Abstract
FAH domain containing protein 1 (FAHD1) acts as oxaloacetate decarboxylase in mitochondria, contributing to the regulation of the tricarboxylic acid cycle. Guided by a high-resolution X-ray structure of FAHD1 liganded by oxalate, the enzymatic mechanism of substrate processing is analyzed in detail. Taking the chemical features of the FAHD1 substrate oxaloacetate into account, the potential inhibitor structures are deduced. The synthesis of drug-like scaffolds afforded first-generation FAHD1-inhibitors with activities in the low micromolar IC50 range. The investigations disclosed structures competing with the substrate for binding to the metal cofactor, as well as scaffolds, which may have a novel binding mode to FAHD1.
1. Introduction
FAH domain containing protein 1 (FAHD1) acts as bifunctional oxaloacetate (OAA) decarboxylase (ODx) [1] and acylpyruvate hydrolase (ApH) [2] in mitochondria. FAHD1 belongs to the broad FAH superfamily of enzymes [3,4]. While its ApH activity is only of minor biological relevance, FAHD1 is involved as ODx in the regulation of the tricarboxylic acid (TCA) cycle flux [5,6]. OAA is not only required for the central citrate synthase reaction in the TCA cycle but also acts as a competitive inhibitor of succinate dehydrogenase and as a cataplerotic metabolite. Our working model refers to a mitochondrial dysfunction associated senescence (MiDAS)-like phenotype [6], where the mitochondrial OAA levels are tightly regulated by FAHD1 activity. In particular, we investigate the role of FAHD1 in human cell metabolism, focusing on metabolic shifts in human cell lines upon FAHD1 knockdown, and we assess the dependence of these cells on FAHD1 for proliferation and survival. Successful experiments will put FAHD1 inhibition into the therapeutic context, to be tested in future efficacy studies in human tissue culture and xenograft models. We anticipate that the elucidation of the role of FAHD1 as a therapeutic target in the treatment of pathologies associated with mitochondrial dysfunction will stimulate translational research with the aim to contribute to new therapeutic strategies for human malignancies.
In the following, we discuss the substrate processing of FAHD1 based on structural information gained from the available X-ray structures of FAHD1. By a theoretical analysis, also taking into consideration the chemical features of the FAHD1 substrates (oxaloacetate and acylpyruvates), valuable proposals for the synthesis of inhibitors could be deduced. Their synthesis afforded the first inhibitors of FAHD1.
The information obtained by comparing the unliganded with the liganded protein models enabled the structural definition of the FAHD1 catalytic center in high resolution. X-ray data suggests a bidentate-binding mode of the 1,2-dicarbonyl motif of the inhibitor to the Mg cofactor and provides a rationale for closure of the lid domain and induction of a catalytically competent dyad (His30-Glu33) [7,8]. However, the structure model of FAHD1 liganded by oxalate does not tell us which residues are key in the enzymatic process, and the essential residues involved in driving the enzymatic reaction remain to be elucidated. Furthermore, no information about lid opening, product release and restauration of the native FAHD1 can be deduced from this data. To gain further mechanistic insights, it is therefore worth to address the central chemical step of both FAHD1 functions (ODx- and ApH) by a theoretical approach.
1.1. The C–C Bond Cleavage Mechanism in ODx- and ApH-Activity of FAHD1
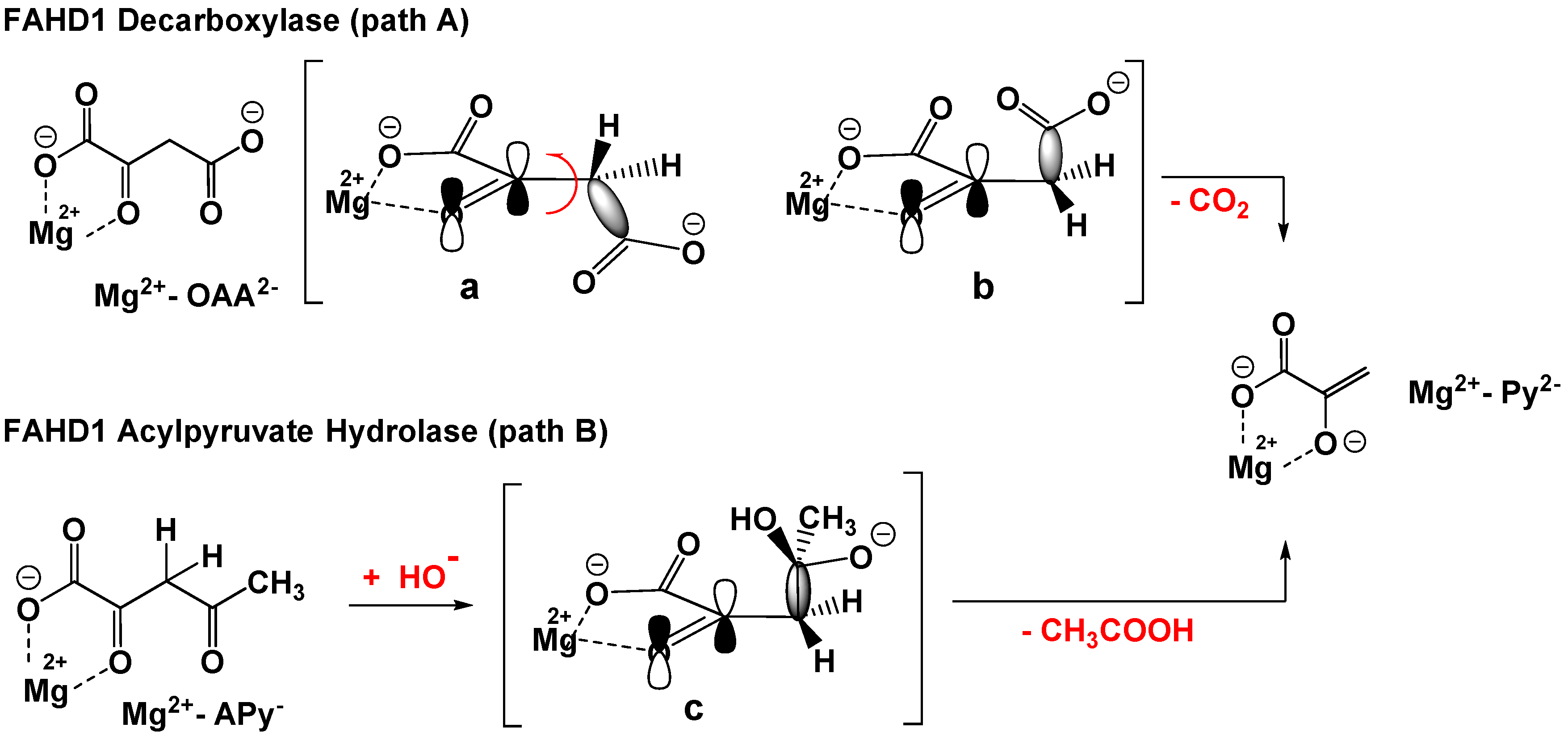
Both the hydrolase and the decarboxylase mechanisms share a common strategic chemical step, namely the scission of an equivalent carbon–carbon single bond in OAA and Ap, respectively. From the experimental data, it can be deduced that the substrates start initial binding to the cofactor in their enol form: Enolized oxaloacetate, as well as acylpyruvates, are planar molecules due to intramolecular resonance-stabilized hydrogen bonding (a comprehensive literature overview of features of oxaloacetate is provided in the Supporting Materials). The C–C σ-bond has no orbital overlap with the C–C double bond of the enol (Figure 1a). The Mg-bound enol of oxaloacetate is therefore not capable for decarboxylation. FAHD1 has to provide residues to isomerize the enol first into the keto form and, subsequently, to adjust a conformation of the molecule that supports a parallel alignment of the σ-bond to the π-bond of the carbonyl (Figure 1b and Figure 2B). This adds an isomerase activity as a third function to FAHD1.
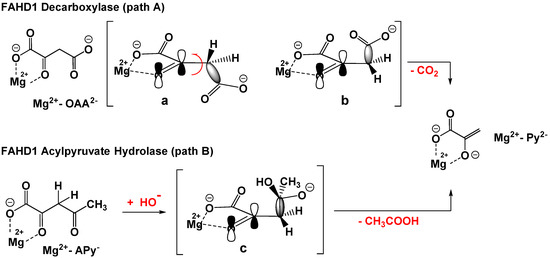
Figure 1.
Orbital model for the ODx and ApH reactions catalyzed by FAHD1. Path A: ODx. Mg2+-OAA2− depicted in two conformations: not competent (a) and competent conformations (b) for decarboxylation and the formation of the pyruvate enolate complex Mg2+-Py2− (bent arrow indicates bond rotation); C–C bond cleavage under CO2 evolution to the Mg2+-complexed enolate of pyruvate (Mg2+-APY2−). Path B: ApH. Nucleophilic addition of hydroxyl to the 2-keto form of OAA2−; (c) intermediate oxyanion already in a productive conformation is shown; C–C bond cleavage under the extrusion of acetic acid and formation of the pyruvate enolate complex Mg2+-Py2−.
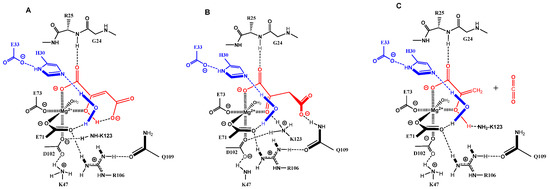
Figure 2.
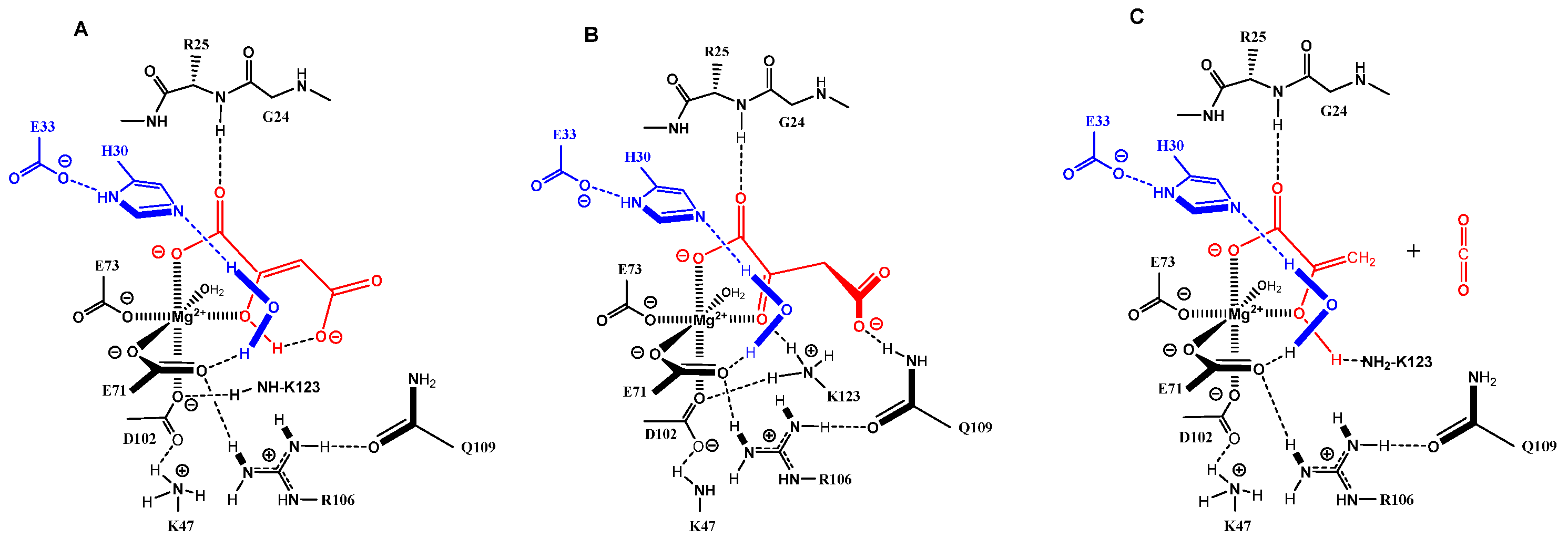
Mg-bound enol form of OAA (red, (A)); Q109 promoted the conformational control of ketonized OAA (red, (B)); quench of the primary enolate of pyruvate by K123 to pyruvate–enol and CO2 release (red, (C)); blue: catalytic dyad E33-H30 and captured water molecule; dotted black lines: hydrogen bonding; filled bonds are in front and point towards the viewer).
A major conclusion of the theoretical analysis of the C–C bond cleavage in the FAHD1 mechanisms is, although the substrates, as well as the mechanisms behind are different, that both pathways lead to the same primary cleavage product, namely pyruvate enolate complexed to Mg2+. It must be taken into account that a pyruvate–enolate complex reveals a more than ten thousand-fold greater stability constant compared to simple carboxylic acids [9], implying a block of lid opening and product release. For the ODx, as well as ApH mechanisms, a quench of the primary enolate to the final enol of pyruvate is proposed to prevent inhibition by the product.
