
Development, Validation, and Application of the LC-MS/MS Method for Determination of 4-Acetamidobenzoic Acid in Pharmacokinetic Pilot Studies in Pigs

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. LC-MS/MS Parameters
2.2. Chromatographic Conditions
2.3. Development of Sample Preparation
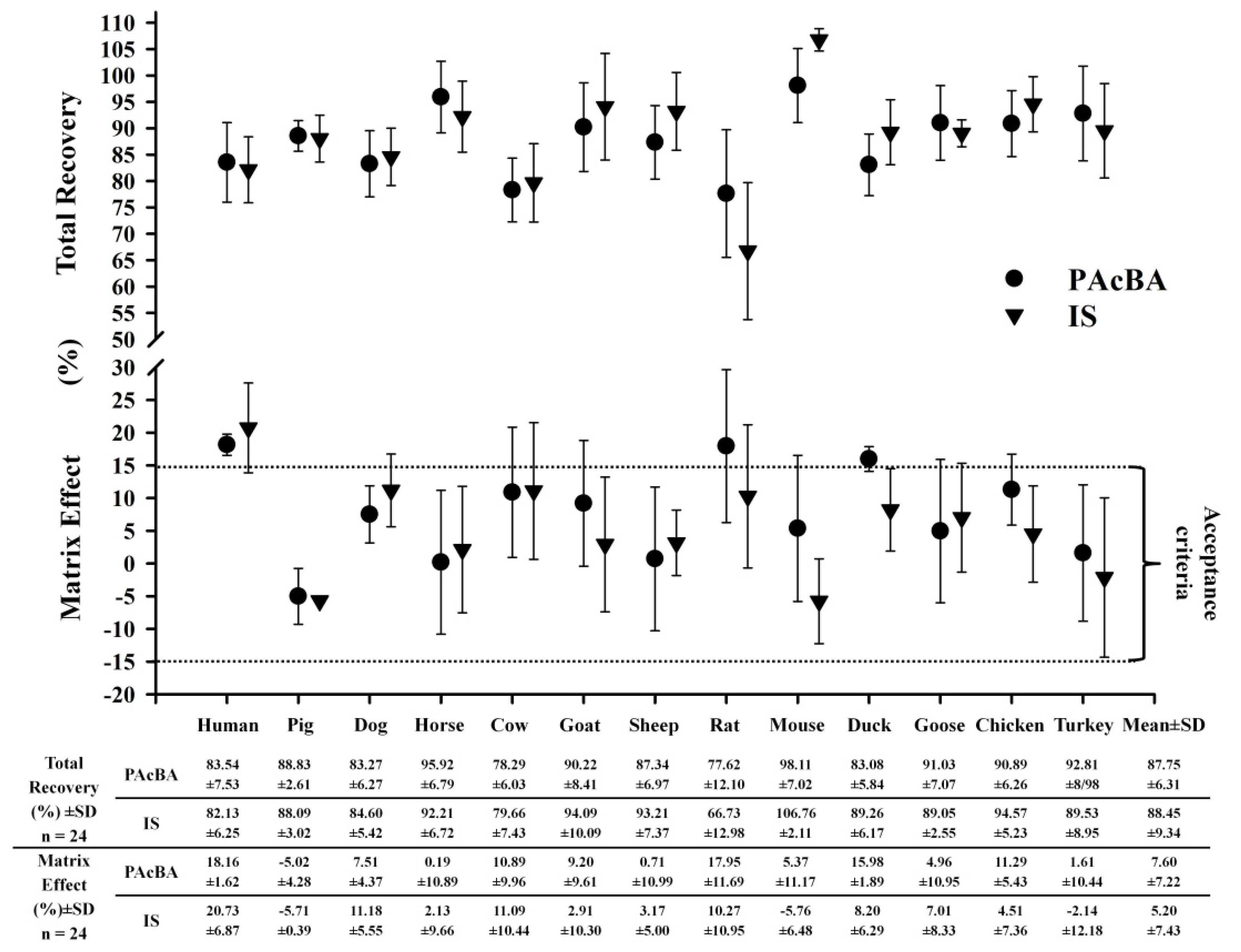
2.4. Validation
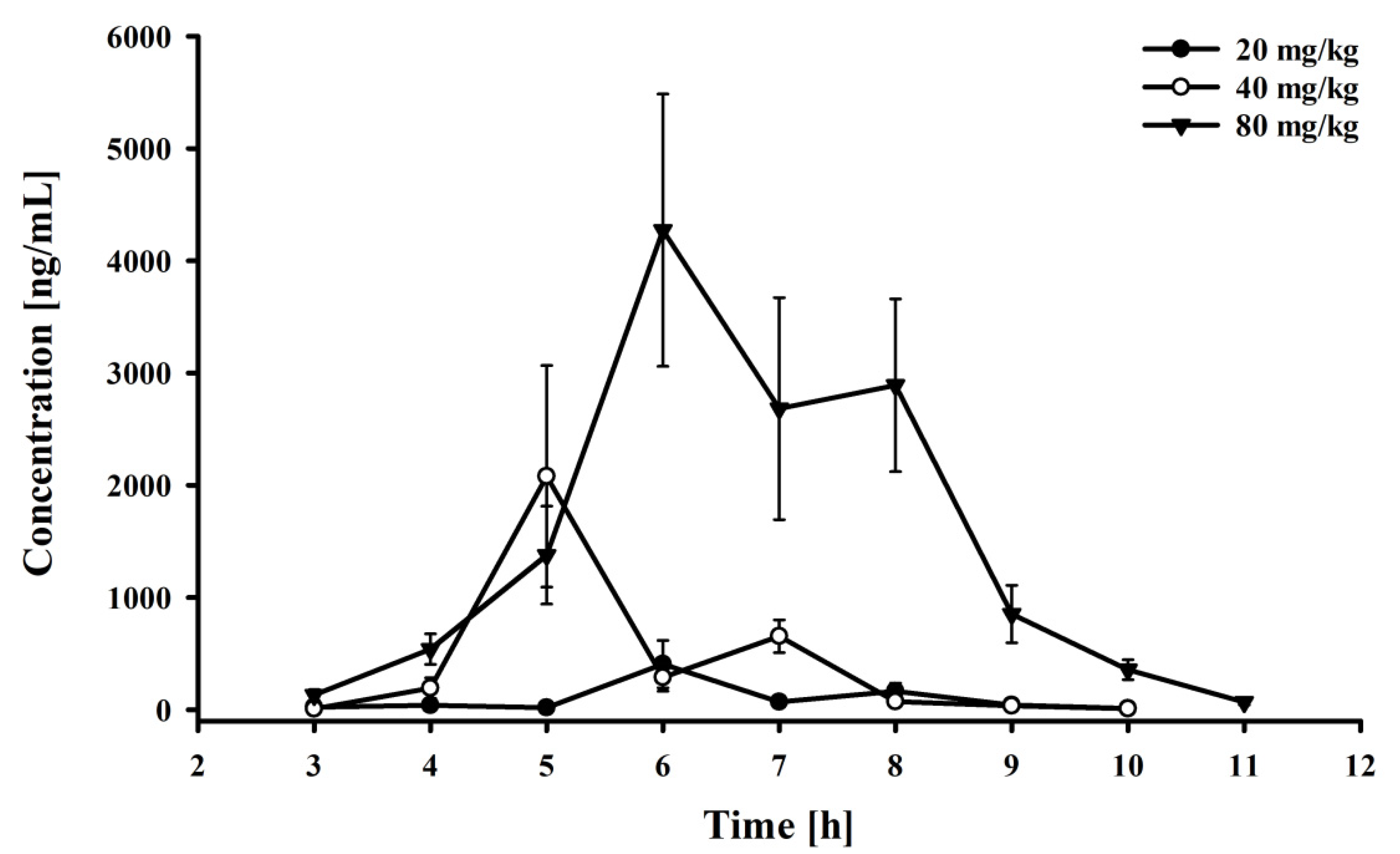
2.5. Pharmacokinetics
3. Materials and Methods
3.1. Animals and Drugs
3.2. Chemicals and Reagents
3.3. Experimental Design
3.4. Chromatography
3.5. Sample Preparation
3.6. Method Validation
3.7. Linearity
3.8. Precision and Accuracy
3.9. Limit of Detection
3.10. Selectivity/Specificity
3.11. Recovery
3.12. Matrix Effect
3.13. Carry Over
3.14. Stability
3.15. Pharmacokinetics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wan, H.; von Lehmann, B.; Riegelman, S. Renal Contribution to Overall Metabolism of Drugs III: Metabolism of p-Aminobenzoic Acid. J. Pharm. Sci. 1972, 61, 1288–1292. [Google Scholar] [CrossRef] [PubMed]
- Campoli-Richards, D.M.; Sorkin, E.M.; Heel, R.C. Inosine pranobex. A preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs 1986, 32, 383–424. [Google Scholar] [CrossRef] [PubMed]
- Majewska, A.; Lasek, W.; Janyst, M.; Młynarczyk, G. Inhibition of adenovirus multiplication by inosine pranobex and interferon α in vitro. Cent. Eur. J. Immunol. 2015, 40, 395–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasek, W.; Janyst, M.; Wolny, R.; Zapała, Ł.; Bocian, K.; Drela, N. Immunomodulatory effects of inosine pranobex on cytokine production by human lymphocytes. Acta Pharm. 2015, 65, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumel, A.S.; Newman, A.S.; O’Daly, J.; Duffy, S.; Grafton, G.; Brady, C.A.; Curnow, J.S.; Barnes, N.M.; Gordon, J. Inosine Acedoben Dimepranol promotes an early and sustained increase in the natural killer cell component of circulating lymphocytes: A clinical trial supporting anti-viral indications. Int. Immunopharmacol. 2017, 42, 108–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Hao, H.; Xie, S.; Wang, X.; Dai, M.; Huang, L.; Yuan, Z. Antibiotic alternatives: The substitution of antibiotics in animal husbandry? Front. Microbiol. 