1. Introduction
Silicones are polymers of broad interest, both on industrial and academic sides. In industry, developments focus on the generation of more and more performing elastomers, widely used in numerous applications, e.g., for their thermal resistance or their innocuity [
1]. In academia, a great deal of work has recently been conducted on (mostly rediscovered) chemistries to generate silicone chains with new functionalities. In the most recent research, we can cite, in particular, aza-Michael addition [
2], thiol-ene click chemistry [
3], and organocatalyzed polymerization and polycondensation (see, e.g., [
4]).
A recurrent domain of development concerns the generation of silicone aqueous emulsions via the ring-opening polymerization of cyclosiloxanes (a recent book published by the Dow company summarizes recent comprehension of silicone dispersions [
5]). Both cationic and anionic catalysts produce silicone dispersions, but the mechanism by which long polymer chains are formed differs, and is still of debate nowadays [
5,
6]. Basically, taking the case of octamethylcyclotetrasiloxane (D
4), polymerizations in emulsion (fast mixing in the presence of excess surfactant), in microsuspension (pre-generation of nanosized monomer droplets, typically by ultrasonication), or microemulsion (thermodynamically stable nanodroplets) follow very different pathways. In all instances, ring opening proceeds to propagate chains. Polymerization is stopped by transfer to water, but silanol chains ends can be reactivated to propagate further. Polycondensation between silanol end-groups also take place, to increase the molar masses. Side reactions that are generally observed in bulk or solution are likely prominent in water: backbiting that gives rise to a variety of cyclosiloxanes with various reactivities, and intermolecular redistributions between chains that enlarge the molar mass distribution. All the difficulty in these systems is to first understand where the different reactions take place (directly in water, at the droplet interface or inside the droplets), and second, at which paces.
Hexamethylcyclotrisiloxane is a monomer of choice when targeting silicone chains with perfectly controlled masses and functionality, as obtained by living anionic polymerization (for a very recent review, see an article just published in this Special Issue [
7]). On the other hand, cationic polymerization of D
3 has been described mostly in the mid-eighties by two groups led by P. Sigwalt and J. Chojnowsky, and not further exploited (a summary of this more than 10 year’s competition can be found in [
8]). Using triflic acid as a superacid, the team of Sigwalt showed that water would retard the polymerization of D
3, but would not inhibit it, albeit at a high catalyst content. To our knowledge, ring-opening cationic polymerization of D
3 in water has hardly been studied. Early on, Weyenberg et al. showed, in a seminal paper, that D
4 or D
3 was easily converted into polymers in the presence of dodecylbenzenesulfonic acid [
9]. They wrote that ‘
polymerization of hexamethylcyclotrisiloxane proceeds at a much faster rate than the cyclotetrasiloxane and, in fact, it is not necessary to pre-emulsify this monomer. Contact of even large crystals of this monomer with DBSA and water at 25 °C gave a quantitative conversion to emulsion polymer within 24 hr’. Hemery et al. have later polymerized D
3, solubilized in toluene, in emulsion by an anionic polymerization process, where they observed fast generation of polymers with an unlikely broad distribution [
10].
Tris(pentafluorophenyl)boron (acronym BCF) was discovered in the early 1960s, and was almost forgotten for 25 years before being rediscovered as a catalyst activator in metallocene-catalyzed olefin polymerization. Its strong Lewis acidity, comparable to those of BF
3.OEt
2, combined with its air stability and water tolerance, has made it a (co-)catalyst of choice for numerous reactions that are summarized in reviews (e.g., [
11]). Since the discovery by Piers et al. that BCF catalyzes the reduction reaction of a silyl ether into alkane in the presence of an hydrogenosilane, this reaction was later patented and published to produce linear silicone chains from alkoxy- and hydrogeno-functionalized silicone molecules [
12]. The so-called Piers–Rubinsztajn reaction was studied in detail, particularly in the team of Professor Chojnowsky in a series of papers explaining the mechanism of catalysis. A precision reaction could thus be performed, starting from model molecules, allowing the generation of complex branched structures with exceptional monodispersity (for a review on this, please see [
13]). For the record, this Lewis acid was also proved to promote a hydrosilylation reaction, albeit in stoichiometric amount, or, more strangely, oligomerization of electron-withdrawing monomers (typically vinyl methylsulfone or acrylonitrile) onto SiH functions through a coordinated ate-type intermediate [
14]. The team of Chojnowsky has shown that D
3 can be open and polymerized by tetramethyldisiloxane (L
2H) and other hydrogenosiloxanes in BCF toluene solution, but polymerization does not proceed in the absence of these molecules [
15].
