
Square-Planar Heteroleptic Complexes of α-Diimine-NiII-Catecholate Type: Intramolecular Ligand-to-Ligand Charge Transfer


 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthetic Procedure for the Complexes 1 and 2
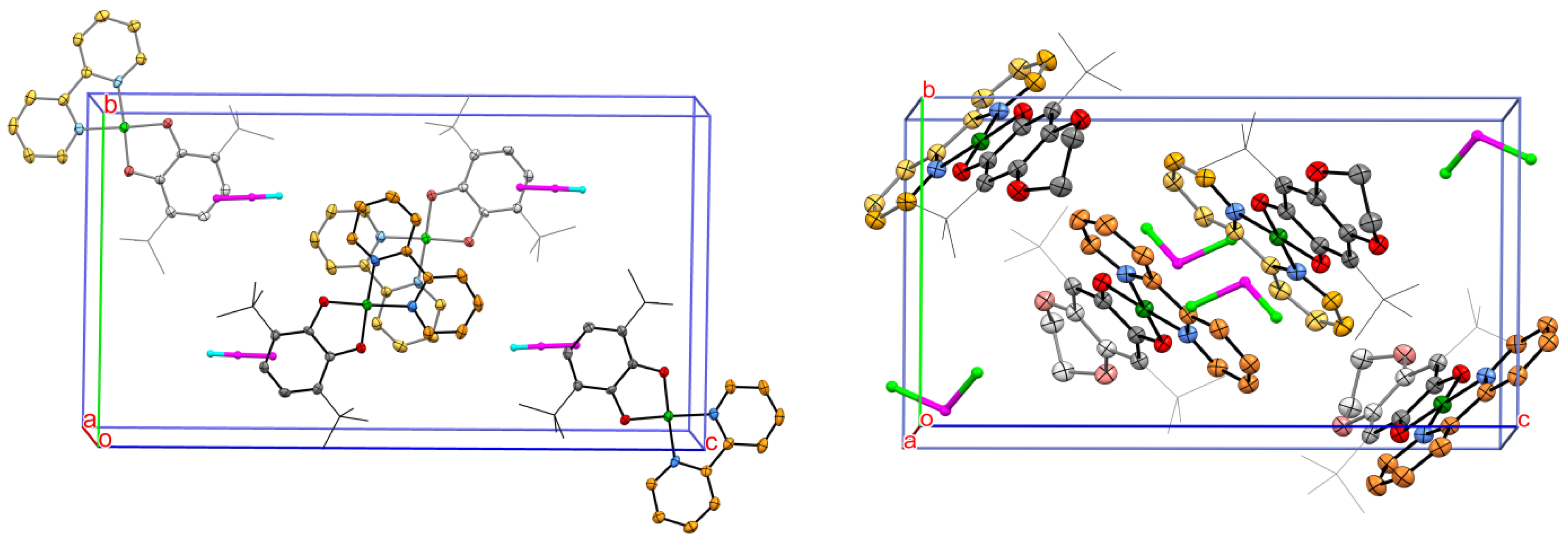
2.2. Molecular Structures of Complexes 1 and 2
2.3. Electrochemical Study of the Complexes 1 and 2
2.4. UV-Vis-NIR Spectroscopy for Complexes 1 and 2
2.5. DFT Calculations
3. Materials and Methods
3.1. Reagents and Methods
3.2. Single-Crystal X-ray Diffraction Studies
3.3. The Synthetic Procedure for the Complexes 1 and 2
3.4. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ward, M.D. Near-infrared electrochromic materials for optical attenuation based on transition-metal coordination complexes. J. Solid State Electrochem. 2005, 9, 778–787. [Google Scholar] [CrossRef]
- Atallah, H.; Taliaferro, C.M.; Wells, K.A.; Castellano, F.N. Photophysics and ultrafast processes in rhenium(I) diimine dicarbonyls. Dalton Trans. 2020, 49, 11565–11576. [Google Scholar] [CrossRef]
- García-Cañadas, J.; Meacham, A.P.; Peter, L.M.; Ward, M.D. A Near-Infrared Electrochromic Window Based on an Sb-Doped SnO2 Electrode Modified with a Ru–Dioxolene Complex. Angew. Chem. Int. Ed. 2003, 42, 3011–3014. [Google Scholar] [CrossRef]
- Mukherjee, S.; Weyhermüller, T.; Bill, E.; Wieghardt, K.; Chaudhuri, P. Tuning of spin transition in radical-containing iron (III) complexes by remote ligand substituents. Inorg. Chem. 2005, 44, 7099–7108. [Google Scholar] [CrossRef]
- Chun, H.; Verani, C.N.; Chaudhuri, P.; Bothe, E.; Bill, E.; Weyhermüller, T.; Wieghardt, K. Molecular and electronic structure of octahedral o-aminophenolato and o-iminobenzosemiquinonato complexes of V (V), Cr (III), Fe (III), and Co (III). Experimental determination of oxidation levels of ligands and metal ions. Inorg. Chem. 2001, 40, 4157–4166. [Google Scholar] [CrossRef]
- Ye, S.; Sarkar, B.; Lissner, F.; Schleid, T.; van Slageren, J.; Fiedler, J.; Kaim, W. Three-Spin System with a Twist: A Bis (semiquinonato) copper Complex with a Nonplanar Configuration at the Copper (II) Center. Angew. Chem. Int. Ed. 2005, 44, 2103–2106. [Google Scholar] [CrossRef] [PubMed]
- Bubrin, M.; Paretzki, A.; Hübner, R.; Beyer, K.; Schwederski, B.; Neugebauer, P.; Zalis, S.; Kaim, W. Probing the Intramolecular Metal-Selenoether Interaction in a Bis (iminosemiquinone) copper (II) Compound. Z. Anorg. Allg. Chem. 2017, 643, 1621–1627. [Google Scholar] [CrossRef] [Green Version]
- Rakshit, R.; Ghorai, S.; Biswas, S.; Mukherjee, C. Effect of ligand substituent coordination on the geometry and the electronic structure of Cu (II)-diradical complexes. Inorg. Chem. 2014, 53, 3333–3337. [Google Scholar] [CrossRef]
- Benedix, R.; Hennig, H.; Kunkely, H.; Vogler, A. Optical ligand-to-ligand charge transfer of Zn(2,2′-bipyridyl)(3,4-toluenedithiolate). Chem. Phys. Lett. 1990, 175, 483–487. [Google Scholar] [CrossRef] [Green Version]