In the ApH mechanism, the generation of the nucleophilic hydroxyl from a conserved (and isolated) water molecule can be performed by the dyad of His-Glu present in reasonable proximity to the catalytically important water in the substrate pocket of FAHD1. The attack of the nucleophile on the pro-chiral carbonyl center of acylpyruvates (C4) follows strictly along the trajectories defined by the orientation of the acyl carbonyl group in the FAHD1 catalytic center [10]. The site of a nucleophilic attack will be under stereospecific control by the enzyme residues interacting with the bound substrate. The striking similarity of both mechanisms is that the oxyanion formed along the acylpyruvase pathway appears at the identical position as the negatively charged carboxylate oxygen in the ODx path. The difference between ODx and ApH mechanisms is the leaving molecule after C–C bond cleavage: carbon dioxide from ODx but acetic acid from the ApH mechanism. The quench of the enolate and lid opening in the ApH mechanism would be supported by the acetic acid.
The ketonization of the OAA enol form (Figure 2A) needs a proton transfer mediated by residues of the catalytic cavity. An enol–hydroxy function shows a more than ten thousand-fold increase in acidity when complexed to magnesium [11]. Lysine K123, which can adopt an equlibrium protonation state in the K43-D102-K123 network, would be able to deprotonate the enol and transfer the proton through the 4-carboxylate to the C3-centered carbanion. However, ketonization alone is not sufficient for decarboxylation. The 2-keto OAA has to adopt a conformation that is not in an energy minimum. A further residue in the catalytic cavity of FAHD1 must exist for control of the conformation of the bound OAA keto form. Through ketonization, the molecule gains rotational freedom around the C2–C3 bond and can achieve the parallel orbital orientation of the C3–C4 bond to the carbonyl π*-orbital by assistance of a residue provided by FAHD1. We assign the carbamoyl group of residue Q109, which is part of the hydrogen bond network E71-R106, the strategic role for the conformational control of bound substrates (Figure 2B). The energy needed for the C3–C4 bond rotation of OAA has been calculated to be 2.4 kcal/mol [12]. The importance of the glutamine residue in the catalytic mechanism has been shown for Cg1458, the prokaryotic analog of FAHD1. Mutation of the corresponding residue almost abolished Cg1458-ODx activity (98%) [13].
Persistent formation of the extremely stable Mg2+–enolate complex [9] can be prohibited in an equilibrium protonation of enolate oxygen through the ammonium group of K123. The products of OAA processing by FAHD1 would be pyruvate enol and carbon dioxide (Figure 2C). However, pyruvate enol in the bidentate-binding mode to Mg2+ still supports the strong hydrogen bonding of the 1-carboxylate to NH of liganded G24. Consequently, the lid of FAHD1 would not open for product release (equivalent to inhibition by the product). At this point of the discussion, it must be added that pyruvate enol is a high-energy molecule storing ~ 7 kcal/moL of free energy relative to the keto form [14]. Taking the energy reservoir present in the pyruvate enol form into account, we propose once more a ketonization of the enol as an additional step prior to the lid opening. The necessary proton transfer could be supported by the conserved water molecule positioned by the E33-H30 dyad or through the D102-K123 network. In the case of the water molecule, the produced hydroxyl would be competent to displace the pyruvate from the magnesium, destroying the stabilization of liganded G24, which would revert to the conformation adopted in the unliganded structure. The lid could open under release of pyruvate as final product. The catalytic center would be restored for the next catalytic cycle upon exposure to the mitochondrial environment (remark: the conserved water molecule would show amphoteric behavior: acidic in the ODx mechanism but nucleophilic in the hydrolase mechanism).
1.2. Design Steps of First Generation of FAHD1 Inhibitors
The analysis of the proposed FAHD1 mechanisms suggests that replacement of the cleavable carbon–carbon bond in FAHD1 substrates by an uncleavable bond, while keeping the Mg2+-binding motif should lead to structures with inhibitory potential (Figure 3).
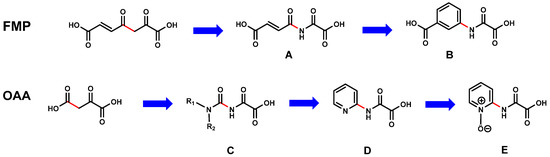
Figure 3.
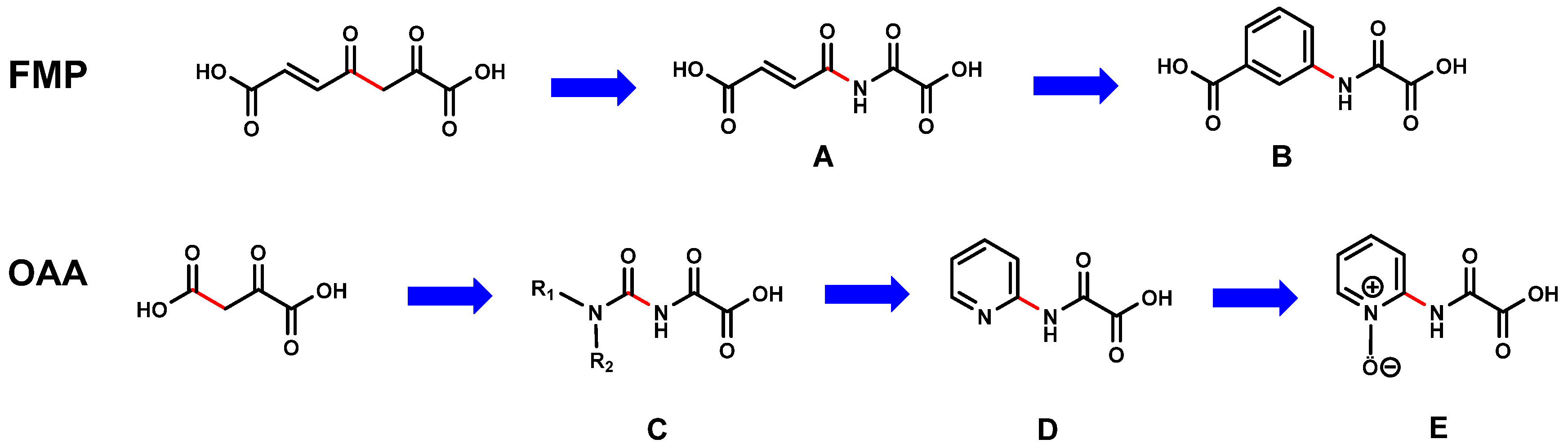
The design principle of the 1st-generation FAHD1 inhibitors. Upper lane: deduction of oxalamic acid (B) starting from substrate fumarylpyruvate (FMP); bottom lane: deduction of N-pyridyl-oxalamic acid (D) and corresponding 1-oxide congener (E) starting from oxaloacetate (OAA); (red: initial C–C bond to be cleaved in FMP and OAA and position of the corresponding bonds in the designed structures (A–E)).
Exchange of the 3-methylene carbon by a nitrogen in fumarylpyruvate (FMP) leads to a flexible and reactive molecule A (depicted in Figure 3 and, also, for the following). The planar enone moiety present in FMP and A can be mimicked with an aromatic ring. The resulting molecule B is resonance-stabilized, capable of Mg2+ binding, and keeps the carboxy function in a similar position as in FMP, respectively. An equivalent consideration as described above can be applied to oxaloacetate (OAA). In case of OAA, the nitrogen exchange of the methylene group leads to a carbamic acid, which is not stable. Substitution by a further nitrogen changes the molecule to a reactive acyl-urea derivative C. As a mimetic of the urea substructure in C, a pyridine ring system can be introduced concomitant with conformational stabilization (D). Oxidation of the pyridine moiety to N-oxide E results in a structure presenting negative charge density in the space where substrate OAA2− positions the carboxyl anion and acylpyruvates their oxyanion intermediate. The deduced scaffolds B, D and E have molecular weights far below 300 and would leave generous room for further SAR studies and optimization of the pharmacological parameters. The proposed molecules B, D and E belong to the class of oxalamic acid derivatives. Such molecules are known and easily accessible by proven experimental procedures.
2. Results and Discussion
2.1. N-Aryl-Oxalamic Acids as FAHD1 Inhibitors
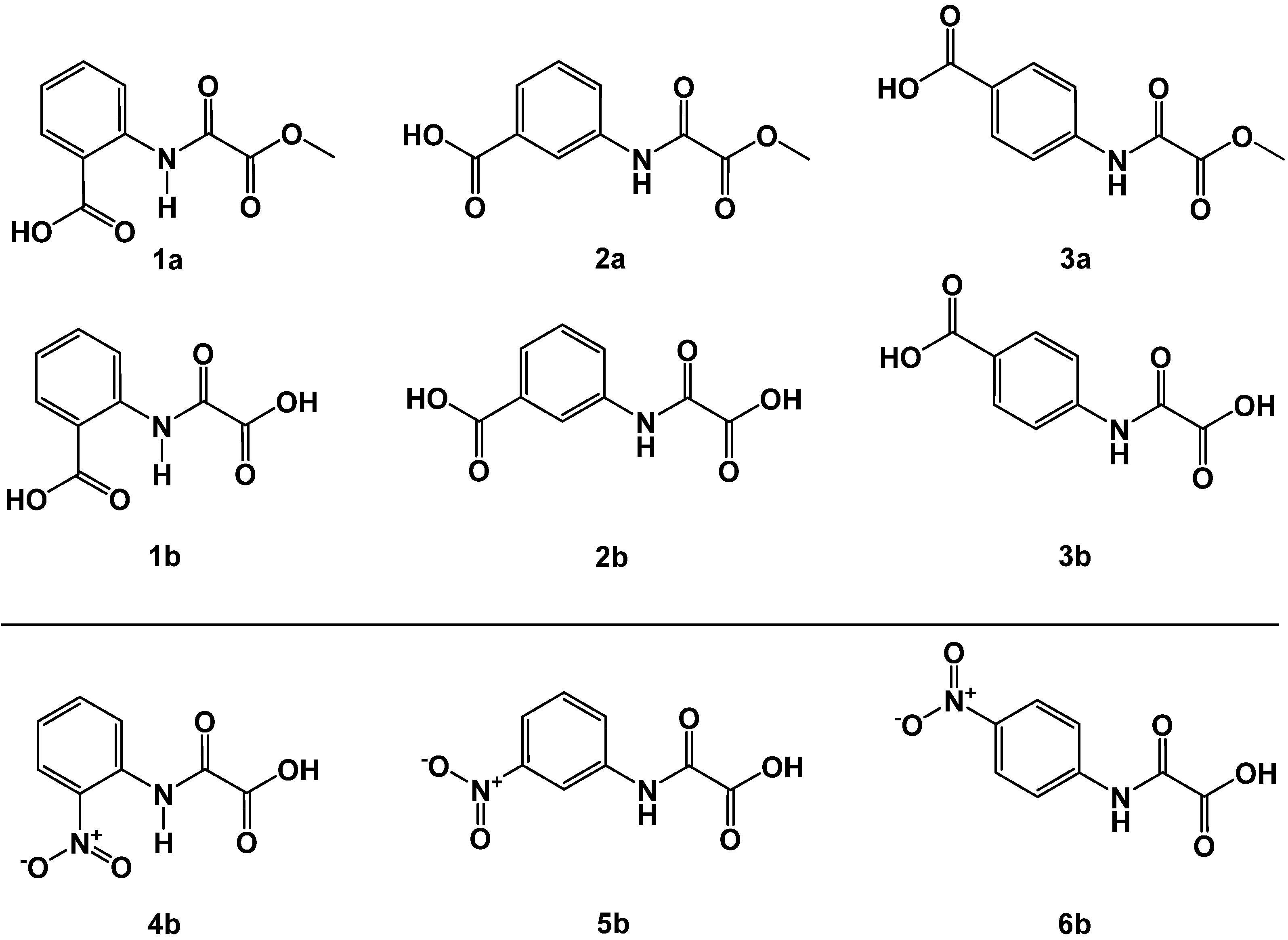
First, we synthesized exemplaric known compounds of scaffold B (Figure 3) derived from anilines functionalized with polar carboxy or nitro groups, which present the charge density into the space of the oxyanion hole in the FAHD1 catalytic cavity. The acylation of amino-substituted benzoic acids yields oxalamic acid methyl esters (1a–3a) in high yields and purity. Analogously, the acylation of nitro-substituted anilines afforded after saponification oxalamic acids 4b–6b. The compounds obtained from the carboxy series revealed instantly a good-to-moderate inhibition of FAHD1-ODx activity (Figure 4 and Table 1).
