
Noscapine Prevents Rotenone-Induced Neurotoxicity: Involvement of Oxidative Stress, Neuroinflammation and Autophagy Pathways

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
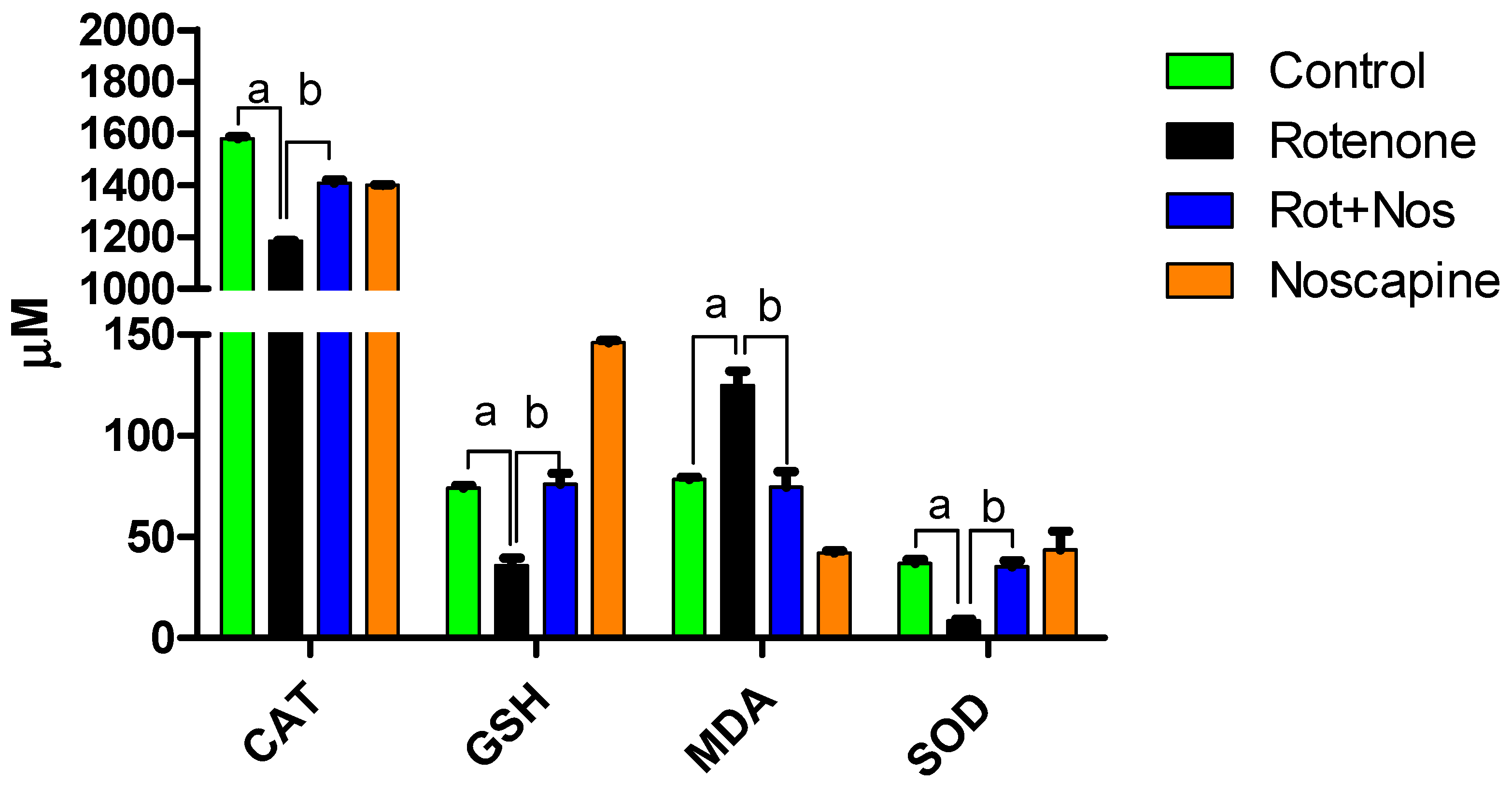
2.1. Noscapine Decreased Rotenone-Induced Oxidative Stress
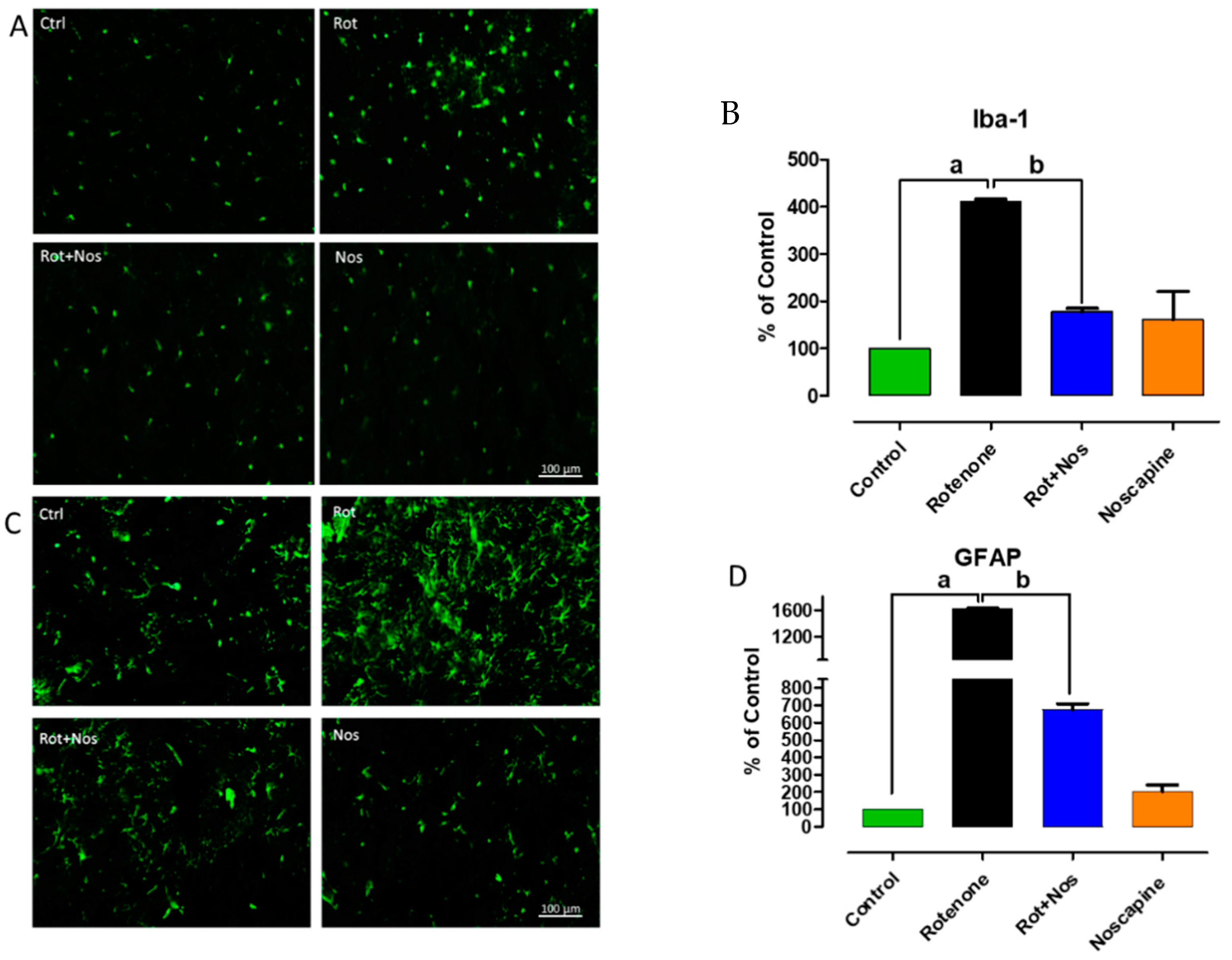
2.2. Noscapine Prevented Rotenone Mediated Neuroinflammation
2.3. Noscapine Prevented Activation of Microglia and Astroglia
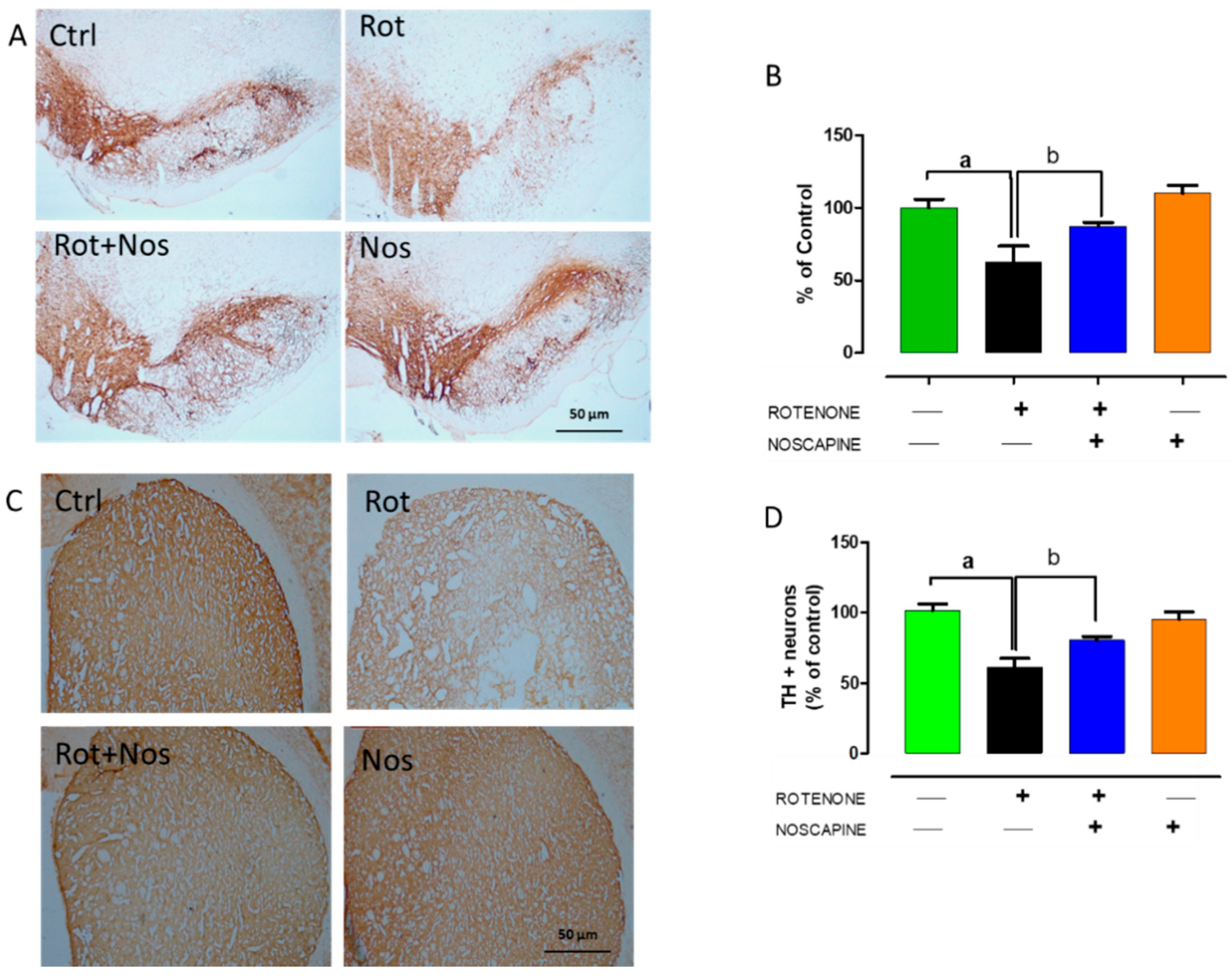
2.4. Noscapine Protected Dopaminergic Neurons against Rotenone Neurotoxicity
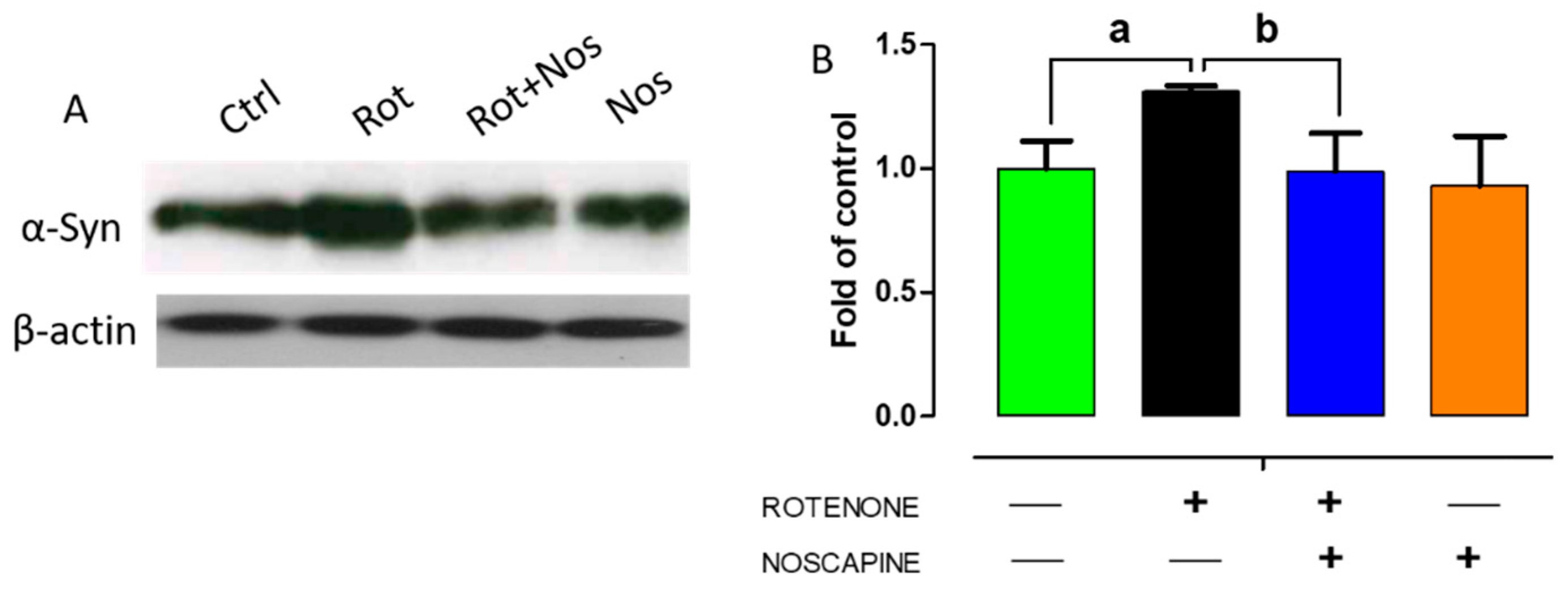
2.5. Noscapine Diminished Over-Expression of α-Synuclein in Experimental Rats
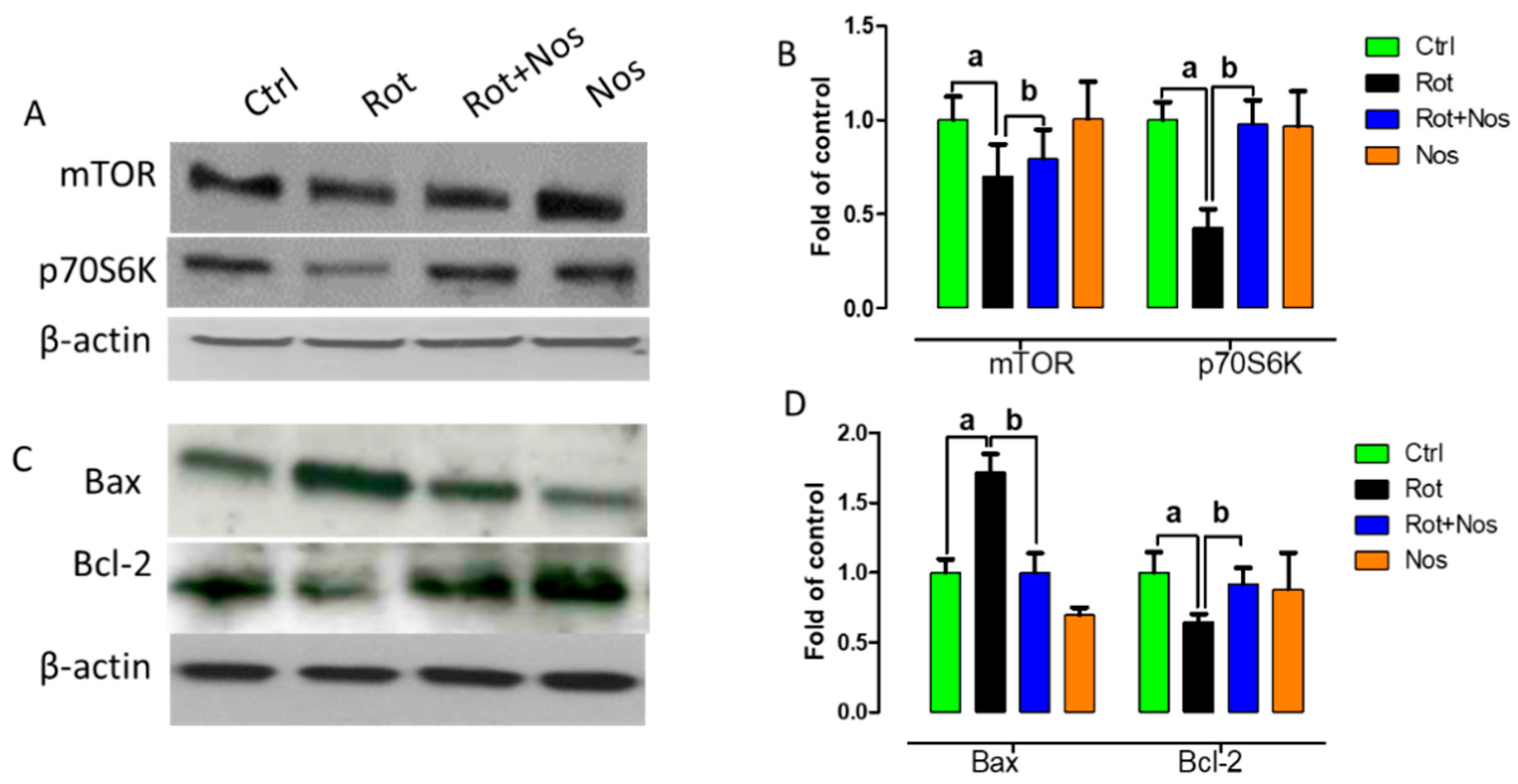
2.6. Noscapine Prevented Neuronal Apoptosis by Restoring mTOR Pathway
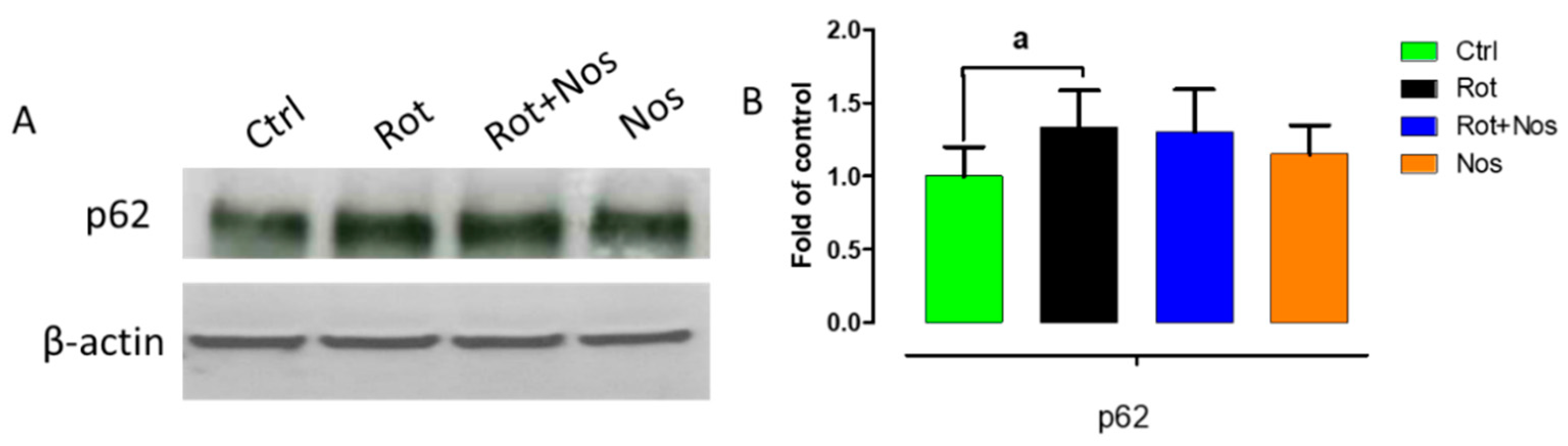
2.7. Effect of Noscapine on Autophagy
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Ethics Statement
4.2. Chemicals and Reagents
4.3. Experimental Design
4.4. Tissue Processing
4.5. Malondialdehyde Assay
4.6. Quantification of Reduced Glutathione
4.7. Assay for Antioxidant Enzyme Activities
4.8. Proinflammatory Cytokines and MMP-9 ELISA Assay
4.9. Assessment of Microglia and Astrocyte Activation by Immunofluorescence Staining
4.10. Quantification of Activated Astrocytes and Microglia in the Striatum
4.11. Immunoblot Analysis
4.12. Immunohistochemistry Analysis
4.13. Assessment of TH-ir Dopaminergic Neurons and TH-ir Dopamine Nerve Fibers Loss
4.14. Protein Estimation
4.15. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Sample Availability
Abbreviations
| PD | Parkinson’s disease |
| ROS | Reactive oxygen species |
| GFAP | Glial fibrillary acidic protein |
| Cox-2 | Cyclooxygenase 2 |
| iNOS | Inducible nitric oxide synthase |
| Rot | Rotenone |
| SOD | Superoxide dismutase |
| CAT | Catalase |
| TNF-α | Tumor necrosis factor-alpha |
| IL-1β | Interleukin-1β |
| Iba-1 | Ionized calcium-binding adapter molecule 1 |
| TH | Tyrosine hydrolase |
| LC3B | Microtubule-associated protein 1 light chain 3 beta |
| mTOR | Mammalian target of rapamycin |
| 4E-BP1 | Eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 |
References