- Cameron, L.A.; Ziller, J.W.; Heyduk, A.F. Near-IR absorbing donor–acceptor ligand-to-ligand charge-transfer complexes of nickel (II). Chem. Sci. 2016, 7, 1807–1814. [Google Scholar] [CrossRef] [Green Version]
- Brasse, M.; Campora, J.; Davies, M.; Teuma, E.; Palma, P.; Alvarez, E.; Sanz, E.; Reyes, M.L. Integrating Catalyst and Co-Catalyst Design in Olefin Polymerization Catalysis: Transferable Dianionic Ligands for the Activation of Late Transition Metal Polymerization Catalysts. Adv. Synth. Catal. 2007, 349, 2111–2120. [Google Scholar] [CrossRef]
- Tahara, K.; Ashihara, Y.; Higashino, T.; Ozawa, Y.; Kadoya, T.; Sugimoto, K.; Ueda, A.; Mori, H.; Abe, M. New π-extended catecholato complexes of Pt(II) and Pd(II) containing a benzothienobenzothiophene (BTBT) moiety: Synthesis, electrochemical behavior and charge transfer properties. Dalton Trans. 2019, 48, 7367–7377. [Google Scholar] [CrossRef]
- BaniKhaled, M.O.; Becker, J.D.; Koppang, M.; Sun, H. Perfluoroalkylation of Square-Planar Transition Metal Complexes: A Strategy To Assemble Them into Solid State Materials with a π–π Stacked Lamellar Structure. Cryst. Growth Des. 2016, 16, 1869–1878. [Google Scholar] [CrossRef]
- Yang, H.; Zhao, Y.; Liu, B.; Su, J.-H.; Fedushkin, I.L.; Wu, B.; Yang, X.-J. Noninnocent ligands: Heteroleptic nickel complexes with α-diimine and 1,2-diketone derivatives. Dalton Trans. 2017, 46, 7857–7865. [Google Scholar] [CrossRef]
- Kramer, W.W.; Cameron, L.A.; Zarkesh, R.A.; Ziller, J.W.; Heyduk, A.F. Donor–Acceptor Ligand-to-Ligand Charge-Transfer Coordination Complexes of Nickel (II). Inorg. Chem. 2014, 53, 8825–8837. [Google Scholar] [CrossRef]
- Bubnov, M.P.; Teplova, I.A.; Druzhkov, N.O.; Fukin, G.K.; Cherkasova, A.V.; Cherkasov, V.K. Catecholato complexes of cobalt and nickel with 1,4-disubstituted-1,4-diazabutadiens-1,3 and 1,2-bis(diphenylphosphino)ethane. J. Chem. Sci. 2015, 127, 527–535. [Google Scholar] [CrossRef] [Green Version]
- Yamada, S.; Matsumoto, T.; Chang, H.C. Impact of Group 10 Metals on the Solvent-Induced Disproportionation of o-Semiquinonato Complexes. Chem. Eur. J. 2019, 25, 8268–8278. [Google Scholar] [CrossRef]
- Kokatam, S.L.; Chaudhuri, P.; Weyhermüller, T.; Wieghardt, K. Molecular and electronic structure of square planar complexes [PdII(tbpy)(LIPN,O)]0, [PdII(tbpy)(LISQN,O)](PF6), and [PdII(tbpy)(LIBQN,O)](PF6)(BF4)·2CH2Cl2: An o-iminophenolato based ligand centered, three-membered redox series. Dalton Trans. 2007, 3, 373–378. [Google Scholar] [CrossRef]
- Best, J.; Sazanovich, I.V.; Adams, H.; Bennett, R.D.; Davies, E.S.; Meijer, A.J.H.M.; Towrie, M.; Tikhomirov, S.A.; Bouganov, O.V.; Ward, M.D. Structure and ultrafast dynamics of the charge-transfer excited state and redox activity of the ground state of mono-and binuclear platinum (II) diimine catecholate and bis-catecholate complexes: A transient absorption, TRIR, DFT, and electrochemical study. Inorg. Chem. 2010, 49, 10041–10056. [Google Scholar] [CrossRef]