2014, 5, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannucci, L.; Krizan, J.; Sima, P.; Stakheev, D.; Caja, F.; Rajsiglova, L.; Horak, V.; Saieh, M. Immunostimulatory properties and antitumor activities of glucans (Review). Int. J. Oncol. 2013, 43, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Kedl, R.M.; Griesgraber, G.W.; Zarraga, I.A.E.; Wightman, P.D. Immunostimulatory compositions and methods of stimulating an immune response. U.S. Patent 7,427,629 B2, 23 September 2008. [Google Scholar]
- Nielsen, P.; Beckett, A.H. The metabolism and excretion in humans of NN-dimethylamino-isopropanol and p-acetamido-benzoic acid after administration of Isoprinosine. J. Pharm. Pharmacol. 1981, 33, 549–550. [Google Scholar] [CrossRef]
- Chan, K.; Miners, J.O.; Birkett, D.J. Direct and simultaneous high-performance liquid chromatographic assay for the determination of p-aminobenzoic acid and its conjugates in human urine. J. Chromatogr. 1988, 426, 103–109. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, Y.; Que, X.T.; Ding, Y.; Yang, L.; Wen, A.; Hang, T. Pharmacokinetic study of inosiplex tablets in healthy Chinese volunteers by hyphenated HPLC and tandem MS techniques. J. Pharm. Anal. 2013, 3, 387–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EMA. Guideline on Bioanalytical Method Validation EMEA/CHMP/EWP/192217/2009; EMA: Amsterdam, The Netherlands, 2011; pp. 1–25. [Google Scholar]
- FDA. Bioanalytical Method Validation. In Guidance for Industry; FDA: Rockwell, MD, USA, 2013; pp. 1–25. [Google Scholar]
- Streeter, D.G.; Pfadenhauer, E.H. Inosiplex: Metabolism and excretion of the dimethylaminoisopropanol and p-acetamidobenzoic acid components in rhesus monkeys. Drug Metab. Dispos. 1984, 12, 199–203. [Google Scholar] [PubMed]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography-mass spectrometry methods: Part II. Anal. Chim. Acta 2015, 870, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography-mass spectrometry methods: Part I. Anal. Chim. Acta 2015, 870, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Gibaldi, M.; Perrier, D. Noncompartmental Analysis Based on Statistical Moment Theory. In Pharmacokinetics, 2nd ed.; Gibaldi, M., Perrier, D., Eds.; Informa Healthcare: New York, NY, USA, 1982; pp. 409–417. [Google Scholar]

