
Structure Solution of Nano-Crystalline Small Molecules Using MicroED and Solid-State NMR Dipolar-Based Experiments

 ,
,
Abstract
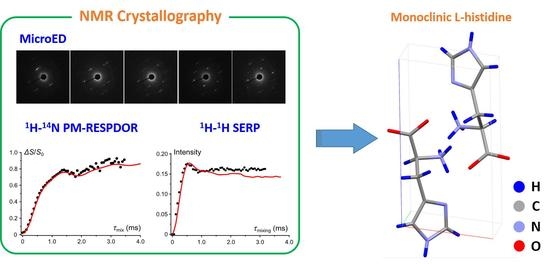
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experiments and Methods
2.1. Sample
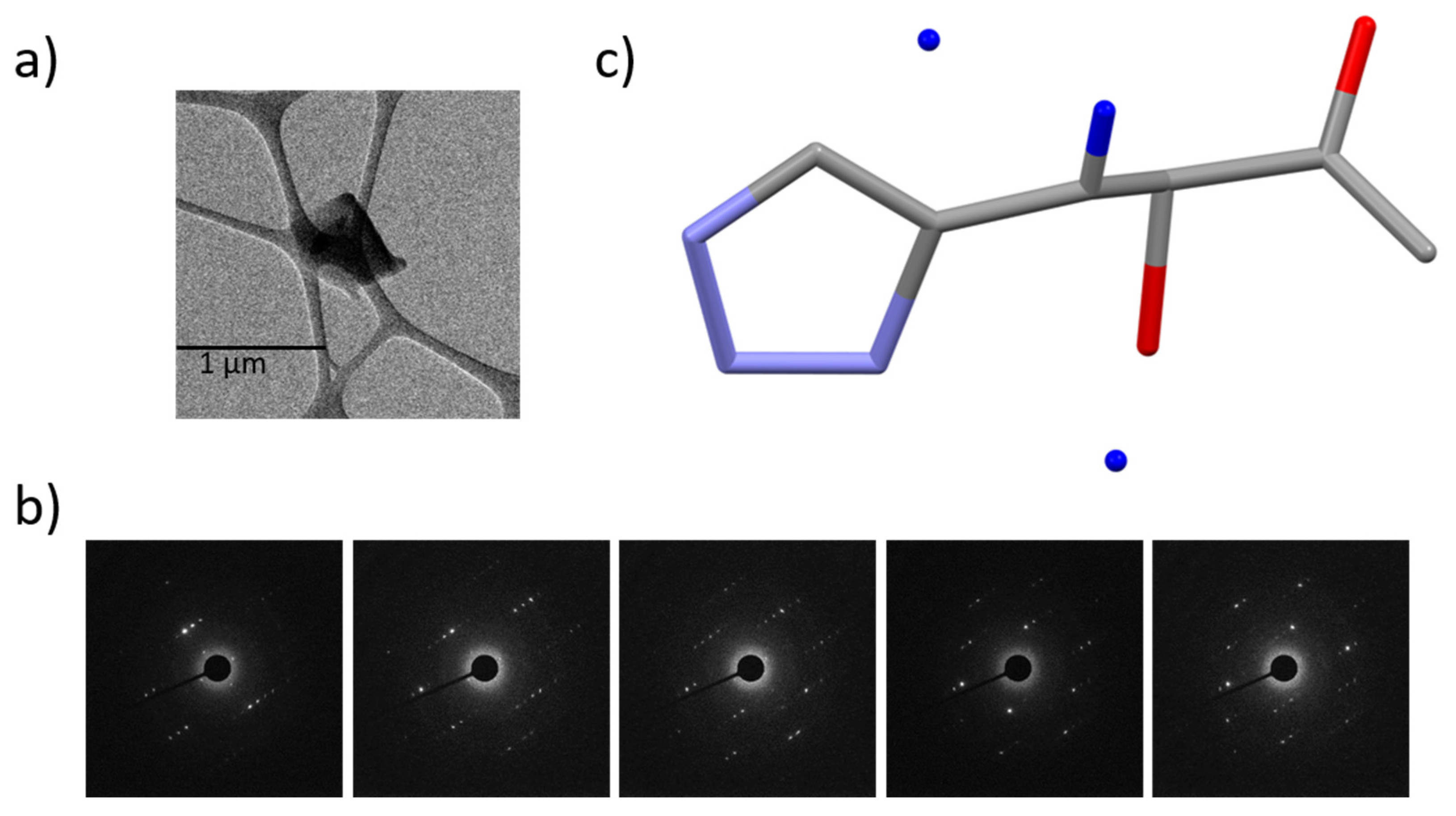
2.2. Three-Dimensional Electron Diffraction Crystallography (MicroED)
2.3. Solid-State Nuclear Magnetic Resonance (NMR)
2.3.1. 1H Double-Quantum (DQ)/1H Single-Quantum (SQ) Correlation
2.3.2. 1H-{13C} Proton-Detected CP-HSQC
2.3.3. 1H-14N Phase-Modulated Rotational-Echo Saturation-Pulse Double-Resonance (PM-S-RESPDOR)
2.3.4. 1H-1H Selective Recoupling of Proton (SERP)
2.4. Spin Dynamics Simulation
2.5. Quantum Chemical (Density Functional Theory (DFT) and GIPAW) Calculation
3. Results and Discussion
3.1. Ambiguities in MicroED Solution
3.2. Spectral Assignments
1H-14N PM-S-RESPDOR Experiment
3.3. 1H-1H SERP Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gemmi, M.; Lanza, A.E. 3D electron diffraction techniques. Acta Crystallographica Section B: Structural Science. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2019, 75, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Mugnaioli, E.; Gorelik, T.; Kolb, U. “Ab initio” structure solution from electron diffraction data obtained by a combination of automated diffraction tomography and precession technique. Ultramicroscopy 2009, 109, 758–765. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Shi, D.; Leslie, A.G.W.; Gonen, T. High-resolution structure determination by continuous-rotation data collection in MicroEDED. Nat. Methods 2014, 11, 927–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán-Afonso, C.; Hong, Y.-L.; Colaux, H.; Iijima, H.; Saitow, A.; Fukumura, T.; Aoyama, Y.; Motoki, S.; Oikawa, T.; Yamazaki, T.; et al. Understanding hydrogen-bonding structures of molecular crystals via electron and NMR nanocrystallography. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Gruene, T.; Wennmacher, J.T.C.; Zaubitzer, C.; Holstein, J.J.; Heidler, J.; Fecteau-Lefebvre, A.; De Carlo, S.; Müller, E.; Goldie, K.N.; Regeni, I.; et al. Rapid Structure Determination of Microcrystalline Molecular Compounds Using Electron Diffraction. Angew. Chem. Int. Ed. 2018, 57, 16313–16317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.G.; Martynowycz, M.W.; Hattne, J.; Fulton, T.J.; Stoltz, B.M.; Rodriguez, J.A.; Nelson, H.M.; Gonen, T. The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent. Sci. 2018, 4, 1587–1592. [Google Scholar] [CrossRef]
- Palatinus, L.; Brázda, P.; Boullay, P.; Perez, O.; Klementová, M.; Petit, S.; Eigner, V.; Zaarour, M.; Mintova, S. Hydrogen positions in single nanocrystals revealed by electron diffraction. Science 2017, 355, 166–169. [Google Scholar] [CrossRef]