- Heinze, K.; Reinhardt, S. Platinum (II) Complexes with Non-Innocent Ligands: Solid-Phase Synthesis, Redox Chemistry and Luminescence. Chem. Eur. J. 2008, 14, 9482–9486. [Google Scholar] [CrossRef]
- Sobottka, S.; Nößler, M.; Ostericher, A.L.; Hermann, G.; Subat, N.Z.; Beerhues, J.; Behr-van der Meer, M.; Suntrup, L.; Albold, U.; Hohloch, S. Tuning PtII-Based Donor–Acceptor Systems through Ligand Design: Effects on Frontier Orbitals, Redox Potentials, UV/Vis/NIR Absorptions, Electrochromism, and Photocatalysis. Chem. Eur. J. 2020, 26, 1314–1327. [Google Scholar] [CrossRef]
- Scattergood, P.A.; Jesus, P.; Adams, H.; Delor, M.; Sazanovich, I.V.; Burrows, H.D.; Serpa, C.; Weinstein, J.A. Exploring excited states of Pt(II) diimine catecholates for photoinduced charge separation. Dalton Trans. 2015, 44, 11705–11716. [Google Scholar] [CrossRef] [Green Version]
- Shavaleev, N.M.; Davies, E.S.; Adams, H.; Best, J.; Weinstein, J.A. Platinum (II) diimine complexes with catecholate ligands bearing imide electron-acceptor groups: Synthesis, crystal structures,(spectro) electrochemical and EPR studies, and electronic structure. Inorg. Chem. 2008, 47, 1532–1547. [Google Scholar] [CrossRef]
- Yang, J.; Kersi, D.K.; Giles, L.J.; Stein, B.W.; Feng, C.; Tichnell, C.R.; Shultz, D.A.; Kirk, M.L. Ligand control of donor–acceptor excited-state lifetimes. Inorg. Chem. 2014, 53, 4791–4793. [Google Scholar] [CrossRef] [PubMed]
- Okabe, N.; Hagihara, K.; Odoko, M.; Muranishi, Y. (2,2′-Bipyridine-κ2N,N′)(2,3-naphthalenediolato-κ2O,O′) palladium(II) and (2,2′-biquinoline-κ2N,N′)(2,3-naphthalenediolato-κ2O,O′) palladium(II). Acta Crystallogr. 2004, C60, m150–m152. [Google Scholar] [CrossRef] [PubMed]
- Okabe, N.; Muranishi, Y.; Aziyama, T. (1,2-Benzenediolato-κ2O,O′)(1,10-phenanthroline-κ2N,N′) palladium(II) dihydrate. Acta Crystallogr. 2003, E59, m936–m938. [Google Scholar] [CrossRef]
- Okabe, N.; Aziyama, T.; Odoko, M. (1,2-Benzenediolato-κ2O,O′)(2,2′-biquinoline-κ2N, N′) palladium (II). Acta Crystallogr. 2005, E61, m2154–m2156. [Google Scholar] [CrossRef]
- Pierpont, C.G. Ligand redox activity and mixed valency in first-row transition-metal complexes containing tetrachlorocatecholate and radical tetrachlorosemiquinonate ligands. Inorg. Chem. 2011, 50, 9766–9772. [Google Scholar] [CrossRef]
- Brown, S.N. Metrical Oxidation States of 2-Amidophenoxide and Catecholate Ligands: Structural Signatures of Metal–Ligand π Bonding in Potentially Noninnocent Ligands. Inorg. Chem. 2012, 51, 1251–1260. [Google Scholar] [CrossRef]
- Poddel’sky, A.I.; Cherkasov, V.K.; Abakumov, G.A. Transition metal complexes with bulky 4,6-di-tert-butyl-N-aryl(alkyl)-o-iminobenzoquinonato ligands: Structure, EPR and magnetism. Coord. Chem. Rev. 2009, 253, 291–324. [Google Scholar] [CrossRef]
- Kaim, W.; Paretzki, A. Interacting metal and ligand based open shell systems: Challenges for experiment and theory. Coord. Chem. Rev. 2017, 344, 345–354. [Google Scholar] [CrossRef]