{kind=link}
{kind=link}
| MS/MS Parameters | Compound | |||
|---|---|---|---|---|
| PAcBA | PAcBA-d3 | |||
| Precursor ions (m/z) | 180.2 | 183.2 | ||
| Product ions (m/z) | 94.0 | 95.0 | ||
| Desolvation gas | nitrogen | |||
| Desolvation gas temperature (°C) | 350 | |||
| Desolvation gas flow (L/h) | 800 | |||
| Collision gas | argon | |||
| Source temperature (°C) | 120 | |||
| Gas cell pirani pressure (mbar) | 3.24 × 10−3 | |||
| Electrospray mode | Positive | |||
| Cone voltage (V) | 30 | |||
| Capillary voltage (kV) | 3 | |||
| Collision energy (eV) | 15 | |||
| Dwel (s) | 0.200 | |||
| Delay (s) | 0.010 | |||
| Retention time window (min) | 6.82–7.35 | |||
| Time (min) | Mobile Phase (%) | Curve | Elution (mL/min) | |
| A | B | |||
| 0.00 | 99 | 1 | 1 | 0.40 |
| 6.00 | 30 | 70 | 6 | 0.40 |
| 7.00 | 0 | 100 | 6 | 0.40 |
| 10.00 | 0 | 100 | 6 | 0.40 |
| 11.50 | 99 | 1 | 6 | 0.40 |
| Parameter | Acceptance Criteria | |
|---|---|---|
| Linearity | Calibration points | At least 75% of calibration points, but not less than 6, should have a deviation (residual) between nominal and back-calculated concentrations of ±15% or less |
| Coefficient of determination (r2) | ||
| Relative residuals (Yi) | ||
| SD of Relative residuals (SYi) | ||
| Stability | Stock and working standard | |
| Autosampler | ||
| Freeze and thaw | ||
| Sample processing temperature | ||
| Precision (RSD or CV) | ||
| Accuracy (Deviation) (for at least 5 points per group/day) | ||
| Limit of detection (LOD) | where | |
| The lowest limit of quantitation (LLOQ) with accuracy and precision | where | |
| Matrix Effect | ||
| Total Recovery | ||
| Selectivity/Specificity | No endogenous peaks in retention time of analyte | |
| Carry Over | Area of carried peaks ≤20% of LLOQ area, and for IS, 5% of its area | |
| Linearity a | r2 | I | II | III | IV | Mean | ||
| 0.9992 | 0.9992 | 0.9975 | 0.9988 | 0.9990 | ||||
| Precision (%) and accuracy (%) | LLOQ | LQC | IQC | MQC | HQC | |||
| Intra-day n = 6; 3 repetitions | Precision | I | 5.22 | 3.64 | 6.17 | 4.87 | 2.81 | |