- Martinez-Martin, P.; Rodriguez-Blazquez, C.; Paz, S.; Forjaz, M.J.; Frades-Payo, B.; Cubo, E.; De Pedro-Cuesta, J.; Lizan, L.; ELEP Group. Parkinson Symptoms and Health Related Quality of Life as Predictors of Costs: A Longitudinal Observational Study with Linear Mixed Model Analysis. PLoS ONE 2015, 10, e0145310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Hussein, S.; Ismail, M. Ageing and Elderly Care in the Arab Region: Policy Challenges and Opportunities. Ageing Int. 2017, 42, 274–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Gomperts, S.N. Lewy body dementias: Dementia with Lewy bodies and Parkinson disease dementia. Contin. Lifelong Learn. Neurol. 2016, 22, 435. [Google Scholar] [CrossRef]
- You, H.; Mariani, L.-L.; Mangone, G.; Nailly, D.L.F.D.; Charbonnier-Beaupel, F.; Corvol, J.-C. Molecular basis of dopamine replacement therapy and its side effects in Parkinson’s disease. Cell Tissue Res. 2018, 373, 111–135. [Google Scholar] [CrossRef]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef]
- Joe, E.-H.; Choi, D.-J.; An, J.; Eun, J.-H.; Jou, I.; Park, S. Astrocytes, microglia, and Parkinson’s disease. Exp. Neurobiol. 2018, 27, 77–87. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Federoff, H.J. Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease. Front. Aging Neurosci. 2017, 9, 176. [Google Scholar] [CrossRef]
- Booth, H.D.; Hirst, W.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Brück, D.; Wenning, G.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274. [Google Scholar] [CrossRef] [Green Version]
- Jayaraj, R.L.; Beiram, R.; Azimullah, S.; Meeran, M.F.N.; Ojha, S.K.; Adem, A.; Jalal, F.Y. Lycopodium Attenuates Loss of Dopaminergic Neurons by Suppressing Oxidative Stress and Neuroinflammation in a Rat Model of Parkinson’s Disease. Molecules 2019, 24, 2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rempe, R.G.; Hartz, A.M.S.; Bauer, B. Matrix metalloproteinases in the brain and blood–brain barrier: Versatile breakers and makers. Br. J. Pharmacol. 2016, 36, 1481–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-J.; Choi, M.-J.; Lee, G.; Gaire, B.P.; Choi, J.W.; Kim, H.-S. Regulation of neuroinflammation by matrix metalloproteinase-8 inhibitor derivatives in activated microglia and astrocytes. Oncotarget 2017, 8, 78677–78690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.; Morris, H.; Schapira, A. Chaperone-mediated autophagy as a therapeutic target for Parkinson disease. Expert Opin. Ther. Targets 2018, 22, 823–832. [Google Scholar] [CrossRef]