- Mao, G.; Song, Y.; Hao, T.; Li, Y.; Xu, T.; Zhang, H.; Jiang, T. Progress in the research of radical anion ligands and their complexes. Chin. Sci. Bull. 2014, 59, 2936–2944. [Google Scholar] [CrossRef]
- Kaim, W. Manifestations of noninnocent ligand behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef] [PubMed]
- Butin, K.P.; Beloglazkina, E.K.; Zyk, N.V. Metal complexes with non-innocent ligands. Russ. Chem. Rev. 2005, 74, 531–553. [Google Scholar] [CrossRef]
- Ghosh, P.; Begum, A.; Herebian, D.; Bothe, E.; Hildenbrand, K.; Weyhermüller, T.; Wieghardt, K. Coordinated o-Dithio-and o-Iminothiobenzosemiquinonate(1−) π-Radicals in [MII(bpy)(L.)](PF6) Complexes. Angew. Chem. Int. Ed. 2003, 42, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Deibel, N.; Schweinfurth, D.; Fiedler, J.; Záliš, S.; Sarkar, B. Isomeric separation in donor–acceptor systems of Pd(II) and Pt(II) and a combined structural, electrochemical and spectroelectrochemical study. Dalton Trans. 2011, 40, 9925–9934. [Google Scholar] [CrossRef] [Green Version]
- Rauth, G.K.; Pal, S.; Das, D.; Sinha, C.; Slawin, A.M.; Woollins, J.D. Synthesis, spectral characterization and electrochemical studies of mixed-ligand complexes of platinum (II) with 2-(arylazo) pyridines and catechols. Single-crystal X-ray structure of dichloro {2-(phenylazo) pyridine} platinum (II). Polyhedron 2001, 20, 363–372. [Google Scholar] [CrossRef]
- Roy, R.; Chattopadhyay, P.; Sinha, C.; Chattopadhyay, S. Synthesis, spectral and electrochemical studies of arylazopyridine complexes of palladium (II) with dioxolenes. Polyhedron 1996, 15, 3361–3369. [Google Scholar] [CrossRef]
- Liu, W.; Heinze, K. Rhenium (I) and platinum (II) complexes with diimine ligands bearing acidic phenol substituents: Hydrogen-bonding, acid–base chemistry and optical properties. Dalton Trans. 2010, 39, 9554–9564. [Google Scholar] [CrossRef]
- Mansuri-Torshizi, H.; Ghadimy, S.; Akbarzadeh, N. Synthesis, characterization, DNA binding and cytotoxic studies of platinum (II) and palladium (II) complexes of the 2, 2’-bipyridine and an anion of 1, 1-cyclobutanedicarboxylic acid. Chem. Pharm. Bull. 2001, 49, 1517–1520. [Google Scholar] [CrossRef] [Green Version]
- Afrasiabi, Z.; Sinn, E.; Chen, J.; Ma, Y.; Rheingold, A.L.; Zakharov, L.N.; Rath, N.; Padhye, S. Appended 1,2-naphthoquinones as anticancer agents 1: Synthesis, structural, spectral and antitumor activities of ortho-naphthaquinone thiosemicarbazone and its transition metal complexes. Inorg. Chim. Acta 2004, 357, 271–278. [Google Scholar] [CrossRef]
- Rebolledo, A.P.; Vieites, M.; Gambino, D.; Piro, O.E.; Castellano, E.E.; Zani, C.L.; Souza-Fagundes, E.M.; Teixiera, L.R.; Batista, A.A.; Beraldo, H. Palladium (II) complexes of 2-benzoylpyridine-derived thiosemicarbazones: Spectral characterization, structural studies and cytotoxic activity. J. Inorg. Biochem. 2005, 99, 698–706. [Google Scholar] [CrossRef]