In this communication, we propose to show some preliminary results on the cationic polymerization of D
3 in excess water. Thanks to our deep knowledge on such processes applied to cyclosiloxanes [
6] and vinyl monomers [
16], we have selected a variety of Bronsted and (water-tolerant) Lewis acids. We particularly show advanced results on the polymerization of D
3 using BCF as a catalyst, and finally propose a brief discussion about a tentative polymerization mechanism.
2. Results and Discussion
2.1. First Screening
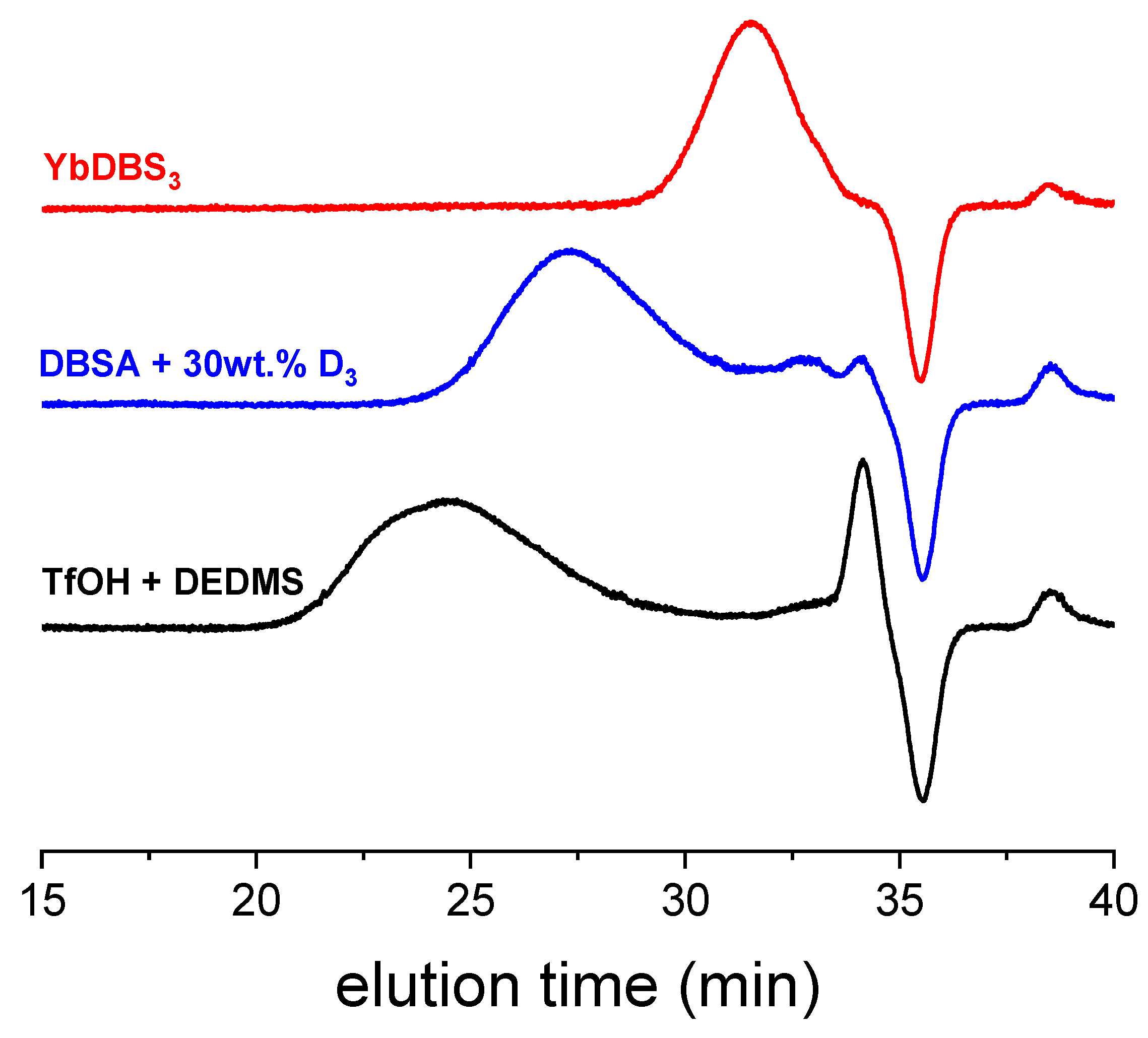
Table 1 summarizes the different results of D
3 polymerization in excess water, using different catalysts. Basically, molar masses and polydispersity, as well as the final contents of polymer, are given here, with selected SEC traces plotted in
Figure 1. All the experiments were conducted at room temperature, in 10 mL vials, using a magnetic agitation (see conditions in
Table 1 footnote). We did not specifically look at the colloidal state of the dispersions here, nor did we follow the kinetics of the reaction.