- Okamoto, K.; Kondo-Okamoto, N. Mitochondria and autophagy: Critical interplay between the two homeostats. Biochim. Biophys. Acta 2012, 1820, 595–600. [Google Scholar] [CrossRef]
- Jackson, M.P.; Hewitt, E.W. Cellular proteostasis: Degradation of misfolded proteins by lysosomes. Essays Biochem. 2016, 60, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Shi, N.; Sun, Y.; Zhang, T.; Sun, X. Therapeutic Effects of Rapamycin on MPTP-Induced Parkinsonism in Mice. Neurochem. Res. 2012, 38, 201–207. [Google Scholar] [CrossRef]
- Kim, D.-K.; Lim, H.-S.; Kawasaki, I.; Shim, Y.-H.; Vaikath, N.N.; El-Agnaf, O.M.A.; Lee, H.-J.; Lee, S.-J. Anti-aging treatments slow propagation of synucleinopathy by restoring lysosomal function. Autophagy 2016, 12, 1849–1863. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Chong, Z.Z.; Shang, Y.C.; Wang, S.; Maiese, K. Shedding new light on neurodegenerative diseases through the mammalian target of rapamycin. Prog. Neurobiol. 2012, 99, 128–148. [Google Scholar] [CrossRef] [Green Version]
- Malagelada, C.; Ryu, E.J.; Biswas, S.C.; Jackson-Lewis, V.; Greene, L.A. RTP801 Is Elevated in Parkinson Brain Substantia Nigral Neurons and Mediates Death in Cellular Models of Parkinson’s Disease by a Mechanism Involving Mammalian Target of Rapamycin Inactivation. J. Neurosci. 2006, 26, 9996–10005. [Google Scholar] [CrossRef] [Green Version]
- Lan, A.-P.; Chen, J.; Zhao, Y.; Chai, Z.; Hu, Y. mTOR signaling in Parkinson’s disease. Neuromol. Med. 2017, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Holmes, G.L.; Stafstrom, C.E. Tuberous Sclerosis Complex and Epilepsy: Recent Developments and Future Challenges. Epilepsia 2007, 48, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2 and autophagy pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://www.nature.com/articles/srep32206#supplementary-information (accessed on 1 December 2020).