- Van Genderen, E.; Clabbers, M.T.B.; Das, P.P.; Stewart, A.; Nederlof, I.; Barentsen, K.C.; Portillo, Q.; Pannu, N.S.; Nicolopoulos, S.; Gruene, T.; et al. Ab initio structure determination of nanocrystals of organic pharmaceutical compounds by electron diffraction at room temperature using a Timepix quantum area direct electron detector. Acta Crystallogr. Sect. A Found. Adv. 2016, 72, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Hattne, J.; Reyes, F.E.; Nannenga, B.L.; Shi, D.; De La Cruz, M.J.; Leslie, A.G.W.; Gonen, T. MicroED data collection and processing. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, U.; Gorelik, T.; Otten, M.T. Towards automated diffraction tomography. Part II-Cell parameter determination. Ultramicroscopy 2008, 108, 763–772. [Google Scholar] [CrossRef]
- Zhang, D.; Oleynikov, P.; Hovmoller, S.; Zou, X. Collecting 3D electron diffraction data by the rotation method. Z. Fuer Krist. 2010, 225, 94–102. [Google Scholar] [CrossRef]
- Shi, D.; Nannenga, B.L.; Iadanza, M.G.; Gonen, T. Three-dimensional electron crystallography of protein microcrystals. eLife 2013, 2, 1–17. [Google Scholar] [CrossRef]
- Yonekura, K.; Kato, K.; Ogasawara, M.; Tomita, M.; Toyoshima, C. Electron crystallography of ultrathin 3D protein crystals: Atomic model with charges. Proc. Natl. Acad. Sci. USA 2015, 112, 3368–3373. [Google Scholar] [CrossRef] [Green Version]
- Clabbers, M.T.B.; Gruene, T.; van Genderen, E.; Abrahams, J.P. Reducing dynamical electron scattering reveals hydrogen atoms. Acta Crystallogr. Sect. A Found. Adv. 2019, 75, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Hynek, J.; Brázda, P.; Rohlíček, J.; Londesborough, M.G.S.; Demel, J. Phosphinic Acid Based Linkers: Building Blocks in Metal–Organic Framework Chemistry. Angew. Chem. Int. Ed. 2018, 57, 5016–5019. [Google Scholar] [CrossRef]
- Brázda, P.; Palatinus, L.; Babor, M. Electron diffraction determines molecular absolute configuration in a pharmaceutical nanocrystal. Science 2019, 364, 667–669. [Google Scholar] [CrossRef]
- Ripmeester, J.A.; Wasylishen, R.E. NMR crystallography. CrystEngComm 2013, 15, 8598. [Google Scholar] [CrossRef]
- Martineau, C.; Senker, J.; Taulelle, F. NMR Crystallography. In Annual Reports on NMR Spectroscopy; Elsevier Ltd.: London, UK, 2014; Volume 82, pp. 1–57. ISBN 9780128001844. [Google Scholar] [CrossRef]
- Mafra, L. Preface. Solid State Nucl. Magn. Reson. 2015, 65, 1. [Google Scholar] [CrossRef]
- Ashbrook, S.E.; McKay, D. Combining solid-state NMR spectroscopy with first-principles calculations-a guide to NMR crystallography. Chem. Commun. 2016, 52, 7186–7204. [Google Scholar] [CrossRef] [Green Version]
- Bryce, D.L.; Taulelle, F. NMR Crystallography. Acta Crystallogr. Sect. C Struct. Chem. 2017, 73, 126–127. [Google Scholar] [CrossRef] [Green Version]
- Bryce, D.L. NMR crystallography: Structure and properties of materials from solid-state nuclear magnetic resonance observables. IUCrJ 2017, 4, 350–359. [Google Scholar] [CrossRef] [Green Version]