The first experiments conducted with triflic acid in large quantities showed that solid D
3 is rapidly consumed to give a totally transparent solution. We could not track the presence of polymer by precipitation of a sample aliquot in excess methanol, nor any other cycles that would have generated oil stains on the vial’s walls. After the addition of a slight quantity of diethoxydimethylsilane (DEDMS, 0.2 eq. of initial D
3) and agitation during 12 h, we observed a precipitate at the bottom of the tube assigned to a polymer of high molar mass (M
n of 180,000 g/mol by SEC). DEDMS introduced in triflic acid aqueous solution in absence of D
3 did not produce a polymer, in agreement with a previous study [
17]. We then concluded that triflic acid opens the cycle to generate water-soluble oligomers that convert into polymer by acid-catalyzed condensation between silanol and ethoxysilane groups. Note that this reaction is very different from the polycondensation reaction of bis-silanol-terminated PDMS long, hydrophobic oligomers that occurs exclusively at droplet interfaces [
18].
Changing a molecular superacid to an acidic surfactant, DBSA, after only 6 h of reaction, we could detect the presence of polymers in the test tubes. SEC curves give molar masses of around 70,000 g/mol, with a larger content of small cycles at a larger D3 content (about 10 wt.% at the end of the polymerization). Since at the time we were looking for cycle-free emulsions, we did not further explore this path; we are currently pursuing some experiments to check how fast and efficient this polymerization is.
We also tested some rare earth Lewis acids (ytterbium and indium chloride salts) combined with sodium dodecylbenzene sulfonate (NaDBS), to generate so-called Lewis acid surfactant complexes (LASCs), as reported before [
16]. Even in large excesses, as tested here, these catalysts produced exclusively oligomers of molar masses around 4500 g/mol. Note that methanol also precipitated the LASC catalyst, so that it cannot be easily separated from the oligomers. According to the price of the catalysts used here, and the short oligomers produced, this alley was not pursued.
2.2. The Case of BCF
The origin of this second set of experiments comes from a study of the condensation reactions of alkoxy- and silanol-functionalized telechelic polymers in water [
17]. When starting from the model molecules, tetramethyldisiloxane and dimethyldimethoxysilane, we observed, the rapid generation of cyclosiloxanes of various sizes, from D
3 to D
7, together with some polymer. The former cycle gradually disappeared with time, whereas the larger ones would accumulate in the reactor. This intriguing observation prompted us to further study the cationic ROP of D
3 in water catalyzed by BCF, in the absence of any other siloxane- or silane-based molecules. Note that we preliminary checked that BCF does not promote D
3 polymerization in toluene overnight (not shown).
A typical experiment consisted of introducing D
3 powder straightaway in a test-tube containing an aqueous solution of the catalyst, at room temperature and under magnetic agitation (see formulation in
Table 2, entry 5).