- Padhye, S.; Afrasiabi, Z.; Sinn, E.; Fok, J.; Mehta, K.; Rath, N. Antitumor metallothiosemicarbazonates: Structure and antitumor activity of palladium complex of phenanthrenequinone thiosemicarbazone. Inorg. Chem. 2005, 44, 1154–1156. [Google Scholar] [CrossRef]
- Okabe, N.; Muranishi, Y.; Aziyama, T. (Benzene-1,2-diolato-κ2O,O′)(di-2-pyridylamine-κ2N,N′) palladium(II) hemihydrate. Acta Crystallogr. 2006, E62, m2778–m2780. [Google Scholar]
- Wang, Y.; Mizubayashi, Y.; Odoko, M.; Okabe, N. (Di-2-pyridylamine-κ2N,N′)(naphthalene-2,3-diolato-κ2O,O′) palladium(II) monohydrate and (di-2-pyridylamine-κ2N,N′)(3-oxidonaphthalene-2-carboxylato-κ2O,O′)palladium(II). Acta Crystallogr. 2005, C61, m67–m70. [Google Scholar] [CrossRef] [Green Version]
- Okabe, N.; Aziyama, T.; Odoko, M. (Benzene-1,2-diolato-κ2O,O′)(2,2′-bipyridine-κ2N,N′) palladium(II). Acta Crystallogr. 2005, E61, m1943–m1945. [Google Scholar] [CrossRef]
- Giribabu, L.; Kanaparthi, R.K.; Velkannan, V. Molecular engineering of sensitizers for dye-sensitized solar cell applications. Chem. Rec. 2012, 12, 306–328. [Google Scholar] [CrossRef]
- Sekar, N.; Gehlot, V.Y. Metal complex dyes for dye-sensitized solar cells: Recent developments. Resonance 2010, 15, 819–831. [Google Scholar] [CrossRef]
- Michaels, H.; Benesperi, I.; Edvinsson, T.; Muñoz-Garcia, A.B.; Pavone, M.; Boschloo, G.; Freitag, M. Copper complexes with tetradentate ligands for enhanced charge transport in dye-sensitized solar cells. Inorganics 2018, 6, 53. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.A.; Besedina, M.A.; Karushev, M.P.; Vasil’ev, V.V.; Timonov, A.M. Photogalvanic and photovoltaic effects in systems based on metal complexes of Schiff bases. Russ. J. Phys. Chem. A 2016, 90, 1088–1094. [Google Scholar] [CrossRef]
- O’regan, B.; Grätzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Grätzel, M. Solar energy conversion by dye-sensitized photovoltaic cells. Inorg. Chem. 2005, 44, 6841–6851. [Google Scholar] [CrossRef]
- Imer, A.G.; Syan, R.H.B.; Gülcan, M.; Ocak, Y.S.; Tombak, A. The novel pyridine based symmetrical Schiff base ligand and its transition metal complexes: Synthesis, spectral definitions and application in dye sensitized solar cells (DSSCs). J. Mater. Sci. Mater. Electron. 2018, 29, 898–905. [Google Scholar] [CrossRef]
- Neuthe, K.; Popeney, C.S.; Bialecka, K.; Hinsch, A.; Sokolowski, A.; Veurmann, W.; Haag, R. Simple NIR complexes and their applicability in dye-sensitized solar cells. Polyhedron 2014, 81, 583–587. [Google Scholar] [CrossRef]
- Gauthier, S.; Caro, B.; Robin-Le Guen, F.; Bhuvanesh, N.; Gladysz, J.A.; Wojcik, L.; Le Poul, N.; Planchat, A.; Pellegrin, Y.; Blart, E.; et al. Synthesis, photovoltaic performances and TD-DFT modeling of push–pull diacetylide platinum complexes in TiO2 based dye-sensitized solar cells. Dalton Trans. 2014, 43, 11233–11242. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, N.; Ho, C.L.; Fu, Y.; Lau, W.S.; Xie, Z.; Wang, L.; Wong, W.Y. Synthesis, characterization, photophysical and photovoltaic properties of new donor–acceptor platinum (II) acetylide complexes. J. Organomet. Chem. 2016, 812, 2–12. [Google Scholar] [CrossRef]