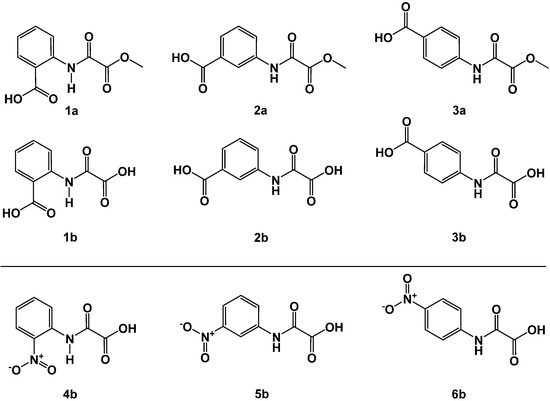
Figure 4.
Oxalamic acids methyl esters 1a–3a and corresponding acids 1b–3b (upper panel); o-, m- and p-nitro-substituted N-phenyl-oxalamic acids 4b–6b (lower panel).

Table 1.
Carboxy substituted N-aryl oxalamic acid derivatives 1a–3b. Nitro-substituted N-aryl oxalamic acids 4b–6b. Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
In the series of methyl esters 1a–3a, we observed a remarkable change in activities as a function of the position of the carboxy substituent. The o-substituted 1a shows only a moderate inhibition (IC50 = 47 µM), whereas the m-substituted congener 2a was already comparable with the only known FAHD1 inhibitor oxalate (IC50 13 µM vs. 7 µM). The p-carboxy-substituted derivative 3a revealed a weak activity.
1b belongs (accordingly the design steps shown in Figure 3) to a fumarylpyruvate (FMP) mimetic. The m-phenyl carboxy group is presented at a similar position as in the FAHD1 substrate FMP, creating an additional interaction not accessible by o- and p-substituted congeners 2a and 3a, respectively. The o-carboxy derivative 1a can be termed as a close mimetic of oxaloacetate. However, a six-membered hydrogen bonding between amide NH and oxygen of the o-carboxylate creates a resonace stabilized flat structure that impairs the presentation of negative charge density to the oxyanion hole of FAHD1. This conformational penalty is obviously romoved upon saponification to the acid 1b, most probably due to electronic repulsion of the corboxy functions. Saponification of the FMP analog 2a to the corresponding acid 2b dit not change the inhibitory activity, suggesting that most of the binding energy of this derivative is carried by the m-substituent. The p-carboxy derivatives 3a and 3b present the N-aryl substituent in a position not covered by substrates OAA or FMP, which explains their reduced activity.
The 2-oxalylamino-benzoic acid 1b is a known competitive inhibitor of protein-tyrosine phosphatase β [15]. The molecule behaved as a phosphate mimetic and has been used as a “minimal unit” mimetic for advanced and selective inhibitors of this enzyme, but 1b apparently could also serve well for FAHD1 inhibitors. The low molecular weight of 1b enables a broad derivatization for the establishment of a structure–activity relationship (SAR) for FAHD1. Somewhat surprising was that oxalamic acid methyl esters 1a–3a kept active. Especially meta-substituted 2a was equally potent copmpared to the ortho-carboxylate presenting 1b. This finding is important, because acid functions are known to impair the cellular uptake. Ester derivatives could act as prodrugs, facilitating membrane crossing (for additional ester derivatives, see Section 2.3). We investigated further substituents on the N-aryl scaffold, such as hydroxy-, methoxy or halogen groups in various positions, but none of the synthesized compounds improved the IC50 below 30 µM (selected results are provided in the Supporting Materials).
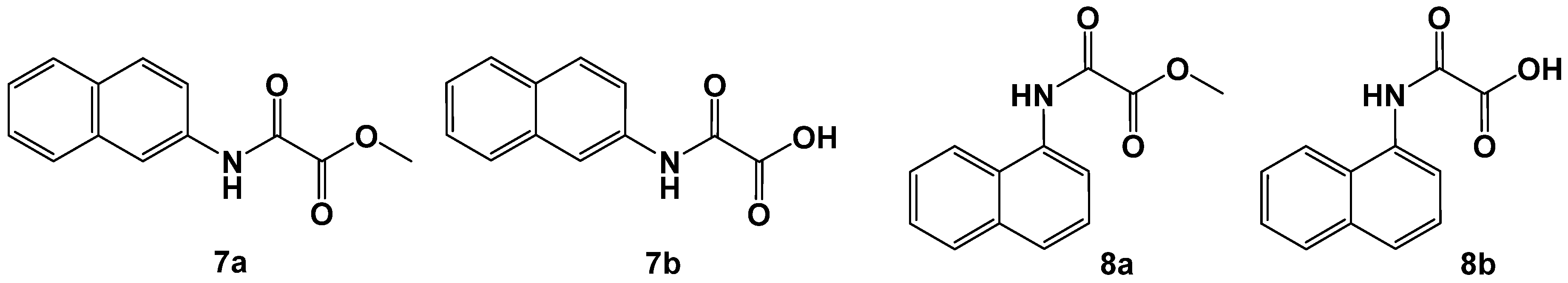
In the next step, we investigated α- and β-naphthylamines as examples for annulated ring systems. It was found that only β-substitution is appropriate for good inhibitors, wheras the α-naphthyl congeners sustain a dramatic loss of activity (Figure 5 and Table 2).
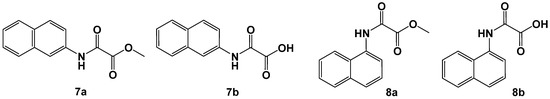
Figure 5.
N-β-naphthyl-oxalamic acid methyl ester (7a) and -acid (7b) and N-α-naphthyl oxalamic acid analogs (8a, 8b).
Table 2.
N-β-naphthyl oxalamic acid derivatives (7a, 7b). N-α-naphthyl oxalamic acid derivatives (8a, 8b). Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams including the fit and standard deviations of the individual experiments (n = 3) are depicted in the Supplementary Materials.
N-β-naphthyl-oxalamic acid methyl ester (7a) and -acid (7b) share a similarity with the m-carboxyl substituted derivatives shown in Figure 4 but still miss the charge presentation. The ester-acid pair 7a and 7b is a rare example where the saponification of the ester to the acid led to a significant loss of activity. The results warrant further investigations of annulation with heterocycles and substitution of the naphthyl moiety by polar functional groups.
Discouraging results were obtained from investigation of compounds where the N-aryl ring was spacered by an alkyl (e.g., N-benzyl or N-cycloalkyl motifs). Such derivatives revealed IC50 > 100 µM (for examples, see the Supporting Materials).
2.2. N-Pyridyl Substituted Oxalamic Acid Derivatives and Their N-Oxides as FAHD1 Inhibitors
In the following, we gave focus to synthesis of pyridine scaffolds (D) and their N-oxides (E), which were deduced from the OAA substrate (Figure 3). Heterocyclic N-oxides have been investigated in numerous medicinal chemistry projects and revealed anticancer, anti-inflammatory, antibacterial, anti-HIV and antiparasitic activities, among many others [16] Additionally, the N-oxide moiety serves as an important molecular recognition motif in biological interactions.
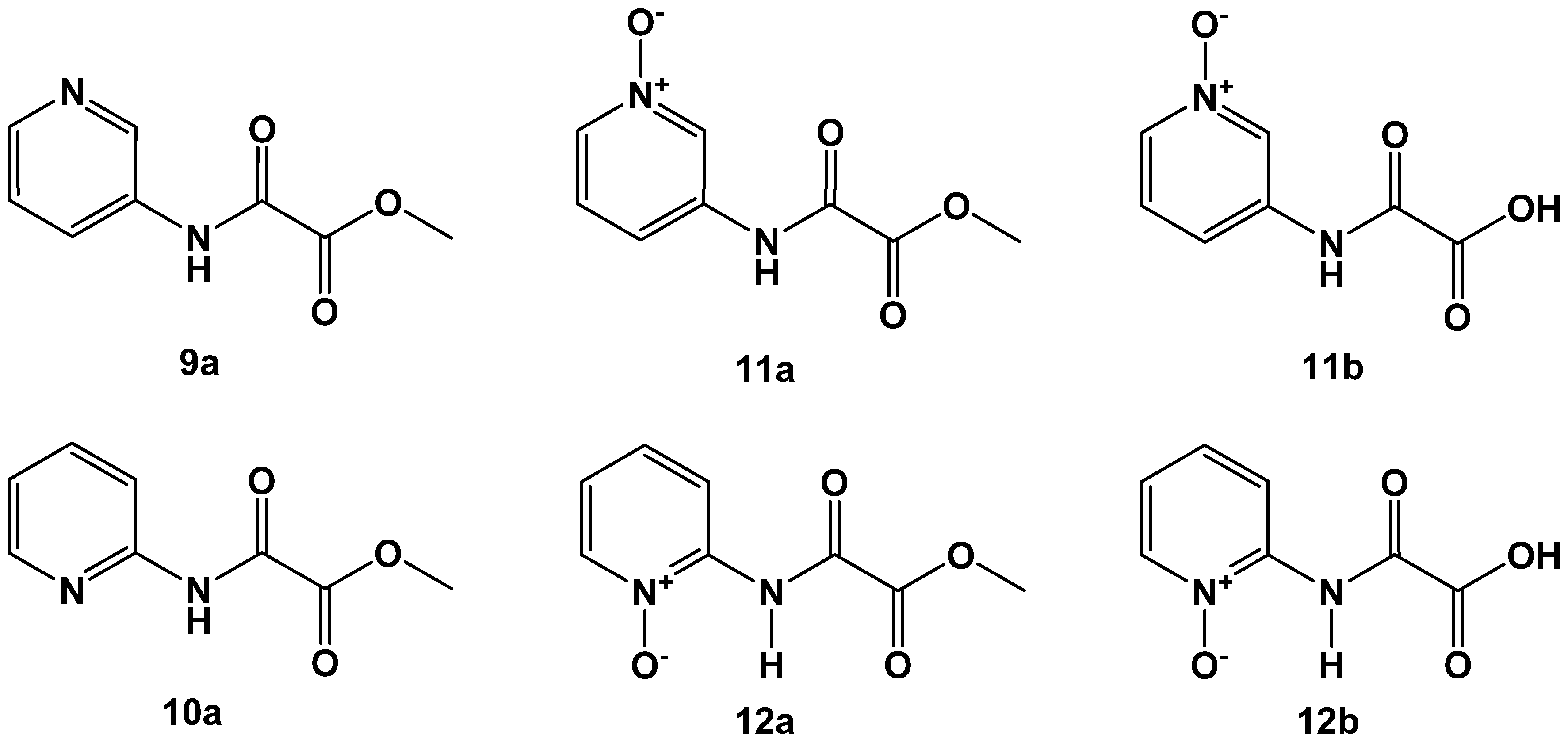
We first synthesized 3-aminopyridine and 2-aminopyridine oxalamic acid methyl esters 9a and 10a (Figure 6). Clean oxidation of the heterocyclic ring nitrogen afforded the corresponding N-oxides 11a and 12a, which were saponified to the acids 11b and 12b. The compounds showed FAHD1 inhibitory activity, as proposed by the design strategy (Table 3). Derivatives 9a, 11a and 11b obtained from the 3-aminopyridine building block turned out to be much superior in FAHD1 inhibitory potency compared to the 2-aminopyridine congeners 10a, 12a and 12b. The 3-aminopyridine-derived compounds can deliver negative charge density towards the space of the oxyanion hole of FAHD1, stabilizing the binding of the inhibitor. In contrast, the 2-aminopyridine congeners present the charge density opposite to the oxyanion hole. Rotation around the C–N bond cannot correct for this obstacle. There is no evidence for a tautomerism from the NMR-spectra of the 2-aminopyridine-derived compounds.
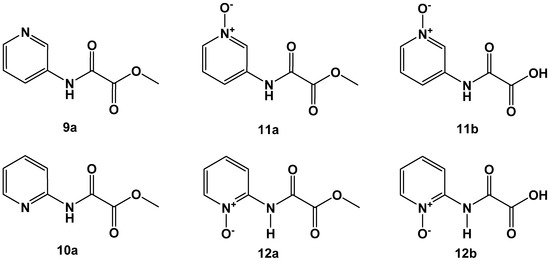
Figure 6.
Oxalamic acid derived from 3-aminopyridine (upper lane) and 2-aminopyridine building blocks (lower lane).
Table 3.