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S.; et al. Rotenone Induction of Hydrogen Peroxide Inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E Pathways, Leading to Neuronal Apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, W.; Lu, Y.; Tian, H.; Duan, C.; Lu, L.; Gao, G.; Wu, X.; Wang, X.; Yang, H. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy 2018, 14, 845–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene–environment interactions. NeuroToxicology 2015, 46, 101–116. [Google Scholar] [CrossRef]
- Sengupta, T.; Vinayagam, J.; Singh, R.; Jaisankar, P.; Mohanakumar, K.P. Plant-Derived Natural Products for Parkinson’s Disease Therapy. Adv. Neurobiol. 2016, 12, 415–496. [Google Scholar] [CrossRef]
- Tomar, V.; Kukreti, S.; Prakash, S.; Madan, J.; Chandra, R. Noscapine and its Analogs as Chemotherapeutic Agent: Current updates. Curr. Top. Med. Chem. 2016, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, M.; Mehrpour, M.; Benaissa, F.; Siadatpour, Z. A preliminary report on the application of noscapine in the treatment of stroke. Eur. J. Clin. Pharmacol. 2003, 59, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Khodarahmi, P.; Rostami, P.; Rashidi, A.; Khodarahmi, I. Anxiolytic effect of noscapine in mice. Pharmacol. Rep. 2006, 58, 568–570. [Google Scholar] [PubMed]
- Landen, J.W.; Hau, V.; Wang, M.; Davis, T.; Ciliax, B.; Wainer, B.H.; Van Meir, E.G.; Glass, J.D.; Joshi, H.C.; Archer, D.R. Noscapine Crosses the Blood-Brain Barrier and Inhibits Glioblastoma Growth. Clin. Cancer Res. 2004, 10, 5187–5201. [Google Scholar] [CrossRef] [Green Version]
- De Maturana, R.L.; Aguila, J.C.; Sousa, A.; Vazquez, N.; Del Río, P.; Aiastui, A.; Gorostidi, A.; De Munain, A.L.; Sanchez-Pernaute, R. Leucine-rich repeat kinase 2 modulates cyclooxygenase 2 and the inflammatory response in idiopathic and genetic Parkinson’s disease. Neurobiol. Aging 2014, 35, 1116–1124. [Google Scholar] [CrossRef]
- He, D.; Huang, B.; Fu, S.; Li, Y.; Ran, X.; Liu, Y.; Chen, G.; Liu, J.; Liu, D. Tubeimoside I Protects Dopaminergic Neurons Against Inflammation-Mediated Damage in Lipopolysaccharide (LPS)-Evoked Model of Parkinson’s Disease in Rats. Int. J. Mol. Sci. 2018, 19, 2242. [Google Scholar] [CrossRef] [Green Version]
- Michel, H.E.; Tadros, M.G.; Esmat, A.; Khalifa, A.; Abdel-Tawab, A.M. Tetramethylpyrazine Ameliorates Rotenone-Induced Parkinson’s Disease in Rats: Involvement of Its Anti-Inflammatory and Anti-Apoptotic Actions. Mol. Neurobiol. 2016, 54, 4866–4878. [Google Scholar] [CrossRef]