- Islam, A.; Sugihara, H.; Hara, K.; Singh, L.P.; Katoh, R.; Yanagida, M.; Takahashi, Y.; Murata, S.; Arakawa, H. New platinum(II) polypyridyl photosensitizers for TiO2 solar cells. New J. Chem. 2000, 24, 343–345. [Google Scholar] [CrossRef]
- Kee, J.W.; Ng, Y.Y.; Kulkarni, S.A.; Muduli, S.K.; Xu, K.; Ganguly, R.; Lu, Y.; Hirao, H.; Soo, H.S. Development of bis (arylimino) acenaphthene (BIAN) copper complexes as visible light harvesters for potential photovoltaic applications. Inorg. Chem. Front. 2016, 3, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Miao, Q.; Gao, J.; Wang, Z.; Yu, H.; Luo, Y.; Ma, T. Syntheses and characterization of several nickel bis (dithiolene) complexes with strong and broad Near-IR absorption. Inorg. Chim. Acta 2011, 376, 619–627. [Google Scholar] [CrossRef]
- Linfoot, C.L.; Richardson, P.; McCall, K.L.; Durrant, J.R.; Morandeira, A.; Robertson, N. A nickel-complex sensitiser for dye-sensitised solar cells. Sol. Energy 2011, 85, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Son, H.J.; Wang, W.; Xu, T.; Liang, Y.; Wu, Y.; Li, G.; Yu, L. Synthesis of fluorinated polythienothiophene-co-benzodithiophenes and effect of fluorination on the photovoltaic properties. J. Am. Chem. Soc. 2011, 133, 1885–1894. [Google Scholar] [CrossRef]
- Bubnov, M.P.; Teplova, I.A.; Cherkasova, A.V.; Baranov, E.V.; Fukin, G.K.; Romanenko, G.V.; Bogomyakov, A.S.; Starikov, A.G.; Cherkasov, V.K.; Abakumov, G.A. Metal-ligand ferromagnetic exchange interactions in heteroligand bis-o-semiquinonato nickel complexes with 2, 2′-dipyridine and 1, 10-phenanthroline. Polyhedron 2019, 158, 262–269. [Google Scholar] [CrossRef]
- Sarkar, B.; Hübner, R.; Pattacini, R.; Hartenbach, I. Combining two non-innocent ligands in isomeric complexes [Pt(pap)mQn]0 (pap = phenylazopyridine, Q = 3,5-di-tert-butyl-benzoquinone). Dalton Trans. 2009, 24, 4653–4655. [Google Scholar] [CrossRef] [PubMed]
- Deibel, N.; Schweinfurth, D.; Hohloch, S.; Fiedler, J.; Sarkar, B. Donor–acceptor systems of Pt(II) and redox-induced reactivity towards small molecules. Chem. Commun. 2012, 48, 2388–2390. [Google Scholar] [CrossRef]
- Okuniewski, A.; Rosiak, D.; Chojnacki, J.; Becker, B. Coordination polymers and molecular structures among complexes of mercury(II) halides with selected 1-benzoylthioureas. Polyhedron 2015, 90, 47–57. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalton Trans. 2007, 90, 955–964. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Reichardt, C., Welton, T., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Gordon, A.; Ford, R. The Chemist’s Companion; Wiley: New York, NY, USA, 1972; p. 143. [Google Scholar]