N-(pyridine-3-yl)oxalamic acid derivatives 9a–11b. N-(pyridine-2-yl) oxalamic acid derivatives 10a–12b. Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
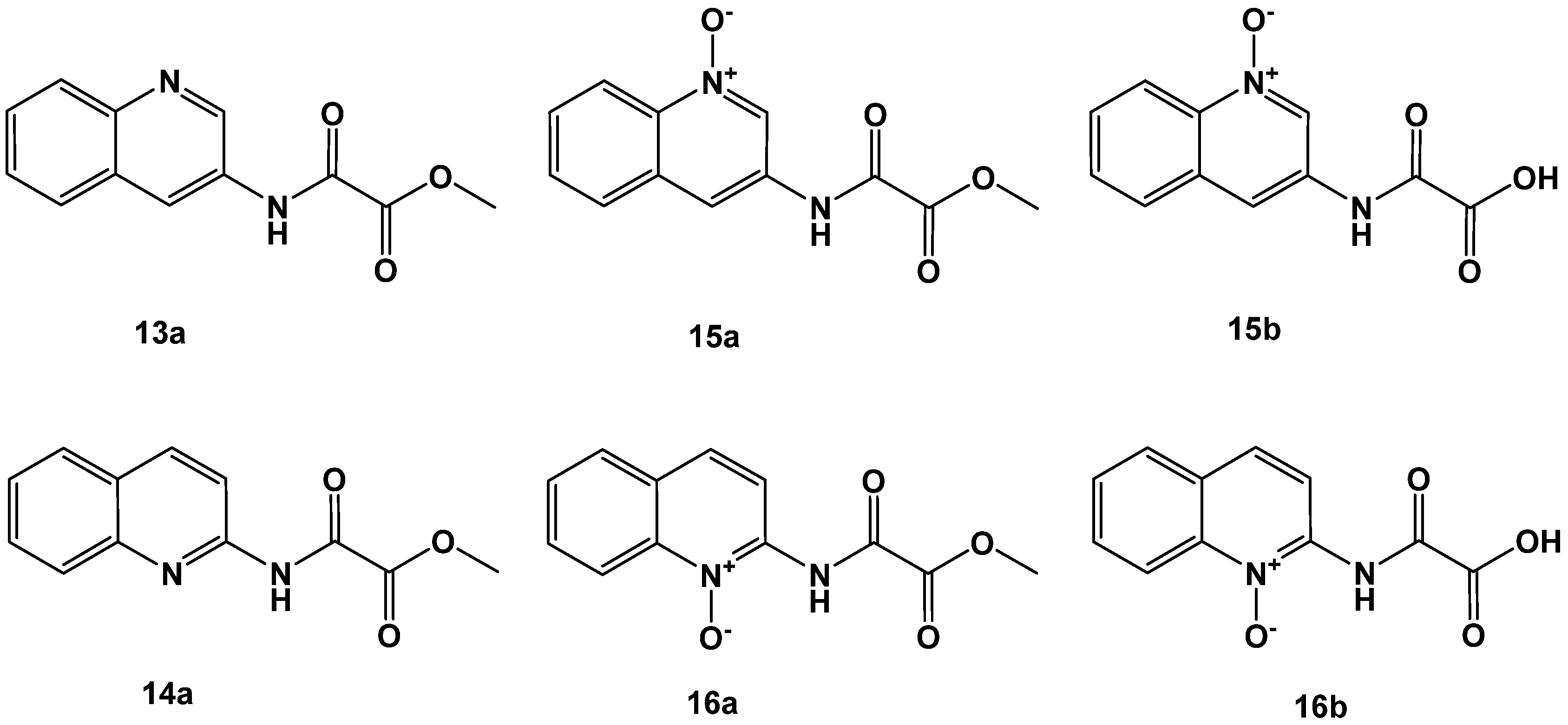
To study the effect of annulation of the pyridine ring, 2-amino or 3-aminoquinoline building blocks were applied in the synthesis. The modification was especially beneficial for the 2-aminoquinolines (14a, 16a and 16b; Figure 7 and Table 4) compared to the nonanullated 2-aminopyridines (10a, 12a and 12b; Figure 6). The 3-aminoquinolines kept good activity with the 3-aminoquinoline N-oxide 15b as the most active representative.
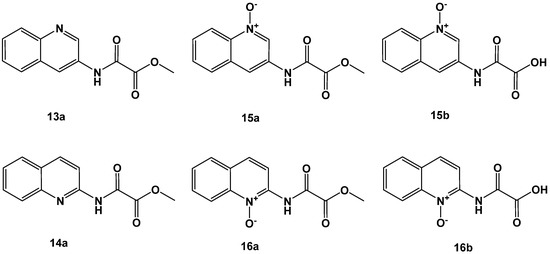
Figure 7.
2-amino-(lower lane) and 3-amino-quinoline derivatives (upper lane).
Table 4.
N-(quinolin-3-yl)oxalamic acid derivatives 13a, 15a and 15b. N-(quinolin-2-yl)oxalamic acid derivatives 14a, 16a and 16b. R Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
2.3. Variations Affecting the 1,2-Dicarbonyl Motif in Oxalamic Acid Derivatives
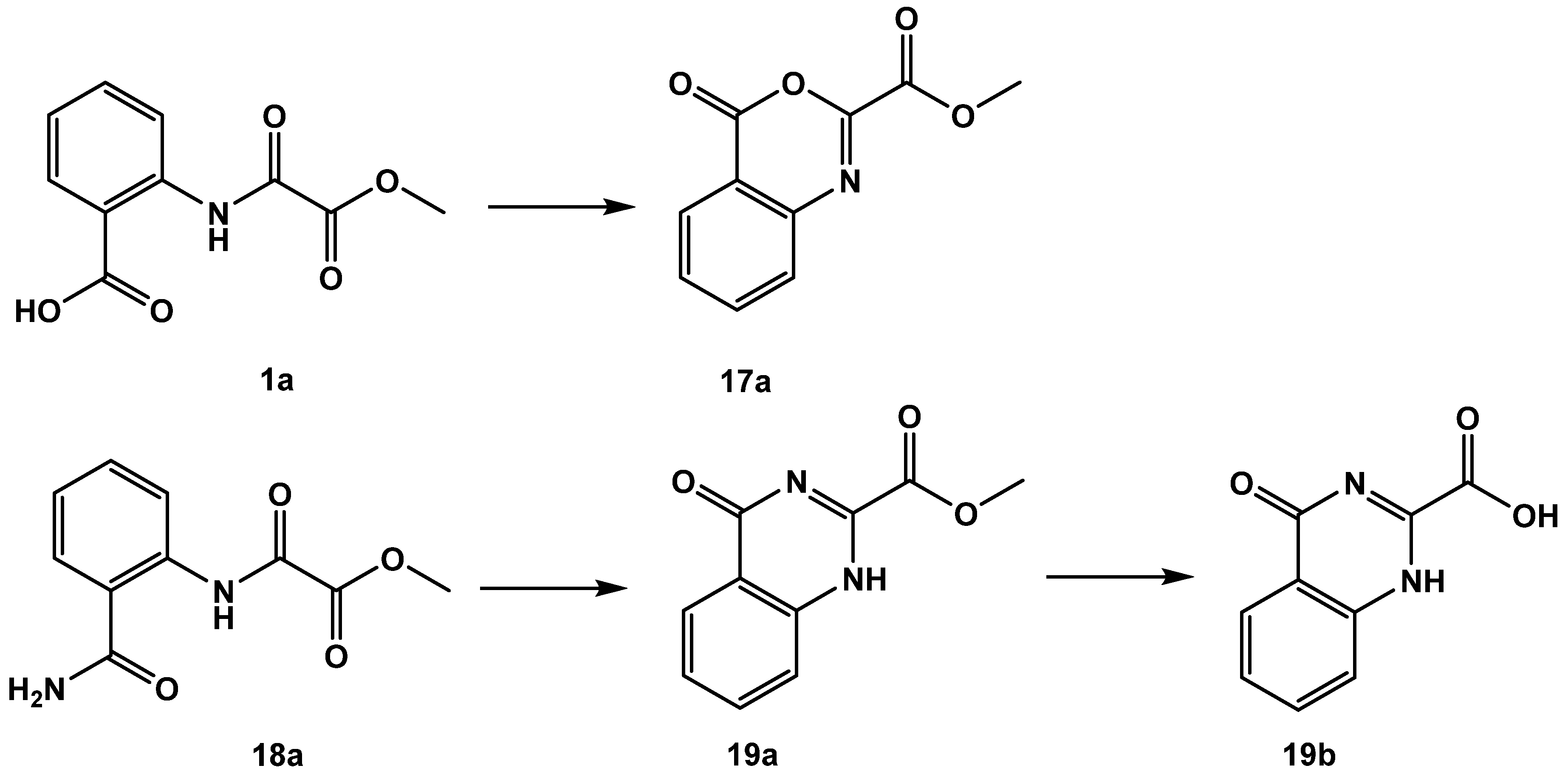
As disclosed by the single molecule X-ray diffraction analysis of FAHD1 with bound oxalate inhibitor, the 1,2-dicarbonyl motif is an essential structural element for bidentate binding to the magnesium cofactor. We explored several modifications in this motif. The oxalamic acid derivative 1a was cyclized at an elevated temperature or upon treatment with acetic acid anhydride to a 4-oxo-4H-benzo[d]-[1,3]oxazine derivative 17a [17]. Analogously, the ortho-amide-bearing methyl ester 18a was cyclized to methyl 4-oxo-1,4-dihydroquinazoline-2-carboxylate 19a [18]. The latter one was saponified to the acid congener 19b (Figure 8). All these small molecules inhibited FAHD1 ODx activity in the low µM IC50 range (Table 5). It must be considered that the highly active 17a is a reactive molecule able to acylate nucleophiles (but, so far, we have no evidence that 17a is modifying residues in the catalytic center of FAHD1).
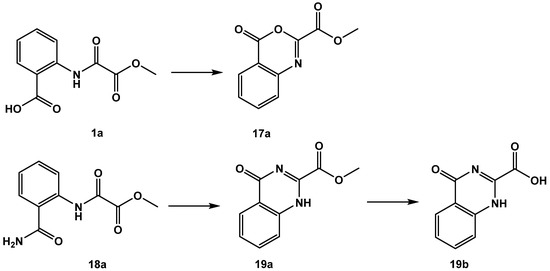
Figure 8.
Products obtained from reactions under participation of the 1,2-dicarbonyl motif.
Table 5.
Modifications of the 1,2-dicarbonyl motif. 4-oxo-4H-benzo[d]-[1,3]oxazine 17a obtained from 1a. Derivatives 19a and 19b obtained from the reaction of the anthranilic acid amide derivative 18a. Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
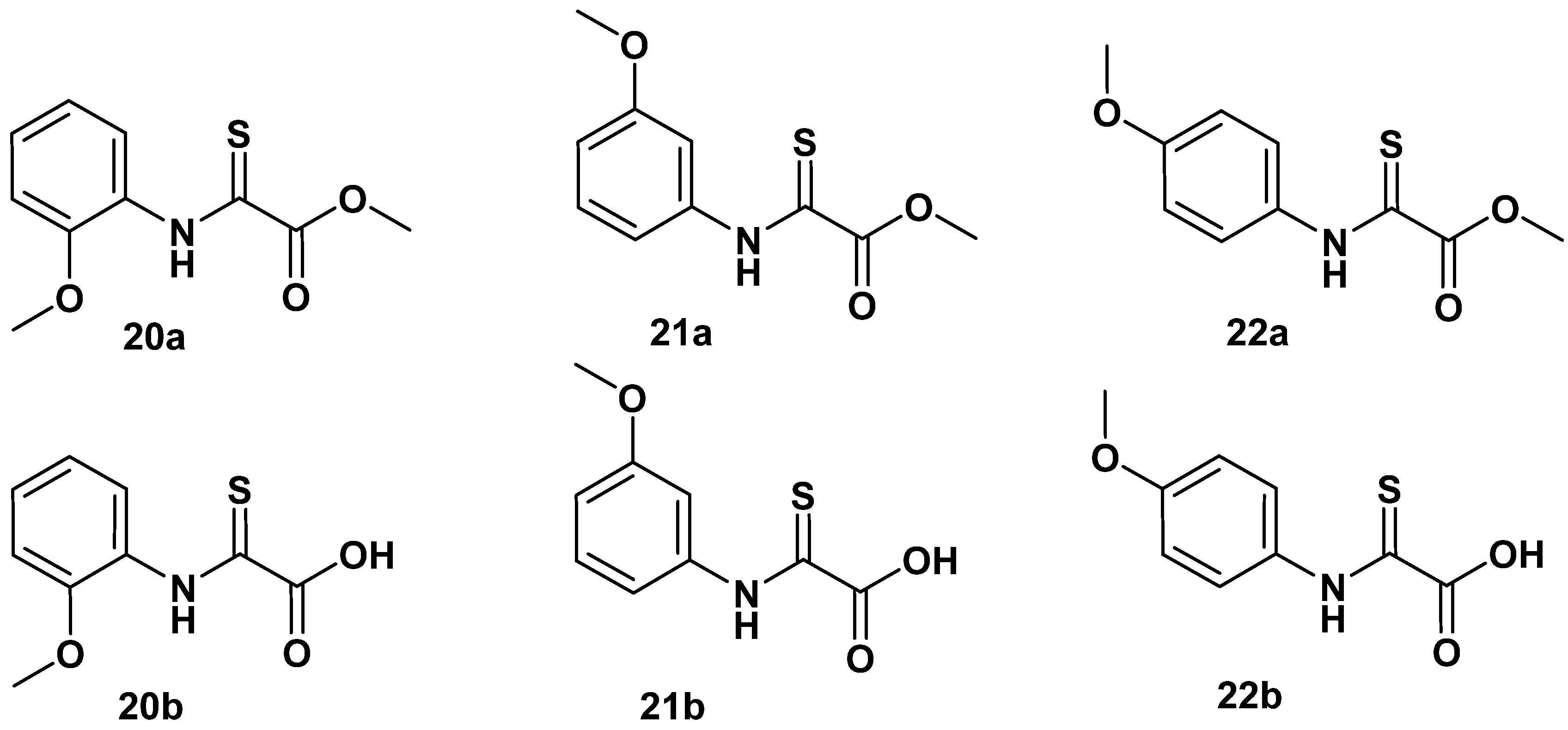
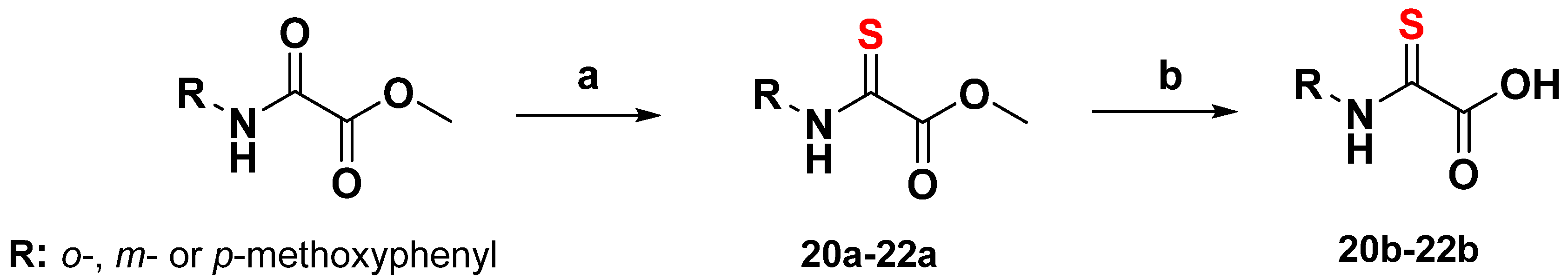
Formation of cyclic 17a is propagated by a nucleophilic attack of 2-amide carbonyl oxygen on the activated ortho-carboxyl group of 1a [17], disclosing a significant polarization of the 2-amide function towards the oxygen atom, which supports strong bidentate binding to the enzyme cofactor and gains stability towards hydrolysis of the amide bond in oxalamic acids. The nucleophilic property of the 2-oxygen in oxalamic acid derivatives inspired us to investigate the reaction with a Lawesson reagent (CAS RN 19172-47-5). We found a clean specific exchange of the 2-carbonyl in oxalamic acid esters to the 2-thio congeners 20a–22a. Saponification of the methyl esters afforded the acid derivatives 20b–22b (Figure 9).
