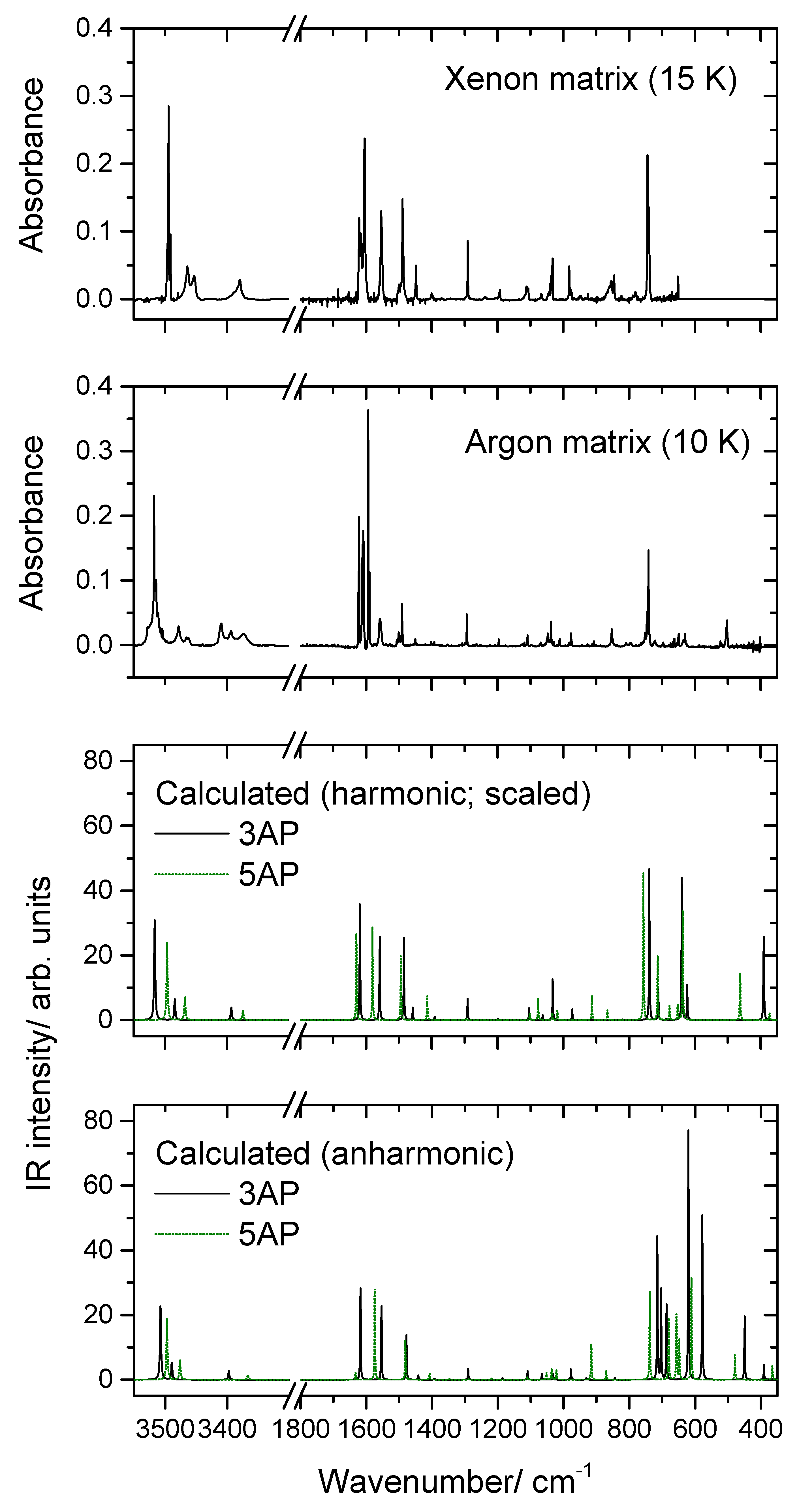
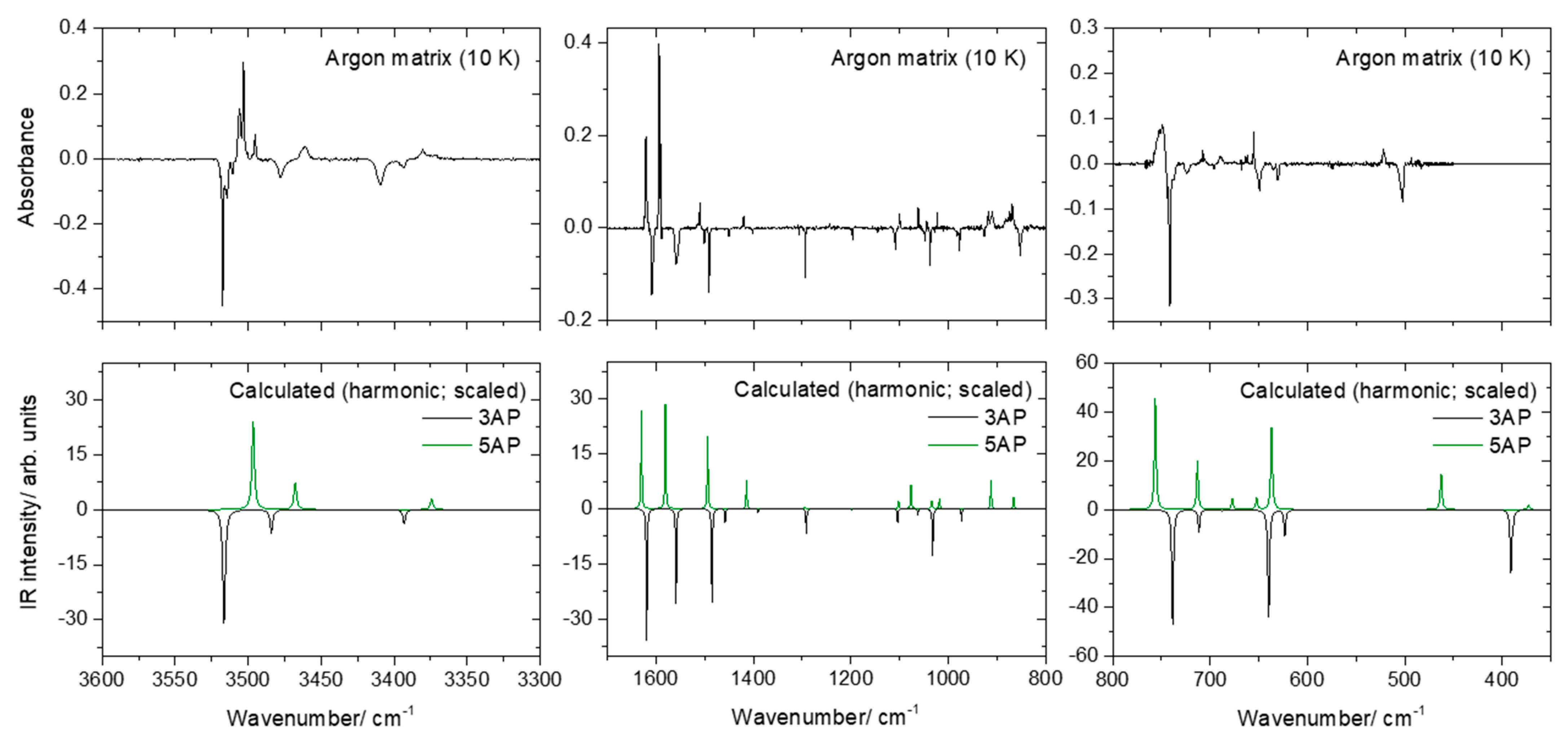
2.2. Infrared Spectra of Matrix-Isolated AP
- (i)
- The first point to note is the fact that several bands appear site split, indicating the existence of multiple trapping sites in both matrices. This is a common feature for matrix isolation IR spectra, and as usual, the bands of higher intensity are those exhibiting more extensive splitting due to the fact that they correspond to more polarized oscillators, which are more sensitive to changes in the local environment.