- Fukin, G.K.; Cherkasov, A.V.; Shurygina, M.P.; Druzhkov, N.O.; Kuropatov, V.A.; Chesnokov, S.A.; Abakumov, G.A. Geometrical and energetical aspects of structure of 3,6-di-tert-butyl-o-benzoquinones. Struct. Chem. 2010, 21, 607–611. [Google Scholar] [CrossRef]
- Bruker. APEX3, SAINT and SADABS; Bruker AXS Inc.: Madison, WI, USA, 2016. [Google Scholar]
- Svetogorov, R.D.; Dorovatovskii, P.V.; Lazarenko, V.A. Belok/XSA diffraction beamline for studying crystalline samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. 2010, D66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallorg. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.; Scalmani, R.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09. Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Shangin, P.G.; Krylova, I.V.; Lalov, A.V.; Kozmenkova, A.Y.; Saverina, E.A.; Buikin, P.A.; Korlyukov, A.A.; Starikova, A.A.; Nikolaevskaya, E.N.; Egorov, M.P.; et al. Supramolecular D/A-layered structures based on germanium complexes with 2,3-dihydroxynaphthalene and N,N-bidentate ligands. RSC Adv. 2021, 11, 21527–21536. [Google Scholar] [CrossRef]
- Arsenyeva, K.V.; Pashanova, K.I.; Trofimova, O.Y.; Ershova, I.V.; Chegerev, M.G.; Starikova, A.A.; Cherkasov, A.V.; Syroeshkin, M.A.; Kozmenkova, A.Y.; Piskunov, A.V. O,N-Heterocyclic germylenes as efficient catalysts for hydroboration and cyanosilylation of benzaldehyde. New J. Chem. 2021, 45, 11758–11767. [Google Scholar] [CrossRef]
- Chemcraft, Version 1.8. 2014. Available online: http://www.chemcraftprog.com (accessed on 20 July 2021).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | 1 | 2 |
|---|---|---|
| Ni(1)–O(1) | 1.814(3) | 1.818(2) |
| Ni(1)–O(2) | 1.815(3) | 1.814(3) |
| Ni(1)–N(1) | 1.882(4) | 1.883(3) |
| Ni(1)–N(2) | 1.891(3) | 1.887(3) |
| O(1)–C(1) | 1.348(5) | 1.368(4) |
| O(2)–C(6) | 1.365(5) | 1.360(4) |
| C(1)–C(2) | 1.422(6) | 1.410(4) |
| C(2)–C(3) | 1.398(6) | 1.408(5) |
| C(3)–C(4) | 1.379(6) | 1.410(5) |
| C(4)–C(5) | 1.405(6) | 1.410(5) |
| C(5)–C(6) | 1.406(6) | 1.400(5) |
| C(1)–C(6) | 1.397(6) | 1.406(5) |
| N(1)–C(15) | 1.352(5) | 1.335(5) |
| N(1)–C(19) | 1.360(5) | 1.357(4) |
| N(2)–C(20) | 1.365(5) | 1.363(4) |
| N(2)–C(24) | 1.333(6) | 1.337(5) |
| Angle | 1 | 2 |
| O(1)–Ni(1)–O(2) | 88.78(14) | 88.43(11) |
| O(1)–Ni(1)–N(1) | 177.57(15) | 178.39(13) |
| O(2)–Ni(1)–N(1) | 93.46(15) | 93.17(13) |
| O(1)–Ni(1)–N(2) | 93.73(16) | 94.75(12) |
| O(2)–Ni(1)–N(2) | 177.49(17) | 176.78(13) |
| N(1)–Ni(1)–N(2) | 84.03(17) | 83.65(13) |
| Compound | E1/2Red.1, V | E1/2Ox.1, V | E1/2Ox.2, V |
|---|---|---|---|
| 1 | −1.86 | −0.21 | −0.03 |
| 2 | −1.86 | −0.54 | −0.21 |
| [Pd] [35] | −1.80 | −0.18 | 0.70 |
| Solvent | 1 | 2 | Shift (2-1) | ||||
|---|---|---|---|---|---|---|---|
| Shift (hyp) | λmax, nm (ε, 103 M−1·cm−1) | E, eV | λmax, nm (ε, 103 M−1·cm−1) | E, eV | Δλ2-1, nm | ΔE2-1, eV | |
| DMF | 580 (3.50) | 2.138 | 618 (3.08) | 2.007 | 38 | 0.131 | |