Even if D
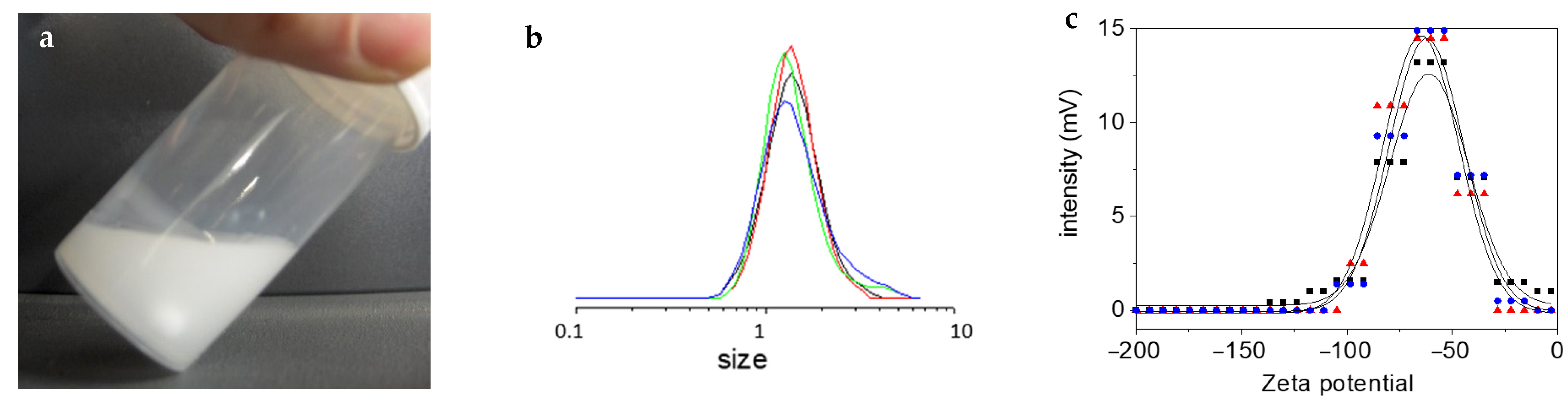
3 first resides as solid chunks at the top of the water phase, it is then gently incorporated with time. A white emulsion (
Figure 2a), of about 1.5 μm in size (
Figure 2b), is generated, and remains almost so until the end of the reaction (we can track a slight enlargement in the size distribution with time, see
Figure 2b). Zeta potential measurements give surface values of about –65 mV (
Figure 2c). We suspect that the silanol groups of the oligomers/polymers protruding at the surface of the droplets stabilize them, as proposed earlier by Vincent et al. [
19] and ourselves [
17]. Only when the content of the silanol groups becomes too small that the polymer precipitates and deposits on the flask wall (
Figure 2a).
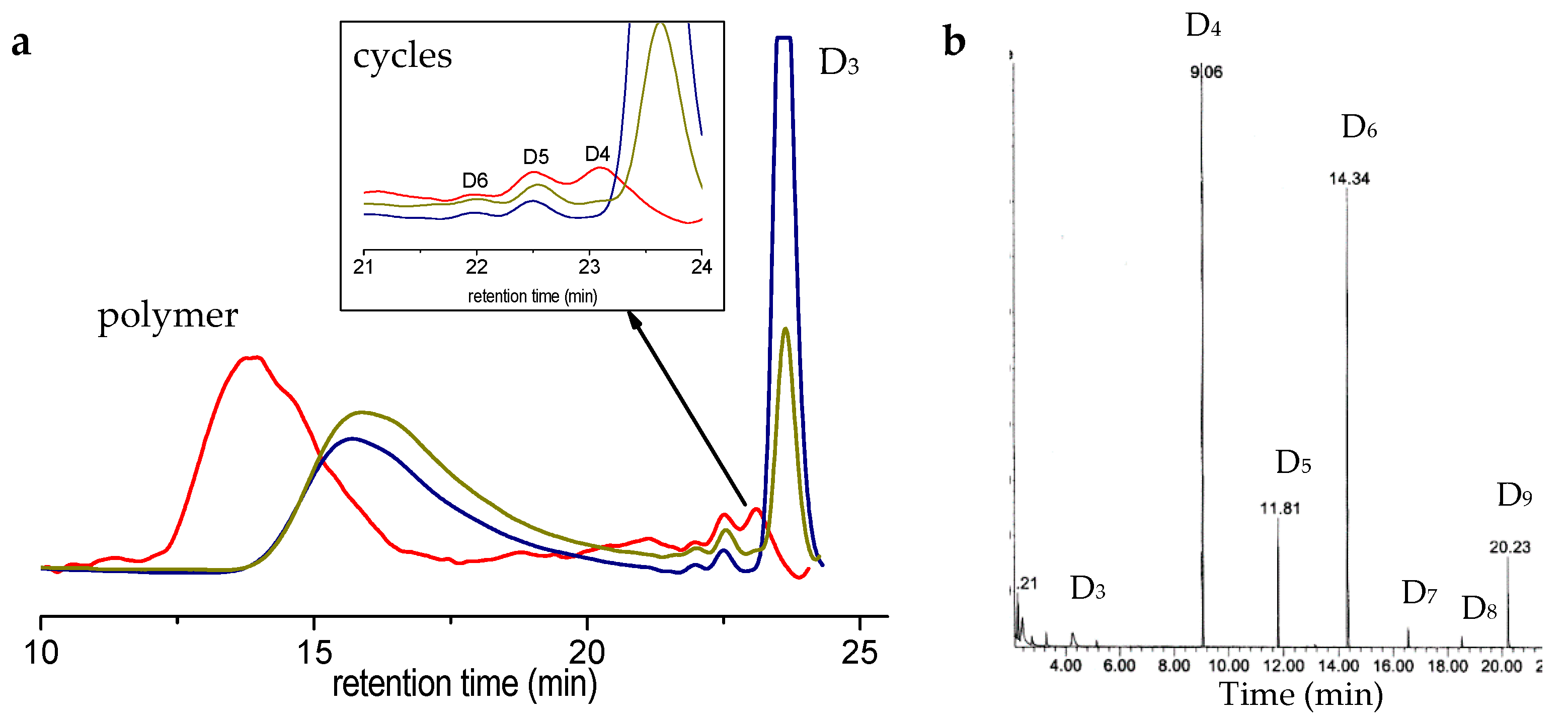
Typical SEC traces are given in
Figure 3a, along with interpretations of it; the average molar masses are reported in
Table 2 at two different reaction times. It can be seen here that polymerization of D
3 occurs quite smoothly, generating polymers of very high molar mass (typically 150,000 g/mol). This is typical of cationic polymerization in emulsion of cyclosiloxanes [
6]. We were not capable of characterizing the chain-end of the polymers of such high molar masses; however, the fact that molar mass increases with time let us think that the polymers chains do not close ends here. Similar results were observed for the system starting from linear precursors [
17].
We can also track, on the SEC trace, a rapid generation of small cycles (typically D
5 and above) which contents grow slowly with time. Intermediate macrocycles are visible on the SEC trace after 3 days of reaction, showing that backbiting and intermolecular redistribution are retarded, but occur in this polymerization system (
Figure 3a). To gain better insight into the course of polymerization, we have injected the intermediate sample in a GC/MS apparatus (
Figure 3b). D
6 and D
9 appear together in larger proportions than other cycles, except for D
4. We observed a similar accumulation of D
3xF cycles building (x = 2,3,4) in the anionic polymerization of D
3F in miniemulsion, before a backbiting reaction occurs extensively and generates intermediate cycles (D
4F, D
5F…), but no macrocycles [
20].
2.3. Introducing Surfactants in the Recipe
As we noted before, the simplest system described above is heterogeneous, i.e., micron-sized droplets are slowly converted into a polymer film, precipitating on the walls of the test tube as a function of time. We tried to use different surfactants to ensure a stabilization of the dispersion throughout the process, while still carrying out the polymerization.