| Accuracy | 4.33 | 2.3 | 3.91 | 3.93 | 2.31 | |||
| Precision | II | 13.71 | 11.37 | 4.45 | 3.45 | 4.65 | ||
| Accuracy | 11.0 | 8.57 | 3.61 | 2.74 | 3.24 | |||
| Precision | III | 13.81 | 5.30 | 3.10 | 2.11 | 3.04 | ||
| Accuracy | 11.0 | 4.47 | 2.38 | 1.43 | 2.49 | |||
| Inter-day n = 18 | Precision | 10.93 | 7.08 | 4.45 | 3.43 | 3.38 | ||
| Accuracy | 8.78 | 5.11 | 3.3 | 2.7 | 2.68 | |||
| LLOQ and LOD c | Concentration b | S/N | ||||||
| LLOQ overall mean n = 18 | 10.00 | 15.69 | ||||||
| LLOQ overall SD n = 18 | 1.09 | 3.95 | ||||||
| LOD overall mean n = 18 | 3.27 | 10.89 | ||||||
| LOD overall SD n = 18 | 1.48 | 6.52 | ||||||
| Carry over | Sample | Peak Area of PAcBA | Peak Area of Mobile Phase | Carry Over (%) | ||||
| Mean | PAcBA | 193,810.4 | 6.27 | 4.69 | ||||
| IS | 24,601.28 | 0 | 0 | |||||
| SD | PAcBA | 2382.238 | 6.93 | 5.19 | ||||
| IS | 218.815 | 0 | 0 | |||||
| Stability | Period (h) | Compound | Decrease/Increase of Quality Control Concentration (%) | |||
|---|---|---|---|---|---|---|
| LQC | IQC | MQC | HQC | |||
| Stock2 °C | 120 | PAcBA | −8.42 | −11.12 | −14.59 | −12.85 |
| IS | −8.74 | −4.17 | −4.99 | −10.21 | ||
| Working standard 2 °C | 72 | PAcBA | −7.64 | 7.81 | 3.72 | 1.71 |
| IS | 8.31 | −2.74 | −4.28 | −11.71 | ||
| 120 | PAcBA | −14.01 | −0.14 | 0.65 | 2.80 | |
| IS | −2.21 | −8.27 | −9.54 | −14.88 | ||
| Autosampler 4 °C | 24 | PAcBA | 2.17 | −0.825 | 0.19 | 1.05 |
| IS | 1.44 | 6.01 | 3.14 | 1.76 | ||
| 48 | PAcBA | 2.03 | 1.14 | 1.35 | 4.31 | |
| IS | 0.60 | −7.85 | −4.09 | −4.53 | ||
| Freeze and thaw −75 °C | 24 | PAcBA | 4.60 | −4.33 | 0.07 | 4.60 |
| IS | −5.17 | 2.04 | −4.63 | −3.09 | ||
| 48 | PAcBA | 4.60 | −3.43 | −1.77 | 1.06 | |
| IS | 9.58 | 4.35 | 5.26 | 1.22 | ||
| 96 | PAcBA | 3.68 | −2.02 | −2.84 | 1.54 | |
| IS | 0.34 | −7.24 | −10.10 | −11.78 | ||
| 1680 | PAcBA | 6.09 | −4.80 | −1.78 | 1.14 | |
| IS | −13.06 | −6.98 | −10.23 | −12.21 | ||
| Sample processing temperature 21 °C | 3 | PAcBA | −3.36 | 0.28 | −0.14 | −0.48 |
| IS | −5.50 | 1.01 | 2.09 | 1.85 | ||
| Pharmacokinetic Parameters | Dose (mg/kg) | ||
|---|---|---|---|
| 20 | 40 | 80 | |
| Mean ± SD | Mean ± SD | Mean ± SD | |