| CH2Cl2 | 610 (4.35) | 2.033 | 685 (4.04) | 1.810 | 75 | 0.223 | |
| THF | 640 (3.43) | 1.938 | 720 (3.18) | 1.722 | 80 | 0.216 | |
| Toluene | 715 (3.57) | 1.734 | 810 (3.38) | 1.531 | 95 | 0.203 | |
| Benzyl alcohol | 580 (3.21) | 2.138 | 636 (2.82) | 1.950 | 56 | 0.188 | |
| Δλhyp, nm | 135 | 192 | |||||
| ΔEhyp, eV | 0.404 | 0.476 | |||||
| Parameter | 1∙CH3CN | 2∙CH2Cl2 |
|---|---|---|
| Formula | C26H31N3NiO2 | C27H32Cl2N2NiO4 |
| Formula weight | 476.25 | 578.15 |
| Temperature (K) | 100(2) | 100(2) |
| Radiation source | microsource | synchrotron |
| wavelength, Å | 0.71073 | 0.74539 |
| colour, habit | blue, needle | green, plate |
| Crystal size, mm | 0.140 × 0.030 × 0.015 | 0.200 × 0.080 × 0.015 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c |
| Unit cell dimentions | ||
| a, Å | 6.8087(13) | 16.283(7) |
| b, Å | 13.788(3) | 9.7500(5) |
| c, Å | 25.089(4) | 17.165(3) |
| β, deg | 90.403(5) | 99.12(3) |
| V, Å3 | 2355.3(8) | 2690.6(13) |
| Z | 4 | 4 |
| density (calcd), g/cm3 | 1.343 | 1.427 |
| Absorption coefficient (mm−1) | 0.851 | 1.078 |
| F(000) | 1008 | 1208 |
| θ range, deg | 2.195–25.058 | 1.328–31.018 |
| Index ranges | −8 ≤ h ≤ 8, −15 ≤ k ≤ 16, −29 ≤ l ≤ 29 | −22 ≤ h ≤ 22, −13 ≤ k ≤ 13, −23 ≤ l ≤ 23 |
| Reflections collected | 15,342 | 31,754 |
| Independent reflections | 4087 [Rint = 0.1175] | 7442 [Rint = 0.1080] |
| Data/restraints/parameters | 4087/0/296 | 7442/0/332 |
| R1, wR2 [I > 2σ(I)] | 0.0599, 0.0963 | 0.0607, 0.1409 |
| R1, wR2 (all data) | 0.1428, 0.1176 | 0.1163, 0.1675 |
| goodness-of-fit on F2 | 0.989 | 1.034 |
| Tmin/Tmax | 0.5897/0.7454 | 0.001/1.000 |
| Extinction coefficient | − | 0.0024(5) |
| Δρmax/Δρmin (e/Å3) | 0.523/−0.492 | 0.679/−1.348 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pashanova, K.I.; Bitkina, V.O.; Yakushev, I.A.; Arsenyev, M.V.; Piskunov, A.V. Square-Planar Heteroleptic Complexes of α-Diimine-NiII-Catecholate Type: Intramolecular Ligand-to-Ligand Charge Transfer. Molecules 2021, 26, 4622. https://doi.org/10.3390/molecules26154622
Pashanova KI, Bitkina VO, Yakushev IA, Arsenyev MV, Piskunov AV. Square-Planar Heteroleptic Complexes of α-Diimine-NiII-Catecholate Type: Intramolecular Ligand-to-Ligand Charge Transfer. Molecules. 2021; 26(15):4622. https://doi.org/10.3390/molecules26154622
Chicago/Turabian StylePashanova, Kira I., Vladlena O. Bitkina, Ilya A. Yakushev, Maxim V. Arsenyev, and Alexandr V. Piskunov. 2021. "Square-Planar Heteroleptic Complexes of α-Diimine-NiII-Catecholate Type: Intramolecular Ligand-to-Ligand Charge Transfer" Molecules 26, no. 15: 4622. https://doi.org/10.3390/molecules26154622
APA StylePashanova, K. I., Bitkina, V. O., Yakushev, I. A., Arsenyev, M. V., & Piskunov, A. V. (2021). Square-Planar Heteroleptic Complexes of α-Diimine-NiII-Catecholate Type: Intramolecular Ligand-to-Ligand Charge Transfer. Molecules, 26(15), 4622. https://doi.org/10.3390/molecules26154622