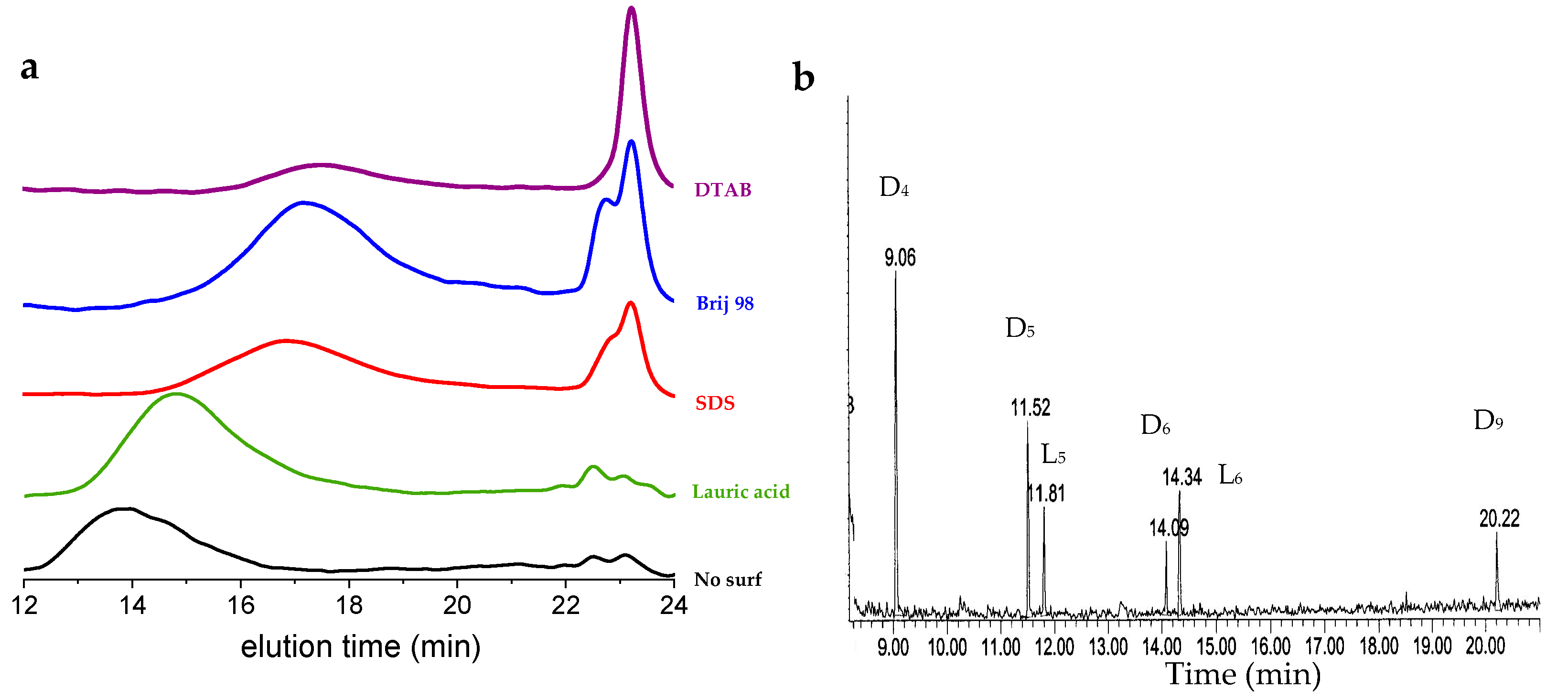
Table 2 summarizes the different trials we made, and
Figure 4 shows the corresponding SEC traces.
Using lauric acid, a translucent dispersion, typical of a microemulsion state, was obtained (average droplet size of 30 nm measured by DLS, not shown). Polymerization appeared to be quite fast, but led to the formation of larger contents of D4 and D5 than without a surfactant (more than 10 wt.%). Lauric acid also allowed rather large molar masses to be produced, around 55,000 g/mol, while keeping the colloidal stability of the dispersion.
With SDS, a similar microemulsion was formed, and polymerization was likely faster than without a surfactant. A polymer of a molar mass of around 15,000 g/mol was produced, together with a large load of small cycles (almost 30 wt.%). The same polymolecularity as for the other rounds was observed, typically around two. DTAB did not produce stable dispersions, and reactions led to a rapid generation of a large load of D4 (above 60%), together with polymers of a low molar mass (13 kg/mol). The fact that only D4 is generated here was not expected, and remains unexplained.
Not shown here is a trial with PVA, where an emulsion was formed, but polymerization did not proceed because of complexation of BCF with the alcohol groups of the dispersant. Brij 98 also complexes the catalyst through the oxygen atoms from ethylene glycol, but does not inhibit polymerization. Molar masses increase with time, together with the content of small cycles from D
4 to macro ones. Looking at the GC/MS of the sample taken after 12 h (
Figure 4b), we can notice the presence of tentatively assigned linear disilanol oligomers, in addition to the same cycles observed before (D
6 and D
9). This confirms the formation of molecular intermediates before they cycle back.
