
Theoretical Study of 2-(Trifluoromethyl)phenothiazine Derivatives with Two Hydroxyl Groups in the Side Chain-DFT and QTAIM Computations


Abstract
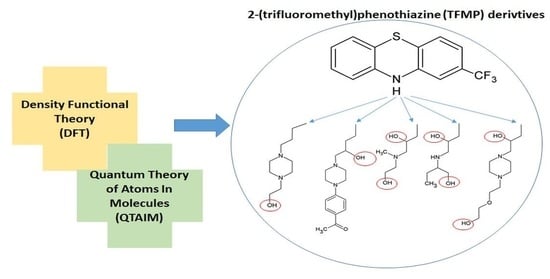
:
1. Introduction
2. Results and Discussion
2.1. DFT Calculations at the Ground State in the Gaseous Phase
2.2. DFT Calculations at the Ground State in the Aqueous Medium
2.3. Quantum Theory of Atoms in Molecules (QTAIM)
3. Materials and Methods
3.1. Compounds
3.2. Computational Details
3.2.1. Chemical Reactions
3.2.2. Thermochemical Parameters
3.2.3. Computational Calculations
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hazhazi, H.; Melkemi, N.; Salah, T.; Bouachrine, M. DFT-based reactivity and combined QSAR, molecular docking of 1,2,4,5-Tetrazine derivatives as inhibitors of Pim-1 kinase. Heliyon 2019, 5, e02451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Mourik, T.; Bühl, M.; Gaigeot, M.P. Density functional theory across chemistry, physics and biology. Philos. Trans. A Math. Phys. Eng. Sci. 2014, 372, 20120488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matta, C.F.; Arabi, A.A. Electron-density descriptors as predictors in quantitative structure--activity/property relationships and drug design. Future Med. Chem. 2011, 3, 969–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Sheikh Ali, A.; Khan, D.; Naqvi, A.; Al-Blewi, F.F.; Rezki, N.; Aouad, M.R.; Hagar, M. Design, Synthesis, Molecular Modeling, Anticancer Studies, and Density Functional Theory Calculations of 4-(1,2,4-Triazol-3-ylsulfanylmethyl)-1,2,3-triazole Derivatives. ACS Omega 2020, 6, 301–316. [Google Scholar] [CrossRef]
- Mary, Y.S.; Mary, Y.S.; Resmi, K.S.; Rad, A.S. Spectroscopic and computational study of chromone derivatives with antitumor activity: Detailed DFT, QTAIM and docking investigations. SN Appl. Sci. 2021, 3, 143. [Google Scholar] [CrossRef]
- Karelou, M.; Kourafalos, V.; Tragomalou, A.P.; Marakos, P.; Pouli, N.; Tsitsilonis, O.E.; Gikas, E.; Kostakis, I.K. Synthesis, Biological Evaluation and Stability Studies of Some Novel Aza-Acridine Aminoderivatives. Molecules 2020, 25, 4584. [Google Scholar] [CrossRef]
- Środa-Pomianek, K.; Michalak, K.; Świątek, P.; Poła, A.; Palko-Łabuz, A.; Wesołowska, O. Increased lipid peroxidation, apoptosis and selective cytotoxicity in colon cancer cell line LoVo and its doxorubicin-resistant subline LoVo/Dx in the presence of newly synthesized phenothiazine derivatives. Biomed. Pharmacother. 2018, 106, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Senthil, K.; Kumaresan, R. A DFT study on the structural, electronic properties and radical scavenging mechanisms of calycosin, glycitein, pratensein and prunetin. Comput. Theor. Chem. 2012, 985, 14–22. [Google Scholar] [CrossRef]
- Lengyel, J.; Rimarčík, J.; Vegánek, A.; Klein, E. On the radical scavenging activity of isoflavones: Thermodynamics of O–H bond cleavage. Phys. Chem. Chem. Phys. 2013, 15, 10895–10903. [Google Scholar] [CrossRef]