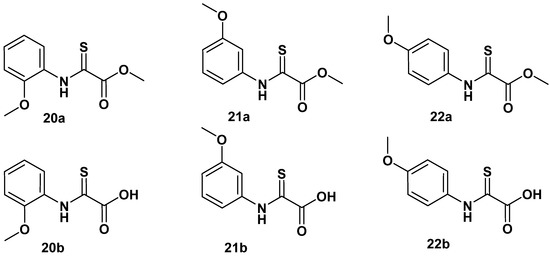
Figure 9.
Selective exchange of 2-oxo function with the Lawesson reagent to 2-thio derivatives.
However, these transformations did not improve the inhibitory activity, as summarized in Table 6.
Table 6.
N-(o-, m-, p-methoxyphenyl)-2-thio oxalamic acid methyl esters (20a–22a and the corresponding acids 21b–22b. Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
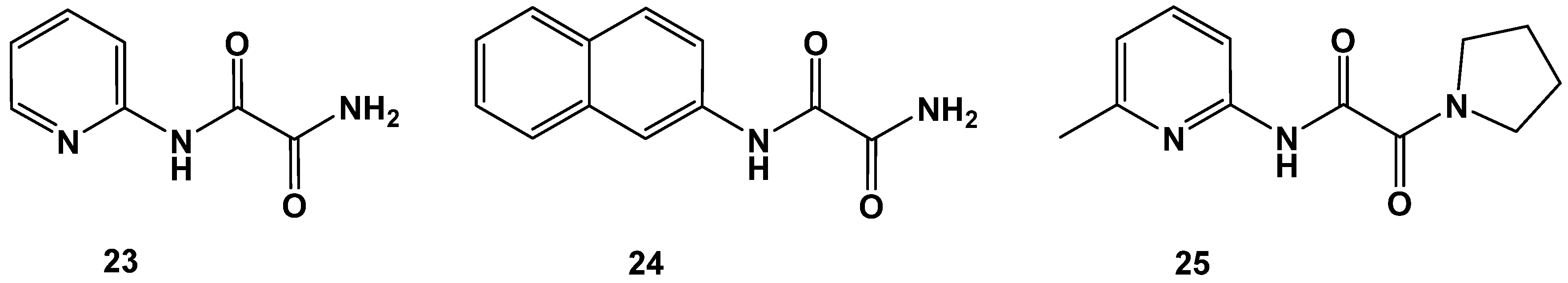
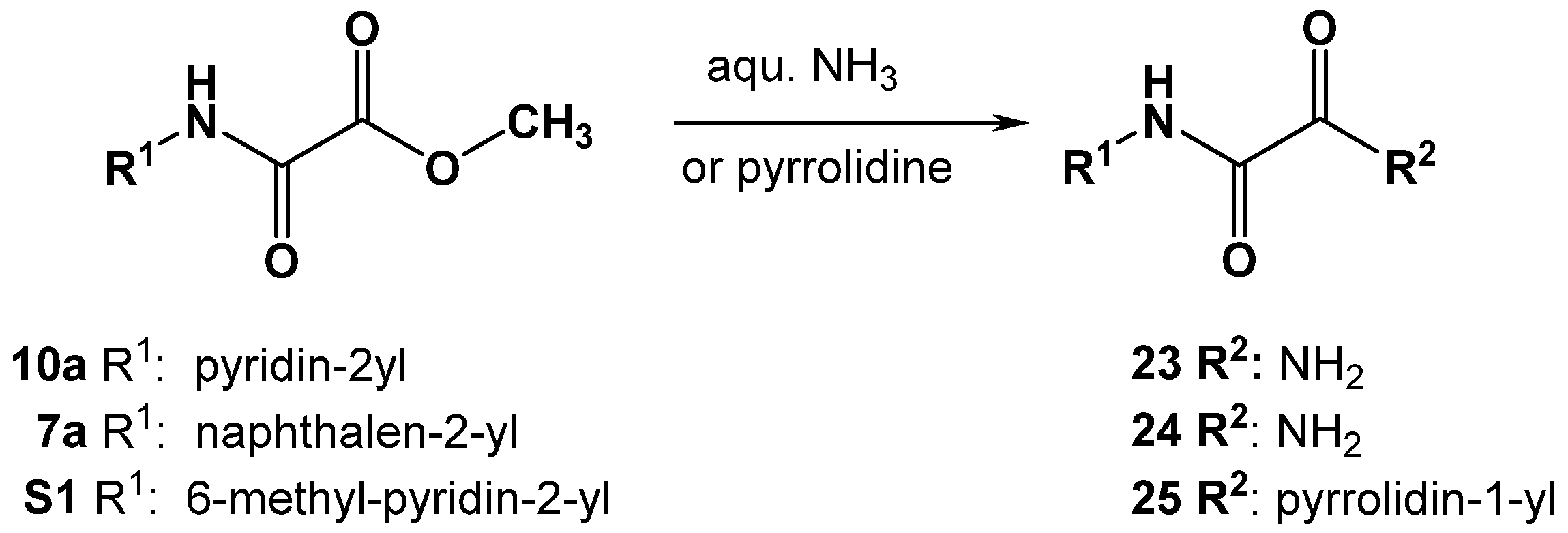
Change of oxalamic esters to oxalamic acid amides was also not a favorable transformation, as revealed by derivatives 23–25. C-terminal cycloalkyl amide 25 was an inferior compound (Figure 10 and Table 7).
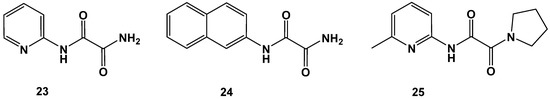
Figure 10.
Examples of oxalamic acid diamides (23–25).
Table 7.
Pyridin-2-yl (23), naphthyl-2-yl (24) and pyrrolidine-1-yl (25) oxalamic acid amides. Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
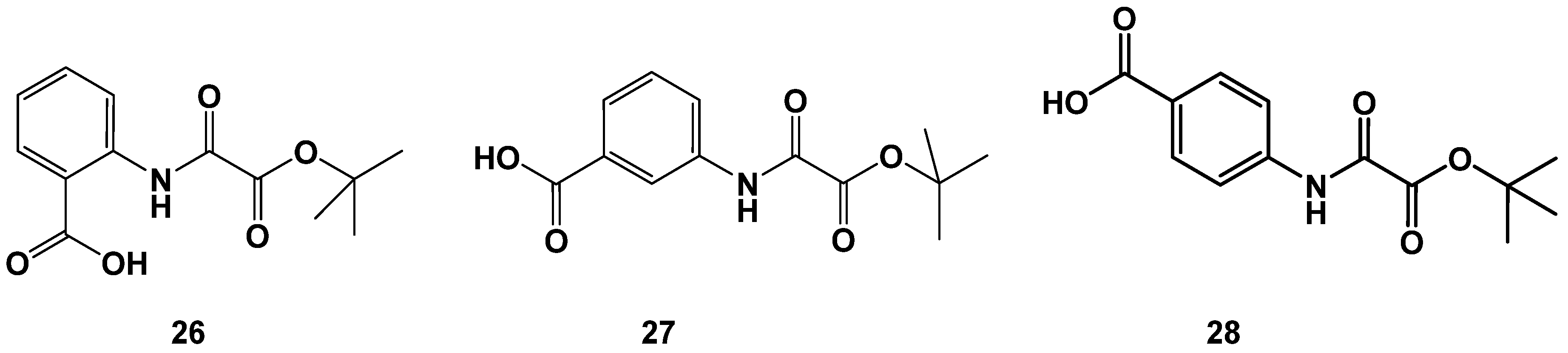
A surprising result became visible when we tested a variation in the terminal ester functionality. Change of methyl esters to tert. butyl esters in the series of ortho-, meta- and para-aminobenzoic acids yielded two highly potent inhibitors 26 and 28, whereas the meta-substitution (27) was not well-tolerated (Figure 11 and Table 8). Comparison of the tert. butyl esters 26–28 with the corresponding methyl esters 1a–3a (see Figure 5 and Table 1) reveals that the modification is beneficial for the methyl esters with lower activity. 1a gained a four-fold and 3a even a six-fold increase in activity, whereas the highly active m-carboxy-substituted 2a sustains a twenty-fold loss in activity upon the change from a methyl to tert. butyl ester.
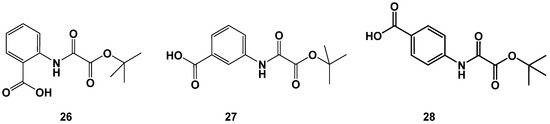
Figure 11.
Identification of highly active tert. butyl esters 26 and 28.
Table 8.
N-(o-, m- or p-carboxy-phenyl) oxalamic acid tert. butyl esters. Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
However, the catalytic center of FAHD1 (as derived from the oxalate ligated FAHD1 X-ray structure) does not feature enough space for the accommodation of a hydrophobic and bulky tert. butyl residue. Especially the activity increase of the p-carboxy-substituted 28, which carries the charge presenting substituent at an unfavorable position (s. 3a, Figure 4), points towards a drastic reorganization within the catalytic center of FAHD1 upon the binding of these structures. These findings need further investigations. The derivatizations of tert. butyl esters were extended from the aminobenzoic acids to other scaffolds and resulted in additional compounds with high inhibitory activity (see the Supporting Materials).
Starting from these observations, and despite the initially negative experience made with variations in the terminal amide region, we explored symmetric oxalamic acid diamides for FAHD1 inhibition.