- Ashbrook, S.E.; Hodgkinson, P. Perspective: Current advances in solid-state NMR spectroscopy. J. Chem. Phys. 2018, 149, 040901. [Google Scholar] [CrossRef] [Green Version]
- Hodgkinson, P. NMR crystallography of molecular organics. Prog. Nucl. Magn. Reson. Spectrosc. 2020, 118–119, 10–53. [Google Scholar] [CrossRef] [PubMed]
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B Condens. Matter Mater. Phys. 2001, 63, 2451011–2451013. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, D.; Parrinello, M. A new ab-initio approach for NMR chemical shifts in periodic systems. J. Phys. Chem. A 2001, 105, 1951–1958. [Google Scholar] [CrossRef]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 1–11. [Google Scholar] [CrossRef]
- Corlett, E.K.; Blade, H.; Hughes, L.P.; Sidebottom, P.J.; Walker, D.; Walton, R.I.; Brown, S.P. Investigating discrepancies between experimental solid-state NMR and GIPAW calculation: N[dbnd]C–N 13C and OH⋯O 1H chemical shifts in pyridinium fumarates and their cocrystals. Solid State Nucl. Magn. Reson. 2020, 108, 101662. [Google Scholar] [CrossRef]
- Harris, R.K.; Joyce, S.A.; Pickard, C.J.; Cadars, S.; Emsley, L. Assigning carbon-13 NMR spectra to crystal structures by the INADEQUATE pulse sequence and first principles computation: A case study of two forms of testosterone. Phys. Chem. Chem. Phys. 2006, 8, 137–143. [Google Scholar] [CrossRef]
- Dračínský, M.; Unzueta, P.; Beran, G.J.O. Improving the accuracy of solid-state nuclear magnetic resonance chemical shift prediction with a simple molecular correction. Phys. Chem. Chem. Phys. 2019, 21, 14992–15000. [Google Scholar] [CrossRef]
- Dračínský, M. Analyzing Discrepancies in Chemical-Shift Predictions of Solid Pyridinium Fumarates. Molecules 2021, 26, 3857. [Google Scholar] [CrossRef]
- Steiner, T.; Majerz, I.; Wilson, C.C. First O−H−N Hydrogen Bond with a Centered Proton Obtained by Thermally Induced Proton Migration. Angew. Chem. Int. Ed. 2001, 40, 2651–2654. [Google Scholar] [CrossRef]
- Mazurek, A.H.; Szeleszczuk, Ł.; Pisklak, D.M. A review on combination of ab initio molecular dynamics and nmr parameters calculations. Int. J. Mol. Sci. 2021, 22, 4378. [Google Scholar] [CrossRef]
- Gan, Z. Measuring multiple carbon-nitrogen distances in natural abundant solids using R-RESPDOR NMR. Chem. Commun. 2006, 4712–4714. [Google Scholar] [CrossRef]
- Chen, L.; Lu, X.; Wang, Q.; Lafon, O.; Trébosc, J.; Deng, F.; Amoureux, J.P. Distance measurement between a spin-1/2 and a half-integer quadrupolar nuclei by solid-state NMR using exact analytical expressions. J. Magn. Reson. 2010, 206, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Q.; Hu, B.; Lafon, O.; Trébosc, J.; Deng, F.; Amoureux, J.P. Measurement of hetero-nuclear distances using a symmetry-based pulse sequence in solid-state NMR. Phys. Chem. Chem. Phys. 2010, 12, 9395–9405. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lafon, O.; Trébosc, J.; Amoureux, J.P. Detailed analysis of the S-RESPDOR solid-state NMR method for inter-nuclear distance measurement between spin-1/2 and quadrupolar nuclei. J. Magn. Reson. 2011, 215, 34–49. [Google Scholar] [CrossRef]