| AUC(0→t) (μg·h/L) | 878.74 ± 372.3 a | 3402.52 ± 1687.26 a | 12,868.1 ± 4896.6 b |
| AUMC(0→t) (μg·h·h/L) | 5935.97 ± 2453 a | 19,015.92 ± 7157.79 a | 88,002.9 ± 38,611.43 b |
| Cmax (ng/mL) | 406.73 ± 211.6 a | 2079.87 ± 787.56 a | 4272.27 ± 1713 b |
| tmax (h) | 6 ± 1 | 5 ± 1.5 | 6 ± 1 |
| Clast (ng/mL) | 12.32 ± 3.89 a | 10.21 ± 2.11 a | 68.11 ± 25.1 b |
| tlast (h) | 11 | 11 | 11 |
| kel (h−1) | 0.49 ± 0.21 | 0.62 ± 0.31 | 0.81 ± 0.37 |
| t1/2kel (h) | 1.42 ± 0.87 | 1.12 ± 0.42 | 0.85 ± 0.29 |
| MRT(0→t) (h) | 6.76 ± 3.21 | 5.59 ± 2.78 | 6.84 ± 3.41 |
| ClB/F (L·h) | 682.8 ± 214.23 a | 352.68 ± 131.2 a,b | 186.51 ± 97.6 b |
| Vdarea/F (L) | 1399.78 ± 731.6 | 568.33 ± 243.65 | 229.56 ± 112.38 |
| kab (h−1) | 0.27 ± 0.11 a | 0.81 ± 0.37 a | 1.96 ± 0.89 b |
| t1/2kab (h) | 2.57 ± 1.21 a | 0.86 ± 0.31 a | 0.36 ± 0.13 b |
| MAT (h) | 3.70 ± 1.7 a | 1.23 ± 0.41 a | 0.51 ± 0.19 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markowska, P.; Procajło, Z.; Wolska, J.; Jaroszewski, J.J.; Ziółkowski, H. Development, Validation, and Application of the LC-MS/MS Method for Determination of 4-Acetamidobenzoic Acid in Pharmacokinetic Pilot Studies in Pigs. Molecules 2021, 26, 4437. https://doi.org/10.3390/molecules26154437
Markowska P, Procajło Z, Wolska J, Jaroszewski JJ, Ziółkowski H. Development, Validation, and Application of the LC-MS/MS Method for Determination of 4-Acetamidobenzoic Acid in Pharmacokinetic Pilot Studies in Pigs. Molecules. 2021; 26(15):4437. https://doi.org/10.3390/molecules26154437
Chicago/Turabian StyleMarkowska, Paulina, Zbigniew Procajło, Joanna Wolska, Jerzy Jan Jaroszewski, and Hubert Ziółkowski. 2021. "Development, Validation, and Application of the LC-MS/MS Method for Determination of 4-Acetamidobenzoic Acid in Pharmacokinetic Pilot Studies in Pigs" Molecules 26, no. 15: 4437. https://doi.org/10.3390/molecules26154437
APA StyleMarkowska, P., Procajło, Z., Wolska, J., Jaroszewski, J. J., & Ziółkowski, H. (2021). Development, Validation, and Application of the LC-MS/MS Method for Determination of 4-Acetamidobenzoic Acid in Pharmacokinetic Pilot Studies in Pigs. Molecules, 26(15), 4437. https://doi.org/10.3390/molecules26154437