2.4. N,N′-Diaryl-Oxalamic Acid Diamides
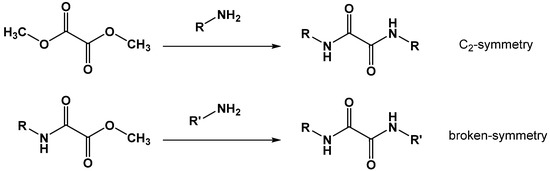
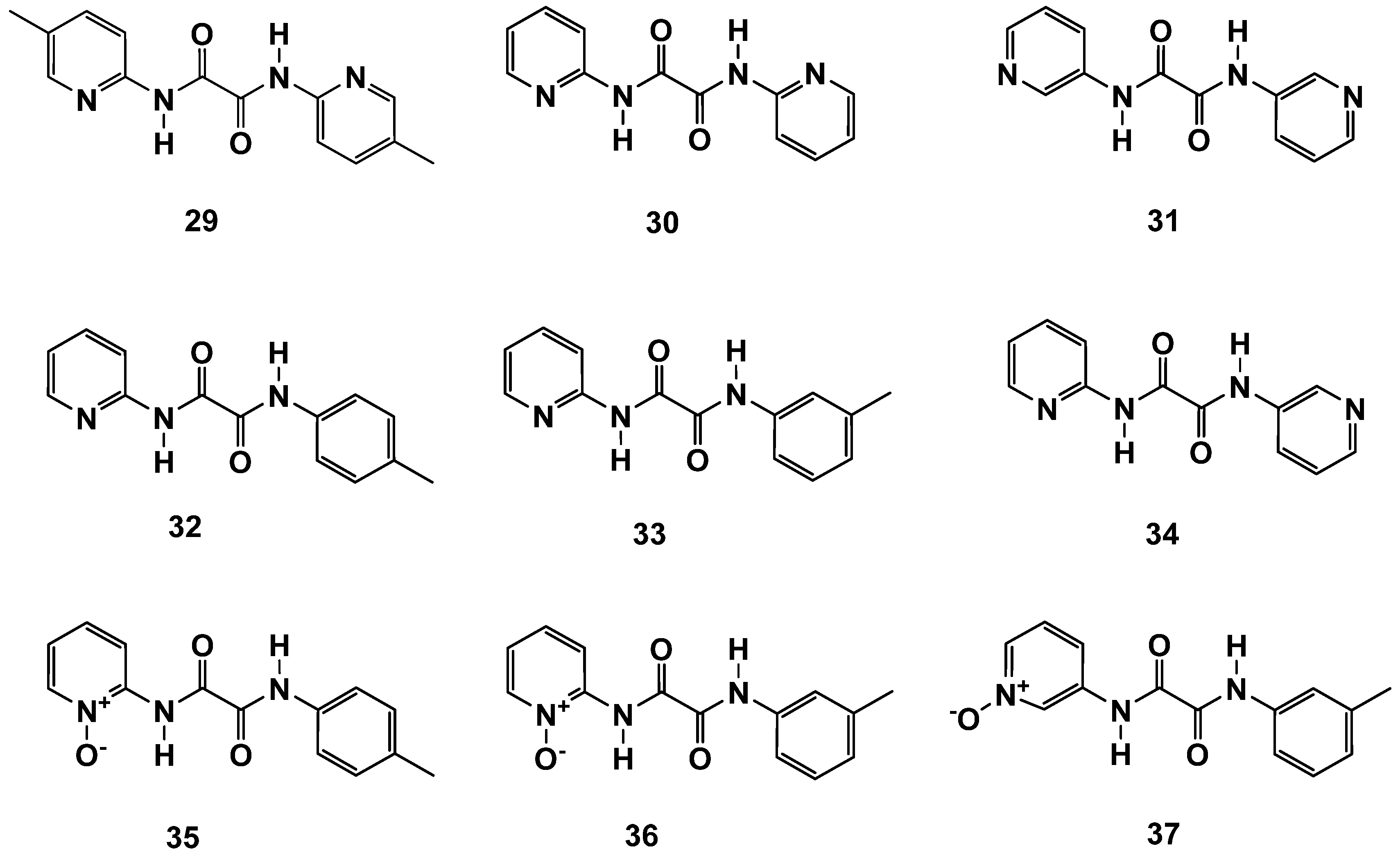
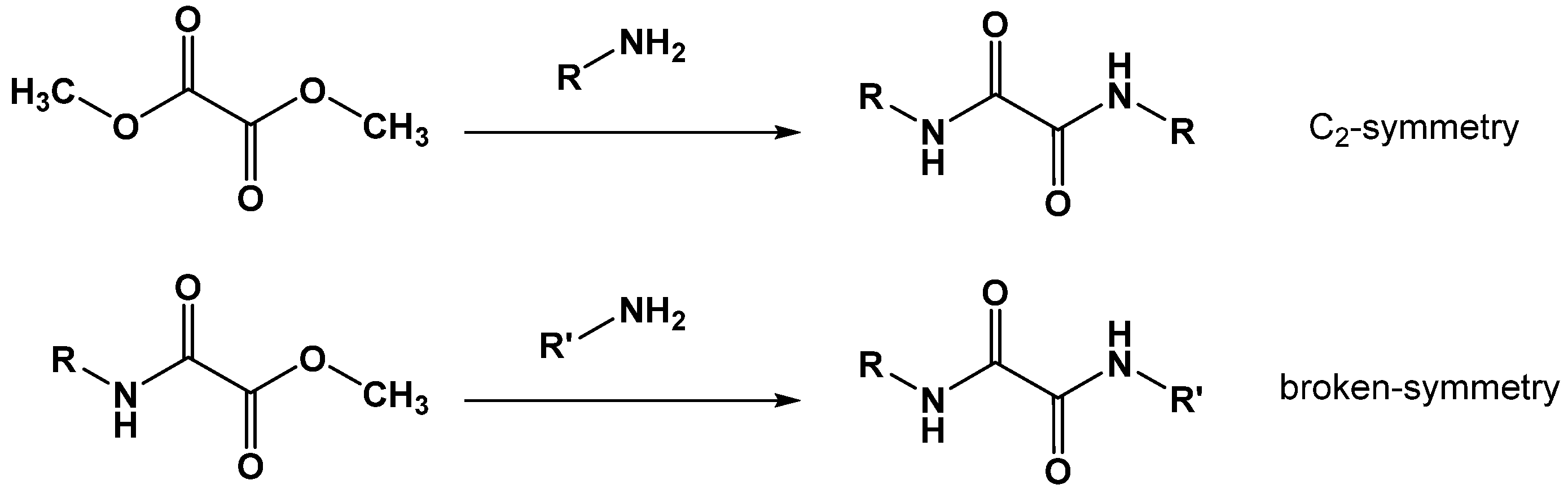
Surprisingly, the synthesis starting with 2-aminopyridine delivered highly active C2-symmetrical compounds (e.g., 29 and 30; Figure 12 and Table 9). Symmetry was not a prerequisite for high activity, as demonstrated by compounds 32 and 33. The C2-symmetrical combination of N,N′-bis-3-aminopyridyl led to a significant loss of activity (31). The combination of N-2-aminopyridyl with N′-3-aminopyridyl (37) even resulted in a total loss of activity. These results point towards a so-far-undisclosed specific interaction of these compounds within FAHD1, which deserves further investigations. The N,N′-diamide scaffold cannot be compared with the compounds described in Section 2.2, which are capable of bidentate binding to the Mg cofactor of FAHD1. The N,N′-diamide molecules adopt a planar structure with the 1,2-carbonyls in s-trans conformation [19] (rotational barrier ~ 12 kcal/mol), which does not support bidentate Mg complexation. The interpretation of the activities may become even more complex when taking into account, that the oxalic acid diamides present recognition motifs for β-sheets or backbone amide bonds (present, e.g., in the exposed FAHD1 lid domain).
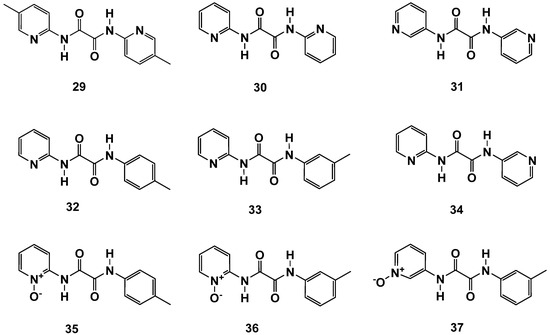
Figure 12.
N,N′-diaryl-oxalamic acid diamides with C2 symmetry and broken symmetry.
Table 9.
C2-symmetric N1,N2-bis(pyridin-2-yl or -3-yl) oxalamides (29–31). N1,N2- diaryl oxalamic acid amides with broken symmetry (32–34). Pyridine 1-oxide derivatives (35–37) obtained from the compounds with broken symmetry. Data fit with GRAPHPAD PRISM 5. Standard error of the fit given in µM. IC50 diagrams, including the fit and standard deviations of the individual experiments (n = 3), are depicted in the Supplementary Materials.
We also synthesized novel N-oxides of N,N′-diamides (35–37) and achieved good results with 2-aminopyridine-based compounds 35 and 36, whereas the 3-aminopyridyl containing derivative 37 was found to be inactive. Obviously, the N′-m-toluidine amide in 37 destroys the activity of acid congener 11b (Figure 6 and Table 3), which represents one of the most active compounds found in this study. In contrast, the 2-aminopyridyl-derived N′-m-toluidine amide 36 gained significant activity compared to its acid congener 12b. The so-far-collected results for the N,N′-diamide scaffold deserve a broad extension of SAR studies with this novel FAHD1 inhibitor scaffold.
N,N′-dipyridyl-containing compounds could also be oxidized to corresponding bis-N,N′-oxides, but the compounds obtained were unfortunately not soluble.
Taking together, the data for tert. butyl esters and the N,N′-diaryl-oxalamic acid diamides, an inhibition mode different to oxalate, can be assumed for these scaffolds. The discovery of the scaffold expands the initially applied design strategy significantly. To clarify the binding mode of these molecules, we initiated already co-crystallization experiments to create a solid basis for the future design of second-generation FAHD1 inhibitors to be active in the nanomolar regime.
3. Materials and Methods
Solvents, reagents and building blocks were purchased from various suppliers and used without further purification. Ethyl acetate and n-hexane (mixture of isomers) were distilled prior use in chromatographic separations.
3.1. Reaction Monitoring and Purification of Compounds
3.1.1. Thin-Layer Chromatography (TLC)
Reaction monitoring was performed by thin-layer chromatography (TLC) on Merck silica gel 60-F254 glass plates or on Macherey & Nagel POLYGRAM SIL G/UV 254 aluminium foils. The plates were developed with mixtures of hexane/ethyl acetate and methanol/ethyl acetate. Compound spots were visualized by UV (254 nm) irradiation or in TLC chamber containing iodine adsorbed on silica gel.
3.1.2. Middle Pressure Liquid Chromatography (MPLC)
Purification of compounds was performed by middle-pressure liquid chromatography (MPLC) on silica gel 60 from Merck (0.040–0.063 µm, 240–400 mesh). The unique home built MPLC system was provided by MoleculeCrafting.HuGs e.U. (Hubert Gstach) and consisted of columns, an FMI pump (Fluid Metering, Inc., Syosset, NY, USA) and an Amersham Superfrac fraction collector.
3.2. Analytical Characterization
3.2.1. High-Resolution Mass Spectroscopy (HRMS)
HRMS measurements were performed on Bruker maxis HD with ESI-q-TOF setup via direct infusion with a flow rate of 180 µL × h−1. ESI parameter: End Plate Offset 500 V, capillary 1500 V, nebulizer 0.4 bar, dry gas 4.0 L × min−1 and dry temperature 200 °C. Tune parameter: Transfer Funnel 1 RF 300.0 Vpp, isCID Energy 0.0 eV, Multipole RF 100.0 Vpp, Quadrupole Ion Energy 2.0 eV and Low Mass m/z 70.00.
3.2.2. Melting Points
Melting points (Mp) were determined with a Bausch & Lomb microscope equipped with a Kofler melting stage and are uncorrected.
3.2.3. Nuclear Magnetic Resonance Spectroscopy (NMR)
NMR spectra were recorded at the NMR Centre of the Faculty of Chemistry, University of Vienna, either on a DRX 600 WB spectrometer (resonance frequencies 600.25 MHz for 1H, 150.93 MHz for 13C) or on an Avance III HDX 700 spectrometer equipped with a quadruple QCIF cryoprobe (700.40 MHz for 1H, 176.12 MHz for 13C), both Bruker instruments (Bruker BioSpin, Rheinstetten, Germany).
The software used for the processing of 1D- (1H, 13C) and 2D- (COSY, HMBC and HSQC) NMR spectra was SpinWorks 4.2.0.0 (copyright 2015, Kirk Marat, University of Manitoba). Coupling constants (J) are given in Hertz (Hz) and refer to the first order interpretation (apparent coupling constants Japp are provided). Assignment of resonances was performed with COSY, HSQC and HMBC, respectively. xJ refers to homonuclear HH-coupling over x bonds. xJCF refers to heteronuclear CF coupling over x bonds. Solvents used for NMR spectroscopy: CDCl3, chloroform-d1 (CAS RN 865-49-6), was filtered through basic, activated aluminium oxide (Sigma Aldrich) prior to use; DMSO-d6, hexadeuterio dimethyl sulfoxide (CAS RN 2206-27-1) was stored over a molecular sieve (4 Å). Chemical shift calibration [20]: CDCl3, 1H δ = 7.26, 13C δ = 77.16; DMSO-d6, 1H δ = 2.50, 13C δ = 39.52 ppm. NMR characterization: br, broad; multiplicity: m, multiplet; s, singlet; d, doublet; t, triplet; q, quaternary or quartet; m [e.g., t, dd, q, tt and sext] notation in brackets describes the overall appearance of the signal pattern; 2D NMR techniques used for the assignment of 1H and 13C resonance signals: HSQC, Heteronuclear Single Quantum Coherence; HMBC, Heteronuclear Multiple Bond Correlation; COSY, Correlation Spectroscopy.