2.4. Proposed Polymerization Scheme
D
3 polymerizes via a cationically catalyzed process in excess water and at room temperature. The initial screening showed that both Bronsted acids and Lewis acids catalyze the reaction, albeit at different paces and for final end results. The fact that triflic acid opens the cycle into small water-soluble oligomers, but does not convert them into polymers, seems to indicate that a condensation reaction of silanols is not likely in these conditions. This is certainly due to the absence of an interface, where this reaction generally takes place [
6,
18]. This also seems to confirm that, in contrast to the previously proposed emulsion polymerization of cyclosiloxane [
5], small hydrophilic oligomers do not chain-extend in water. It would be worth in the future to mix together D
3, DEDMS, and a non-ionic surfactant from the first place, with a view of generating a stable latex while gaining high molar mass silicone polymer.
DBSA and BCF catalysis holds the following comparable features: fast polymerization, large molar mass polymers, and a fair load of small cycles, as expected from such cyclosiloxane cationic process. This most likely shows that BCF acts here principally as a Bronsted catalyst. Ring opening, one to two condensation steps, and back-cyclization, together with true ring-opening polymerization, take place here. When adding lauric acid to the BCF system, an acidic surfactant that is too weak to participate to the reaction, the results match perfectly with the DBSA-catalyzed ones, as follows: molar masses of typically 55 to 70 kg/mol, stable dispersions and cycle contents of 10 wt.%. It would be interesting to introduce both DBSA and BCF in the recipe, to see if synergy occurs; we plan to conduct such process soon.
Introducing other surfactants that could interact together with BCF led to more complex results. In all the cases, larger contents of cycles were observed, and polymerization was generally faster than in the absence of surfactants. Molar masses were rather low, around 15 kg/mol, which may be due to the larger content of water inside the monomer droplets, due to the presence of excess D
4 (this cycle is more polar than silicone chains). The type of dispersions depend on the content and nature of the surfactant, but this was not studied in detail here.
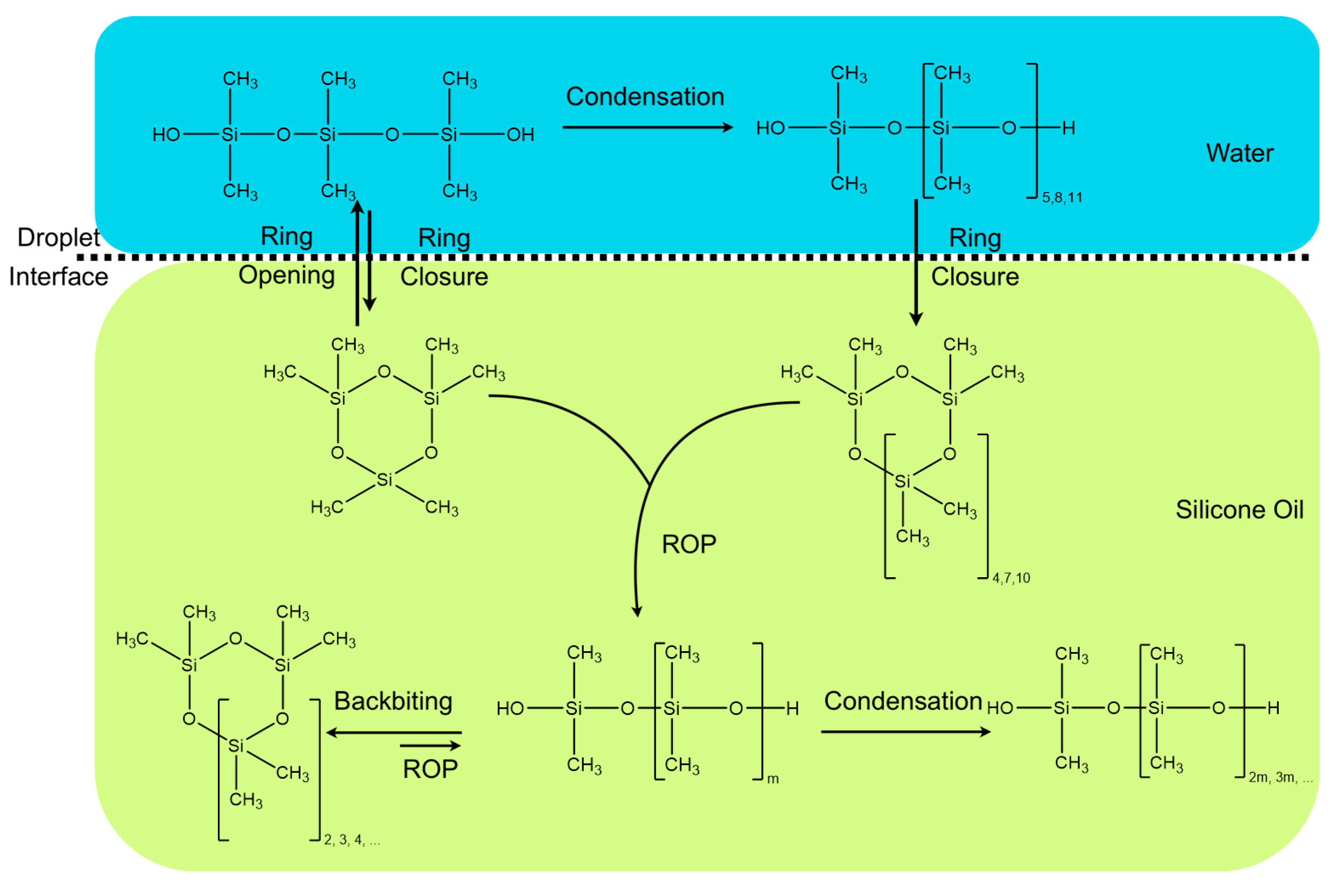
Figure 5 summarizes the reaction taking place in this process for the exemplary case of BCF.
3. Materials and Methods