- (ii)
- As already mentioned, somehow surprisingly the anharmonic calculations fail to reproduce properly the observed spectra in the region between 700 and 500 cm−1. The frequencies are also not well predicted, but the estimation of the intensities is very much in error, being by far more intense than the observed ones. Interestingly, the agreement between the calculated harmonic (scaled) frequencies and intensities and the observed spectra, in this spectral region, is much better, describing fairly well the observations. The reason(s) for the failure of the anharmonic calculations in predicting appropriately this spectral region must be scrutinized in a dedicated investigation where several molecules have to be considered, but this lays outside the scope of the present study. Nevertheless, the problem seems to be related to the description of the amine moiety (or, at least, an amine fragment directly bound to an aromatic ring) since most of the vibrations giving rise to bands in this spectral region have major contributions from modes of this molecular fragment, in particular the γ(NH2) rocking mode (in line with this reasoning, a strong underestimation of the intensity of the δ(NH2) vibration of 5AP by the anharmonic calculations shall also be noticed; see Table 3).
- (iii)
- Among all the calculated frequencies, those obtained for g(NH) mode are the ones predicted as more deviated from the experimental frequencies. In this case, the anharmonic calculations provide considerably better results than the harmonic ones (even after scaling these latter using the same scale factor as for all other vibrations absorbing below 1800 cm−1) but still underestimate the observed frequencies by approximately 10% (~50 cm−1). In this case, however, one can attribute the disagreement as being mostly determined by interactions between the N–H fragment and the matrix host that make the movement of the hydrogen atom out of the plane of the ring more difficult (i.e., they lead to a larger force constant for this oscillator in the matrix than in the free molecule). Two different (possibly acting simultaneously) types of interactions can be considered: on the one side, packing-related interactions, which can be expected to favor the planarity of the molecule (hence increasing the energy of the system upon the hydrogen atom movement to out of the plane of the ring), and, on the other side, H-bond like interactions, where the N–H group appears as donor and the host matrix atoms as acceptors (this type of interaction is well-known for molecules bearing an O–H moiety [21,22,23] but can also be expected to occur in the case of an N–H group).