- Duong, N.T.; Rossi, F.; Makrinich, M.; Goldbourt, A.; Chierotti, M.R.; Gobetto, R.; Nishiyama, Y. Accurate 1H-14N distance measurements by phase-modulated RESPDOR at ultra-fast MAS. J. Magn. Reson. 2019, 308, 106559. [Google Scholar] [CrossRef]
- Duong, N.T.; Gan, Z.; Nishiyama, Y. Selective 1H-14N Distance Measurements by 14N Overtone Solid-State NMR Spectroscopy at Fast MAS. Front. Mol. Biosci. 2021, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Duong, N.T.; Raran-Kurussi, S.; Nishiyama, Y.; Agarwal, V. Quantitative 1H–1H Distances in Protonated Solids by Frequency-Selective Recoupling at Fast Magic Angle Spinning NMR. J. Phys. Chem. Lett. 2018, 9, 5948–5954. [Google Scholar] [CrossRef]
- Duong, N.T.; Raran-Kurussi, S.; Nishiyama, Y.; Agarwal, V. Can proton-proton recoupling in fully protonated solids provide quantitative, selective and efficient polarization transfer? J. Magn. Reson. 2020, 317, 106777. [Google Scholar] [CrossRef] [PubMed]
- Potnuru, L.R.; Duong, N.T.; Ahlawat, S.; Raran-Kurussi, S.; Ernst, M.; Nishiyama, Y.; Agarwal, V. Accuracy of 1H-1H distances measured using frequency selective recoupling and fast magic-angle spinning. J. Chem. Phys. 2020, 153, 084202. [Google Scholar] [CrossRef]
- Oikawa, T.; Okumura, M.; Kimura, T.; Nishiyama, Y. Solid-state NMR meets electron diffraction: Determination of crystalline polymorphs of small organic microcrystalline samples. Acta Crystallogr. Sect. C Struct. Chem. 2017, 73, 219–228. [Google Scholar] [CrossRef]
- Kabsch, W. research papers XDS research papers. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Crystallogr. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Agarwal, V.; Zhang, R. Efficient symmetry-based γ-encoded DQ recoupling sequences for suppression of t1-noise in solid-state NMR spectroscopy at fast MAS. Solid State Nucl. Magn. Reson. 2021, 114, 101734. [Google Scholar] [CrossRef]
- Marion, D.; Ikura, M.; Tschudin, R.; Bax, A. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J. Magn. Reson. 1989, 85, 393–399. [Google Scholar] [CrossRef]
- Brinkmann, A.; Kentgens, A.P.M. Proton-selective O-17-H-1 distance measurements in fast magic-angle-spinning solid-state NMR spectroscopy for the determination of hydrogen bond lengths. J. Am. Chem. Soc. 2006, 128, 14758–14759. [Google Scholar] [CrossRef] [Green Version]
- Nimerovsky, E.; Gupta, R.; Yehl, J.; Li, M.; Polenova, T.; Goldbourt, A. Phase-modulated LA-REDOR: A robust, accurate and efficient solid-state NMR technique for distance measurements between a spin-1/2 and a quadrupole spin. J. Magn. Reson. 2014, 244, 107–113. [Google Scholar] [CrossRef]