- Amić, D.; Stepanić, V.; Lučić, B.; Marković, Z.; Marković, J.M.D. PM6 study of free radical scavenging mechanisms of flavonoids: Why does O–H bond dissociation enthalpy effectively represent free radical scavenging activity? J. Mol. Model. 2013, 19, 2593–2603. [Google Scholar] [CrossRef]
- Farmanzadeh, D.; Najafi, M. Antioxidant activity of aminothiazol hydroxycoumarin derivatives. J. Theor. Comput. Chem. 2013, 12, 1350058. [Google Scholar] [CrossRef]
- Wang, G.; Xue, Y.; An, L.; Zheng, Y.; Dou, Y.; Zhang, L.; Liu, Y. Theoretical study on the structural and antioxidant properties of some recently synthesised 2,4,5-trimethoxy chalcones. Food Chem. 2015, 171, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Messaadia, L.; Bekkar, Y.; Benamira, M.; Lahmar, H. Predicting the antioxidant activity of some flavonoids of Arbutus plant: A theoretical approach. Chem. Phys. Impact 2020, 1, 100007. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Anderson, S.G.; Duke, A.J. Quantum Topology of Molecular Charge Distributions. J. Am. Chem. Soc. 1979, 101, 1389–1395. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, Version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Grabowski, S.J. Ab Initio Calculations on Conventional and Unconventional Hydrogen Bonds-Study of the Hydrogen Bond Strength. J. Phys. Chem. A 2001, 105, 10739–10746. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental Electron Density Analysis of Mn2(CO)10: Metal-Metal and Metal-Ligand Bond Characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Hansen, P.E.; Kamounah, F.S.; Saeed, B.A.; MacLachlan, M.J.; Spanget-Larsen, J. Intramolecular Hydrogen Bonds in Normal and Sterically Compressed o-Hydroxy Aromatic Aldehydes. Isotope Effects on Chemical Shifts and Hydrogen Bond Strength. Molecules 2019, 24, 4533. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.; Lukes, V.; Ilcin, M. DFT/B3LYP study of tocopherols and chromans antioxidant action energetics. Chem. Phys. 2007, 336, 51–57. [Google Scholar] [CrossRef]
- Marković, Z.; Milenković, D.; Ðorović, J.; Marković, J.M.D.; Stepanić, V.; Lučić, B.; Amić, D. Free radical scavenging activity of morin 2′-O- phenoxide anion. Food Chem. 2012, 135, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- DiLabio, G.A.; Pratt, D.A.; LoFaro, A.D.; Wright, J.S. Theoretical study of X-H bond energetics (X = C, N, O, S): Application to substituent effects, gas phase acidities, and redox potentials. J. Phys. Chem. A 1999, 103, 1653–1661. [Google Scholar] [CrossRef]
- Fifen, J.J.; Nsangou, M.; Dhaouadi, Z.; Motapon, O.; Jaidane, N. Solvent effect on the antioxidant activity of 3,4-dihydroxyphenylpyruvic acid: DFT and TD-DFT studies. Comput. Theor. Chem. 2011, 966, 232–243. [Google Scholar] [CrossRef]
- Rimarčík, J.; Lukeš, V.; Klein, E.; Ilčin, M. Study of the solvent effect on the enthalpies of homolytic and heterolytic N-H bond cleavage in p-phenylenediamine and tetracyano-p-phenylenediamine. J. Mol. Struct. (Theochem) 2010, 952, 25–30. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Bartmess, J.E. Thermodynamics of the electron and the proton. J. Phys. Chem. 1994, 98, 6420–6424. [Google Scholar] [CrossRef]