- (iv)
- A final note shall be made regarding the origin of the band observed in the argon matrix at 853 cm−1. This band can be partially due to the γ(CH)a mode of 3AP (calculated intensity ~1 km mol−1; see Table 2), but the observed intensity appears to be too large for this to be the sole contribution to the intensity of the band. It can be tentatively suggested that the intense (~6 km mol−1) combination mode predicted by the anharmonic calculations at 833 cm−1 also contributes to the observed band. This combination mode is associated with the vibration whose fundamental transition gives rise to the intense band observed at 649 cm−1 (a mixed vibration with major contributions from γ(NH2) and a torsion of the ring) plus the torsion of the amine group (τ(NH2, whose fundamental was predicted at 223 cm−1 by the anharmonic calculations).
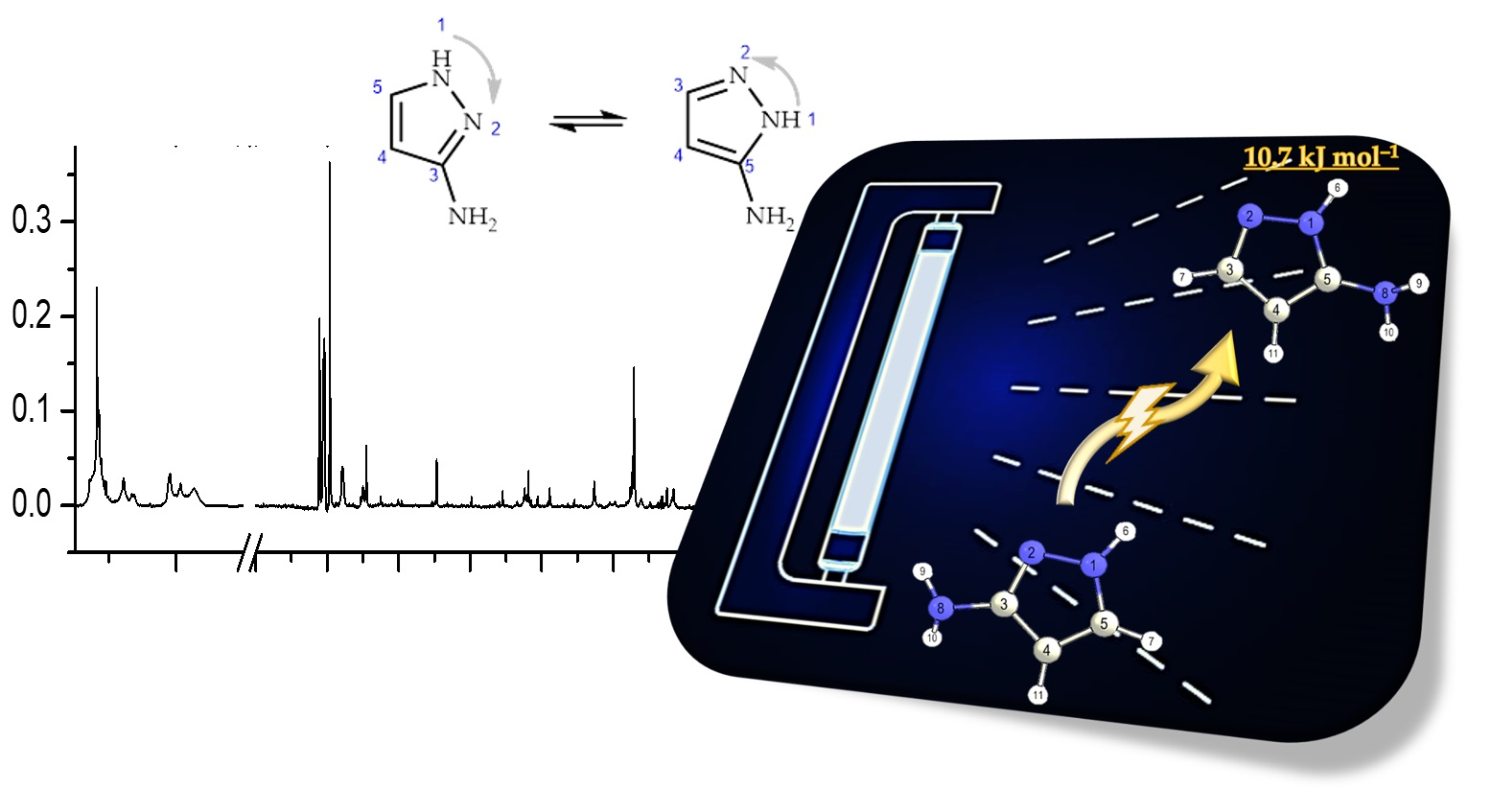
2.3. UV-Induced Phototautomerization
3. Materials and Methods
3.1. Experimental Details
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mert, S.; Kasimogullari, R.; Ok, S. A Short Review on Pyrazole Derivatives and Their Applications. J. Postdr. Res. 2014, 2, 64–72. [Google Scholar]
- Castillo, J.C.; Portilla, J. Recent advances in the synthesis of new pyrazole derivatives. Targets Heterocycl. Syst. 2018, 22, 194–223. [Google Scholar] [CrossRef]
- Kumari, S.; Paliwal, S.; Chauhan, R. Synthesis of Pyrazole Derivatives Possessing Anticancer Activity: Current Status. Synth. Commun. 2014, 44, 1521–1578. [Google Scholar] [CrossRef]
- Anwar, H.F.; Elnagdi, M.H. Recent developments in aminopyrazole chemistry. Arkivoc 2009, 2009, 198–250. [Google Scholar] [CrossRef] [Green Version]
- Fichez, J.; Busca, P.; Prestat, G. Recent Advances in Aminopyrazoles Synthesis and Functionalization. Targets Heterocycl. Syst. 2017, 21, 322–347. [Google Scholar] [CrossRef]
- Lim, F.P.L.; Tan, K.C.; Luna, G.; Tiekink, E.R.; Dolzhenko, A.V. A new practical synthesis of 3-amino-substituted 5-aminopyrazoles and their tautomerism. Tetrahedron 2019, 75, 2314–2321. [Google Scholar] [CrossRef]
- Shaabani, A.; Nazeri, M.T.; Afshari, R. 5-Amino-pyrazoles: Potent reagents in organic and medicinal synthesis. Mol. Divers. 2018, 23, 751–807. [Google Scholar] [CrossRef]
- Emelina, E.E.; Petrov, A.A.; Filyukov, D.V. Structure and tautomerism of 4-substituted 3(5)-aminopyrazoles in solution and in the solid state: NMR study and Ab initio calculations. Russ. J. Org. Chem. 2014, 50, 412–421. [Google Scholar] [CrossRef]
- Dorn, H. Tautomerie und Nomenklatur der? Pyrazolone? und Amino-pyrazole. J. Prakt. Chem. 1973, 315, 382–418. [Google Scholar] [CrossRef]