- Wen, Z.; Zhang, J.; Tang, P.; Tu, N.; Wang, K.; Wu, G. Overexpression of miR-185 inhibits autophagy and apoptosis of dopaminergic neurons by regulating the AMPK/mTOR signaling pathway in Parkinson’s disease. Mol. Med. Rep. 2018, 17, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Giacoppo, S.; Bramanti, P.; Mazzon, E. Triggering of inflammasome by impaired autophagy in response to acute experimental Parkinson’s disease: Involvement of the PI3K/Akt/mTOR pathway. Neuroreport 2017, 28, 996. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell. Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J.; Chen, L.; Zhang, P.-P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janda, E.; Isidoro, C.; Carresi, C.; Mollace, V. Defective Autophagy in Parkinson’s Disease: Role of Oxidative Stress. Mol. Neurobiol. 2012, 46, 639–661. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef]
- Saggu, H.; Cooksey, J.; Dexter, D.; Wells, F.R.; Lees, A.; Jenner, P.; Marsden, C.D. A Selective Increase in Particulate Superoxide Dismutase Activity in Parkinsonian Substantia Nigra. J. Neurochem. 1989, 53, 692–697. [Google Scholar] [CrossRef]
- Yoritaka, A.; Hattori, N.; Mori, H.; Kato, K.; Mizuno, Y. An immunohistochemical study on manganese superoxide dismutase in Parkinson’s disease. J. Neurol. Sci. 1997, 148, 181–186. [Google Scholar] [CrossRef]
- Cacabelos, R.; Carrera, I.; Fernández-Novoa, L.; Alejo, R.; Corzo, L.; Rodríguez, S.; Alcaraz, M.; Nebril, L.; Casas, A.; Fraile, C. Parkinson’s Disease: New solutions to old problems. EuroEspes J. 2017, 11, 74–96. [Google Scholar]
- Yuan, H.; Zhang, Z.-W.; Liang, L.-W.; Shen, Q.; Wang, X.-D.; Ren, S.-M.; Ma, H.-J.; Jiao, S.-J.; Liu, P. Treatment strategies for Parkinson’s disease. Neurosci. Bull. 2010, 26, 66–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Extra Pharmacopoeia. Nat. Cell Biol. 1937, 139, 780. [CrossRef]
- Idänpään-Heikkilä, J.E.; Jalonen, K.; Vartiainen, A. Evaluation of the Antitussive Effect of Noscapine and Codeine on Citric Acid Cough in Guinea-Pigs. Acta Pharmacol. Toxicol. 2009, 25, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Empey, D.W.; Laitinen, L.A.; Young, G.A.; Bye, C.E.; Hughes, D.T.D. Comparison of the antitussive effects of codeine phosphate 20 mg, dextromethorphan 30 mg and noscapine 30 mg using citric acid-induced cough in normal subjects. Eur. J. Clin. Pharmacol. 1979, 16, 393–397. [Google Scholar] [CrossRef]