- Bizarro, M.M.; Costa Cabral, B.J.; dos Santos, R.M.B.; Simões, J.A.M. Substituent effects on the O–H bond dissociation enthalpies in phenolic compounds: Agreements and controversies. Pure Appl. Chem. 1999, 71, 1249–1256. [Google Scholar] [CrossRef] [Green Version]
- Parker, V.D. Homolytic bond (H-A) dissociation free energies in solution. Applications of the standard potential of the (H+/H∙) couple. J. Am. Chem. Soc. 1992, 114, 7458–7462. [Google Scholar] [CrossRef]
- Wright, J.S.; Johnson, E.R.; DiLabio, G.A. Predicting the activity of phenolic antioxidants: Theoretical method, analysis of substituent effects, and application to major families of antioxidants. J. Am. Chem. Soc. 2001, 123, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Jeding, I.; Evans, P.J.; Akanmu, D.; Dexter, D.; Spencer, J.D.; Aruoma, O.I.; Jenner, P.; Halliwell, B. Characterization of the potential antioxidant and pro-oxidant actions of some neuroleptic drugs. Biochem. Pharmacol. 1995, 49, 359–365. [Google Scholar] [CrossRef]
- Dos Santos, C.G.; Silva, A.L.; Souza, F.L.; Lanfredi, A.J.; Di Mascio, P.; Nascimento, O.R.; Rodrigues, T.; Nantes, I.L. UV-light effects on cytochrome c modulated by the aggregation state of phenothiazines. PLoS ONE 2013, 8, e76857. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FLU | APh-FLU | MAE-TPR | ABu-TPR | HEE-FLU | |
|---|---|---|---|---|---|
| Gaseous Phase | |||||
| BDE (1) | 99.1 | – | 92.1 | 98.5 | 98.0 |
| BDE (2) | – | 98.3 | 91.4 | 97.0 | 96.8 |
| IP | 155.6 | 156.5 | 161.5 | 160.6 | 158.2 |
| PDE (1) | 257.7 | – | 244.9 | 252.2 | 254.1 |
| PDE (2) | – | 251.6 | 244.2 | 250.7 | 252.9 |
| PA (1) | 369.6 | – | 340.9 | 356.5 | 360.0 |
| PA (2) | – | 348.7 | 341.5 | 349.6 | 355.8 |
| ETE (1) | 43.7 | – | 65.5 | 56.3 | 52.3 |
| ETE (2) | – | 59.4 | 64.2 | 61.6 | 55.3 |
| Aqueous Medium | |||||
| BDE (1) | 94.3 | – | 86.0 | 94.3 | 93.8 |
| BDE (2) | – | 92.8 | 84.6 | 90.6 | 93.1 |
| IP | 83.5 | 86.7 | 87.6 | 87.4 | 86.7 |
| PDE (1) | 53.3 | – | 38.7 | 47.1 | 47.4 |
| PDE (2) | – | 46.4 | 37.2 | 43.5 | 46.7 |
| PA (1) | 64.1 | – | 55.1 | 64.0 | 60.8 |
| PA (2) | – | 60.1 | 53.3 | 59.2 | 60.9 |
| ETE (1) | 72.6 | – | 71.2 | 70.5 | 73.3 |
| ETE (2) | – | 72.9 | 71.6 | 71.6 | 72.5 |
| ∆BDE | – | – | 1.4 | 3.7 | 0.7 |
| ∆PDE | – | – | 1.5 | 3.6 | 0.7 |
| ∆PA | – | – | 1.8 | 4.8 | 0.1 |
| Compound | Group | Bond Path between Atoms | |||||
|---|---|---|---|---|---|---|---|
| Gaseous Phase | |||||||
| FLU | –OH(1) | O51–H52 | 0.36103 | −2.47444 | −0.76510 | 0.07325 | −0.69185 |
| APh-FLU | –OH(2) | O19–H34 | 0.35820 | −2.46741 | −0.76065 | 0.07190 | −0.68875 |
| MAE-TPR | –OH(1) | O26–H28 | 0.34402 | −1.79570 | −0.57436 | 0.06272 | −0.51164 |
| –OH(2) | O27–H34 | 0.34110 | −1.78820 | −0.56957 | 0.06126 | −0.50831 | |
| ABu-TPR | –OH(1) | O28–H44 | 0.36031 | −2.46190 | −0.76294 | 0.07373 | −0.68921 |
| –OH(2) | O19–H37 | 0.36192 | −2.48677 | −0.76654 | 0.07242 | −0.69412 | |