- Gonzalez, E.; Faure, R.; Vincent, E.-J.; Espada, M.; Elguero, J. Etude en Résonance Magnétique Nucléaire du Carbone-13 de Quelques Aminopyrazoles. Sur le Problème de la Détermination des Constantes d’Equilibre Tautomère. Magn. Reson. Chem. 1979, 12, 587–592. [Google Scholar] [CrossRef]
- Puello, J.Q.; Obando, B.I.; Foces-Foces, C.; Infantes, L.; Claramunt, R.M.; Cabildo, P.; Jiménez, J.; Elguero, J. Structure and tautomerism of 3(5)-amino-5(3)-arylpyrazoles in the solid state and in solution: An X-ray and NMR study. Tetrahedron 1997, 53, 10783–10802. [Google Scholar] [CrossRef]
- Marín-Luna, M.; Alkorta, I.; Elguero, J. A theoretical study of the gas phase (proton affinity) and aqueous (pKa) basicity of a series of 150 pyrazoles. New J. Chem. 2015, 39, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Catalan, J.; Menéndez, M.; Laynez, J.; Claramunt, R.M.; Bruix, M.; De Mendoza, J.; Elguero, J. Basicity of azoles. VII. Basicity ofC-aminopyrazoles in relation to tautomeric and protonation studies. J. Heterocycl. Chem. 1985, 22, 997–1000. [Google Scholar] [CrossRef]
- Jarończyk, M.; Dobrowolski, J.C.; Mazurek, A.P. Theoretical studies on tautomerism and IR spectra of pyrazole derivatives. J. Mol. Struct. THEOCHEM 2004, 673, 17–28. [Google Scholar] [CrossRef]
- Chermahini, A.N.; Teimouri, A. Theoretical studies on proton transfer reaction of 3(5)-substituted pyrazoles. J. Chem. Sci. 2014, 126, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Secrieru, A.; O’Neill, P.M.; Cristiano, M.L.S. Revisiting the Structure and Chemistry of 3(5)-Substituted Pyrazoles. Molecules 2019, 25, 42. [Google Scholar] [CrossRef] [Green Version]
- Kruszewski, J.; Krygowski, T. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Cyrański, M.K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1420. [Google Scholar] [CrossRef]
- Cyrański, M.K. Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev. 2005, 105, 3773–3811. [Google Scholar] [CrossRef]
- Ildiz, G.O.; Nunes, C.M.; Kuş, N.; Fausto, R. FTIR investigation of the O–H··Xe interaction in simple carboxylic acids in solid xenon. J. Chem. Phys. 2012, 137, 64309. [Google Scholar] [CrossRef] [PubMed]
- Lopes, S.; Domanskaya, A.; Fausto, R.; Räsänen, M.; Khriachtchev, L. Formic and acetic acids in a nitrogen matrix: Enhanced stability of the higher-energy conformer. J. Chem. Phys. 2010, 133, 144507. [Google Scholar] [CrossRef] [Green Version]