- Spina, M.B.; Cohen, G. Dopamine turnover and glutathione oxidation: Implications for Parkinson disease. Proc. Natl. Acad. Sci. USA 1989, 86, 1398–1400. [Google Scholar] [CrossRef] [Green Version]
- Przedborski, S. Neuroinflammation and Parkinson’s disease. Hum. Hypothal.-Middle Posterior Reg. 2007, 83, 535–551. [Google Scholar] [CrossRef]
- Przedborski, S.; Chen, Q.; Vila, M.; Giasson, B.I.; Djaldatti, R.; Vukosavic, S.; Souza, J.M.; Jackson-Lewis, V.; Lee, V.M.; Ischiropoulos, H. Oxidative post-translational modifications of alpha-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J. Neurochem. 2001, 76, 637–640. [Google Scholar] [CrossRef]
- Ara, J.; Przedborski, S.; Naini, A.B.; Jackson-Lewis, V.; Trifiletti, R.R.; Horwitz, J.; Ischiropoulos, H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc. Natl. Acad. Sci. USA 1998, 95, 7659–7663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, D.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Taylor, J.M.; Main, B.S.; Crack, P. Neuroinflammation and oxidative stress: Co-conspirators in the pathology of Parkinson’s disease. Neurochem. Int. 2013, 62, 803–819. [Google Scholar] [CrossRef] [PubMed]
- Ferger, A.I.; Campanelli, L.; Reimer, V.; Muth, K.N.; Merdian, I.; Ludolph, A.C.; Witting, A. Effects of mitochondrial dysfunction on the immunological properties of microglia. J. Neuroinflamm. 2010, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Pittman, R.N.; Messam, C.A.; Mills, J.C. Asynchronous Death as a Characteristic Feature of Apoptosis. In Cell Death and Diseases of the Nervous System; Springer: Berlin/Heidelberg, Germany, 1999; pp. 29–43. [Google Scholar]
- Hunot, S.; Dugas, N.; Faucheux, B.; Hartmann, A.; Tardieu, M.; Debre, P.; Agid, Y.; Dugas, B.; Hirsch, E.C. FcepsilonRII/CD23 is expressed in Parkinson’s disease and induces, in vitro, production of nitric oxide and tumor necrosis factor-alpha in glial cells. J. Neurosci. 1999, 19, 3440–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Duke, D.C.; Moran, L.B.; Pearce, R.K.B.; Graeber, M.B. The medial and lateral substantia nigra in Parkinson’s disease: mRNA profiles associated with higher brain tissue vulnerability. Neurogenetics 2007, 8, 83–94. [Google Scholar] [CrossRef]