- Nimerovsky, E.; Makrinich, M.; Goldbourt, A. Analysis of large-anisotropy-spin recoupling pulses for distance measurement under magic-angle spinning NMR shows the superiority and robustness of a phase modulated saturation pulse. J. Chem. Phys. 2017, 146, 124202. [Google Scholar] [CrossRef]
- Makrinich, M.; Nimerovsky, E.; Goldbourt, A. Pushing the limit of NMR-based distance measurements–retrieving dipolar couplings to spins with extensively large quadrupolar frequencies. Solid State Nucl. Magn. Reson. 2018, 92, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Makrinich, M.; Sambol, M.; Goldbourt, A. Distance measurements between carbon and bromine using a split-pulse PM-RESPDOR solid-state NMR experiment. Phys. Chem. Chem. Phys. 2020, 22, 21022–21030. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.; Rasmussen, J.T.; Nielsen, N.C. SIMPSON: A general simulation program for solid-state NMR spectroscopy. J. Magn. Reson. 2000, 147, 296–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tošner, Z.; Andersen, R.; Stevensson, B.; Edén, M.; Nielsen, N.C.; Vosegaard, T. Computer-intensive simulation of solid-state NMR experiments using SIMPSON. J. Magn. Reson. 2014, 246, 79–93. [Google Scholar] [CrossRef]
- Juhl, D.W.; Tošner, Z.; Vosegaard, T. Versatile NMR simulations using SIMPSON. Annu. Rep. NMR Spectrosc. 2020, 100, 1–59. [Google Scholar] [CrossRef]
- Varini, N.; Ceresoli, D.; Martin-Samos, L.; Girotto, I.; Cavazzoni, C. Enhancement of DFT-calculations at petascale: Nuclear Magnetic Resonance, Hybrid Density Functional Theory and Car-Parrinello calculations. Comput. Phys. Commun. 2013, 184, 1827–1833. [Google Scholar] [CrossRef] [Green Version]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Buongiorno Nardelli, M.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Ceresoli, D. Pseudopotentials. Available online: https://sites.google.com/site/dceresoli/pseudopotentials (accessed on 30 June 2021).




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duong, N.T.; Aoyama, Y.; Kawamoto, K.; Yamazaki, T.; Nishiyama, Y. Structure Solution of Nano-Crystalline Small Molecules Using MicroED and Solid-State NMR Dipolar-Based Experiments. Molecules 2021, 26, 4652. https://doi.org/10.3390/molecules26154652
Duong NT, Aoyama Y, Kawamoto K, Yamazaki T, Nishiyama Y. Structure Solution of Nano-Crystalline Small Molecules Using MicroED and Solid-State NMR Dipolar-Based Experiments. Molecules. 2021; 26(15):4652. https://doi.org/10.3390/molecules26154652
Chicago/Turabian StyleDuong, Nghia Tuan, Yoshitaka Aoyama, Katsumi Kawamoto, Toshio Yamazaki, and Yusuke Nishiyama. 2021. "Structure Solution of Nano-Crystalline Small Molecules Using MicroED and Solid-State NMR Dipolar-Based Experiments" Molecules 26, no. 15: 4652. https://doi.org/10.3390/molecules26154652