Hexamethylcyclotrisiloxane (D3, 95%) was either kindly given by Bluestar Silicones (1st set of experiments) or purchased from ABCR (2nd set of experiments). Triflic acid (reagent grade, 98%), 4-DBSA (mixture of isomers, >95%), DTAB (>98%), SDS (98%) and lauric acid (≥98%) were all purchased from Sigma-Aldrich (Saint Louis, MO, USA). Brij 98 (hydroxyl titration: 50 to 65 mg KOH/g) came from ACROS organics (The Hague, Netherlands). Tris(pentafluorophenyl)borane (B(C6F5)3, purity 97%) was obtained from Lancaster (Watd Hill, MA, USA).
Here, two series of experiments were performed at different times and locations. In the first set of experiments, size exclusion chromatography, SEC, was carried out using a Malvern Viscotek (Malvern, UK) GPC Max apparatus equipped with three Shodex columns (KF-804, -805, and -806). Detection systems were a refractive index and differential viscometry detectors. Toluene (HPLC grade, provided by Sigma-Aldrich) was eluted at 1 mL/min with diisopropylethylamine as flow marker [
21]. In the second set of experiments, a Spectra Physics (Andover, MA, USA) apparatus with two PL gel columns (5 µm particles size, 300 mm length, with the following two pores sizes: one with 50 Å and one with 100 Å) and a Styragel HR2 column (7.8 mm internal diameter × 300 mm length) were used. An SP8430 differential refractometer achieved the detection. The toluene was eluted at a flow rate of 0.8 mL/min using diethylether as a flow marker. In both systems, the temperature for the SEC column set and the detector chamber was 35 °C to ensure stable baselines, high chromatographic efficiency, and consistent results. The standards used to calibrate the SEC were polystyrene standards.
Gas chromatography coupled with a mass spectrometer (GC/MS) was done on a 6890 N apparatus from Agilent Technologies (Santa Clara, CA, USA), equipped with an electrospray mass detector Agilent 5973 N and an apolar capillary column HP5-MS 30 m × 0.25 mm (stationary phase made of a film of diphenyldimethylpolysiloxane 5%, 0.25 µm). Conditions used were as follows: initial temperature 45 °C during 2 min, temperature ramp of 2 °C/min up to 50 °C then 10 °C/min up to final temperature, 250 °C, set during 10 min. Peak integration were corrected with factors inherent of each silicone species, according to the procedure published elsewhere [
22].
Particle sizes were determined by dynamic diffraction of a laser beam on a Nanotrac NPA 250 device (Microtrac Inc., Montgomeryville, PA, USA), typically in a size range between 8 nm and 6.54 µm. The light dispersed by the particles entails a Doppler effect, due to Brownian motion. The Microtrac® Windows Software amplified, filtered, and mathematically treated this signal is to produce a size distribution.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}