- Watson, M.B.; Richter, F.; Lee, S.K.; Gabby, L.; Wu, J.; Masliah, E.; Effros, R.B.; Chesselet, M.-F. Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp. Neurol. 2012, 237, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Lücking, C.; Brice, A. Alpha-synuclein and Parkinson’s disease. Cell. Mol. Life Sci. CMLS 2000, 57, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R.; van der Brug, M. Cell systems and the toxic mechanism(s) of α-synuclein. Exp. Neurol. 2008, 209, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javed, H.; Azimullah, S.; Khair, S.B.A.; Ojha, S.; Haque, M.E. Neuroprotective effect of nerolidol against neuroinflammation and oxidative stress induced by rotenone. BMC Neurosci. 2016, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mochizuki, H.; Goto, K.; Mori, H.; Mizuno, Y. Histochemical detection of apoptosis in Parkinson’s disease. J. Neurol. Sci. 1996, 137, 120–123. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Saha, A.R.; Ninkina, N.N.; Hanger, D.; Anderton, B.H.; Davies, A.M.; Buchman, V.L. Induction of neuronal death by α-synuclein. Eur. J. Neurosci. 2000, 12, 3073–3077. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Rah, J.; Choi, S.H.; Shin, J.K.; Min, K.; Kim, H.; Park, C.H.; Kim, S.; Kim, E.; Lee, S.; et al. α-Synuclein regulates neuronal survival via Bcl-2 family expression and PI3/Akt kinase pathway. FASEB J. 2002, 16, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, O. Apoptotic mechanisms and antiapoptotic therapy in the MPTP model of Parkinson’s disease. Toxicol. Lett. 2003, 139, 135–151. [Google Scholar] [CrossRef]
- Crino, P.B. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol. Med. 2011, 17, 734–742. [Google Scholar] [CrossRef] [PubMed]
- LaSarge, C.L.; Danzer, S.C. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front. Mol. Neurosci. 2014, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.J.; Reis, G.; Kang, H.; Gingras, A.-C.; Sonenberg, N.; Schuman, E.M. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc. Natl. Acad. Sci. USA 2002, 99, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-H.; Guo, F.; Shelburne, J.; Watkins, S.; Chu, C.T. Localization of Phosphorylated ERK/MAP Kinases to Mitochondria and Autophagosomes in Lewy Body Diseases. Brain Pathol. 