3.3. Inhibitor Assay on the 96-Well Plate
The target inhibitor class features UV absorption in the region around 255 nm, where oxaloacetate is absorbing as well, while the absorbance of the product pyruvate is negligible at this wavelength. It can be observed that lower concentrations of OAA display an initial raise of the OD 255 nm signal. The steady state is typically reached at about 5 min. This observation can be attributed to an increase of the OAA-enol form, indicating that not all OAA introduced into the assay mixture was already in the enol form. The assay starts with 100% bound enol form of OAA. From our literature survey of metal catalyzed OAA decarboxylation, we can conclude that the rate determining step of the decarboxylation is slow ketonization. The enol form of OAA is not prone to decarboxylation (as suggested by theory). We speculate that our designed inhibitors compete with the transient enol form of OAA.
In a direct assay on the 96-well plate (Table 10), ODx activity of FAHD1 and its inhibition is performed as follows: We used a freshly prepared 20-mM OAA solution in ddH2O and any dilutions of an original 100-mM inhibitor solution (dilution “0”), which we refer to as dilution “0” to “6” that cover the range of mM down to nM inhibition. We used a 40-µM solution of recombinant protein [21], which was freshly prepared from working solutions of about 1 µg/µL, as determined via the Pierce™ BCA Protein Assay Kit (Thermo). Three rows on a 96-well plate (wells labeled 1–12) are used for one inhibitor in order to have triplets (n = 3). Well 1 was used to check for the auto-decarboxylation of OAA. Well 2 was used to check for possible reactions of the inhibitor with OAA. Well 3 was used to check for FAHD1 stability in the presence of higher inhibitor concentrations. Well 4 was used to check for the inhibitor stability and OD signal over time. Well 5 was used to measure the uninhibited FAHD1-specific activity. Wells 6–12 were used for titrations of the inhibitor.
Table 10.
Pipetting scheme for a direct ODx assay on the 96-well plate, to screen for IC50 values of FAHD1 in vitro.
3.4. General Synthetic Procedures
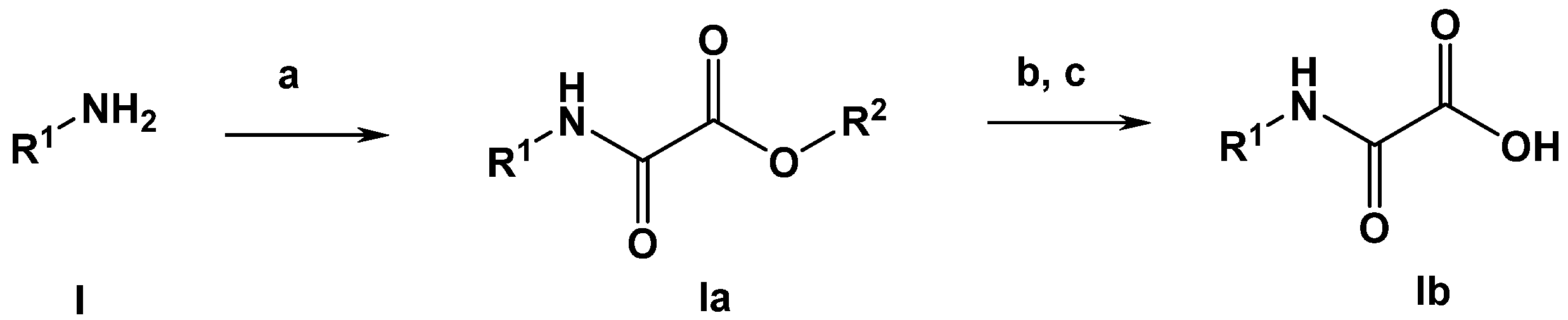
Oxalamic acid derivatives can be obtained by kown methods. The generally applid reaction path is depicted in Scheme 1.
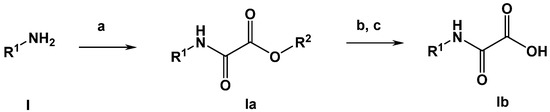
Scheme 1.
General synthesis of oxalamic acid derivatives (R2: methyl or tert. -butyl). Conditions: (a) methyl or tert. butyl 2-chloro-2-oxoacetate, triethylamine, DCM or DMF; (b) 2N NaOH, ethanol, 2N HCl or (c) K2CO3, H2O-MeOH; 2N HCl for 4b–6b.
3.4.1. General Synthesis of Oxalamic Acid Derivatives
To a solution of the desired amine (0.5 mmol) in N,N-dimethylformamide (dichloromethane for anthranilic acid) (10 mL) was added 70 µL of triethylamine (0.5 mmol). The solution was cooled to 0 °C. To the stirred mixture, a solution of methyl 2-chloro-2-oxoacetate (CAS RN. 5781-53-3; 1.1 equivalents) in 10 mL of dichloromethane was added dropwise within 10 min. The reaction mixture was stirred at room temperature for 3 h. Subsequently, the reaction mixture was diluted with 50 mL of ethyl acetate. The organic layer was extracted three times with 20 mL of HCl (2N), dried over Na2SO4 and evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel using ethyl acetate/n-hexane (2:1) as the eluent. The crystalline product obtained was finally dried in vacuo.
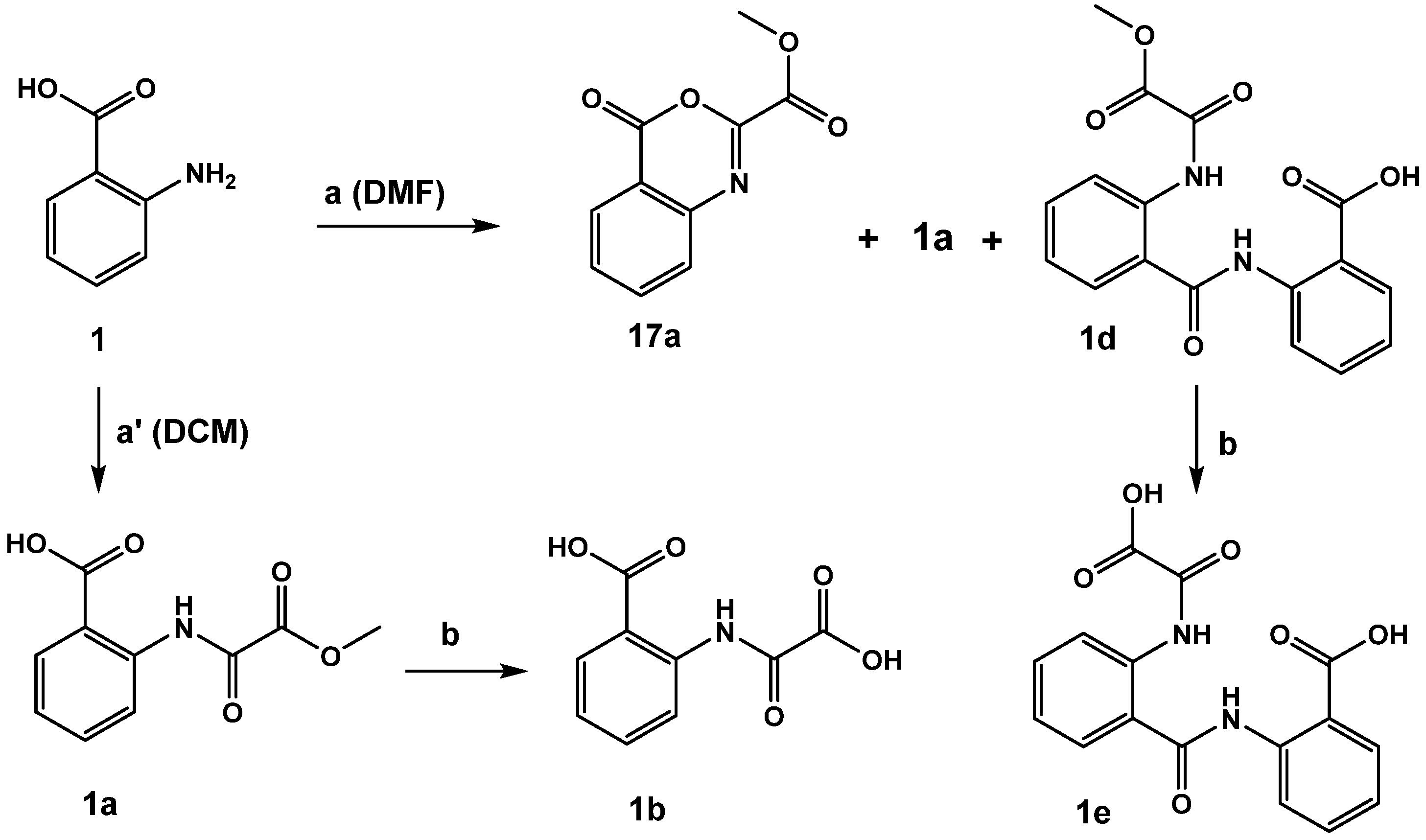
The acylation of anthranilic acid 1 turned out to be solvent-dependent (Scheme 2). Reaction performed in DCM led in 98% yield to the desired methyl ester product 1a, wherase the reaction in DMF resulted in a complex mixture containing the desired product 1a as a minor component, methyl 4-oxo--benzo[d][1,3]oxazine-2-carboxylate (17a) as the major product and little of 2-(2-(2-methoxy-2-)-benzamido)benzoic acid 1d. By-product 1d formed upon reaction of 17a with unreacted starting material 1. Saponification of 1d yielded the corresponding diacid 1e (both compounds revealed moderate inhibitory activity).
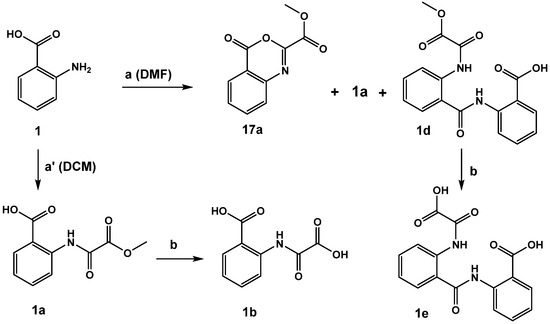
Scheme 2.
Conditions: (a) methyl 2-chloro-2-oxoacetate, triethylamine, DCM or DMF. (b) 2N NaOH, ethanol, 2N HCl.
3.4.2. Saponification of Methyl Ester Derivatives
To a solution of a methyl ester derivativ in 5 mL of methanol was added NaOH (2N, 2eq.). The resulting mixture was stirred for 10 min and finally quenched with HCl (2N). The mixture obtained was extracted three times with ethyl acetate. The organic phases were combined, dried over Na2SO4 and evaporated. The crystalline product obtained was dried in vacuo.
3.4.3. General Synthesis of 2-Thiooxoacetate Derivatives
Lawesson’s reagent (CAS RN 19172-47-5) (0.20 mmol) was added to a solution of a methyl ester derivate (S2, S3, or S4) (0.40 mmol) in toluene (2 mL). The reaction mixture was stirred at 75 °C for 7 h. The crude mixture was concentrated under reduced pressure, and the product was purified by column chromatography (n-hexane/ethyl acetate 10:1). The crystalline product (20a, 21a, 22a) was dried in vacuo before being dissolved in ethanol (5 mL) and saponified with NaOH (2N, 2eq.). The resulting mixture was stirred for 3 h and, finally, quenched with HCl (2N). The product obtained was extracted several times with ethyl acetate. The organic layers were combined, dried over Na2SO4 and evaporated. The crystalline product (20b, 21b and 22b) obtained was dried in vacuo (yield: 90–95%) (Scheme 3).
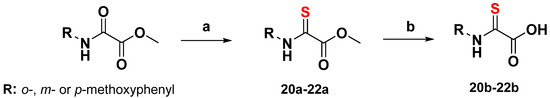
Scheme 3.
Conditions: (a) Lawesson’s reagent, toluene; (b) 2N NaOH, ethanol; 2N HCl.
3.4.4. Synthesis of Pyridine and Quinoline N-Oxides
To a solution of the corresponding oxalamic acid ester (methyl or tert. butyl) in dichloromethane, 1.1 equivalents of m-chloroperoxybenzoic acid (m-CPBA) were added. The reaction mixture was stirred overnight at room temperature. The solvent was removed under reduced pressure. To the obtained reaction mixture, cold di-isopropyl ether (DIPE) was added. The obtained heterogeneous slurry was vigorously stirred for 2 h. Subsequently, the formed crystals were collected by filtration and washed three times with cold DIPE. The crystals were dried under reduced pressure. (Note: some of the compound’s form defined crystalline 1:1 clathrate with 3-chlorobenzoic acid).