- Kus, N.; Sharma, A.; Peña, I.; Bermúdez, M.C.; Cabezas, C.; Alonso, J.L.; Fausto, R. Conformers of β-aminoisobutyric acid probed by jet-cooled microwave and matrix isolation infrared spectroscopic techniques. J. Chem. Phys. 2013, 138, 144305. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, M.P.; Sandford, S.A.; Allamandola, L.J. The Infrared Spectra of Nitriles and Related Compounds Frozen in Ar and H2O. Astrophys. J. 1997, 476, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Bégué, D.; Qiao, G.; Wentrup, C. Nitrile Imines: Matrix Isolation, IR Spectra, Structures, and Rearrangement to Carbodiimides. J. Am. Chem. Soc. 2012, 134, 5339–5350. [Google Scholar] [CrossRef]
- Jacox, E., III; Milligan, D.E. Infrared Study of the Reactions of CH2 and NH with C2H2 and C2H4 in Solid Argon. J. Am. Chem. Soc. 1963, 85, 278–282. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Krishnan, R.S.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Peng, C.; Schlegel, H.B. Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States. Isr. J. Chem. 1993, 33, 449–454. [Google Scholar] [CrossRef]
- Adamo, C.; Jacquemin, D. The calculations of excited-state properties with Time-Dependent Density Functional Theory. Chem. Soc. Rev. 2013, 42, 845–856. [Google Scholar] [CrossRef]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2014, 16, 14334–14356. [Google Scholar] [CrossRef] [PubMed]
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. J. Chem. Phys. 2005, 122, 014108. [Google Scholar] [CrossRef] [PubMed]
- Bloino, J.; Barone, V. A second-order perturbation theory route to vibrational averages and transition properties of molecules: General formulation and application to infrared and vibrational circular dichroism spectroscopies. J. Chem. Phys. 2012, 136, 124108. [Google Scholar] [CrossRef]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. Phys. Chem. Chem. Phys. 2014, 16, 1759–1787. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Bloino, J.; Guido, C.A.; Lipparini, F. A fully automated implementation of VPT2 Infrared intensities. Chem. Phys. Lett. 2010, 496, 157–161. [Google Scholar] [CrossRef]
- Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. 2021. Available online: https://www.chemcraftprog.com (accessed on 14 July 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | 3AP | Parameter | 5AP |
|---|---|---|---|
| C3–N8 | 1.395 | C5–N8 | 1.399 |
| C3–N2 | 1.329 | C3–N2 | 1.326 |
| C5–N1 | 1.353 | C5–N1 | 1.359 |
| N1–N2 | 1.359 | N1–N2 | 1.360 |
| C3–C4 | 1.421 | C3–C4 | 1.415 |
| C4–C5 | 1.379 | C4–C5 | 1.384 |
| N1–H6 | 1.006 | N1–H6 | 1.007 |
| N8–H9 | 1.012 | N8–H9 | 1.013 |
| N8–H10 | 1.010 | N8–H10 | 1.011 |
| C3–N8–H9 | 112.6 | C5–N8–H9 | 114.1 |
| C3–N8–H10 | 114.2 | C5–N8–H10 | 112.4 |
| H9–N8–H10 | 111.5 | H9–N8–H10 | 109.9 |
| N2–C3–N8–H9 | 15.1 | N1–C5–N8–H9 | 59.2 |
| N2–C3–N8–H10 | 143.6 | N1–C5–N8–H10 | −174.8 |
| Experimental | Calculated (Harmonic) | Calculated (Anharmonic) | ||||