| HEE-FLU | –OH(1) | O34–H35 | 0.36657 | −2.50894 | −0.77544 | 0.07410 | −0.70134 |
| –OH(2) | O25–H48 | 0.35682 | −2.45795 | −0.75789 | 0.07170 | −0.68619 | |
| Aqueous Medium | |||||||
| FLU | –OH(1) | O51–H52 | 0.35724 | −2.45912 | −0.75797 | 0.07160 | −0.68637 |
| APh-FLU | –OH(2) | O19–H34 | 0.35485 | −2.45107 | −0.75399 | 0.07061 | −0.68339 |
| MAE-TPR | –OH(1) | O26–H28 | 0.36003 | −2.05718 | −0.66478 | 0.07523 | −0.58953 |
| –OH(2) | O27–H34 | 0.35527 | −2.03688 | −0.65751 | 0.07415 | −0.58337 | |
| ABu-TPR | –OH(1) | O19–H37 | 0.36096 | −2.48715 | −0.76507 | 0.07164 | −0.69343 |
| –OH(2) | O28–H44 | 0.35644 | −2.45112 | −0.75599 | 0.07161 | −0.68439 | |
| HEE-FLU | –OH(1) | O34–H35 | 0.36475 | −2.51694 | −0.77333 | 0.07205 | −0.70128 |
| –OH(2) | O25–H48 | 0.35250 | −2.43066 | −0.74919 | 0.07076 | −0.67843 | |
| Derivative | Atoms | d (Å) | EHB (kcal × mol−1) | Group | |||||
|---|---|---|---|---|---|---|---|---|---|
| Gaseous phase | |||||||||
| FLU | N38–H52 | 2.29 | – | – | – | – | – | – | – |
| APh-FLU | N18–H34 | 2.22 | −4.6 | OH(2) | 0.02037 | 0.07216 | −0.01472 | 0.01638 | 0.00166 |
| MAE-TPR | N22–H28 | 2.44 | – | – | – | – | – | – | – |
| N22–H34 | 2.32 | – | – | – | – | – | – | – | |
| ABu-TPR | N18–H44 | 2.24 | – | – | – | – | – | – | – |
| N14–H37 | 2.40 | – | – | – | – | – | – | – | |
| HEE-FLU | N28–H35 | 4.51 | – | – | – | – | – | – | – |
| N22–H48 | 2.18 | −4.9 | OH(2) | 0.02164 | 0.07484 | −0.01566 | 0.01718 | 0.00152 | |
| O31–H35 | 2.45 | – | – | – | – | – | – | – | |
| Aqueous medium | |||||||||
| FLU | N38–H52 | 2.23 | – | – | – | – | – | – | – |
| APh-FLU | N18–H34 | 2.17 | −5.0 | OH(2) | 0.02211 | 0.07577 | −0.01598 | 0.01746 | 0.00148 |
| MAE-TPR | N22–H28 | 2.46 | – | – | – | – | – | – | – |
| N22–H34 | 2.25 | – | – | – | – | – | – | – | |
| ABu-TPR | N18–H44 | 2.19 | −4.8 | OH(1) | 0.02138 | 0.07363 | −0.01535 | 0.01688 | 0.00153 |
| N14–H37 | 2.39 | – | – | – | – | – | – | – | |
| HEE-FLU | N28–H35 | 4.54 | – | – | – | – | – | – | – |
| N22–H48 | 2.11 | −5.7 | OH(2) | 0.02495 | 0.08212 | −0.01834 | 0.01944 | 0.00110 | |
| O31–H35 | 2.48 | – | – | – | – | – | – | – | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poła, A.; Palko-Łabuz, A.; Środa-Pomianek, K. Theoretical Study of 2-(Trifluoromethyl)phenothiazine Derivatives with Two Hydroxyl Groups in the Side Chain-DFT and QTAIM Computations. Molecules 2021, 26, 5242. https://doi.org/10.3390/molecules26175242
Poła A, Palko-Łabuz A, Środa-Pomianek K. Theoretical Study of 2-(Trifluoromethyl)phenothiazine Derivatives with Two Hydroxyl Groups in the Side Chain-DFT and QTAIM Computations. Molecules. 2021; 26(17):5242. https://doi.org/10.3390/molecules26175242
Chicago/Turabian StylePoła, Andrzej, Anna Palko-Łabuz, and Kamila Środa-Pomianek. 2021. "Theoretical Study of 2-(Trifluoromethyl)phenothiazine Derivatives with Two Hydroxyl Groups in the Side Chain-DFT and QTAIM Computations" Molecules 26, no. 17: 5242. https://doi.org/10.3390/molecules26175242