3.4.5. Synthesis of Oxalamic Acid Amides
Oxalamic acid amides were prepared by aminolysis of N-aryl oxalamic acid methyl esters (Scheme 4).
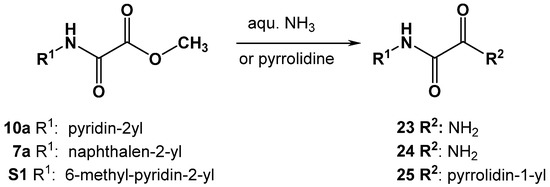
Scheme 4.
Synthesis of oxalamic acid amide derivatives 23, 24 and 25.
To a solution of oxalamic acid methyl ester (10a, 7a or S1) in 5 mL of toluene, the corresponding amine was added (aqueous ammonia for 23, 24 or 1.1 equivalents of pyrrolidine for 25). The reaction mixture was stirred at room temperature in a sealed vessel. The reaction status was monitored by TLC. Toluene was removed under reduced pressure. Subsequently, the obtained oily residue was diluted with ethyl acetate. The organic phase was washed three times with deionized water. The organic layer was dried over Na2SO4 and evaporated under reduced pressure. The crystalline product (23–25) obtained was dried in vacuo (yields: 45–60%).
3.4.6. Synthesis of N,N′-Diaryl-Oxalamic Acid Diamides
N,N′-diaryl-oxalamic acid diamides with C2-symmetry can be achieved in one step by reacting an amine scaffold with oxalic acid dimethyl ester (DIMOX) (method A). Nonsymmetric N,N′-diaryl-oxalamic acid diamides are best obtained by aminolysis of N-aryl-oxalamic acid methyl esters with an N′-amine building block (method B) (Scheme 5).
Scheme 5.
Synthesis of oxalamic acid amide derivatives 29–37.
Synthesis of Oxalic Acid Diamides by Aminolysis of DIMOX
Typical example for method A: Synthesis of N1,N2-di(pyridin-2-yl)oxalamide (30).
Two equivalents of pyridin-2-amine and one equivalent of dimethyl oxalate (DIMOX) were dissolved in DMF by slight heating. The homogeneous solution obtained was heated to 120 °C and kept under stirring in a nitrogen atmosphere overnight. The reaction mixture was cooled to room temperature. Subsequently, the reaction mixture was diluted with ethyl acetate. The organic phase was washed three times with brine, dried over Na2SO4 and filtered. The solvent was removed on a rotary evaporator under reduced pressure. The pure product 30 was obtained by MPLC separation (stationary phase: silica gel; eluent: appropriate mixture of n-hexane and ethyl acetate); Yield: 44% (the reactions did not go to full completion; for the analytical characterization of compound 30, see the Supplementary Materials).
Synthesis of Oxalic Acid Diamides by Aminolysis of Methyl Esters
Typical example for method B: Synthesis of N1-(pyridin-2-yl)-N2-(m-tolyl)oxalamide (33).
Methyl ester 10 and 1.1 eq. of m-toluidine were dissolved in DMF. The solution obtained was heated to 110 °C and stirred under nitrogen atmosphere overnight. The reaction mixture was cooled to room temperature and diluted with ethyl acetate. The organic phase was washed three times with brine, dried over Na2SO4 and filtered. The solvent was removed on a rotary evaporator under reduced pressure. The products were purified by MPLC separation (stationary phase: silica gel; eluent: appropriate mixture of n-hexane and ethyl acetate). Yield: 50% (for the analytical characterization of compound 33, see the Supporting Materials).
4. Conclusions
We addressed the central chemical step of both FAHD1 functions (ODx- and ApH) by a theoretical approach. The results obtained from this approach verified a mechanism-based design concept of first-generation FAHD1 inhibitors. Replacement of the common scissile C–C bond in FAHD1 substrates by a simple carbon–nitrogen bond, together with the negative charge density presentation in the appropriate space of the catalytic cavity, turned out to be successful for the identification of the first generation of FAHD1 inhibitors. As a surprising result from the synthesis efforts, we identified the molecules that suggested a novel binding mode for inhibition of FAHD1. Valuable proposals for the synthesis of inhibitors could be deduced, and co-crystallization and STD-NMR experiments with selected compounds have been initiated.
We anticipate that a successful elucidation of the role of FAHD1 as a therapeutic target will stimulate translational research, with the aim to contribute to new therapeutic strategies for human malignancies. The current attempts to develop small molecules with the ability to modulate FAHD1 catalytic activity aim at translational strategies to fine-tune FAHD1 activity in physiological and pathological conditions. We will continue to synthesize and analyze FAHD1 novel and more potent inhibitor candidates, starting from the structure of existing inhibitors with the one-digit micromolar functional IC50 described in this work. After screening for binding affinity, the most promising molecules will be used for the first experiments with selected human cell lines expressing FAHD1, which will be treated with the most active inhibitors (and their methyl esters to enhance cell permeability) in different concentrations. We anticipate that the inhibition of FAHD1 over longer periods will induce the mitochondrial dysfunction in human cells. Such a model would provide a new important research tool and serve as the basis for new therapeutic molecules for human malignancies.
Supplementary Materials
The following are available online. A comprehensive discussion of the “many faces” of FAHD1 substrate oxaloacetate (OAA), additional compounds (S5-S22) investigated for FAHD1 inhibition (Figure S4), all analytical data for compounds synthesized, as well as titration curves for compounds discussed in the manuscript.
Author Contributions
Conceptualization, A.K.H.W., P.J.-D. and H.G.; Data curation, A.K.H.W. and S.v.G.; Funding acquisition, K.R.L. and P.J.-D.; Investigation, R.W., P.K. and M.P.E.; Methodology, A.K.H.W. and H.G.; Project administration, K.R.L. and P.J.-D.; Resources, P.J.-D.; Software, A.K.H.W., J.R.L., S.v.G. and S.M.; Supervision, A.K.H.W., K.R.L., P.J.-D. and H.G.; Validation, S.M.; Visualization, J.R.L.; Writing—original draft, A.K.H.W. and H.G.; Writing—review and editing, A.K.H.W., P.J.-D. and H.G. All authors have read and agreed to the published version of the manuscript.
Funding
A.K.H.W. and P.J-D. were supported by a grant from the Austrian Science Fund (FWF), P315820.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
The authors thank Hanspeter Kählig, Susanne Felsinger and Ricarda Ofenschüssel from the NMR Centre of the Faculty of Chemistry, University of Vienna for measuring the NMR spectra and Daniel Dobusch and Judith Wackerlig from the Division of Pharmaceutical Sciences, University of Vienna for providing the HRMS Data. We thank MoleculeCrafting.HuGs e.U. for providing the laboratory infrastructure, supervision and financial support.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds described in the manuscript and in supplementary materials are available from the corresponding authors.
References
- Pircher, H.; von Grafenstein, S.; Diener, T.; Metzger, C.; Albertini, E.; Taferner, A.; Unterluggauer, H.; Kramer, C.; Liedl, K.R.; Jansen-Dürr, P. Identification of FAH Domain-containing Protein 1 (FAHD1) as Oxaloacetate Decarboxylase. J. Biol. Chem. 2015, 290, 6755–6762. [Google Scholar] [CrossRef] [Green Version]
- Pircher, H.; Straganz, G.D.; Ehehalt, D.; Morrow, G.; Tanguay, R.M.; Jansen-Dürr, P. Identification of Human Fumarylacetoacetate Hydrolase Domain-containing Protein 1 (FAHD1) as a Novel Mitochondrial Acylpyruvase. J. Biol. Chem. 2011, 286, 36500–36508. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.K.H.; Loeffler, J.R.; Liedl, K.R.; Gstach, H.; Jansen-Dürr, P. The fumarylacetoacetate hydrolase (FAH) superfamily of enzymes: Multifunctional enzymes from microbes to mitochondria. Biochem. Soc. Trans. 2018, 46, 295–309. [Google Scholar] [CrossRef]
- Hong, H.; Seo, H.; Park, W.; Kim, K.K.-J. Sequence, structure and function-based classification of the broadly conserved FAH superfamily reveals two distinct fumarylpyruvate hydrolase subfamilies. Environ. Microbiol. 2020, 22, 270–285. [Google Scholar] [CrossRef]
- Weiss, A.K.H.; Albertini, E.; Holzknecht, M.; Cappuccio, E.; Dorigatti, I.; Krahbichler, A.; Damisch, E.; Gstach, H.; Jansen-Dürr, P. Regulation of cellular senescence by eukaryotic members of the FAH superfamily—A role in calcium homeostasis? Mech. Ageing Dev. 2020, 190, 111284. [Google Scholar] [CrossRef] [PubMed]
- Etemad, S.; Petit, M.; Weiss, A.K.H.; Schrattenholz, A.; Baraldo, G.; Jansen-Dürr, P. Oxaloacetate decarboxylase FAHD1—A new regulator of mitochondrial function and senescence. Mech. Ageing Dev. 2019, 177, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.K.H.; Naschberger, A.; Loeffler, J.R.; Gstach, H.; Bowler, M.W.; Holzknecht, M.; Cappuccio, E.; Pittl, A.; Etemad, S.; Dunzendorfer-Matt, T.; et al. Structural basis for the bi-functionality of human oxaloacetate decarboxylase FAHD1. Biochem. J. 2018, 475, 3561–3576. [Google Scholar] [CrossRef]
- Weiss, A.K.H.; Naschberger, A.; Cappuccio, E.; Metzger, C.; Mottes, L.; Holzknecht, M.; von Velsen, J.; Bowler, M.W.; Rupp, B.; Jansen-Dürr, P. Structural and functional comparison of fumarylacetoacetate domain containing protein 1 in human and mouse. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [Green Version]
- Tate, S.S.; Grzybowski, A.K.; Datta, S.P. 266. The stability constants of the magnesium complexes of the keto and enol isomers of oxaloacetic acid at 25°. J. Chem. Soc. 1964, 1381–1389. [Google Scholar] [CrossRef]
- Creighton, D.J.; Rose, I.A. Studies on the mechanism and stereochemical properties of the oxalacetate decarboxylase activity of pyruvate kinase. J. Biol. Chem. 1976, 251, 61–68. [Google Scholar] [CrossRef]
- Tate, S.S.; Grzybowski, A.K.; Datta, S.P. 265. The acid dissociations of the keto and enol isomers of oxaloacetic acid at 25°. J. Chem. Soc. 1964, 1372–1380. [Google Scholar] [CrossRef]
- Mulholland, A.J.; Richards, W.G. Calculations on the substrates of citrate synthase-I. Oxaloacetate. J. Mol. Struct. 1998, 429, 13–21. [Google Scholar] [CrossRef]
- Ran, T.; Gao, Y.; Marsh, M.; Zhu, W.; Wang, M.; Mao, X.; Xu, L.; Xu, D.; Wang, W. Crystal structures of Cg1458 reveal a catalytic lid domain and a common catalytic mechanism for the FAH family. Biochem. J. 2012, 449, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Kresge, A.J. Generation and study of enols and other reactive species. Pure Appl. Chem. 1991, 63, 213–221. [Google Scholar] [CrossRef]
- Andersen, H.S.; Iversen, L.F.; Jeppesen, C.B.; Branner, S.; Norris, K.; Rasmussen, H.B.; Møller, K.B.; Møller, N.P. 2-(oxalylamino)-benzoic acid is a general, competitive inhibitor of protein-tyrosine phosphatases. J. Biol. Chem. 2000, 275, 7101–7108. [Google Scholar] [CrossRef] [Green Version]
- Mfuh, A.M.; Larionov, O.V. Heterocyclic N-Oxides—An Emerging Class of Therapeutic Agents. Curr. Med. Chem. 2015, 22, 2819–2857. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.M. Synthesis and reactions of 2-hetero-4H-3,1-benzoxazin-4-ones. J. Heterocycl. Chem. 2000, 37, 1369–1388. [Google Scholar] [CrossRef]
- Liao, B.-L.; Pan, Y.-J.; Zhang, W.; Pan, L.-W. Four Natural Compounds Separated from Folium Isatidis: Crystal Structures and Antibacterial Activity. Chem. Biodivers. 2018, 15, e1800152. [Google Scholar] [CrossRef]
- Zhang, W.-J.; Wang, F.; Zhang, G.-L.; Xiao, X. A triclinic polymorph with Z = 3 of N, N ′-bis(2-pyridyl)oxamide. Acta Crystallogr. Sect. E Struct. Rep. Online 2011, 67, o972–o973. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Weiss, A.K.H.; Holzknecht, M.; Cappuccio, E.; Dorigatti, I.; Kreidl, K.; Naschberger, A.; Rupp, B.; Gstach, H.; Jansen-Dürr, P. Expression, Purification, Crystallization, and Enzyme Assays of Fumarylacetoacetate Hydrolase Domain-Containing Proteins. J. Vis. Exp. 2019, e59729. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).