|---|---|---|---|---|---|---|
| Approximate Description | Ar Matrix | Xe Matrix | ν | IIR | ν | IIR |
| ν(NH) | 3519.0 | 3496.0 | 3516.4 | 97.6 | 3507.3 | 73.9 |
| 3517.5 | 3494.0 | |||||
| 3515.5 | 3491.0 | |||||
| 3514.0 | ||||||
| 3511.5 | ||||||
| 3510.5 | ||||||
| ν(NH2)a | 3478.0 | 3464.5 | 3484.1 | 20.4 | 3489.1 | 16.4 |
| ν(NH2)s | 3409.0 | 3379.5 | 3393.1 | 12.3 | 3397.4 | 8.6 |
| 3393.5 | ||||||
| ν(CH)s | 3182.5 | 3181.0 (?) | 3114.6 | 0.4 | 3137.5 | 1.4 |
| ν(CH)a | 3151.0 | 3157.0 | 3097.7 | 1.4 | 3118.2 | 2.3 |
| δ(NH2) | 1611.0 | 1604.5 | 1618.8 | 118.9 | 1617.6 | 90.6 |
| ν(C4C5) − ν(C3N2) | 1557.0 | 1554.0 | 1558.5 | 80.8 | 1553.5 | 71.8 |
| ν(C3C4) − ν(C3N8) | 1500.5 | 1489.0 | 1484.8 | 82.1 | 1477.7 | 44.5 |
| 1491.0 | 1486.5 | |||||
| δ(NH) + ν(C5N1) | 1450.0 | 1448.0 | 1458.0 | 12.1 | 1441.7 | 4.3 |
| ν(C4C5) + ν(C3N2) − ν(C3C4) | 1401.5 | 1400.0 | 1390.9 | 3.5 | 1392.5 | 0.8 |
| δ(C5H) + δ(C4H) + δ(NH) | 1294.0 | 1291.0 | 1291.4 | 20.9 | 1290.0 | 11.0 |
| ν(NN) | 1196.5 | 1195.0 | 1198.6 | 1.5 | 1185.9 | 1.7 |
| 1193.0 | ||||||
| tw(NH2) + ν(C5N1) | 1109.0 | 1112.0 | 1104.2 | 11.9 | 1109.4 | 8.5 |
| 1106.0 | ||||||
| δ(C4H) + tw(NH2) | 1048.5 | 1068.0 | 1062.8 | 5.1 | 1065.9 | 6.0 |
| δ(C5H) + ν(NN) | 1037.5 | 1041.5 | 1032.4 | 40.1 | 1031.8 | 5.6 |
| 1036.5 | ||||||
| 1034.5 | ||||||
| 1032.5 | ||||||
| ν(C3C4) + δ(ring-NNC3) | 982.0 | 982.0 | 973.1 | 10.4 | 977.7 | 10.4 |
| 977.0 | 978.0 | |||||
| 976.0 | 975.0 | |||||
| δ(ring-NNC5) | 927.0 | 925.0 | 922.0 | 0.1 | 930.7 | 1.7 |
| γ(CH)a | 853.0(?) | n.obs. | 820.3 | 0.4 | 843.9 | 1.8 |
| γ(CH)s + γ(NH2) + τ(ring) | 742.0 | 741.0 | 738.6 | 147.4 | 715.0 | 139.3 |
| 741.0 | 739.0 | |||||
| γ(CH)s − γ(NH2) | 723.0 | 718.0 | 711.5 | 29.2 | 703.5 | 87.6 |
| ν(C3N8) + δ(ring-CCN2) | 696.0 | 694.5 | 687.8 | 1.3 | 686.9 | 74.7 |
| γ(NH2) + τ(ring) | 649.0 | 651.0 | 639.9 | 140.0 | 620.4 | 245.1 |
| τ(ring) + γ(NH) | 635.0 | n.i. | 623.4 | 34.5 | 578.1 | 160.4 |
| 630.5 | ||||||
| γ(NH) | 503.1 | n.i. | 391.0 | 79.6 | 449.5 | 61.6 |
| δ(C3N8) | n.i. | n.i. | 388.6 | 9.2 | 390.6 | 15.0 |
| γ(C3N8) | n.i. | n.i. | 299.8 | 17.0 | 328.7 | 28.1 |
| τ(NH2) | n.i. | n.i. | 241.0 | 39.3 | 223.2 | 36.0 |
| Experimental | Calculated (Harmonic) | Calculated (Anharmonic) | ||||
|---|---|---|---|---|---|---|
| Approximate Description | Ar Matrix | Xe Matrix | ν | IIR | ν | IIR |
| ν(NH) | 3505.0 | 3485.0 | 3496.6 | 76.0 | 3497.1 | 59.6 |
| 3503.0 | 3479.0 | |||||
| 3495.5 | ||||||
| ν(NH2)a | 3461.0 | 3452.0 | 3467.7 | 23.7 | 3476.1 | 19.0 |
| ν(NH2)s | 3380.0 | 3358.0 | 3374.3 | 9.4 | 3366.9 | 4.9 |
| 3371.5 | ||||||
| ν(CH)s | 3163.0 | 3176.0 (?) | 3106.7 | 1.4 | 3129.6 | 2.0 |