2006, 13, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.; Gibson, S.B. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J. Cell Sci. 2007, 120, 4155–4166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant α-Synuclein Confers Toxicity to Neurons in Part through Inhibition of Chaperone-Mediated Autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Beal, M.F.; Thomas, B. Autophagy in neurodegenerative disorders: Pathogenic roles and therapeutic implications. Trends Neurosci. 2010, 33, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xilouri, M.; Vogiatzi, T.; Stefanis, L. Alpha-synuclein degradation by autophagic pathways: A potential key to Parkinson’s Disease pathogenesis. Autophagy 2008, 4, 917–919. [Google Scholar] [CrossRef] [Green Version]
- Mader, B.J.; Pivtoraiko, V.N.; Flippo, H.M.; Klocke, B.J.; Roth, K.; Mangieri, L.; Shacka, J.J. Rotenone Inhibits Autophagic Flux Prior to Inducing Cell Death. ACS Chem. Neurosci. 2012, 3, 1063–1072. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Rojas, J.P.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- McCloy, R.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial inhibition of Cdk1 in G2phase overrides the SAC and decouples mitotic events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayaraj, R.L.; Beiram, R.; Azimullah, S.; M. F., N.M.; Ojha, S.K.; Adem, A.; Jalal, F.Y. Noscapine Prevents Rotenone-Induced Neurotoxicity: Involvement of Oxidative Stress, Neuroinflammation and Autophagy Pathways. Molecules 2021, 26, 4627. https://doi.org/10.3390/molecules26154627
Jayaraj RL, Beiram R, Azimullah S, M. F. NM, Ojha SK, Adem A, Jalal FY. Noscapine Prevents Rotenone-Induced Neurotoxicity: Involvement of Oxidative Stress, Neuroinflammation and Autophagy Pathways. Molecules. 2021; 26(15):4627. https://doi.org/10.3390/molecules26154627
Chicago/Turabian StyleJayaraj, Richard L., Rami Beiram, Sheikh Azimullah, Nagoor Meeran M. F., Shreesh K. Ojha, Abdu Adem, and Fakhreya Yousuf Jalal. 2021. "Noscapine Prevents Rotenone-Induced Neurotoxicity: Involvement of Oxidative Stress, Neuroinflammation and Autophagy Pathways" Molecules 26, no. 15: 4627. https://doi.org/10.3390/molecules26154627