| ν(CH)a | 3145.0 | 3146.0 | 3083.3 | 6.8 | 3103.4 | 8.3 |
| δ(NH2) | 1620.5 | 1615.5 | 1629.6 | 84.5 | 1632.7 | 7.3 |
| ν(C4C5) − ν(C5N8) | 1593.5 | 1586.5 | 1580.4 | 90.3 | 1574.1 | 88.8 |
| 1589.5 | ||||||
| ν(C5N1) + ν(C3N2) | 1510.5 | 1499.5 | 1493.8 | 64.9 | 1481.3 | 40.0 |
| δ(NH) + ν(C5N1) | 1420.0 | 1401.0 | 1414.1 | 24.2 | 1407.6 | 6.4 |
| ν(C3N2) + ν(C3C4)+δ(C4H) | 1353.0 (?) | n.obs. | 1352.5 | 0.6 | 1346.9 | 0.8 |
| δ(C3H) + δ(NH) | n.obs. | n.obs. | 1294.2 | 1.7 | 1289.4 | 0.6 |
| δ(C3H) + δ(C4H) | 1243.0 (?) | 1242.0 (?) | 1219.5 | 0.7 | 1219.0 | 1.4 |
| tw(NH2) + ν(C5N1) | 1100.5 | n.obs. | 1102.2 | 6.6 | 1110.7 | 2.4 |
| ν(NN) + τω(NH2) | 1063.0 | 1057.5 | 1076.9 | 20.7 | 1052.4 | 7.2 |
| δ(C3H)–δ(C4H)+ν(C3C4) | 1045.0 | n.obs. | 1034.2 | 7.2 | 1036.2 | 11.3 |
| 1041.0 | ||||||
| ν(C4C5) + ν(C5N1) | 1023.5 | 1006.0 (?) | 1018.4 | 9.2 | 1021.8 | 9.4 |
| δ(ring-NNC3) | 919.0 | 898.5 | 913.0 | 24.1 | 915.6 | 35.5 |
| 917.0 | ||||||
| 910.0 | ||||||
| γ(C3H) | 888.5 | 855.0 | 886.3 | 9.9 | 870.2 | 8.6 |
| 881.5 | 845.0 | |||||
| 875.0 | ||||||
| 872.0 | ||||||
| 869.0 | ||||||
| 867.5 | ||||||
| γ(C4H) | 752.2 | 730.0 | 756.4 | 143.6 | 737.9 | 86.9 |
| γ(NH2) − γ(C4H) | 707.0 | 702.0 | 713.1 | 62.5 | 680.3 | 63.0 |
| δ(ring-CCN1) + ν(C5N8) | 690.0 | 668.0 | 677.3 | 14.8 | 656.7 | 66.7 |
| τ(ring) | 663.5 | 660.0 (?) | 652.3 | 15.5 | 648.0 | 39.6 |
| t(ring) + γ(NH) | 655.5 | 655.0 (?) | 636.9 | 106.8 | 611.0 | 99.1 |
| γ(NH) | 523.0 | n.i. | 462.8 | 47.9 | 478.8 | 25.4 |
| δ(CN8) | n.i. | n.i. | 373.0 | 6.6 | 365.2 | 14.1 |
| γ(CN8) | n.i. | n.i. | 275.9 | 7.3 | 277.3 | 9.5 |
| τ(NH2) | n.i. | n.i. | 138.2 | 45.3 | 125.2 | 37.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Secrieru, A.; Lopes, S.; Cristiano, M.L.S.; Fausto, R. Structure and IR Spectra of 3(5)-Aminopyrazoles and UV-Induced Tautomerization in Argon Matrix. Molecules 2021, 26, 4299. https://doi.org/10.3390/molecules26144299
Secrieru A, Lopes S, Cristiano MLS, Fausto R. Structure and IR Spectra of 3(5)-Aminopyrazoles and UV-Induced Tautomerization in Argon Matrix. Molecules. 2021; 26(14):4299. https://doi.org/10.3390/molecules26144299
Chicago/Turabian StyleSecrieru, Alina, Susy Lopes, Maria L. S. Cristiano, and Rui Fausto. 2021. "Structure and IR Spectra of 3(5)-Aminopyrazoles and UV-Induced Tautomerization in Argon Matrix" Molecules 26, no. 14: 4299. https://doi.org/10.3390/molecules26144299
APA StyleSecrieru, A., Lopes, S., Cristiano, M. L. S., & Fausto, R. (2021). Structure and IR Spectra of 3(5)-Aminopyrazoles and UV-Induced Tautomerization in Argon Matrix. Molecules, 26(14), 4299. https://doi.org/10.3390/molecules26144299